

Abstract
A significant barrier to the success of engineered tissues is the inadequate transport of nutrients and gases to, and waste away from, cells within the constructs, after implantation. Generation of microtubular networks by endothelial cells in engineered constructs to mimic the in vivo transport scheme is essential for facilitating tissue survival by promoting the in vitro formation of microvessels that integrate with host microvasculature, after implantation. Previously, we reported that select pressures stimulate endothelial proliferation involving protubulogenic molecules such as fibroblast growth factor-2 (FGF-2) and vascular endothelial growth factor-C (VEGF-C). Based on this, we investigated fluid pressure as a selective modulator of early tubulogenic activity with the intent of assessing the potential utility of this mechanical stimulus as a tissue-engineering control parameter. For this purpose, we used a custom pressure system to expose two-dimensional (2D) and three-dimensional (3D) cultures of endothelial cells to static pressures of 0 (controls), 20, or 40 mmHg for 3 days. Compared to controls, 2D endothelial cultures exposed to 20, but not 40 mmHg, exhibited significantly (p<0.05) enhanced cell growth that depended on VEGF receptor-3 (VEGFR-3), a receptor for VEGF-C. Moreover, endothelial cells grown on microbeads and suspended in 3D collagen gels under 20 mmHg, but not 40 mmHg, displayed significantly (p<0.05) increased sprout formation. Interestingly, pressure-dependent proliferation and sprout formation occurred in parallel with pressure-sensitive upregulation of VEGF-C and VEGFR-3 expression and were sensitive to local FGF-2 levels. Collectively, the results of the present study provided evidence that early endothelial-related tubulogenic activity depends on local hydrostatic pressure levels in the context of local growth factor conditions. In addition to relevance to microvascular diseases associated with interstitial hypertension (e.g., cancer and glaucoma), these findings provided first insight into the potential utility of hydrostatic pressure as a fine-tune control parameter to optimize microvascularization of tissue-engineering constructs in the in vitro setting before their implantation.
Introduction
The United Network for Organ Sharing, as of October of 2011, reported that >112,000 patients are on waiting lists for transplantation surgery. A shortage of tissues due to such large demands and the limited availability of the donor material point to a critical need for engineered tissues that duplicate the complex organization of biological matrices.1 A significant barrier to the success of these constructs is inadequate passive transport of nutrients and gases to resident cells. Past efforts to formulate tissue-engineering strategies relied on utilizing fluid flow or matrix compression to facilitate delivery of nutrients to, and promote removal of waste away from, cells within synthetic tissue constructs to ensure their survival postimplantation. However, their sustainability after insertion in vivo has been suboptimal with only thin (<2-mm thick) constructs2 exhibiting viability.
Contemporary approaches to promote survival of engineered constructs after implantation involve the in vitro pre-establishment of microvessel networks within synthetic tissues that will eventually incorporate into, and mimic, the in vivo convective transport scheme. The importance of microvessel networks to the viability of tissues is exemplified by the 200-μm diffusion limit of gases (e.g., O2 and CO2) in biological matrices.2 Dense capillary networks are essential for supplying nutrients and oxygen to cells in tissues (native or engineered). Moreover, dense lymphatic networks prevent accumulation of metabolic wastes by facilitating their efficient transport away from interstitial cells. Growth of functional microtube networks within engineered constructs is, therefore, an optimal design outcome.
In addition to other functions (e.g., hemostasis, intercellular transport, and vasoactivity), endothelial cells initiate and orchestrate microvessel formation (i.e., tubulogenesis) related to the growth of microvascular (i.e., angiogenesis) or lymphatic (lymphangiogenesis) capillaries. These processes are similar in that early events include cell invasion into the interstitium, proliferation, and morphogenesis into multicellular sprouts that eventually develop into tubes and self-assemble into microvessel networks capable of transporting bulk fluid within their lumens.3–7 However, despite advances in our understanding of endothelial tubulogenesis, there are still limitations related to optimizing the formation of microvessel networks in engineered tissues. One possible explanation is that past efforts to control tubulogenic processes have occurred with limited consideration for the cellular mechanoenvironment.
The endothelial phenotype is a consequence of its mechanoenvironment as evidenced by the reported effects of fluid shear and solid matrix stresses on a multitude of endothelial processes (i.e., vasomotor activity, barrier function, and inflammation), including tubulogenesis.8 Recognition of the critical role of mechanotransduction in endothelial biology resulted in the formulation of novel bioreactor-preconditioning strategies9–12 that, in addition to relying on flow and matrix compression to promote convective transport, simultaneously focused on controlling fluid shear13–15 and solid16–18 stresses within the three-dimensional (3D) matrices to mechanobiologically influence resident cells. The goal was to stimulate the release of tubulogenic molecules, in situ, to act in an autocrine or paracrine fashion to upregulate capillary and/or lymphatic formation by endothelial cells incorporated in the engineered constructs.
The use of pressure as a bioreactor parameter for modulating endothelial tubulogenesis however has received little attention despite its ubiquitous presence in in vivo tissues and the ease with which it can be applied in vitro without the influence of fluid flow and substrate deformation. Static pressures of 80–170 mmHg influence endothelial processes, including vasocontractility,19,20 hemostasis,21 and barrier function.22,23 Interestingly, pressures also control angiogenic processes with effects on proliferation24–28 and expression of tubulogenic molecules, for example, integrin αv,29 promatrix metalloproteinase-1,26 fibroblast growth factor-2 (FGF-2),24,29–31 vascular endothelial growth factor-C (VEGF-C), interleukin-8, tissue plasminogen activator,32 and von Willebrand factor.33 Interestingly, while stretch34,35 and fluid flow36 alter expression of VEGF-A,37 neither of these upregulate VEGF-C expression. Pressure is thus a unique stimulus of endothelial tubulogenic activity that affords a distinct level of control for tissue-engineering endeavors.
Based on this evidence, we predicted that hydrostatic pressure modulates the capacity of endothelial cells to form microvessels. As such, we investigated pressure as a trigger for endothelial tubulogenic activity as part of our initial efforts to assess its utility as a control parameter for mechanobiological preconditioning of engineered tissues. For these studies, we used bovine aortic endothelial cells (BAEC) based on their extensive use as a model endothelial cell line for vascular research, including those related to angiogenesis.38,39 As a starting point, we exposed cells to 0- (i.e., atmospheric; controls), 20- (within the physiologic range of up to 30 mmHg for the microcirculation or interstitium), and 40- (a supraphysiological level) mmHg hydrostatic pressures consistent with prior studies,24,26,28,29 including our own.23,27,31,32 Moreover, we further explored the involvement of FGF-2 as well as VEGF receptor-3 (VEGFR-3) and its ligand, VEGF-C, which was previously shown to exhibit pressure-sensitive expression.32 The use of BAEC was advantageous for this purpose, since these cells express both VEGF-C40 and VEGFR-338,41,42 and do not require exogenous growth factors for baseline activity. Finally, we assessed early events of endothelial tubulogenesis using an established microcarrier bead model39,43,44 of physiologic tubulogenesis (in a collagen matrix) permissive for quantifying sprout formation, a prerequisite for capillary growth involving matrix invasion and proliferation.43
Materials and Methods
Cells
BAEC (Cell Applications) were cultured in the Dulbecco's modified Eagle's medium (DMEM; HyClone) containing 10% fetal bovine serum (FBS; HyClone) and 1% penicillin-streptomycin-l-glutamine solution (HyClone) under standard incubator conditions (i.e., humidified 37°C, 5% CO2/95% air). Cells of passage 3–15 were used for experiments.
Substrates
Experiments with BAEC in a two-dimensional (2D) culture format were conducted on polystyrene substrates (Falcon multiwell plates) precoated with 0.2% gelatin (Sigma-Aldrich). Experiments using 3D cell cultures were carried out on commercially available Cytodex®3 microcarrier beads (GE Healthcare) prepared as described.39,43,44
Pressure exposure
BAEC were exposed to 0- (i.e., control), 20-, or 40-mmHg sustained hydrostatic pressures (above atmospheric) using a custom pressure system (Fig. 1) consisting of a compressed gas cylinder attached to two water vessels (in series) that supplied humidified 5% CO2/95% air to a polycarbonate chamber assembly containing cell preparations. Hydrostatic pressure levels in the pressure chamber assembly were adjusted manually to targeted levels using the 5% CO2/95% air source in conjunction with the first water vessel setup in a vertical fluid column configuration. During experiments, the fluid layer over the cells on rigid substrates remained stationary, since the gas phase over supernatants was pressurized; the cells were thus exposed to a normal stress equal to the applied pressure plus the height of the culture medium (minimized to ≤2-mmHg hydrostatic head). The control chamber passively received gas from the pressure chamber through low-flow resistance tubing and expelled it to the atmosphere. Controls were thus BAEC maintained under atmospheric pressure, but otherwise similar experimental conditions. Pressures in both chambers were monitored in real time with strain gauge transducers (Statham) linked to a signal conditioner (Validyne) interfaced to a Dell computer through LabView Signals Express data acquisition software (National Instruments). We also monitored CO2 levels using a DATEX monitor that sampled the atmosphere in the control chamber. Chambers were kept at 37°C with a temperature-controlled incubator.
FIG. 1.

Schematic representation of the laboratory system for exposing endothelial cell cultures to static pressure regimes of interest. Cell preparations were exposed to pressure regimes characterized by a hydrostatic pressure established by adjusting the depth of the air tube in the cylindrical fluid column attached to the compressed gas tank.
Crystal violet uptake
Cells on 2D substrates were exposed to the prescribed pressures in the absence and presence of 1–2.5 ng/mL FGF-2, 2.5 ng/mL VEGF-A, or 0–20 μM MAZ51 (Sigma-Aldrich), a small-molecule inhibitor of VEGFR-3 (a high-affinity receptor for VEGF-C).45 After experiments, BAEC were fixed with 2% paraformaldehyde/0.5% glutaraldehyde (Electron Microscopy Sciences) at 4°C and stained with 0.5% (w/v) crystal violet (Sigma-Aldrich) in 20% methanol (Fisher) as described.27 Crystal violet uptake was quantified by spectrophotometry (Biotek) at 570 nm as a measure of cell density. We showed time-dependent changes in crystal violet uptake to be a sensitive measure of proliferation when compared to BrdU uptake.27
Immunofluorescence
Populations of BAEC in 2D gelatin-coated Petri dishes (Falcon) were exposed to the prescribed pressures for 3 days, enzymatically released from substrates, and immediately fixed with 0.25% p-formaldehyde in 0.1 M sodium phosphate. Fixed cells were labeled with mouse monoclonal antibodies to human VEGFR-3 (Millipore; cross-reactivity with bovine antigen) in 1% bovine serum albumin (BSA; Sigma-Aldrich) at room temperature for 1 h and subsequently stained with fluorescent-labeled goat anti-mouse IgG secondary antibodies conjugated to Alexa-Fluor®488 according to standard procedures. Cellular VEGF-C was labeled with goat anti-human VEGF-C (Santa Cruz Biotechnologies; cross-reactivity with bovine antigen) and stained with donkey anti-goat IgG conjugated to Alexa-Fluor488, both in staining buffers with 0.1% saponin. After labeling, cells were rinsed of excess unbound antibodies, resuspended in 0.5% BSA, and analyzed using an LSR II flow cytometer (Becton-Dickinson). For each sample, at least 10,000 cells were acquired and analyzed using FACsDiva (Becton-Dickinson). Histograms of antigen-specific fluorescence intensity (related to the numbers of antibodies bound to cells) were generated for each sample. The mean fluorescence intensity was used to quantify expression levels.
Tubulogenesis assay
The tubulogenesis assay using collagen hydrogels was adapted from reported procedures.43 Microcarrier beads were seeded at 1×106 cells/2,500 beads in a culture medium, incubated in a standard incubator with gentle agitation for 4 h, rinsed, and cultured overnight in fresh medium. Collagen gel solutions were generated by combining the following reagents at 4°C: 10 parts 200 mM l-glutamine (Invitrogen), 327 parts rat-tail collagen I (Becton-Dickinson), 67 parts 0.1 N sodium hydroxide, 54 parts 0.53 N sodium bicarbonate, 100 parts 10×DMEM, and 443 parts deionized water (reagents from Sigma-Aldrich unless otherwise indicated). This solution was then mixed with ¼ volume of the culture medium. Gel solutions with endothelialized beads were generated using the same formulation, but with medium containing endothelialized beads at concentrations to achieve 15–20 beads/well of a 24-well plate. Gels were formed by allowing an acellular collagen layer to begin to, but not completely, polymerize at 37°C for 5 min and then overlaying a second layer containing endothelialized beads (Fig. 2) followed by incubation at 37°C for 30 min. This procedure ensured that the endothelialized beads were suspended within a homogeneous gel matrix.
FIG. 2.

Schematic representation depicting the preparation of the Cytodex bead assay to assess endothelial tubulogenic activity. Endothelial tube-like sprout formation was assessed using a collagen gel bead assay prepared as described in this illustration.
For experiments, beads in polymerized gels were immersed in an equal volume of the culture medium with and without 2.5 ng/mL FGF-2 (Sigma-Aldrich) or 2.5 ng/mL VEGF-A (Sigma-Aldrich). After experiments, gels were fixed with 2% paraformaldehyde/0.5% glutaraldehyde for 24 h at 4°C. Images of sprouts emanating from cell layers were captured at the central focal plane of the beads using an Olympus IX71 microscope with a 10× objective (110× magnification) interfaced to Simple PCI software. Endothelial sprouts (linear, multicellular structures at least 50 μm in length43) were manually counted and measured using ImageJ (NIH) software.
Statistical analyses
Data were expressed as mean±standard error. Activity of BAEC in the absence or presence of biochemical stimulation under atmospheric (control) pressures was assessed using raw values to ascertain the baseline behavior. The means of these experimental treatments were compared using Student's t-test with p<0.05 delineating significant differences. Bonferroni's corrected p-values were used for multiple comparisons. Responses of BAEC to pressure were normalized to those of matched controls (i.e., cells under atmospheric pressure, but otherwise similar experimental conditions) and expressed as fold change to account for any affects due to the influence of exogenous chemicals on baseline activity. Significant fold changes were determined with a one-sample t-test using a fold change=1 for control values and a p<0.05 denoting a significant difference from this threshold value. Different pressure treatments were compared using Student's t-test with Bonferroni's adjustment for multiple comparisons; p<0.05 denoted a significant difference.
Results
Endothelial cell proliferation is pressure magnitude dependent
Relative to cells maintained under control (i.e., 2 mmHg above atmospheric) pressure conditions, BAEC exposed to 20 mmHg, but not 40 mmHg, for 3 days exhibited significant (p<0.05) increases in cell densities (Fig. 3). Notably, pressure-sensitive proliferation of BAEC exhibited a different pattern of dependence in the presence of FGF-2. Under 1 ng/mL FGF-2, BAEC exposed to both 20 and 40 mmHg for 3 days displayed significantly enhanced population growth relative to matched controls (Fig. 3). Interestingly, BAEC stimulated with 1 ng/mL FGF-2 under control conditions did not exhibit enhanced growth after 3 days (Fig. 3). In fact, we determined the minimum FGF-2 concentration required to stimulate the growth of BAEC to be somewhere between 1 and 2.5 ng/mL (Fig. 3). The growth of BAEC is thus pressure sensitive in the context of the surrounding FGF-2 environment.
FIG. 3.

Endothelial growth is function of local hydrostatic pressure levels and extracellular levels of fibroblast growth factor-2 (FGF-2). (A) Endothelial cell populations were maintained under atmospheric (control) pressure conditions as well as exposed to 20- and 40-mmHg hydrostatic pressures above atmospheric in the absence (white bars) and presence (dark bars) of 1 ng/mL FGF-2 for 3 days. Crystal violet uptake of pressurized cells was normalized to those of match cell preparations maintained under atmospheric (control) pressure, but otherwise similar experimental (i.e., with or without FGF-2) conditions, and expressed as fold change. Bars in (A) are mean fold change±standard error; n=5 independent experiments. #p<0.05 compared to control levels assigned a value of 1 (dashed line) using one-sample t-test. (B) Growth of bovine aortic endothelial cells (BAEC) under 1–2.5 ng/mL FGF-2 was determined for cells under control pressure conditions to define the baseline influence of this growth factor (i.e., positive control). Cell densities were determined by spectroscopic analysis (at 570 nm) of crystal violet uptake for each growth factor condition and compared using paired Student's t-test in conjunction with Bonferroni's adjustment for multiple comparisons; *p<0.017. Bars in (B) are mean values±standard error; n=5 to 6 independent experiments.
VEGFR-3 mediates pressure-sensitive endothelial cell proliferation
To investigate the possibility that VEGF-C participates in the proliferative responses of BAEC to pressure in line with our previous report,32 we blocked VEGFR-3 activity using the VEGFR-3 antagonist, MAZ51.45 Under control conditions, endothelial cell growth for 3 days was reduced dose dependently (R2=0.92; p<0.01) with increasing concentrations (0–20 μM) of MAZ51 (Fig. 4). In terms of pressure exposure, MAZ51 at concentrations ⩾1 μM impaired the proliferative responses of BAEC exposed to 20 mmHg for 3 days (Fig. 4).
FIG. 4.

Pressure-sensitive endothelial proliferative responses depend on VEGR-3 activity. (A) We defined the relationship between MAZ51 and baseline endothelial proliferation under atmospheric pressures using linear regression analyses. Points in (A) are cell densities (reflected by absorbance of crystal violet at 570 nm)±standard error. MAZ51 concentration dependence of cell densities was assessed by linear regression. (B) We also examined the influence of 0–20 μM MAZ51 on the growth response of BAEC to 20-mmHg-pressure exposure. Crystal violet uptake of pressurized cells was normalized to those of cells maintained under atmospheric (control) pressure, but otherwise similar experimental (i.e., MAZ51) conditions, and expressed as fold change. Bars in (B) are mean±standard error; n=5. #p<0.05 compared to levels of matched control assigned a value of 1 (dashed line) using one-sample t-test.
VEGF-C and VEGFR-3 protein expression is pressure sensitive
Under all conditions, BAEC exhibited baseline expression levels of VEGF-C and VEGFR-3 (Fig. 5). BAEC exposed to 20 mmHg exhibited enhanced expression levels of VEGFR-3 and VEGF-C relative to those of matched controls (Fig. 6). Exposure of cells to 40 mmHg for 3 days had no effect on VEGF-C expression levels (Fig. 6). Interestingly, BAEC exposed to 40 mmHg exhibited a small, but significant, increase in VEGFR-3 expression relative to cells under control pressure, but significantly (p<0.05) less than cells exposed to 20 mmHg.
FIG. 5.

Pressure upregulates cellular levels of VEGF-C and membrane expression of its high-affinity receptor, VEGFR-3. (A, B) Histograms of fluorescence intensity, that is, for either VEGF-C (A) or VEGFR-3 (B), were plotted for unlabeled BAEC (no-stain; filled curve) and cells labeled only with the appropriate species-specific secondary antibodies, Alexa®488 conjugates (dotted line) as well as for control cells (thin black line), and cells exposed to 20 mmHg (thick black line) for 3 days and detected with antigen-specific primary antibodies. Mean fluorescence intensities reflecting bound antibodies for either VEGF-C (C) or VEGFR-3 (D) on BAEC exposed to either 20 or 40 mmHg (20 or 40, respectively) were normalized to those of matched controls (dashed line) and expressed as fold change. Bars in (C) and (D) are mean±standard error; n=4 to 6. #p<0.05 compared to control levels (dashed line) assigned a value of 1 (dashed line) using one-sample t-test. *p<0.05 compared using Student's t-test.
FIG. 6.
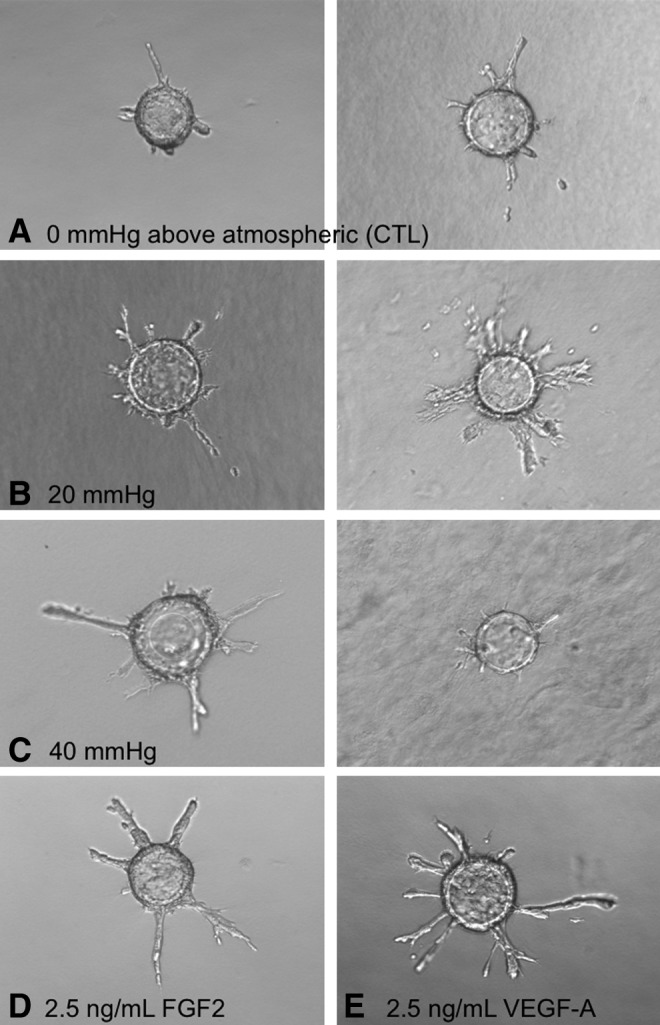
BAEC on microcarrier beads exhibited sprout formation in response to exposure to 20-, but not 40-mmHg hydrostatic pressures. Images of BAEC on Cytodex beads suspended in collagen gels and maintained under atmospheric (control) pressure conditions (A) as well as exposed to either 20- (B) or 40- (C) mmHg hydrostatic pressures for 3 days; 2 representative images are provided for each pressure condition. Positive controls for these experiments were bead preparations of BAEC (1 image each) maintained under control pressures in the presence of either 2.5 ng/mL FGF-2 (D) or 2.5 ng/mL VEGFA (E) for 3 days. Images acquired under 110× magnification.
Endothelial sprout formation is pressure sensitive
BAEC on microcarrier beads in collagen gels under all conditions exhibited the capacity to form sprouts, including under the influence of 2.5 ng/mL FGF-2 or 2.5 ng/mL VEGF-A, both of which served as positive controls (Figs. 6 and 7). The majority of the tube-like sprouts were of lengths >75 μm (Fig. 7). The value of 75 μm was chosen, since it represented one-half of the average diameters of the Cytodex beads.
FIG. 7.

Exposure to 20-, but not 40-mmHg hydrostatic pressures significantly enhanced sprout formation of BAEC. BAEC on Cytodex beads suspended in collagen gels for 3 days were assessed for their ability to form sprouts (A), including those that were >75 μm (B) in response to protubulogenic stimulation by 2.5 ng/mL FGF2 (white bars) and 2.5 ng/mL VEGF-A (bars with a diagonal line pattern). Bars in (A) and (B) represent mean tube numbers±standard error; n=9. *p<0.017 using paired t-test with Bonferroni's adjustment. For pressure experiments, the total number of sprouts (C) as well as the number of sprouts extending 75 μm (D) from endothelial monolayers after exposure to either 20 or 40 mmHg for 3 days were manually counted per bead for 15 beads, normalized to those of matched controls, and expressed as fold change. Bars in (C) and (D) are mean fold change±standard error; n=4 to 5. #p<0.05 compared to control levels assigned a value of 1 (dashed line) using one-sample t-test.
Exposure of BAEC to 20-, but not 40-mmHg hydrostatic pressures for 3 days significantly upregulated sprout formation relative to matched controls (Fig. 7). Moreover, the number of sprouts extending at least 75 μm from the bead surface was significantly (p<0.05) increased under 20 mmHg. As was the case for proliferation of BAEC, pressure-sensitive sprout formation exhibited a different pattern of dependence in the presence of FGF-2. In this case, BAEC exposed to both 20 and 40 mmHg in the presence of 2.5 ng/mL FGF-2 exhibited significantly enhanced sprout formation (Fig. 8). Interesting, only BAEC on beads exposed to 20, but not 40 mmHg, in the presence of FGF-2 exhibited significantly enhanced numbers of sprouts with lengths >75 μm (Fig. 8).
FIG. 8.

Pressure-dependent endothelial sprout formation was sensitive to extracellular levels of FGF-2. Representative images of BAEC on Cytodex beads in collagen gels exposed to either 20- (B) or 40-mmHg (D) static pressures in the presence of 2.5 ng/mL FGF-2 for 3 days. Images of time-matched controls are provided for each pressure level (A and C). The total number of sprouts (E) as well as the number of sprouts extending 75 μm (F) from endothelial monolayers under pressure stimulation were manually counted per bead for 15 beads and normalized to those of controls. Vertical bars in (E) and (F) are mean fold changes in sprout formation±standard error; n=4 to 5. #p<0.05 compared to levels of matched controls assigned a value of 1 using one-sample t-test.
Discussion
The present study provided first evidence that hydrostatic pressure is a magnitude-dependent modulator of endothelial tubulogenesis. From a tissue-engineering perspective, controlled upregulation of endothelial tube formation is beneficial as a strategy to establish microvessels within a construct before implantation with the expectation that it will rapidly integrate with host tissues. Proposed schemes for mechanobiological preconditioning of 3D constructs have included using fluid flow to stimulate tubulogenic activity of endothelial cells lining the lumens of vessel-like constructs or residing within 3D matrices containing growth factors.10,11 These models, however, are limited by the extent to which the flow is generated within the interstitium and by the generation of complex, nonuniform mechanical (shear, pressure, and tensile) stress distributions that obscure the contributions of each stress component to the observed tubulogenic responses. The advantage of using pressure as a mechanobiological stimulus is the potential to stimulate endothelial cells throughout a 3D matrix with the only source of variation resulting from their depths in the constructs. As such, our study considered a new approach for vascularizing tissues of thicknesses beyond the current 2-mm limitation,2 the single-most challenging issue facing the tissue-engineering industry.1
Evidence that indicates an important role for hydrostatic pressure in regulating microvessel formation is derived from studies of aberrant tubulogenic activity during significant pathologies. A common denominator of these conditions is an altered pressure environment for endothelial cells. In hypertensive pulmonary arteries, vascular remodeling elicits formation of plexiform lesions that involves misguided angiogenesis arising from an altered endothelial phenotype.46 Ocular hypertension (>21 mmHg) is a clinical feature for neovascular glaucoma involving a typical angiogenic and, possibly even, lymphangiogenic activity.47–49 Tumor metastasis correlates with interstitial hypertension (pressures>50 mmHg50) and enhanced angio-/lymphangiogenic51 activity for bone,52,53 intestinal,54 and lung55 cancers. Interestingly, in vitro stimulation of osteosarcoma cells with 50 mmHg elicits expression of the prolymphangiogenic factor, VEGF-C.52,53 This pressure is in the range of that which enhanced endothelial proliferation, FGF-2 signaling, and VEGF-C expression in culture.27,31,32 Collectively, these links between pressure and tubulogenic activity point to the potential of using similar pressure ranges to promote microvascularization in engineered constructs.
Along these lines, the hydrostatic pressures used in the present study influenced cell growth in a magnitude-dependent fashion (Fig. 3) consistent with past reports for bovine aortic,56 calf pulmonary artery,24,30 human umbilical vein,29 and immortalized human aortic (HAoEC) endothelium.26 Reportedly, static pressures ranging from 2–150 mmHg enhance endothelial proliferation, while further elevations up to 200 mmHg lead to reduced mitotic rates.26,28,56 Notably, proliferation rates of BAEC in our study were unaffected by exposure to 40 mmHg (Fig. 3), which is within the pressure range reported to be mitogenic for BAEC56 and transformed HAoEC.26 However, the proliferative responses of BAEC to 40 mmHg by Sumpio et al.56 required at least 7 days of pressure exposure; we pressurized our cells for only 3 days. Moreover, the discrepancy between the pressure-sensitive proliferative activity of our BAEC and immortalized HAoEC26 is likely due to genetic variations arising from differences in species or transformation status. Despite such variations, the effect of pressure on endothelial growth appears to be most potent after slight to moderate pressure elevations, but is gradually reduced and eventually blocked as pressures rise.
Reportedly, the proliferative responses of human and bovine endothelium to 2–11-mmHg hydrostatic pressure levels require autocrine FGF-2 activity via release from cytosolic stores.27,31 Moreover, the proliferative responses of endothelial cells under a 40-mmHg mean cyclic pressure require exogenous FGF-2 activity and occur with concomitant phosphorylation of FGFR2.31 Our observations that FGF-2 stimulation was necessary for BAEC exposed to 40-mmHg static pressures to exhibit a growth response (Fig. 3) further suggested the capability of this growth factor to influence pressure-sensitive tubulogenic activity, particularly since BAEC under 20 mmHg did not require FGF-2 to exhibit a proliferative response.
We also demonstrated the potential utility of pressure as a mechanobiological stimulus of tubulogenesis by linking enhanced growth of BAEC under 20 mmHg to the activity of VEGFR-3 (Fig. 4), the high-affinity receptor for VEGF-C and VEGF-D. It is important to note that MAZ51 is capable of blocking VEGFR-2-related cell activity, but it does so at concentrations ≥20 μM.45,57 Blockade of the proliferative responses of BAEC to 20-mmHg pressures at concentrations between 1–20 μM (Fig. 3) is in line with the effective range of MAZ51 on VEGFR-3 activity45 and is consistent with results from our earlier work showing that the proliferative responses of endothelial cells to cyclic pressure occurred in parallel with enhanced VEGF-C transcription and required VEGF-C activity.32 It, however, is also possible that VEGF-D plays a role in the growth response of BAEC to 20-mmHg-pressure exposure, since MAZ51 antagonizes the tyrosine kinase activity of VEGFR-3 resulting from ligand binding. However, whereas BAEC have been shown to express VEGF-C in the present study (Fig. 5) and by others,40 there has been, to our knowledge, no reports that demonstrate these cells express VEGF-D. Moreover, we previously excluded the possibility that pressure-sensitive proliferation by human endothelial cells involved VEGF-D.32
Notably, the fact that VEGF-C is upregulated in BAEC by exposure to 20 mmHg, but not 40 mmHg (Fig. 5), is consistent with the observed pattern of pressure-sensitive proliferation for the BAEC used in this study (Fig. 3). Moreover, the magnitude-dependent effect of pressure on growth of BAEC extended down to the receptor level where we observed a similar pattern of pressure dependence on VEGFR-3 expression (Fig. 5). Although both pressure levels elicited a significant increase in VEGFR-3 relative to atmospheric pressure controls, the 20-mmHg regime exhibited more potency than the 40-mmHg stimulus (Fig. 5). Collectively, these results demonstrated that VEGF-C and VEGFR-3 play key roles in pressure-sensitive endothelial cell proliferation. Moreover, this regulatory mechanism appears to act in an autocrine fashion, since no other cells were present in our experimental system.
Together, the results of the present study (Figs. 3–5) and our prior data27,31,32 suggest that the effects of pressure on endothelial cells converge on a putative FGF-2- and VEGF-C-signaling axis. In line with this possibility, FGF-2 and VEGF-C both regulate tubulogenesis by endothelial cells under atmospheric pressure conditions.58 In fact, exogenous FGF-2 and VEGF-C synergistically enhance in vitro angiogenic tube formation by BAEC.38 Interestingly, our prior evidence27,31,32 suggests that pressure-sensitive FGF-2 activity is upstream of enhanced VEGF-C transcription. This is consistent with evidence that FGF-2 upregulates VEGF-C in an in vitro setting59 as well as during coronary vasculogenesis in the embryonic myocardium.60 The involvement of FGF-2 and VEGF-C in pressure-sensitive endothelial tubulogenesis is therefore consistent with a putative sequential interaction.
The current paradigm describing tubulogenesis points to endothelial invasion into subluminal tissues, followed by their assembly into sprouts, which mature into tubes.7,61 Sprouts form as linear multicellular structures having an invasive tip cell and trunk cells that are proliferative and capable of forming lumens. Endothelial sprouts, however, lack a lumen and typically form within 2–3 days of stimulation.7 Sprout formation under all conditions tested in this study occurred within this time frame. Moreover, we did not detect morphological indicators of lumens in these structures. Our failure to detect lumens however agrees with literature evidence, indicating that assembly of lumen-containing vessels requires the paracrine actions of other cells (e.g., fibroblasts and pericytes).61,62 Inclusion of additional cell types in co-culture with our BAEC, however, would have expanded the present study beyond our intended scope, since examining the responses of cocultures would have required an analysis of the pressure responses of each cell type in isolation to gain useful data related to existing interactions. We thus focused on early cell function-based events of tubulogenesis with the endothelial cells serving as the principle initiators of this process.
In this regard, the observed robust tubulogenic responses of BAEC to the 20, but not the 40 mmHg-pressure stimulus (Figs. 6 and 7) was in line with a similar magnitude-dependent effect on cell proliferation (Fig. 3) as well as on VEGF-C and VEGFR-3 expression (Fig. 5). The lack of any synergistic or additive effect of FGF-2 on sprout formation (Fig. 7), as well as on cell growth (Fig. 3), responses of BAEC exposed to 20 mmHg suggested that either (i) FGF-2 was not involved in pressure-sensitive tubulogenic activity, or (ii) the influence of this pressure on sprouting activity reached threshold levels above which FGF-2 had no effect. In support of the latter, our consistent observations that FGF-2 promoted a detectable pressure-sensitive increase in endothelial sprouting (Fig. 8) under 40 mmHg, as was the case for cell proliferation (Fig. 4), suggested the involvement of this growth factor in endothelial mechanotransduction of pressure as previously reported.24,29,31 Notably, the combination of 40 mmHg and FGF-2 favored initiation of sprout formation rather than extension beyond 75 μm (Fig. 8). This is consistent with the significant, but small, increase in proliferation afforded by exposure of BAEC to 40 mmHg in the presence of FGF-2 (Fig. 4) and the dependence of sprout projection on proliferation.7,39
It is important to point out that the observed pressure effects occurred in the absence of any flow, since static pressures were applied to the incompressible supernatant layer. We also do not expect the applied pressures to have deformed the porous, fully hydrated collagen gel matrix. Moreover, it should be emphasized that our study focused on the potential use of pressure as an approach to microvascularize synthetic tissue constructs. The model of BAEC used in this study provided an opportunity to do so while investigating the involvement of VEGF-C and VEGFR-3. Although VEGFR-3 expression has largely been associated with lymphatic-derived endothelium, numerous investigators have shown that this receptor is expressed by blood endothelium, including BAEC.38,41,63 For example, Tammela et al.64 reported VEGFR-3 expression by blood endothelial cells at the tips of developing sprouts. Notably, their findings in conjunction with the results of the present study are in line with a role for pressure in early endothelial tubulogenic activity related to sprout formation. Future work, however, is needed to validate our approach in human endothelial cells considering that the end application is the engineering of replacement human tissues.
Finally, it is unclear whether pressure is angiogenic or lymphangiogenic, since FGF-2 and VEGF-C are capable of upregulating both processes. Interestingly, low doses of FGF-2 elicit lymphangiogenesis, while high concentrations promote angiogenesis.65 This is interesting in light of the influence of low FGF-2 concentrations on endothelial responsiveness to 40 mmHg even in the presence of high serum concentrations (Figs. 4 and 8), which is known to attenuate growth factor effects,43 as well as our prior work showing that pressure facilitates FGF-2/FGFR2 interactions and enhances VEGF-C transcription. Further work is necessary to resolve this issue by fully characterizing, at the molecular level, the pressure-dependent tubulogenic phenotype.
In summary, the salient finding of our study is that early endothelial tubulogenic activity exhibits pressure sensitivity. It remains to be determined whether pressure-sensitive sprouting leads to the eventual establishment of lumen-containing tubular networks. However, our results did provide novel evidence substantiating pressure as a stimulus of endothelial sprouting, one of the earliest events in tube formation. In addition to general implications related to microvascular pathobiology (e.g., glaucoma and cancer), our study serves as a first step to verify the potential utility of pressure as a fine-tune control parameter to optimize microvessel network formation in the in vitro setting by rapidly initiating early endothelial events. Such a parameter may serve as a powerful tissue-engineering tool to be incorporated in mechanobiological preconditioning schemes to vascularize tissue constructs so as to improve their in vivo viability.
Acknowledgments
This work was supported by the University of Kentucky Research Support Fund (PI: H. Shin, Ph.D.), an AHA Beginning-Grant-in-Aid (PI: H. Shin), an NSF-Kentucky EPSCoR Bioengineering Initiative grant (PI: R. Andrews, Ph.D.), an NIH R01 grant (HL086644; PI: M. Fannon), and a Research to Prevent Blindness grant (PI: M. Fannon). We thank S. Lai-Fook, Ph.D., and E. Bruce, Ph.D., of the Center for Biomedical Engineering at the University of Kentucky for their technical and material assistance with setting up the pressure system. We also thank X. Lei for repeating some final replicate experiments and Xiaoyan Zhang, M.Sc., for flow cytometric analyses.
Disclosure Statement
No competing financial interests exist.
References
- 1.Loffredo F. Lee R.T. Therapeutic vasculogenesis: it takes two. Circ Res. 2008;103:128. doi: 10.1161/CIRCRESAHA.108.180604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Griffith C.K. Miller C. Sainson R.C. Calvert J.W. Jeon N.L. Hughes C.C., et al. Diffusion limits of an in vitro thick prevascularized tissue. Tissue Eng. 2005;11:257. doi: 10.1089/ten.2005.11.257. [DOI] [PubMed] [Google Scholar]
- 3.Swartz M.A. Skobe M. Lymphatic function, lymphangiogenesis, and cancer metastasis. Microsc Res Tech. 2001;55:92. doi: 10.1002/jemt.1160. [DOI] [PubMed] [Google Scholar]
- 4.Holopainen T. Bry M. Alitalo K. Saaristo A. Perspectives on lymphangiogenesis and angiogenesis in cancer. J Surg Oncol. 2011;103:484. doi: 10.1002/jso.21808. [DOI] [PubMed] [Google Scholar]
- 5.Folkman J. Tumor angiogenesis: a possible control point in tumor growth. Ann Intern Med. 1975;82:96. doi: 10.7326/0003-4819-82-1-96. [DOI] [PubMed] [Google Scholar]
- 6.Carmeliet P. Conway E.M. Growing better blood vessels. Nat Biotechnol. 2001;19:1019. doi: 10.1038/nbt1101-1019. [DOI] [PubMed] [Google Scholar]
- 7.Bayless K.J. Davis G.E. Sphingosine-1-phosphate markedly induces matrix metalloproteinase and integrin-dependent human endothelial cell invasion and lumen formation in three-dimensional collagen and fibrin matrices. Biochem Biophys Res Commun. 2003;312:903. doi: 10.1016/j.bbrc.2003.11.017. [DOI] [PubMed] [Google Scholar]
- 8.Chien S. Mechanotransduction and endothelial cell homeostasis: the wisdom of the cell. Am J Physiol Heart Circ Physiol. 2007;292:H1209. doi: 10.1152/ajpheart.01047.2006. [DOI] [PubMed] [Google Scholar]
- 9.Freed L.E. Guilak F. Guo X.E. Gray M.L. Tranquillo R. Holmes J.W., et al. Advanced tools for tissue engineering: scaffolds, bioreactors, and signaling. Tissue Eng. 2006;12:3285. doi: 10.1089/ten.2006.12.3285. [DOI] [PubMed] [Google Scholar]
- 10.Helm C.L. Zisch A. Swartz M.A. Engineered blood and lymphatic capillaries in 3-D VEGF-fibrin-collagen matrices with interstitial flow. Biotechnol Bioeng. 2007;96:167. doi: 10.1002/bit.21185. [DOI] [PubMed] [Google Scholar]
- 11.Kannan R.Y. Salacinski H.J. Sales K. Butler P. Seifalian A.M. The roles of tissue engineering and vascularisation in the development of micro-vascular networks: a review. Biomaterials. 2005;26:1857. doi: 10.1016/j.biomaterials.2004.07.006. [DOI] [PubMed] [Google Scholar]
- 12.Melero-Martin J.M. De Obaldia M.E. Kang S.Y. Khan Z.A. Yuan L. Oettgen P., et al. Engineering robust and functional vascular networks in vivo with human adult and cord blood-derived progenitor cells. Circ Res. 2008;103:194. doi: 10.1161/CIRCRESAHA.108.178590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Swartz M.A. Boardman K.C., Jr The role of interstitial stress in lymphatic function and lymphangiogenesis. Ann N Y Acad Sci. 2002;979:197. doi: 10.1111/j.1749-6632.2002.tb04880.x. discussion 29–34. [DOI] [PubMed] [Google Scholar]
- 14.Ng C.P. Helm C.L. Swartz M.A. Interstitial flow differentially stimulates blood and lymphatic endothelial cell morphogenesis in vitro. Microvasc Res. 2004;68:258. doi: 10.1016/j.mvr.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 15.Goldman J. Conley K.A. Raehl A. Bondy D.M. Pytowski B. Swartz M.A., et al. Regulation of lymphatic capillary regeneration by interstitial flow in skin. Am J Physiol Heart Circ Physiol. 2007;292:H2176. doi: 10.1152/ajpheart.01011.2006. [DOI] [PubMed] [Google Scholar]
- 16.Shiu Y.T. Weiss J.A. Hoying J.B. Iwamoto M.N. Joung I.S. Quam C.T. The role of mechanical stresses in angiogenesis. Crit Rev Biomed Eng. 2005;33:431. doi: 10.1615/critrevbiomedeng.v33.i5.10. [DOI] [PubMed] [Google Scholar]
- 17.Krishnan L. Underwood C.J. Maas S. Ellis B.J. Kode T.C. Hoying J.B., et al. Effect of mechanical boundary conditions on orientation of angiogenic microvessels. Cardiovasc Res. 2008;78:324. doi: 10.1093/cvr/cvn055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Davis G.E. Camarillo C.W. Regulation of endothelial cell morphogenesis by integrins, mechanical forces, and matrix guidance pathways. Exp Cell Res. 1995;216:113. doi: 10.1006/excr.1995.1015. [DOI] [PubMed] [Google Scholar]
- 19.Hishikawa K. Nakaki T. Marumo T. Suzuki H. Kato R. Saruta T. Pressure enhances endothelin-1 release from cultured human endothelial cells. Hypertension. 1995;25:449. doi: 10.1161/01.hyp.25.3.449. [DOI] [PubMed] [Google Scholar]
- 20.Hishikawa K. Nakaki T. Suzuki H. Saruta T. Kato R. Transmural pressure inhibits nitric oxide release from human endothelial cells. Eur J Pharmacol. 1992;215:329. doi: 10.1016/0014-2999(92)90051-5. [DOI] [PubMed] [Google Scholar]
- 21.Silverman M.D. Waters C.R. Hayman G.T. Wigboldus J. Samet M.M. Lelkes P.I. Tissue factor activity is increased in human endothelial cells cultured under elevated static pressure. Am J Physiol. 1999;277:C233. doi: 10.1152/ajpcell.1999.277.2.C233. [DOI] [PubMed] [Google Scholar]
- 22.Muller-Marschhausen K. Waschke J. Drenckhahn D. Physiological hydrostatic pressure protects endothelial monolayer integrity. Am J Physiol Cell Physiol. 2008;294:C324. doi: 10.1152/ajpcell.00319.2007. [DOI] [PubMed] [Google Scholar]
- 23.Shin H.Y. Bizios R. Gerritsen M.E. Cyclic pressure modulates endothelial barrier function. Endothelium. 2003;10:179. doi: 10.1080/10623320390237883. [DOI] [PubMed] [Google Scholar]
- 24.Acevedo A.D. Bowser S.S. Gerritsen M.E. Bizios R. Morphological and proliferative responses of endothelial cells to hydrostatic pressure: role of fibroblast growth factor. J Cell Physiol. 1993;157:603. doi: 10.1002/jcp.1041570321. [DOI] [PubMed] [Google Scholar]
- 25.Hasel C. Durr S. Bauer A. Heydrich R. Bruderlein S. Tambi T., et al. Pathologically elevated cyclic hydrostatic pressure induces CD95-mediated apoptotic cell death in vascular endothelial cells. Am J Physiol Cell Physiol. 2005;289:C312. doi: 10.1152/ajpcell.00107.2004. [DOI] [PubMed] [Google Scholar]
- 26.Kato S. Sasaguri Y. Azagami S. Nakano R. Hamada T. Arima N., et al. Ambient pressure stimulates immortalized human aortic endothelial cells to increase DNA synthesis and matrix metalloproteinase 1 (tissue collagenase) production. Virchows Arch. 1994;425:385. doi: 10.1007/BF00189576. [DOI] [PubMed] [Google Scholar]
- 27.Shin H.Y. Gerritsen M.E. Bizios R. Regulation of endothelial cell proliferation and apoptosis by cyclic pressure. Ann Biomed Eng. 2002;30:297. doi: 10.1114/1.1458595. [DOI] [PubMed] [Google Scholar]
- 28.Vouyouka A.G. Powell R.J. Ricotta J. Chen H. Dudrick D.J. Sawmiller C.J., et al. Ambient pulsatile pressure modulates endothelial cell proliferation. J Mol Cell Cardiol. 1998;30:609. doi: 10.1006/jmcc.1997.0625. [DOI] [PubMed] [Google Scholar]
- 29.Schwartz E.A. Bizios R. Medow M.S. Gerritsen M.E. Exposure of human vascular endothelial cells to sustained hydrostatic pressure stimulates proliferation. Involvement of the alphaV integrins. Circ Res. 1999;84:315. doi: 10.1161/01.res.84.3.315. [DOI] [PubMed] [Google Scholar]
- 30.Salwen S.A. Szarowski D.H. Turner J.N. Bizios R. Three-dimensional changes of the cytoskeleton of vascular endothelial cells exposed to sustained hydrostatic pressure. Med Biol Eng Comput. 1998;36:520. doi: 10.1007/BF02523225. [DOI] [PubMed] [Google Scholar]
- 31.Shin H.Y. Schwartz E.A. Bizios R. Gerritsen M.E. Receptor-mediated basic fibroblast growth factor signaling regulates cyclic pressure-induced human endothelial cell proliferation. Endothelium. 2004;11:285. doi: 10.1080/10623320490904205. [DOI] [PubMed] [Google Scholar]
- 32.Shin H.Y. Smith M.L. Toy K.J. Williams P.M. Bizios R. Gerritsen M.E. VEGF-C mediates cyclic pressure-induced endothelial cell proliferation. Physiol Genomics. 2002;11:245. doi: 10.1152/physiolgenomics.00068.2002. [DOI] [PubMed] [Google Scholar]
- 33.Starke R.D. Ferraro F. Paschalaki K.E. Dryden N.H. McKinnon T.A. Sutton R.E., et al. Endothelial von Willebrand factor regulates angiogenesis. Blood. 2011;117:1071. doi: 10.1182/blood-2010-01-264507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Suzuma I. Hata Y. Clermont A. Pokras F. Rook S.L. Suzuma K., et al. Cyclic stretch and hypertension induce retinal expression of vascular endothelial growth factor and vascular endothelial growth factor receptor-2: potential mechanisms for exacerbation of diabetic retinopathy by hypertension. Diabetes. 2001;50:444. doi: 10.2337/diabetes.50.2.444. [DOI] [PubMed] [Google Scholar]
- 35.Zheng W. Seftor E.A. Meininger C.J. Hendrix M.J. Tomanek R.J. Mechanisms of coronary angiogenesis in response to stretch: role of VEGF and TGF-beta. Am J Physiol Heart Circ Physiol. 2001;280:H909. doi: 10.1152/ajpheart.2001.280.2.H909. [DOI] [PubMed] [Google Scholar]
- 36.Gan L. Miocic M. Doroudi R. Selin-Sjogren L. Jern S. Distinct regulation of vascular endothelial growth factor in intact human conduit vessels exposed to laminar fluid shear stress and pressure. Biochem Biophys Res Commun. 2000;272:490. doi: 10.1006/bbrc.2000.2663. [DOI] [PubMed] [Google Scholar]
- 37.Ferrara N. Role of vascular endothelial growth factor in regulation of physiological angiogenesis. Am J Physiol Cell Physiol. 2001;280:C1358. doi: 10.1152/ajpcell.2001.280.6.C1358. [DOI] [PubMed] [Google Scholar]
- 38.Pepper M.S. Mandriota S.J. Jeltsch M. Kumar V. Alitalo K. Vascular endothelial growth factor (VEGF)-C synergizes with basic fibroblast growth factor and VEGF in the induction of angiogenesis in vitro and alters endothelial cell extracellular proteolytic activity. J Cell Physiol. 1998;177:439. doi: 10.1002/(SICI)1097-4652(199812)177:3<439::AID-JCP7>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 39.Nakatsu M.N. Hughes C.C. An optimized three-dimensional in vitro model for the analysis of angiogenesis. Methods Enzymol. 2008;443:65. doi: 10.1016/S0076-6879(08)02004-1. [DOI] [PubMed] [Google Scholar]
- 40.Kumar R. Harris-Hooker S. Sanford G.L. The expression of growth factors and their receptors in retinal and endothelial cells cocultured in the rotating bioreactor. Ethn Dis. 2008;18:S2–44. [PubMed] [Google Scholar]
- 41.Persaud K. Tille J.C. Liu M. Zhu Z. Jimenez X. Pereira D.S., et al. Involvement of the VEGF receptor 3 in tubular morphogenesis demonstrated with a human anti-human VEGFR-3 monoclonal antibody that antagonizes receptor activation by VEGF-C. J Cell Sci. 2004;117:2745. doi: 10.1242/jcs.01138. [DOI] [PubMed] [Google Scholar]
- 42.Tille J.C. Wang X. Lipson K.E. McMahon G. Ferrara N. Zhu Z., et al. Vascular endothelial growth factor (VEGF) receptor-2 signaling mediates VEGF-C(deltaNdeltaC)- and VEGF-A-induced angiogenesis in vitro. Exp Cell Res. 2003;285:286. doi: 10.1016/s0014-4827(03)00053-3. [DOI] [PubMed] [Google Scholar]
- 43.Dietrich F. Lelkes P.I. Fine-tuning of a three-dimensional microcarrier-based angiogenesis assay for the analysis of endothelial-mesenchymal cell co-cultures in fibrin and collagen gels. Angiogenesis. 2006;9:111. doi: 10.1007/s10456-006-9037-x. [DOI] [PubMed] [Google Scholar]
- 44.Nehls V. Drenckhahn D. A novel, microcarrier-based in vitro assay for rapid and reliable quantification of three-dimensional cell migration and angiogenesis. Microvasc Res. 1995;50:311. doi: 10.1006/mvre.1995.1061. [DOI] [PubMed] [Google Scholar]
- 45.Kirkin V. Thiele W. Baumann P. Mazitschek R. Rohde K. Fellbrich G., et al. MAZ51, an indolinone that inhibits endothelial cell and tumor cell growth in vitro, suppresses tumor growth in vivo. Int J Cancer. 2004;112:986. doi: 10.1002/ijc.20509. [DOI] [PubMed] [Google Scholar]
- 46.Tuder R.M. Voelkel N.F. Angiogenesis and pulmonary hypertension: a unique process in a unique disease. Antioxid Redox Signal. 2002;4:833. doi: 10.1089/152308602760598990. [DOI] [PubMed] [Google Scholar]
- 47.Fong A. Lee G. Reducing vision loss in chronic eye disease. Aust Fam Physician. 2009;38:774. [PubMed] [Google Scholar]
- 48.Lynch S.S. Cheng C.M. Bevacizumab for neovascular ocular diseases. Ann Pharmacother. 2007;41:614. doi: 10.1345/aph.1H316. [DOI] [PubMed] [Google Scholar]
- 49.Yucel Y.H. Johnston M.G. Ly T. Patel M. Drake B. Gumus E., et al. Identification of lymphatics in the ciliary body of the human eye: a novel “uveolymphatic” outflow pathway. Exp Eye Res. 2009;89:810. doi: 10.1016/j.exer.2009.08.010. [DOI] [PubMed] [Google Scholar]
- 50.Boucher Y. Jain R.K. Microvascular pressure is the principal driving force for interstitial hypertension in solid tumors: implications for vascular collapse. Cancer Res. 1992;52:5110. [PubMed] [Google Scholar]
- 51.Cao Y. Why and how do tumors stimulate lymphangiogenesis? Lymphat Res Biol. 2008;6:145. doi: 10.1089/lrb.2008.1007. [DOI] [PubMed] [Google Scholar]
- 52.Park H.R. Min K. Kim H.S. Jung W.W. Park Y.K. Expression of vascular endothelial growth factor-C and its receptor in osteosarcomas. Pathol Res Pract. 2008;204:575. doi: 10.1016/j.prp.2008.01.015. [DOI] [PubMed] [Google Scholar]
- 53.Nathan S.S. Huvos A.G. Casas-Ganem J.E. Yang R. Linkov I. Sowers R., et al. Tumor interstitial fluid pressure may regulate angiogenic factors in osteosarcoma. J Orthop Res. 2008;26:1520. doi: 10.1002/jor.20633. [DOI] [PubMed] [Google Scholar]
- 54.Aurello P. Rossi Del Monte S. D'Angelo F. Cicchini C. Ciardi A. Bellagamba R., et al. Vascular endothelial growth factor C and microvessel density in gastric carcinoma: correlation with clinicopathological factors. Our experience and review of the literature. Oncol Res. 2009;17:405. doi: 10.3727/096504009788912525. [DOI] [PubMed] [Google Scholar]
- 55.Arinaga M. Noguchi T. Takeno S. Chujo M. Miura T. Uchida Y. Clinical significance of vascular endothelial growth factor C and vascular endothelial growth factor receptor 3 in patients with nonsmall cell lung carcinoma. Cancer. 2003;97:457. doi: 10.1002/cncr.11073. [DOI] [PubMed] [Google Scholar]
- 56.Sumpio B.E. Widmann M.D. Ricotta J. Awolesi M.A. Watase M. Increased ambient pressure stimulates proliferation and morphologic changes in cultured endothelial cells. J Cell Physiol. 1994;158:133. doi: 10.1002/jcp.1041580117. [DOI] [PubMed] [Google Scholar]
- 57.Lin C.I. Chen C.N. Huang M.T. Lee S.J. Lin C.H. Chang C.C., et al. Lysophosphatidic acid up-regulates vascular endothelial growth factor-C and lymphatic marker expressions in human endothelial cells. Cell Mol Life Sci. 2008;65:2740. doi: 10.1007/s00018-008-8314-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cao R. Eriksson A. Kubo H. Alitalo K. Cao Y. Thyberg J. Comparative evaluation of FGF-2-, VEGF-A-, and VEGF-C-induced angiogenesis, lymphangiogenesis, vascular fenestrations, and permeability. Circ Res. 2004;94:664. doi: 10.1161/01.RES.0000118600.91698.BB. [DOI] [PubMed] [Google Scholar]
- 59.Jih Y.J. Lien W.H. Tsai W.C. Yang G.W. Li C. Wu L.W. Distinct regulation of genes by bFGF and VEGF-A in endothelial cells. Angiogenesis. 2001;4:313. doi: 10.1023/a:1016080321956. [DOI] [PubMed] [Google Scholar]
- 60.Lavine K.J. White A.C. Park C. Smith C.S. Choi K. Long F., et al. Fibroblast growth factor signals regulate a wave of Hedgehog activation that is essential for coronary vascular development. Genes Dev. 2006;20:1651. doi: 10.1101/gad.1411406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Koh W. Stratman A.N. Sacharidou A. Davis G.E. In vitro three dimensional collagen matrix models of endothelial lumen formation during vasculogenesis and angiogenesis. Methods Enzymol. 2008;443:83. doi: 10.1016/S0076-6879(08)02005-3. [DOI] [PubMed] [Google Scholar]
- 62.Ucuzian A.A. Greisler H.P. In vitro models of angiogenesis. World J Surg. 2007;31:654. doi: 10.1007/s00268-006-0763-4. [DOI] [PubMed] [Google Scholar]
- 63.Jenny B. Harrison J.A. Baetens D. Tille J.C. Burkhardt K. Mottaz H., et al. Expression and localization of VEGF-C and VEGFR-3 in glioblastomas and haemangioblastomas. J Pathol. 2006;209:34. doi: 10.1002/path.1943. [DOI] [PubMed] [Google Scholar]
- 64.Tammela T. Zarkada G. Nurmi H. Jakobsson L. Heinolainen K. Tvorogov D., et al. VEGFR-3 controls tip to stalk conversion at vessel fusion sites by reinforcing notch signalling. Nat Cell Biol. 2011;13:1202. doi: 10.1038/ncb2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chang L.K. Garcia-Cardeña G. Farnebo F. Fannon M. Chen E.J. Butterfield C., et al. Dose-dependent response of FGF-2 for lymphangiogenesis. Proc Natl Acad Sci U S A. 2004;101:11658. doi: 10.1073/pnas.0404272101. [DOI] [PMC free article] [PubMed] [Google Scholar]