

Abstract

Adenosine A1 receptor (A1AR) agonists have antinociceptive effects in multiple preclinical models of acute and chronic pain. Although numerous A1AR agonists have been developed, clinical applications of these agents have been hampered by their cardiovascular side effects. Herein we report a series of novel A1AR agonists, some of which are structurally related to adenosine 5′-monophosphate (5′-AMP), a naturally occurring nucleotide that itself activates A1AR. These novel compounds potently activate A1AR in several orthogonal in vitro assays and are subtype selective for A1AR over A2AAR, A2BAR, and A3AR. Among them, UNC32A (3a) is orally active and has dose-dependent antinociceptive effects in wild-type mice. The antinociceptive effects of 3a were completely abolished in A1AR knockout mice, revealing a strict dependence on A1AR for activity. The apparent lack of cardiovascular side effects when administered orally and high affinity (Ki of 36 nM for the human A1AR) make this compound potentially suitable as a therapeutic.
INTRODUCTION
The adenosine A1 receptor (A1AR) belongs to a family of four G protein-coupled receptors that includes A1AR, A2AAR, A2BAR, and A3AR.1 A1AR is expressed in the highest density in the brain, adipose tissue, kidney, and heart atria and to a lower extent in ventricles, lung, pancreas, liver, and GI tract.2–4 A1AR is also expressed in nociceptive dorsal root ganglia neurons.5 A1AR and A3AR couple to inhibitory Gi/Go proteins, which inhibit adenylate cyclase activity, thus reducing cellular levels of cyclic adenosine monophosphate (cAMP). On the other hand, A2AAR and A2BAR couple to stimulatory Gs proteins and stimulate adenylate cyclase activity.6 Adenosine receptors also modulate phospholipase C, influencing inositol triphosphate and Ca2+ release from internal stores.7 In addition, adenosine receptors regulate potassium and calcium channels.8 Many pathophysiological states are associated with changes in adenosine levels, including asthma, neurodegenerative disorders, psychosis, anxiety, and chronic inflammatory disease.9–12 A1AR has been linked to a variety of human conditions. For example, adenosine is used clinically to treat supraventricular tachycardia and targets A1AR in the atrioventicular node of the heart. This clinical application prompted development of selective A1AR agonists as antiarrythmic agents.13 Moreover, adenosine can reduce allodynia and hyperalgesia associated with chronic pain in rodents and humans.14–19 Ectonucleotidases that dephosphorylate adenosine 5′-monophosphate (5′-AMP) to adenosine have potent, long-lasting, and entirely A1AR-dependent thermal and mechanical antinociceptive effects when administered intrathecally.20–22 Lastly, Goldman and co-workers found that A1AR mediates the local antinociceptive effects of acupuncture, suggesting that peripheral activation of A1ARs can contribute to analgesia.23
We recently found that 5′-AMP can directly activate A1AR without being hydrolyzed to adenosine.24 In view of this finding, we explored the structure–activity relationships (SAR) of 5′-AMP analogues with respect to their A1AR agonist activity in our studies below. Furthermore, clinical applications of the available A1AR agonists have been hampered by their cardiovascular side effects,15,25 and few orally bioavailable compounds are known.11,12,26 Herein we report a novel series of potent and selective A1AR agonists, which are orally active in a mouse model of acute thermal nociception and lack apparent cardiovascular side effects.
RESULTS AND DISCUSSION
Chemistry
Isopropylidene-protected 2-chlorocyclopentyl adenosine 1a was synthesized according to the literature procedure,27 reacted with methyl, ethyl, and tert-butyl N,N-diisopropylphosphoramidite in the presence of tetrazole, and subsequently treated with hydrogen peroxide to produce phosphate diesters 2a–c (Scheme 1). Isopropylidene protecting groups were then removed using p-TsOH in methanol to give 3a, 3b, and 3c. Compound 3c was treated with TFA to cleave the tert-butyl protecting groups, and the resulting free phosphate was converted to its disodium salt 3d by NaOH titration. Treatment of tert-butyl phosphate diester 2c with TFA gave isopropylidene-protected phosphate 4. The phosphate functionality in 4 was then converted to the dichloride via oxalyl chloride treatment, and the product reacted with glycine ethyl ester and phenethyl alcohol to produce compounds 3e and 3f, respectively, following pTsOH-assisted isopropylidene deprotection.
Scheme 1.

Synthesis of Compounds 3a–fa
Pd/C and acetic acid-mediated dechlorination of 2a followed by isopropylidene deprotection under standard conditions gave 5a (Scheme 2). Isopropylidene-protected N6-cyclopentyl adenosine 6 was synthesized according to published procedures28 and reacted with tert-butyl N,N-diisopropylphosporamidite to give intermediate 7, which was then taken through a sequence similar to above to arrive at the disodium salt 5c.
Scheme 2.

Synthesis of Compounds 5a–ca
We also developed a protocol for solvent-free coupling of 2,6-dichloropurine (8) and β-d-ribofuranose 1,2,3,5-tetraacetate (9) catalyzed by p-TsOH, carried out in a microwave reactor to furnish 2,6-dichloropurineriboside triacetate (10) (Scheme 3). Treatment of 10 with 2-amino-3-phenylpropanol in ethanol overnight gave intermediate 11. Subsequently, acyl protecting groups in 11 were removed by methanolic ammonia treatment to give 12a as a mixture of two diastereoisomers inseparable by column chromatography. Reaction of 11 with methyl and tert-butyl N,N-diisopropylphosporamidite followed by addition of hydrogen peroxide produced phosphate diesters, and the acyl protecting groups were similarly removed via methanolic ammonia treatment to furnish the desired products 12b and 12c. TFA treatment of 12b followed by titrating sodium hydroxide into a methanolic solution of the resulting phosphate yielded the desired disodium salt 12d as a mixture of two diastereoisomers.
Scheme 3.

Synthesis of Compounds 12a–da
Reaction of 10 with (1R,2R)-2-aminocyclopentanol produced intermediate 13, which was phosphorylated using methyl and benzyl phosphoramidite to furnish 14a and 14b, respectively, after deprotection (Scheme 4). Benzyl protecting groups in 14b were removed via Pd-mediated hydrogenation, and the resulting phosphate was treated with NaOH to give the desired phosphate disodium salt, compound 14c.
Scheme 4.

Synthesis of Compounds 14a–ca
Biological Evaluation
The newly synthesized or commercially available ligands were initially tested in a cAMP accumulation assay, which measures inhibition of isoproterenolor forskolin-stimulated cAMP accumulation in human embryonic kidney 293T (HEK293T) cells transiently transfected with human A1AR. Efficacies of the tested compounds were compared to 2-chloro-N6-cyclopentyladenosine (3), a potent full agonist of A1AR.29 All test compounds except 3b and 14a were full agonists at the highest concentration tested in this assay (Table 1). We previously reported that 5′-AMP is an A1AR agonist,24 consistent with other studies30–32 showing that A1AR can be activated by adenosine analogues with large and negatively charged groups at the 5′-position.
Table 1.
cAMP Accumulation Assay Results

| ||
|---|---|---|
| compd | R | EC50 (μM)a |
| adenosine | – | 0.039 |
| 2′-AMP | – | 0.49 |
| 3′-AMP | – | 0.44 |
| 5′-AMP | – | 0.50 |
| 5 | H | 0.0063b |
| 5c | PO(OH)2 | 0.10 |
| 5a | PO(OMe)2 | 4.4 |
| 3 | H | 0.014/0.0046b |
| 3d | PO(OH)2 | 0.072 |
| 3a | PO(OMe)2 | 1.4 |
| 3b | PO(OEt)2 | > 10 |
| 3f | PO(OCH2CH2Ph)2 | 6.1 |
| 3e | PO(NHCH2CO2Et)2 | 0.53 |
| 12a | H | 0.55 |
| 12d | PO(OH)2 | 1.5 |
| 12b | PO(OMe)2 | 5.4 |
| 14 | H | 0.0059b |
| 14c | PO(OH)2 | 0.12b |
| 14a | PO(OMe)2 | >10b |
EC50 values are the average of at least two independent experiments with SD values that are 3-fold less than the average.
Forskolin substituted for isoproterenol stimulation.
Interestingly, we found that adenosine 2′-monophosphate (2′-AMP) and adenosine 3′-monophosphate (3′-AMP) were also full agonists of A1AR with similar potencies (EC50 values of 0.49 μM and 0.44 μM, respectively) (Table 1). This was not due to hydrolysis to adenosine for the following reasons: (1) Adenosine gives a characteristic “bimodal” response in the HEK293T cAMP assay due to its activation of endogenous Gs-coupled A2AAR in addition to the Gi-coupled ectopically expressed A1AR.24 If AMP hydrolysis to adenosine were occurring, we would expect to see a bimodal response for 2′-, 3′-, and 5′-AMP. However, we did not observe a bimodal response for 2′-, 3′-, or 5′-AMP (data not shown). (2) Inclusion of α,β-methylene adenosine 5′-diphosphate (αβ-met-ADP), a potent inhibitor of ecto-5′-nucleotidase (NT5E/CD73),33 did not significantly alter the potencies of 2′-AMP or 3′-AMP (Table S1, Supporting Information).
Recent X-ray crystallography studies using A2AAR have identified positively charged histidine residues deep within the agonist binding pocket, which are conserved in A1AR.34,35 One of these residues, His278 in TM7, is proximal to the 2′ and 3′ hydroxyl groups in the ribose moiety of adenosine. Thus, a phosphate group at the 2′ or 3′ positions could form a stabilizing charge–charge interaction with His278, explaining this novel activity of 2′- and 3′-AMP.
N6 substitution in the purine moiety has been shown to improve binding affinities of ligands to A1AR.29 In our cAMP accumulation assay, N6-cyclopentyl adenosine (5) was indeed more potent than adenosine (EC50: 0.0063 μM versus 0.039 μM) (Table 1). The corresponding 5′-monophosphate derivative 5c was less potent (EC50 = 0.10 μM) than compound 5. This result is consistent with the potency changes from adenosine to 5′-AMP. Although the conversion of 5′-phosphate (5c) to 5′-phosphate dimethyl ester (5a) reduced A1AR agonist potency, this dimethyl ester modification could potentially increase oral bioavailability of this series.
The 2-chloro-N6-cyclopentyl adenosine subseries was also explored (Table 1). Compound 3 and its 5′-monophosphate analogue 3d were equal to or slightly more potent than their corresponding des-chloro analogues (5 and 5c). The 2-chloro-N6-cyclopentyl adenosine 5′-phosphate dimethyl ester 3a, which was designed to improve oral bioavailability, was a full agonist in this assay with an EC50 value of 1.4 μM. The potency rank for 5′-alcohol, phosphate, and phosphate dimethyl ester is consistent for both the 2-chloro and des-chloro subseries. These SAR results further support our recent finding that 5′-AMP is a full agonist of A1AR.24 We next explored the SAR at the 5′ position by examining diethyl and diphenethyl esters 3b and 3f as well as phosphate diamide 3e. Interestingly, phosphate diamide 3e was slightly more potent than dimethyl ester 3a. On the other hand, diethyl ester 3b was significantly less potent than 3a while diphenethyl ester 3f was equal to or slightly less potent than 3a. These SAR findings suggest that modifications to the ester or amide groups are tolerated in general, resulting in full agonists of A1AR with moderate potencies with the exception of compound 3b.
Two additional subseries which contain a phosphate or phosphate dimethyl ester-modified hydroxyl moiety in the N6-substituent of the purine (compounds 12b, 12d, 14a, and 14c) were examined (Table 1). Consistent with the previously published results,24,36–38 alcohols 12a and GR-79236 (14) were potent A1AR agonists in the cAMP accumulation assay (EC50 = 0.55 μM and 0.0059 μM). The corresponding phosphates 12d and 14c were in general less potent (EC50 = 1.5 μM and 0.12 μM) than the alcohols 12a and 14, but still showed significant agonist activity. The N6 position projects toward the solvent-exposed extracellular face of A1AR, so it is not surprising that a negatively charged phosphate group is tolerated at this position. The phosphate diesters 12b and 14a (EC50 = 5.4 μM and EC50 > 10 μM) were generally less potent than the phosphates 12d and 14c, a SAR trend similar to that observed in the two subseries described above.
We then selected compounds 3d (a representative phosphate) and 3a (a representative phosphate ester) and tested them in an orthogonal functional assay. This assay utilizes chimeric G proteins to visualize human A1AR activation in real-time and at a single cell resolution by measuring Ca2+ mobilization (Figure 1).24 The Emax of 3d and 3a were normalized to the Emax of adenosine in this assay. Both 3d and 3a were potent full agonists of A1AR with EC50 values of 0.021 μM and 0.52 μM, respectively. Therefore, using two distinct and complementary assay platforms, we confirmed that compounds 3d and 3a directly activated A1AR-mediated signaling.
Figure 1.

Compounds 3d and 3a are full agonists of A1AR in a Ca2+ mobilization assay. 3d: EC50 = 0.021 μM; and 3a: EC50 = 0.52 μM. Adenosine (EC50 = 1.41 μM) was used as a positive control. 27–50 cells per condition. All data are presented as mean ± standard error.
Compounds 3a and 3d were next evaluated in a radioligand binding assay to determine their binding affinities to human A1AR using tritiated compound 3 as the radioligand. Consistent with their functional activities in the cAMP accumulation and Ca2+ mobilization assays, compounds 3a and 3d had high affinities for the human A1AR with Ki values of 36 nM and 25 nM, respectively (Figure 2).
Figure 2.

Compounds 3a and 3d have high binding affinities to human A1AR. Concentration–response curves of 3a and 3d in the radioligand binding assay using tritiated compound 3 as the radioligand, and 5′-N-ethylcarboxamidoadenosine (NECA) as a positive control (data not shown).
To evaluate subtype selectivity (selectivity for A1AR over A2AAR, A2BAR, and A3AR) of compounds 3a and 3d, we tested 3a and 3d in human A2AAR, A2BAR, and A3AR radioligand binding assays (Table 2). Both compounds had no binding affinity to A2BAR (<10% inhibition at 50 μM and 10 μM, respectively). In addition, compounds 3a and 3d were >100-fold selective for A1AR over A2AAR and >10-fold selective for A1AR over A3AR. Compounds 3a and 3d are “compound 3-like” agonists, containing both 2-chloro and N6-cyclopentyl groups, which convey strong subtype selectivity for A1AR. Compound 3 itself displays >100-fold selectivity for A1AR over A2AAR and A2BAR.29,39 As expected, compounds 3a and 3d retained this A1AR selectivity after modification at the 5′ position. A3AR is known to be a low-affinity adenosine receptor,40 so the modestly decreased binding affinity of 3a and 3d at A3AR is not surprising.
Table 2.
Subtype Selectivity of Compounds 3a and 3d Assessed Using Radioligand Binding Assays
| binding affinity (Ki (nM)) |
binding affinity ratio |
|||||
|---|---|---|---|---|---|---|
| compound | A1 | A2A | A3 | % inhibition/[concentration] at A2B | A2A/A1 | A3/A1 |
| 3a | 36 | 4700 | 450 | 7%/[50 μM] | 130 | 12 |
| 3d | 25 | 1600 | 1300 | 0%/[10 μM] | 640 | 52 |
Having established that compounds 3a and 3d are potent and subtype selective A1AR agonists, we next evaluated the antinociceptive effects of 3a in mice. We focused on 3a for in vivo studies because of its enhanced potential to demonstrate oral activity.41 Notably, we found that oral administration of 3a dose-dependently inhibited noxious thermal sensitivity (Figure 3A and 3B). Significant effects were observed even at a low dose (50 nmol/kg). The effects of 3a were similar to 5 (Figure 3C and 3D), which was used as a positive control in this mouse model. In addition, intrathecal (IT) or intravenous (IV) injection of 3a produced marked and dose-dependent inhibition of noxious thermal sensitivity (Figure S1, Supporting Information), similar to oral administration. Importantly, the thermal antinociceptive effect of 3a was completely abolished in the A AR knockout (A1AR–/–) mice even at a high dose (5 nmol, IT injection) (Figure 4), strongly demonstrating that the biological effects of 3a are A1AR-mediated. Taken together, these results reveal that compound 3a is an orally active compound with potent A1AR-mediated antinociceptive activity in mice. It is worth noting that carboxylic acid esters are degraded rapidly in vivo by carboxylesterases; however, the corresponding alkyl esters of phosphates are typically metabolically stable.41 Therefore, it is unlikely that the observed in vivo effects of the phosphate ester 3a are due to hydrolysis of both alkyl esters to produce 3d or the dephosphorylated product 3.
Figure 3.

Oral administration of compound 3a dose-dependently decreases thermal sensitivity in wild-type mice. (A) Compound 3a and (C) compound 5 were orally administrated at 50, 500, or 5000 nmol/kg immediately after determination of the baseline time point. Paw withdrawal latency was assessed at the indicated times, using Hargreaves apparatus to deliver noxious thermal stimulus. Ten C57BL/6 male mice per dose group. t tests were conducted relative to the corresponding vehicle time point. (B, D) Area under curve (AUC) analysis of data shown in (A, C). Five hour AUC measurements were calculated relative to baseline paw withdrawal latency for each mouse and averaged for each treatment condition. All data are presented as mean ± standard error. * p < 0.05, ** p < 0.005, *** p < 0.0005.
Figure 4.
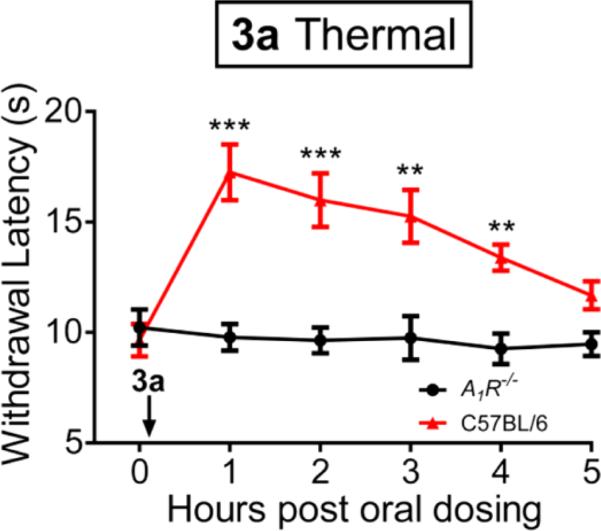
Effects of 3a on thermal sensitivity in wild-type mice are completely abolished in A1AR–/– mice. Time course of effects of 3a on on thermal paw withdrawal latency. 3a (5 nmol) was administered via intrathecal injection immediately after determination of the baseline time point. Thermal paw withdrawal latency in wild type and A1AR–/– mice were monitored using Hargreaves apparatus. Ten C57BL/6 male mice per group. All data are presented as mean ± standard error. t tests were conducted relative to the baseline time point. ** p < 0.005. *** p < 0.0005.
Clinical applications of the currently available A1AR agonists are hampered by their cardiovascular side effects.32 To determine if compound 3a affected cardiovascular function, we monitored heart rate and body temperature in wild-type and A1AR–/– mice following oral administration (5 was administered as a positive control). At the highest dose tested (5000 nmol/kg, oral administration), compound 3a had negligible effects on heart rate and body temperature in wild-type and A1AR–/– mice (Figure 5A and 5C). On the other hand, compound 5 elicited a statistically significant decrease in heart rate and body temperature which lasted for 4 to 6 h in wild-type mice (Figure 5B and 5D) but not in A1AR–/– mice. Compound 5 caused a modest increase in heart rate in A1AR–/– mice, possibly reflecting known off-target activation of stimulatory A2AR.42,43 Collectively, our results indicate that our novel A1AR agonist 3a has potent antinociceptive effects but minimal to no cardiovascular side effects when administered orally at a high 5000 nmol/kg dose. In contrast, the same high dose of 5 has antinociceptive effects and significant cardiovascular side effects. These data in turn suggest 3a has a large therapeutic window and uniquely lacks cardiovascular side-effects that are associated with other A1AR agonists such as compound 5.
Figure 5.

Compound 3a does not have long-lasting effects on heart rate or body temperature while compound 5 causes a significant decrease in heart rate and body temperature in wild-type mice. (A, C) Effects of 3a on (A) heart rate and (C) body temperature in wild-type (red) and A1AR–/– (black) mice. (B, D) Effects of 5 on (B) heart rate and (D) body temperature in wild-type (red) and A1AR–/– (black) mice. Compounds were orally administered at 5000 nmol/kg immediately before telemetry recording began. Eight C57BL/6 male mice per group for A1AR–/– body temperature measurements, and six male mice per group for all other conditions. Red bar: p < 0.05.
CONCLUSIONS
In summary, we designed and synthesized a novel series of A1AR agonists which possess marked potency in several orthogonal in vitro assays and are subtype selective for A1AR over A2AAR, A2BAR, and A3AR. Surprisingly, our SAR studies revealed that the addition of a phosphate or, in some cases a phosphate ester group, to the 5′, 3′, 2′, or N6 moiety of adenosine is tolerated. These findings reveal that A1AR can be activated by diverse natural and non-natural nucleotide and nucleoside analogues. Among our novel A1AR agonists, compound 3a had potent, dose-dependent, and A1AR-dependent antinociceptive activity in mice via oral administration. The apparent lack of cardiovascular side effects in vivo and nanomolar affinity at human A1AR makes this novel compound potentially suitable for treating pain and other physiological processes that are modulated by A1AR activation.
EXPERIMENTAL SECTION
General Procedure for Chemical Synthesis
HPLC data of all compounds were acquired using an Agilent 6110 Series system with the UV detector set to 220 nm. Samples were injected (<10 μL) onto an Agilent Eclipse Plus 4.6 × 50 mm, 1.8 μM, C18 column at room temperature. Mobile phases consisting of H2O + 0.1% acetic acid (A) and MeOH + 0.1% acetic acid (B) were used. A linear gradient from 10% to 100% B during 5.0 min was used followed by 100% B for another 2 min with a flow rate of 1.0 mL/min. Mass spectra (MS) data were acquired in positive ion mode using an Agilent 6110 single quadrupole mass spectrometer with an electrospray ionization (ESI) source. High-resolution mass spectra (HRMS) were acquired using a Shimadzu LCMS-IT-TOF time-of-flight mass spectrometer. Nuclear magnetic resonance (NMR) spectra were recorded on a Varian Mercury spectrometer at 400 MHz for proton (1H NMR) and 100 MHz for carbon (13C NMR); chemical shifts are reported in ppm (δ) relative to the solvent peaks.44 Preparative HPLC was performed using an Agilent Prep 1200 series with the UV detector set to 220 nm. Samples were injected onto a Phenomenex Luna 75 × 30 mm, 5 μM, C-18 column at room temperature. Mobile phases consisting of H2O + 0.1% TFA (A) and MeOH (B) were used at a flow rate of 30 mL/min. A linear gradient from 10% to 100% B in 17.0 min was followed by 100% B for another 3 min. Adenosine, 5′-AMP, 3′-AMP, 2′-AMP, 3, 5, and 14 were purchased from Sigma-Aldrich. All synthesized compounds have >95% purity by the above analytical HPLC method unless specific purities are noted below.
Preparation of ((2R,3S,4R,5R)-5-(2-Chloro-6-(cyclopentylamino)-9H-purin-9-yl)-3,4-dihydroxytetrahydrofuran-2-yl) Phosphates (3a,b)
To a solution of 2-chloro-N6-cyclopentyl-2′,3′-O-(1-methyl-ethylidene)-(9-Cl)adenosine (1) (0.73 mmol) in CH2Cl2 (15 mL) were added the appropriate phosphoramidite (1.5 mmol) and tetrazole (2.4 mmol). The reaction mixture was heated at 60 °C for 2 h and cooled to 0 °C, and hydrogen peroxide (0.3 mL, of 30% w/w solution) was added dropwise. The reaction mixture was stirred for 1 h at rt, diluted with CH2Cl2 (100 mL), and washed with 10% aqueous sodium metabisulfite (25 mL × 2), saturated aqueous sodium bicarbonate (25 mL × 2), water (25 mL × 2), and brine (25 mL × 2). The solvent was then removed under reduced pressure, and the residue was purified by preparative HPLC to afford the TFA salts of 2a,b. To a solution of phosphates 2a,b (0.32 mmol) in methanol (10 mL) was then added p-toluenesulfonic acid (0.03 mmol), the mixture was heated at 60 °C for 2 h, and the solvent was removed under reduced pressure. The residues were then purified by reverse phase preparative HPLC to afford the TFA salts of the title compounds.
((2R,3S,4R,5R)-5-(2-Chloro-6-(cyclopentylamino)-9H-purin-9-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methyl Dimethyl Phosphate (3a)
(22%) white solid. 1H NMR (300 MHz, DMSO-d6): δ 8.30 (s, 1H), 5.86 (d, J = 5.0 Hz, 1H), 5.75–5.50 (m, 1H), 4.58–4.48 (m, 1H), 4.50–4.35 (m, 1H), 4.30–4.40 (m, 4H), 3.72–3.53 (m, 6H), 2.11–1.89 (m, 2H), 1.89–1.51 (m, 6H); 13C NMR (101 MHz, DMSO-d6) δ 155.12, 153.78, 149.95, 139.89, 118.88, 87.92, 83.01, 82.94, 73.54, 70.35, 67.33, 54.64, 54.58, 54.52, 52.14, 33.45, 32.27, 23.93; HRMS calcd for C17H25ClN5O7P + H: 478.1253; found: 478.1243 [M + H]+.
((2R,3S,4R,5R)-5-(2-Chloro-6-(cyclopentylamino)-9H-purin-9-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methyl Diethyl Phosphate (3b)
(22%) white solid. 1H NMR (400 MHz, CD3OD): δ 8.21 (s, 1H), 5.97 (d, J = 4.3 Hz, 1H), 4.72–4.62 (m, 1H), 4.50 (brs, 1H), 4.44 (t, J = 5.2 Hz, 1H), 4.38–4.33 (m, 1H), 4.32–4.26 (m, 1H), 4.26–4.20 (m, 1H), 4.15–4.02 (m, 4H), 2.18–2.01 (m, 2H), 1.92–1.74 (m, 2H), 1.73–1.52 (m, 4H), 1.37–1.24 (m, 6H); HRMS calcd for C19H29ClN5O7P + H: 506.1566; found: 506.1551 [M + H]+.
Sodium ((2R,3S,4R,5R)-5-(2-Chloro-6-(cyclopentylamino)-9H-purin-9-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methyl Phosphate (3d)
To a solution of 1 (300 mg, 0.73 mmol) in CH2Cl2 (15 mL) were added tert-buytl N,N-diisopropylphosphoramidite (405 mg, 1.5 mmol) and tetrazole (5 mL of 0.45 M solution in ACN, 2.4 mmol), and the reaction mixture was heated at 60 °C for 2 h. The temperature was decreased to 0 °C, and hydrogen peroxide (0.3 mL, of 30% w/w solution) was added. The reaction mixture was stirred for 1 h at rt, CH2Cl2 (100 mL) was added, and the organic layer was washed with 10% sodium metabisulfite (25 mL × 2), saturated sodium bicarbonate (25 mL × 2), water (25 mL × 2), and brine (25 mL × 2). The solvent was removed under reduced pressure and purified by preparative HPLC to afford the TFA salt of compound 2c (268 mg, 61%). To a solution of phosphate 2c (44 mg, 0.09 mmol) in methanol (10 mL) was added p-toluenesulfonic acid (5 mg, 0.03 mmol), and the mixture was heated at 60 °C for 2 h. The solvent was removed under reduced pressure, trifluoroacetic acid (0.3 mL, 2.63 mmol) was added in CH2Cl2 (10 mL), and the reaction mixture was stirred for 1 h at rt. The solvent was removed under reduced pressure, and the residue was purified by preparative HPLC to afford the TFA salt of compound 3d. The phosphate was then dissolved in methanol (5 mL), NaOH (0.90 mmol, 36 mg) in water (5 mL) was added, and the mixture was stirred for 30 min at rt. The solvent was removed under reduced pressure to afford the title compound 3d (18 mg, 42% yield) as a white solid. 1H NMR (300 MHz, CD3OD): δ 8.47 (s, 1H), 6.02 (d, J = 5.7 Hz, 1H), 4.65–4.55 (m, 1H), 4.55–4.45 (m, 1H), 4.41–4.35 (m, 1H), 4.25–4.20 (m, 1H), 4.12–4.05(m, 3H), 2.23–2.07 (m, 2H), 1.86–1.69 (m, 7H); HRMS calcd for C15H21ClN5O7P + H: 450.0945; found: 450.0911 [M + H]+.
Ethyl 2-(((((2R,3S,4R,5R)-5-(2-Chloro-6-(cyclopentylamino)-9H-purin-9-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methoxy)((2-oxo-2-ethyloxyethyl)amino)phosphoryl) amino)acetate (3e)
To a solution of phosphate 4 (150 mg, 310 mmol) in CH2Cl2 (5 mL) were added oxalyl chloride (155 mg, 1.23 mmol) and DMF (1 drop), and the mixture was stirred at rt for 2 h. The solvent was removed under reduced pressure, and the residue was dissolved in DMF (10 mL) followed by addition of N,N-diisopropylethylamine (121 mg, 0.92 mmol) and ethyl 2-aminoacetate (119 mg, 0.78 mmol). The reaction mixture was then stirred for 4 h at rt, and the solvent was removed under reduced pressure. The residue was dissolved in water, extracted with ethyl acetate (3 × 50 mL), and dried over Na2SO4. The solvent was removed under reduced pressure, the residue was dissolved in methanol (10 mL), p-toluenesulfonic acid (1 mg, 0.009 mmol) was added, and the mixture was heated at 60 °C for 2 h. The solvent was removed under reduced pressure, and the residue was purified by preparative HPLC to afford the TFA salt of the title compound 3e (15 mg, 7%) as a white solid. 1H NMR (400 MHz, CDCl3): δ 8.54 (s, 1H), 7.39–7.26 (m, 1H), 6.06 (s, 1H), 5.62–4.82 (m, 5H), 4.66–4.45 (m, 3H), 4.40–4.20 (m, 2H), 4.16–4.05 (m, 3H), 3.75–3.60 (m, 4H), 2.16–2.05 (m, 2H), 1.84–1.44 (m, 5H), 1.23 (m, 6H); HRMS calcd for C23H35ClN7O9P + H: 620.1995; found: 620.2008 [M + H]+.
((2R,3S,4R,5R)-5-(2-Chloro-6-(cyclopentylamino)-9H-purin-9-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methyl Diphenethyl Phosphate (3f)
To a solution of phosphate 4 (150 mg, 310 mmol) in CH2Cl2 (5 mL) were added oxalyl chloride (155 mg, 1.23 mmol) and DMF (1 drop), and the reaction mixture was stirred for 2 h at rt. The solvent was then removed under reduced pressure, and the residue was dissolved in DMF (10 mL) followed by addition of N,N-diisopropylethylamine (121 mg, 0.92 mmol) and phenylethyl alcohol (119 mg, 0.78 mmol). The reaction mixture was stirred at rt for 4 h, and the solvent was removed under reduced pressure. The residue was dissolved in water, extracted with ethyl acetate (3 × 50 mL), dried over Na2SO4, filtered, and concentrated in vacuo. To this were added methanol (10 mL) and p-toluenesulfonic acid (1 mg), and the mixture was heated at 60 °C for 2 h. The solvents were removed under reduced pressure, and the residue was purified by preparative HPLC to provide the TFA salt of the title compound 3f (15 mg, 8%) as a white solid. 1H NMR (400 MHz, CDCl3): δ 7.93 (s, 1H), 7.36–7.07 (m, 10H), 6.20 (brs, 1H), 5.89 (d, J = 4.5 Hz, 1H), 4.56 (brs, 1H), 4.49–4.43 (m, 1H), 4.39–4.35 (m, 1H), 4.30–4.25 (m, 1H), 4.17–4.03 (m, 4H), 4.03–3.95 (m, 1H), 2.98–2.76 (m, 5H), 2.22–2.04 (m, 2H), 1.87–1.61 (m, 4H), 1.60–1.45 (m, 2H), 1.39–1.13 (m, 2H); HRMS calcd for C31H37ClN5O7P + H: 658.2192; found: 658.2173 [M + H]+.
((3aR,4R,6R,6aR)-6-(2-Chloro-6-(cyclopentylamino)-9H-purin-9-yl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)methyl Dihydrogen Phosphate (4)
To a solution of phosphate 3c (150 mg, 0.25 mmol) in CH2Cl2 (10 mL) was added TFA (0.3 mL, 2.63 mmol) in CH2Cl2 (10 mL), and the reaction mixture was stirred for 1 h at rt. The solvent was removed under reduced pressure to afford the title compound 4 (93 mg, 76% yield) as a white solid, carried on to the next step without further purification.
((2R,3S,4R,5R)-5-(6-(Cyclopentylamino)-9H-purin-9-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methyl Dimethyl Phosphate (5a)
To a solution of phosphate 2a (600 mg, 1.16 mmol) in methanol (50 mL) were added 10% Pd/C (300 mg, 50% w/w) and acetic acid (50 mL), and the reaction flask was fitted with a hydrogen balloon. The reaction mixture was then stirred for 6 h at rt and filtered through Celite. The solvent was removed under reduced pressure, the residue was dissolved in methanol (10 mL), and p-toluenesulfonic acid (17 mg, 0.16 mmol) was added. The reaction mixture was heated at 60 °C for 2 h, the solvent was removed under reduced pressure, and the residue was purified via preparative HPLC to afford the TFA salt of the title compound 5a (200 mg, 38% yield) as a white solid. 1H NMR (300 MHz, DMSO-d6): δ 8.41 (brs, 1H), 8.30 (s, 1H), 5.86 (d, J = 5.2 Hz, 1H), 4.61 (t, J = 5.4 Hz, 1H), 4.35–4.05 (m, 2H), 3.70–3.62 (m, 6H), 2.05–1.91 (m, 2H), 1.81–1.55 (m, 7 H); HRMS calcd for C17H26N5O7P + H: 444.1643; found: 444.1630 [M + H]+.
Sodium ((2R,3S,4R,5R)-5-(6-(Cyclopentylamino)-9H-purin-9-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methyl Phosphate (5c)
To a solution of 6 (281 mg 0.75 mmol) in CH2Cl2 (15 mL) were added tert-buytl-N,N-diisopropylphosphoramidite (405 mg, 1.5 mmol) and tetrazole (5 mL of 0.45 M solution in ACN, 2.4 mmol). The reaction mixture was heated at 60 °C for 2 h and then cooled to 0 °C, and hydrogen peroxide (0.3 mL, of 30% w/w solution) was added dropwise. The reaction mixture was then stirred for an additional 1 h, diluted with CH2Cl2 (100 mL), and washed with 10% sodium metabisulfite (25 mL × 2), saturated sodium bicarbonate (25 mL × 2), water (25 mL × 2), and brine (25 mL × 2). The solvent was removed under reduced pressure to afford compound 7 (217 mg, 51% yield), which was used in the following steps without further purificaiton. To a solution of phosphate 7 (145 mg, 0.26 mmol) in methanol (10 mL) was added p-toluenesulfonic acid (28 mg, 0.03 mmol), and the reaction mixture was heated at 60 °C for 2 h. The solvent was removed under reduced pressure, TFA (0.3 mL, 2.63 mmol) in CH2Cl2 (10 mL) was added, and the reaction mixture was stirred at rt for an additional 1 h. The solvent was again removed under reduced pressure, and the residue was purified by preparative HPLC to afford the TFA salt of the title compound. To a solution of the TFA salt (50 mg, 0.12 mmol) in methanol (5 mL) was added NaOH (9.6 mg, 0.24 mmol) in water (5 mL), and the reaction mixture was stirred for 30 min at rt. The solvent was removed under reduced pressure to afford the title compound 5c (55 mg, 99% yield) as a white solid. 1H NMR (300 MHz, DMSO-d6): δ 8.52 (s, 1H), 8.30 (s, 1H), 7.47 (d, J = 6.1, 1H), 7.17 (d, J = 6.0, 1H), 5.95 (d, J = 6.0 Hz, 1H), 5.48 (brs, 2H), 4.55 (t, J = 6.0, 2.4 Hz, 1H), 4.40 (m, 1H), 4.29 (m, 1H), 2.19–1.92 (m, 2H), 1.80–1.54 (m, 7H); MS(ESI) m/z 460.4[M - H]–
(2R,3R,4R,5R)-2-(Acetoxymethyl)-5-(2,6-dichloro-9H-purin-9-yl)-tetrahydrofuran-3,4-diyl Diacetate (10)
2,6-Dichloro-9H-purine (2.38 g, 12.6 mmol), (2S,3R,4R,5R)-5-(acetoxymethyl)-tetrahydrofuran-2,3,4-triyl triacetate (4.00 g, 12.6 mmol) and p-toluenesulfonic acid monohydrate (10 mg, 0.06 mmol) were ground to a smooth consistency using a mortar and pestle. The powder was transferred to a microwave vial and heated in the microwave reactor for 5 min at 100 °C at a 50 W power setting. The resulting brown oil was dissolved in methanol (25 mL) and stirred for 2 h. The precipitate was collected and washed with methanol to afford the title compound 10 (2.87 g, 51%) as a tan solid. 1H NMR (300 MHz, DMSO): 8.92 (s, 1 H), 6.32 (d, J = 5.0 Hz, 1 H), 5.90 (t, J = 5.4 Hz, 1H), 5.62 (t, J = 5.5 Hz, 1H), 4.46–4.25 (m, 4H), 2.11(s, 1H), 2.05(s, 1H), 2.02(s, 1H); MS(ESI) m/z 447 [M + H]+.
(2R,3R,4R,5R)-2-(Acetoxymethyl)-5-(2-chloro-6-((1-hydroxy-3-phenylpropan-2-yl)amino)-9H-purin-9-yl)tetrahydrofuran-3,4-diyl Diacetate (11)
To a solution of 2-amino-3-phenylpropanol (67.6 mg, 0.45 mmol) in ethanol (10 mL) were added 2,6-dichloropurine riboside (10) (100 mg, 0.22 mmol) and triethylamine (25 mg, 0.25 mmol). The reaction mixture was stirred at 70 °C for 12 h and partitioned between ethyl acetate (100 mL) and water (50 mL). The aqueous layer was extracted with ethyl acetate (100 mL × 2). The combined organics were washed with brine (10 mL) and dried over Na2SO4. The solvent was removed under reduced pressure, and the residue was purified by flash column chromatography on silica gel (50% EtOAc/hexanes) to afford the title compound 11 (clear oil) as an inseparable mixture of diastereomers (242 mg, 86%). 1H NMR (400 MHz, CDCl3): δ 7.84–7.82 (m, 1H), 7.24–7.13 (m, 5H), 6.10–6.07 (m, 1H), 5.73–5.70 (m, 1H), 5.56–5.52 (m, 1H), 4.70–4.49 (m, 2H), 4.34–4.28 (m, 3H), 3.90–3.60 (m, 2H), 3.03–2.90 (m, 2H), 2.12–1.96 (m, 9H); 13C NMR (126 MHz, CDCl3) δ 170.4, 169.7, 169.6, 169.5, 169.4, 154.8, 138.1, 137.9, 129.4, 128.5, 126.5, 85.6, 85.7, 70.6, 67.1, 63.1, 62.4, 53.8, 37.2, 37.1, 20.8, 20.7, 20.6, 20.5, 20.5, 20.4; MS(ESI) m/z 562 [M + H]+.
(2R,3R,4S,5R)-2-(2-Chloro-6-((1-hydroxy-3-phenylpropan-2-yl)-amino)-9H-purin-9-yl)-5-(hydroxymethyl)tetrahydrofuran-3,4-diol (12a)
To acetate 11 (65 mg, 0.116 mmol) was added methanolic ammonia (30 mL) at 0 °C, the reaction vessel was sealed, and the reaction mixture was stirred for 5 h at rt. The solvent was then removed under reduced pressure, and the residue was purified by preparative HPLC to afford the TFA salt of the title compound 12a (30 mg, 60%) (white solid) as an inseparable mixture of diastereomers. 1H NMR (400 MHz, DMSO): δ 8.42–8.34 (m, 1H), 8.08 (d, J = 8.3 Hz, 1H), 7.35–7.20 (m, 4H), 7.17–7.07 (m, 1H), 5.80 (d, J = 5.8 Hz, 1H), 4.55–4.34 (m, 2H), 4.15–4.06 (m, 1H), 3.98–3.89 (m, 1H), 3.65 (dd, J = 12.0, 4.0 Hz, 1H), 3.60–3.42 (m, 3H), 3.04–2,71 (m, 2H); 13C NMR (126 MHz, DMSO) δ 158.91, 158.54, 155.91, 155.44, 153.49, 149.90, 140.22, 139.95, 139.65, 139.46, 129.49, 128.73, 128.53, 126.36, 118.89, 117.72, 87.86, 87.64, 86.16, 74.06, 70.81, 63.59, 62.82, 61.80, 56.27, 54.36, 40.58, 40.37, 40.16, 39.95, 39.74, 39.53, 39.32, 37.64, 36.71, 31.11.
Preparation of 2-((2-Chloro-9-(3,4-dihydroxy-5-(hydroxymethyl)-tetrahydrofuran-2-yl)-9H-purin-6-yl)amino)-3-phenylpropyl Phosphates (12b,c)
To a solution of 11 (0.35 mmol) in CH2Cl2 (5 mL) were added the appropriate phosphoramidite (0.70 mmol) and tetrazole (0.45 M solution in THF, 2.22 mL, 1.02 mmol), and the mixture was heated for 2 h at 60 °C. The reaction was then cooled to 0 °C, H2O2 (0.2 mL of 30% solution, 0.225 mmol) was added, and the reaction mixture was stirred for an additional 1 h. The reaction mixture was diluted with CH2Cl2 (100 mL) and washed with 10% aq sodium metabisulfite (20 mL), sat. aq sodium bicarbonate (20 mL), water (20 mL), and brine (20 mL). The solvent was removed under reduced pressure, and the residue was purified by flash column chromatography on silica gel (10% methanol/CH2Cl2) to afford the acetate. To the acetate (0.135 mmol) was added methanolic ammonia (20 mL) at 0 °C, the reaction vessel was sealed, and the reaction mixture was stirred for 5 h at rt. The solvent was then removed under reduced pressure, and the residue was purified by preparative HPLC to afford compounds 12b,c.
2-((2-Chloro-9-(3,4-dihydroxy-5-(hydroxymethyl)-tetrahydrofuran-2-yl)-9H-purin-6-yl)amino)-3-phenylpropyl Dimethyl Phosphate (12b)
(38%) (clear oil) 1H NMR (400 MHz, CDCl3): δ 8.28 (d, J = 51.4 Hz, 1H), 7.98 (dd, J = 23.7, 7.3 Hz, 1H), 7.35–7.15 (m, 5H), 5.83 (m, 1H), 4.79 (d, J = 29.4 Hz, 2H), 4.40 (s, 1H), 4.33–4.20 (m, 2H), 4.18–4.05 (m, 1H), 4.01–3.88 (m, 1H), 3.83–3.64 (m, 6H), 3.14–2.94 (m, 3H); 13C NMR (126 MHz, CDCl3) δ 160.9, 160.6, 155.7, 154.0, 148.5, 139.9, 139.6, 136.8, 129.4, 128.8, 127.1, 116.8, 116.5, 116.0, 113.9, 91.6, 87.9, 74.2, 72.1, 71.8, 67.9, 62.9, 62.6, 55.0, 52.5, 36.7; MS(ESI) m/z 544 [M + H]+.
Di-tert-butyl (2-((2-Chloro-9-(3,4-dihydroxy-5-(hydroxymethyl)-tetrahydrofuran-2-yl)-9H-purin-6-yl)amino)-3-phenylpropyl) Phosphate (12c)
(61%) (white solid). 1H NMR (400 MHz, CD3OD): δ 8.18 (s, 1H), 7.27–7.10 (m, 5H), 5.90 (m, 1H), 4.78 (br s, 1H), 4.64–4.56 (m, 1H), 4.26–4.20 (m, 1H), 4.15–4.05 (m, 2H), 4.03–3.90 (m, 1H), 3.85–3.80 (m, 1H), 3.74–3.65 (m, 1H), 3.30–3.22 (m, 2H), 3.02–2.90 (m, 2H) 1.37 (s, 9H), 1.33 (s, 9H); 13C NMR (101 MHz, CD3OD) δ 156.39, 155.20, 150.68, 141.78, 138.93, 130.40, 129.50, 129.39, 127.68, 127.51, 91.03, 90.97, 87.85, 84.64, 84.56, 75.55, 75.47, 72.35, 72.33, 68.23, 68.17, 63.31, 63.30, 53.35, 49.64, 49.43, 49.28, 49.21, 49.07, 49.00, 48.79, 48.57, 48.36, 37.93, 30.13, 30.09, 30.09, 30.05; HRMS calcd for C27H39ClN5O8P + H: 628.2298; found: 628.2289 [M + H]+.
Sodium 2-((2-Chloro-9-((2R,3R,4S,5R)-3,4-dihydroxy-5-(hydroxymethyl)tetrahydrofuran-2-yl)-9H-purin-6-yl)amino)-3-phenylpropyl Phosphate (12d)
To a solution of phosphate 12c (100 mg, 0.159 mmol) in CH2Cl2 (16 mL) was added trifluoroacetic acid (0.2 mL, 1.75 mmol), and the reaction mixture was stirred at rt for 1 h. The solvent was then removed under reduced pressure, and the residue was purified by preparative HPLC to afford the TFA salt of the title compound. The phosphate salt was then dissolved in methanol (5 mL), NaOH (9.6 mg, 0.24 mmol) in water (5 mL) was added, and the reaction mixture was stirred for 30 min at rt. The solvent was then removed under reduced pressure to afford the title compound 12d (55 mg, 99%) as a white solid. 1H NMR (400 MHz, CD3OD): δ 8.16 (s, 1H), 7.22–7.14 (m, 2H), 7.14–7.06 (m, 2H), 7.06–6.98 (m, 1H), 5.80–5.76 (m, 1H), 4.64 (br s, 1H), 4.52 (t, J = 5.5 Hz, 1H), 4.22–4.16 (m, 1H), 4.08–3.91 (m, 3H), 3.80–3.72 (m, 1H), 3.64–3.60 (m, 1H), 3.22–3.15 (m, 2H), 3.00–2.82 (m, 2H); 13C NMR (126 MHz, D2O) δ 155.7, 153.7, 148.6, 140.3, 138.4, 129.9, 128.1, 126.5, 118.8, 88.6, 85.9, 73.7, 70.7, 65.5, 61.7, 53.4, 37.6; HRMS calcd for C19H23ClN5O8P + H: 516.1051; found: 516.1019 [M + H]+.
((2R,3R,4R,5R)-2-(Acetoxymethyl)-5-(2-chloro-6-(((1R,2R)-2-hydroxycyclopentyl)amino)-9H-purin-9-yl)tetrahydrofuran-3,4-diyl Diacetate (13)
To a solution of (1R,2R)-2-aminocyclopentanol (346 mg, 3.42 mmol) in ethanol (3 mL) were added 2,6-dichloropurine riboside (10) (305 mg, 0.68 mmol) and triethylamine (686 mg, 6.80 mmol). The reaction mixture was heated at 70 °C for 12 h and partitioned between ethyl acetate (100 mL) and water (50 mL). The aqueous layer was extracted with ethyl acetate (100 mL × 2). The combined organics were washed with brine (10 mL) and dried over Na2SO4. The solvent was removed under reduced pressure, and the residue was purified by flash column chromatography on silica gel (50% EtOAc/hexanes) to afford the title compound 13 (clear oil) (323 mg, 92%). 1H NMR (400 MHz, CDCl3): δ 8.30 (s, 1H), 6.90 (s, 1H), 5.80–5.77 (m, 1H), 5.57–5.54 (m, 1H), 4.48–4.46 (m, 1H), 4.44–4.38 (m, 2H), 4.15–4.01 (m, 1H), 3.47–3.40 (m, 2H), 3.32–2.89 (m, 2H), 2.19 (s, 3H), 2.15–2.12 (m, 5H), 2.09–2.07 (m, 4H), 2.13–2.08 (m, 1H), 2.05–1.65 (m, 2H), MS(ESI) m/z 5.12 [M + H]+.
(1R,2R)-2-((2-Chloro-9-((2R,3R,4S,5R)-3,4-dihydroxy-5-(hydroxymethyl)tetrahydrofuran-2-yl)-9H-purin-6-yl)amino)-cyclopentyl Dimethyl Phosphate (14a)
To a solution of acetate 13 (60 mg, 0.12 mmol) in CH2Cl2 (5 mL) were added dimethyl diisopropylphosphoramidite (45 mg, 0.23 mmol) and tetrazole (0.45 M solution in THF, 0.83 mL, 0.37 mmol), and the mixture was heated at 60 °C for 2 h. The reaction was then cooled to 0 °C, H2O2 (0.1 mL of 30% solution, 0.12 mmol) was added, and the reaction mixture was stirred for an additional 1 h. The reaction was then diluted with CH2Cl2 (100 mL) and washed with 10% sodium metabisulfite, saturated sodium bicarbonate, water, and brine. The solvent was removed under reduced pressure, and the residue was purified by flash column chromatography on silica gel (10% methanol/CH2Cl2) to afford the phosphate as a clear oil. To the phosphate (60 mg, 0.097 mmol) was then added methanolic ammonia (30 mL) at 0 °C, the reaction vessel was sealed, and the reaction mixture was stirred for 5 h at rt. The solvent was removed under reduced pressure, and the residue was purified by flash column chromatography (10% EtOAc/hexanes) to afford the title compound 14a (white solid) (18 mg, 40%). 1H NMR (400 MHz, CD3OD): δ 8.30 (s, 1H), 5.92 (d, J = 6.0 Hz, 1H), 4.88–4.87 (m, 1H), 4.85–4.75 (m, 1H), 4.74–4.60 (m, 2H), 4.33–4.31 (m, 1H), 4.15 (q, J = 2.8 Hz, 1H), 3.89 (dd, J = 12.5, 2.7 Hz, 1H), 3.80–3.69 (m, 6H), 2.31–2.23 (m, 1H), 2.20–2.03 (m, 1H), 2.00–1.83 (m, 3H), 1.80–1.62 (m, 1H); HRMS calcd for C17H25ClN5O8P + H: 494.1208; found: 494.1219 [M + H]+.
(2R,3R,4R,5R)-2-(Acetoxymethyl)-5-(2-chloro-6-((2-((di-tert-butoxyphosphoryl)oxy)cyclopentyl)amino)-9H-purin-9-yl)-tetrahydrofuran-3,4-diyl Diacetate (14b)
To a solution of 13 (429 mg, 0.838 mmol) in CH2Cl2 (5 mL) were added dibenzyl diisopropylphosphoramidite (579 mg, 1.68 mmol) and tetrazole (0.45 M solution in THF, 6.0 mL, 2.68 mmol) and the reaction mixture was heated for 2 h at 60 °C. The temperature was lowered to 0 °C, H2O2 (0.5 mL of 30% solution, 0.19 mmol) was added, and the reaction mixture was stirred for an additional 1 h. The reaction was then diluted with CH2Cl2 (100 mL) and washed with 10% aq sodium metabisulfite (30 mL), saturated aq sodium bicarbonate (30 mL), water (30 mL), and brine (30 mL). The solvent was removed under reduced pressure, and the residue was purified by flash column chromatography on silica gel (10% methanol/CH2Cl2) to afford the title compound 14b (clear oil) (100 mg, 15%). 1H NMR (400 MHz, CDCl3): δ 8.01 (s, 1H), 7.35–7.11 (m, 10H), 6.09 (d, J = 5.5 Hz, 1H), 5.65 (t, J = 5.5 Hz, 2H), 5.49–5.46 (m, 1H), 5.01–4.89 (m, 4H), 4.80–4.70 (m, 1H), 4.65–4.50 (m, 1H), 4.43–4.36 (m, 1H), 4.35–4.36 (m, 2H), 2.25–2.15 (m, 1H), 2.13–2.04 (m, 6H), 2.01 (s, 3H), 1.99–1.90 (m, 1H), 1.86–1.67 (m, 3H), 1.63–1.44 (m, 1H); MS(ESI) m/z 772 [M + H]+.
Sodium (1R,2R)-2-((2-Chloro-9-((2R,3R,4S,5R)-3,4-dihydroxy-5-(hydroxymethyl)tetrahydrofuran-2-yl)-9H-purin-6-yl)amino)-cyclopentyl Phosphate (14c)
To a solution of phosphate 14b (32 mg, 0.038 mmol) in methanol (10 mL) was added Pd–C (10%, 30 mg), and the reaction mixture was stirred for 12 h at rt in a pressure vessel under 60 psi of hydrogen gas. After 12 h, the mixture was filtered through Celite to remove the catalyst. The solvent was removed under reduced pressure, and 6 N methanolic ammonia (25 mL) was added at 0 °C. The reaction was again sealed in a pressure vessel and stirred for 12 h at rt. The volatiles were removed under reduced pressure, and the residue was purified via preparative HPLC chromatography to give the TFA salt of the title compound. NaOH (13.5 mg, 0.34 mmol) in water (5 mL) was added, and the reaction mixture was stirred for 30 min at rt. The solvent was then removed under reduced pressure to afford the disodium salt of the title compound 14c (80 mg, 99%) as a white solid. 1H NMR (400 MHz, D2O): δ 8.10 (s, 1H), 5.83 (d, J = 5.5 Hz, 1H), 4.72–4.69 (m, 1H), 4.46–4.30 (m, 1H), 4.30–4.24 (m, 1H), 4.24–4.06 (m, 2H), 3.82–3.62 (m, 2H), 2.17–2.07 (m, 1H), 2.05–1.90 (m, 1H), 1.77–1.53 (m, 3H), 1.46–1.29 (m, 1H); HRMS calcd for C15H21ClN5O8P + H: 466.0895; found: 466.0862 [M + H]+.
cAMP Accumulation Assay
cAMP determinations were made using a modified GloSensor Luciferase detection system (Promega). Low passage, subconfluent HEK293T/17 cells (ATCC CRL-11268) were grown in Dulbecco's Modified Eagle's Medium without phenol red (Gibco #31053) and supplemented with 10% fetal bovine serum (Hyclone ‘Characterized’ #SH30071.03). Cells were reverse transfected by spotting a calcium phosphate DNA complex mixture containing 12.5 ng each of GloSensor 22F plasmid (Promega #E2301) and human adenosine A1 receptor plasmid (Adora1, GenBank accession #AY136746, Missouri S&T Clone Collection (www.cdna.org) in 25 mM HEPES at pH 7.1, 140 mM sodium chloride, 0.75 mM disodium monophosphate, and 250 mM calcium chloride. Cells were immediately added at a density of 20 000 cells per well using a Multidrop 384 (Titertek) to 384-well white, clear-bottom tissue culture plates (Corning #3707). Cell plates were incubated for 24 h at 37 °C and 5% CO2. Sixteen-point, 1:3 dilution curves of test compounds in the presence or absence of 10 μM final αβ-met-ADP (Sigma #M3763) were brought up to 8× final concentration in Hanks’ Balanced Salt Solution (Gibco #14175) supplemented with 2 mM HEPES, pH 7.4, and immediately added to the cell plates with a Multimek automated liquid handling device (Nanoscreen, Charleston, SC). Following a 10 min incubation at room temperature, 100 μM final 3-isobutyl-1-methylxanthine (Sigma) and 175 nM (–)-isoproterenol bitartrate (Sigma) or 1.5 μM forskolin (Tocris) were added by Multimek. Seven minutes later, GloSensor cAMP reagent (Promega #E1291) containing 0.1% final luciferin and supplemented with 0.3% final NP40 (Tergitol, Sigma #NP40S) to permeabilize the cells was added by Multimek along with a final 5 μL addition of 100% ethanol (Decon Laboratories) to eliminate bubbles. Luminescence was read on an Envision platereader (Perkin-Elmer) for 15 min. Data from approximately 95% of the maximal response for isoproterenol or forskolin (10–14 min post GloSensor reagent addition) were normalized for scale to 100% response equivalent to the response of 1 μM 3 and 0% response equal to the response from isoproterenol or forskolin alone. Normalized data were fit in GraphPad Prism with four parameter curves for EC50 determinations. Because HEK293T cells endogenously express A2AR,45,46 the cAMP response for adenosine and 5, known agonists of both A1AR (Gi coupled) and A2AR (Gs coupled), was bimodal.24 Concentrations below 1 μM were used to calculate potencies for adenosine, 5, 14, and 14c, against the A1 receptor.
Ca+ Mobilization
Assays of Ca+ mobilization followed a published procedure.24
Radioligand Binding
Radioligand binding assays were performed by Cerep France http://www.cerep.fr/
Behavior Assay
All procedures and behavioral experiments involving vertebrate animals were approved by the Institutional Animal Care and Use Committee at the University of North Carolina at Chapel Hill. C57BL/6 mice were purchased from Jackson Laboratories. A1AR–/–47,48 mice were backcrossed to C57BL/6 mice (Jackson) for 12 generations. Male mice, 2–4 months old, were acclimated to the testing room, equipment, and experimenter for 1–3 days before behavioral testing. Noxious thermal sensitivity was measured by heating one hindpaw with a Plantar Test apparatus (IITC) following the Hargreaves method.49 The radiant heat source intensity was calibrated so that a paw-withdrawal reflex was evoked in ~10 s, on average, in wild-type C57BL/6 mice. Cutoff time was 20 s. One measurement was taken from each paw at the indicated time points to determine paw withdrawal latency. Compounds (10 mM, dissolved in DMSO) were diluted in 0.9% saline then administered to unanesthetized mice via acute intrathecal injection (5 μL, direct lumbar puncture method50), intravenous tail vein injection, or oral gavage. The final DMSO concentration was 5% or less.
Telemetry
Data Sciences International ETA-F20 transmitters were implanted as follows: A 2 cm midline abdominal incision was made in anesthetized mice. The transmitter was placed intraabdominally on top of the intestines, parallel with the long axis of the body and the two leads pointing caudally. A large (14 gauge) needle was used to pass through the abdominal muscles on either side of the incision. The leads were passed through the lumen of the needle, one on each side, and the needle was withdrawn. The leads were placed (positive by the xiphoid and negative on the right pectoral) and anchored in place. The abdomen was closed with absorbable sutures and the skin with nonabsorbable sutures.
Supplementary Material
ACKNOWLEDGMENTS
We thank Dr. Mauricio Rojas at the UNC Mouse Cardiovascular Models Core for implanting telemetry devices, Dr. Lindsey Ingerman James for critical reading the experimental section and review of spectra, and Dr. Bryan Roth and the National Institute of Mental Health, Psychoactive Drug Screen Program, for assay support. We also acknowledge the assistance of Adam Cheely for his contributions in the development of the assay techniques. The research described here was supported by a grant from NINDS (R01NS067688).
ABBREVIATIONS USED
- A1AR
Adenosine A1 receptor
- A2AAR
adenosine A2A receptor
- A2BAR
adenosine A2B receptor
- A3AR
adenosine A3 receptor
- cAMP
cyclic adenosine monophosphate
- 5′-AMP
adenosine 5′-monophosphate
- SAR
structure–activity relationships
- 2′-AMP
adenosine 2′-monophosphate
- 3′-AMP
adenosine 3′-monophosphate
- αβ-met-ADP
α,β-methylene adenosine 5′-diphosphate
- NT5E
ecto-5′-nucleotidase/CD73
- IT
intrathecal
- IV
intravenous
Footnotes
ASSOCIATED CONTENT
Supporting Information
Cyclic AMP responses for 2′-, 3′-, and 5′-AMP in absence or presence of αβ-met-ADP. Effects of compound 3a in the antinociceptive mouse model via IT and IV administrations. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
REFERENCES
- 1.Fredholm BB, Arslan G, Halldner L, Kull B, Schulte G, Wasserman W. Structure and function of adenosine receptors and their genes. Naunyn-Schmiedeberg's Arch. Pharmacol. 2000;362:364–374. doi: 10.1007/s002100000313. [DOI] [PubMed] [Google Scholar]
- 2.Swanson TH, Drazba JA, Rivkees SA. Adenosine A1 receptors are located predominantly on axons in the rat hippocampal formation. J. Comp. Neurol. 1995;363:517–531. doi: 10.1002/cne.903630402. [DOI] [PubMed] [Google Scholar]
- 3.Saura CA, Mallol J, Canela EI, Lluis C, Franco R. Adenosine deaminase and A1 adenosine receptors internalize together following agonist-induced receptor desensitization. J. Biol. Chem. 1998;273:17610–17617. doi: 10.1074/jbc.273.28.17610. [DOI] [PubMed] [Google Scholar]
- 4.Middlekauff HR, Rivkees SA, Raybould HE, Bitticaca M, Goldhaber JI, Weiss JN. Localization and functional effects of adenosine A1 receptors on cardiac vagal afferents in adult rats. Am. J. Physiol. 1998;274:H441–447. doi: 10.1152/ajpheart.1998.274.2.H441. [DOI] [PubMed] [Google Scholar]
- 5.Schulte G, Robertson B, Fredholm BB, DeLander GE, Shortland P, Molander C. Distribution of antinociceptive adenosine A1 receptors in the spinal cord dorsal horn, and relationship to primary afferents and neuronal subpopulations. Neuroscience. 2003;121:907–916. doi: 10.1016/s0306-4522(03)00480-9. [DOI] [PubMed] [Google Scholar]
- 6.Proctor WR, Dunwiddie TV. Pre- and postsynaptic actions of adenosine in the in vitro rat hippocampus. Brain Res. 1987;426:187–190. doi: 10.1016/0006-8993(87)90441-0. [DOI] [PubMed] [Google Scholar]
- 7.Johansson SM, Lindgren E, Yang JN, Herling AW, Fredholm BB. Adenosine A1 receptors regulate lipolysis and lipogenesis in mouse adipose tissue-interactions with insulin. Eur. J. Pharmacol. 2008;597:92–101. doi: 10.1016/j.ejphar.2008.08.022. [DOI] [PubMed] [Google Scholar]
- 8.Boison D. Adenosine as a neuromodulator in neurological diseases. Curr. Opin. Pharmacol. 2008;8:2–7. doi: 10.1016/j.coph.2007.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mantell S, Jones R, Trevethick M. Design and application of locally delivered agonists of the adenosine A(2A) receptor. Expert. Rev. Clin. Pharmacol. 2010;3:55–72. doi: 10.1586/ecp.09.57. [DOI] [PubMed] [Google Scholar]
- 10.Castillo CA, Albasanz JL, Leon D, Jordan J, Pallas M, Camins A, Martin M. Age-related expression of adenosine receptors in brain from the senescence-accelerated mouse. Exp. Gerontol. 2009;44:453–461. doi: 10.1016/j.exger.2009.04.006. [DOI] [PubMed] [Google Scholar]
- 11.Jacobson KA, van Galen PJ, Williams M. Adenosine receptors: pharmacology, structure−activity relationships, and therapeutic potential. J. Med. Chem. 1992;35:407–422. doi: 10.1021/jm00081a001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Poulsen SA, Quinn RJ. Adenosine receptors: new opportunities for future drugs. Bioorg. Med. Chem. 1998;6:619–641. doi: 10.1016/s0968-0896(98)00038-8. [DOI] [PubMed] [Google Scholar]
- 13.Gao ZG, Jacobson KA. Emerging adenosine receptor agonists. Expert Opin. Emerg. Drugs. 2007;12:479–492. doi: 10.1517/14728214.12.3.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hayashida M, Fukuda K, Fukunaga A. Clinical application of adenosine and ATP for pain control. J. Anesth. 2005;19:225–235. doi: 10.1007/s00540-005-0310-8. [DOI] [PubMed] [Google Scholar]
- 15.Zylka MJ. Pain-relieving prospects for adenosine receptors and ectonucleotidases. Trends. Mol. Med. 2011;17:188–196. doi: 10.1016/j.molmed.2010.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sawynok J, Liu XJ. Adenosine in the spinal cord and periphery: release and regulation of pain. Prog. Neurobiol. 2003;69:313–340. doi: 10.1016/s0301-0082(03)00050-9. [DOI] [PubMed] [Google Scholar]
- 17.Eisenach JC, Rauck RL, Curry R. Intrathecal, but not intravenous adenosine reduces allodynia in patients with neuropathic pain. Pain. 2003;105:65–70. doi: 10.1016/s0304-3959(03)00158-1. [DOI] [PubMed] [Google Scholar]
- 18.Eisenach JC, Curry R, Hood DD. Dose response of intrathecal adenosine in experimental pain and allodynia. Anesthesiology. 2002;97:938–942. doi: 10.1097/00000542-200210000-00028. [DOI] [PubMed] [Google Scholar]
- 19.Belfrage M, Segerdahl M, Arner S, Sollevi A. The safety and efficacy of intrathecal adenosine in patients with chronic neuropathic pain. Anesth. Analg. 1999;89:136–142. doi: 10.1097/00000539-199907000-00023. [DOI] [PubMed] [Google Scholar]
- 20.Sowa NA, Voss MK, Zylka MJ. Recombinant ecto-5′-nucleotidase (CD73) has long lasting antinociceptive effects that are dependent on adenosine A1 receptor activation. Mol. Pain. 2010;6:20. doi: 10.1186/1744-8069-6-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sowa NA, Vadakkan KI, Zylka MJ. Recombinant mouse PAP has pH-dependent ectonucleotidase activity and acts through A(1)-adenosine receptors to mediate antinociception. PLoS One. 2009;4:e4248. doi: 10.1371/journal.pone.0004248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zylka MJ, Sowa NA, Taylor-Blake B, Twomey MA, Herrala A, Voikar V, Vihko P. Prostatic acid phosphatase is an ectonucleotidase and suppresses pain by generating adenosine. Neuron. 2008;60:111–122. doi: 10.1016/j.neuron.2008.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goldman N, Chen M, Fujita T, Xu Q, Peng W, Liu W, Jensen TK, Pei Y, Wang F, Han X, Chen JF, Schnermann J, Takano T, Bekar L, Tieu K, Nedergaard M. Adenosine A1 receptors mediate local anti-nociceptive effects of acupuncture. Nat. Neurosci. 2010;13:883–888. doi: 10.1038/nn.2562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rittiner JE, Korboukh I, Hull-Ryde EA, Jin J, Janzen WP, Frye SV, Zylka MJ. AMP Is an Adenosine A1 Receptor Agonist. J. Biol. Chem. 2012;287:5301–5309. doi: 10.1074/jbc.M111.291666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Curros-Criado MM, Herrero JF. The antinociceptive effects of the systemic adenosine A1 receptor agonist CPA in the absence and in the presence of spinal cord sensitization. Pharmacol., Biochem. Behav. 2005;82:721–726. doi: 10.1016/j.pbb.2005.11.014. [DOI] [PubMed] [Google Scholar]
- 26.Klotz KN. Adenosine receptors and their ligands. Naunyn-Schmiedeberg's Arch. Pharmacol. 2000;362:382–391. doi: 10.1007/s002100000315. [DOI] [PubMed] [Google Scholar]
- 27.Franchetti P, Cappellacci L, Vita P, Petrelli R, Lavecchia A, Kachler S, Klotz KN, Marabese I, Luongo L, Maione S, Grifantini M. N6-Cycloalkyl- and N6-bicycloalkyl-C5′(C2′)-modified adenosine derivatives as high-affinity and selective agonists at the human A1 adenosine receptor with antinociceptive effects in mice. J. Med. Chem. 2009;52:2393–2406. doi: 10.1021/jm801456g. [DOI] [PubMed] [Google Scholar]
- 28.Mathew SC, Ghosh N, By Y, Berthault A, Virolleaud MA, Carrega L, Chouraqui G, Commeiras L, Condo J, Attolini M, Gaudel-Siri A, Ruf J, Parrain JL, Rodriguez J, Guieu R. Design, synthesis and biological evaluation of a bivalent micro opiate and adenosine A1 receptor antagonist. Bioorg. Med. Chem. Lett. 2009;19:6736–6739. doi: 10.1016/j.bmcl.2009.09.112. [DOI] [PubMed] [Google Scholar]
- 29.Klotz KN, Hessling J, Hegler J, Owman C, Kull B, Fredholm BB, Lohse MJ. Comparative pharmacology of human adenosine receptor subtypes - characterization of stably transfected receptors in CHO cells. Naunyn-Schmiedeberg's Arch. Pharmacol. 1998;357:1–9. doi: 10.1007/pl00005131. [DOI] [PubMed] [Google Scholar]
- 30.Schenone S, Brullo C, Musumeci F, Bruno O, Botta M. A1 receptors ligands: past, present and future trends. Curr. Top. Med. Chem. 2010;10:878–901. doi: 10.2174/156802610791268729. [DOI] [PubMed] [Google Scholar]
- 31.Fredholm BB, AP IJ, Jacobson KA, Klotz KN, Linden J. International Union of Pharmacology. XXV. Nomenclature and classification of adenosine receptors. Pharmacol. Rev. 2001;53:527–552. [PMC free article] [PubMed] [Google Scholar]
- 32.Elzein E, Zablocki J. A1 adenosine receptor agonists and their potential therapeutic applications. Expert. Opin. Investig. Drugs. 2008;17:1901–1910. doi: 10.1517/13543780802497284. [DOI] [PubMed] [Google Scholar]
- 33.Naito Y, Lowenstein JM. 5′-Nucleotidase from rat heart membranes. Inhibition by adenine nucleotides and related compounds. Biochem. J. 1985;226:645–651. doi: 10.1042/bj2260645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xu F, Wu H, Katritch V, Han GW, Jacobson KA, Gao ZG, Cherezov V, Stevens RC. Structure of an agonist-bound human A2A adenosine receptor. Science. 2011;332:322–327. doi: 10.1126/science.1202793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lebon G, Warne T, Edwards PC, Bennett K, Langmead CJ, Leslie AG, Tate CG. Agonist-bound adenosine A2A receptor structures reveal common features of GPCR activation. Nature. 2011;474:521–525. doi: 10.1038/nature10136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Daly JW, Padgett W, Thompson RD, Kusachi S, Bugni WJ, Olsson RA. Structure-activity relationships for N6-substituted adenosines at a brain A1-adenosine receptor with a comparison to an A2-adenosine receptor regulating coronary blood flow. Biochem. Pharmacol. 1986;35:2467–2481. doi: 10.1016/0006-2952(86)90042-0. [DOI] [PubMed] [Google Scholar]
- 37.Qu X, Cooney G, Donnelly R. Short-term metabolic and haemodynamic effects of GR79236 in normal and fructose-fed rats. Eur. J. Pharmacol. 1997;338:269–276. doi: 10.1016/s0014-2999(97)81930-9. [DOI] [PubMed] [Google Scholar]
- 38.Muller CE. Adenosine receptor ligands-recent developments part I. Agonists. Curr. Med. Chem. 2000;7:1269–1288. doi: 10.2174/0929867003374101. [DOI] [PubMed] [Google Scholar]
- 39.Linden J, Thai T, Figler H, Jin X, Robeva AS. Characterization of human A(2B) adenosine receptors: radioligand binding, western blotting, and coupling to G(q) in human embryonic kidney 293 cells and HMC-1 mast cells. Mol. Pharmacol. 1999;56:705–713. [PubMed] [Google Scholar]
- 40.Bozarov A, Wang YZ, Yu JG, Wunderlich J, Hassanain HH, Alhaj M, Cooke HJ, Grants I, Ren T, Christofi FL. Activation of adenosine low-affinity A3 receptors inhibits the enteric short interplexus neural circuit triggered by histamine. Am. J. Physiol. Gastrointest. Liver Physiol. 2009;297:G1147–1162. doi: 10.1152/ajpgi.00295.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hecker SJ, Erion MD. Prodrugs of phosphates and phosphonates. J. Med. Chem. 2008;51:2328–2345. doi: 10.1021/jm701260b. [DOI] [PubMed] [Google Scholar]
- 42.Cappellacci L, Franchetti P, Vita P, Petrelli R, Lavecchia A, Costa B, Spinetti F, Martini C, Klotz KN, Grifantini M. 5′-Carbamoyl derivatives of 2′-C-methyl-purine nucleosides as selective A1 adenosine receptor agonists: affinity, efficacy, and selectivity for A1 receptor from different species. Bioorg. Med. Chem. 2008;16:336–353. doi: 10.1016/j.bmc.2007.09.035. [DOI] [PubMed] [Google Scholar]
- 43.Vittori S, Lorenzen A, Stannek C, Costanzi S, Volpini R, AP IJ, Kunzel JK, Cristalli G. N-cycloalkyl derivatives of adenosine and 1-deazaadenosine as agonists and partial agonists of the A(1) adenosine receptor. J. Med. Chem. 2000;43:250–260. doi: 10.1021/jm9911231. [DOI] [PubMed] [Google Scholar]
- 44.Gottlieb HE, Kotlyar V, Nudelman A. NMR Chemical Shifts of Common Laboratory Solvents as Trace Impurities. J. Org. Chem. 1997;62:7512–7515. doi: 10.1021/jo971176v. [DOI] [PubMed] [Google Scholar]
- 45.Inbe H, Watanabe S, Miyawaki M, Tanabe E, Encinas JA. Identification and characterization of a cell-surface receptor, P2Y15, for AMP and adenosine. J. Biol. Chem. 2004;279:19790–19799. doi: 10.1074/jbc.M400360200. [DOI] [PubMed] [Google Scholar]
- 46.Cooper J, Hill SJ, Alexander SP. An endogenous A2B adenosine receptor coupled to cyclic AMP generation in human embryonic kidney (HEK 293) cells. Br. J. Pharmacol. 1997;122:546–550. doi: 10.1038/sj.bjp.0701401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hua X, Erikson CJ, Chason KD, Rosebrock CN, Deshpande DA, Penn RB, Tilley SL. Involvement of A1 adenosine receptors and neural pathways in adenosine-induced bronchoconstriction in mice. Am. J. Physiol. Lung Cell Mol. Physiol. 2007;293:L25–32. doi: 10.1152/ajplung.00058.2007. [DOI] [PubMed] [Google Scholar]
- 48.Johansson B, Halldner L, Dunwiddie TV, Masino SA, Poelchen W, Gimenez-Llort L, Escorihuela RM, Fernandez-Teruel A, Wiesenfeld-Hallin Z, Xu XJ, Hardemark A, Betsholtz C, Herlenius E, Fredholm BB. Hyperalgesia, anxiety, and decreased hypoxic neuroprotection in mice lacking the adenosine A1 receptor. Proc. Natl. Acad. Sci. U.S.A. 2001;98:9407–9412. doi: 10.1073/pnas.161292398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hargreaves K, Dubner R, Brown F, Flores C, Joris J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain. 1988;32:77–88. doi: 10.1016/0304-3959(88)90026-7. [DOI] [PubMed] [Google Scholar]
- 50.Fairbanks CA. Spinal delivery of analgesics in experimental models of pain and analgesia. Adv. Drug Delivery Rev. 2003;55:1007– 1041. doi: 10.1016/s0169-409x(03)00101-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.