

Abstract
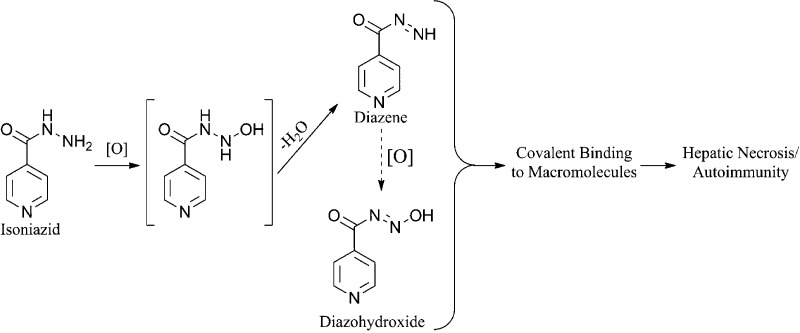
Isoniazid (INH) is associated with serious liver injury and autoimmunity. Classic studies in rats indicated that a reactive metabolite of acetylhydrazine is responsible for the covalent binding and toxicity of INH. Studies in rabbits suggested that hydrazine might be the toxic species. However, these models involved acute toxicity with high doses of INH, and INH-induced liver injury in humans has very different features than such animal models. In this study, we demonstrated that a reactive metabolite of INH itself can covalently bind in the liver of mice and also to human liver microsomes. Covalent binding also occurred in rats, but it was much less than that in mice. We were able to trap the reactive metabolite of INH with N-α-acetyl-l-lysine in incubations with human liver microsomes. This suggests that the reactive intermediate of INH that leads to covalent binding is a diazohydroxide rather than a radical or carbocation because those reactive metabolites would be too reactive to trap in this way. Treatment of mice or rats with INH for up to 5 weeks did not produce severe liver injury. The alanine transaminase assay (ALT) is inhibited by INH, and other assays such as glutamate and sorbitol dehydrogenase (SDH) were better biomarkers of INH-induced liver injury. High doses of INH (200 and 400 mg/kg/day) for one week produced steatosis in rats and an increase in SDH, which suggests that it can cause mitochondrial injury. However, steatosis was not observed when INH was given at lower doses for longer periods of time to either mice or rats. We propose that covalent binding of the parent drug can contribute to INH-induced hepatotoxicity and autoimmunity. We also propose that these are immune-mediated reactions, and there are clinical data to support these hypotheses.
Introduction
Although it can cause serious liver injury including liver failure, because of its efficacy, INH remains a mainstay for the treatment of tuberculosis.1 Classic studies done more than 3 decades ago indicated that the hepatotoxicity of INH was due to the bioactivation of acetylhydrazine (AcHz), a metabolite of INH.2 Specifically, a single large dose of INH (300 mg/kg) produced no hepatic necrosis, while six oral doses of 100 mg/kg each produced little necrosis and then only when rats were pretreated with phenobarbital.2,3 In contrast, acetylisoniazid (AcINH) treatment produced more necrosis, especially when rats were pretreated with phenobarbital, and the extent of necrosis was even greater when AcHz was administered instead of AcINH.2 There was covalent binding of the acetyl group in the liver when rats were treated with 14C-acetyl-labeled AcINH, but no binding was observed when the aromatic ring of AcINH was labeled.2,4 Lastly, it was shown that pretreatment of rats with an acyl amidase inhibitor (bis-p-nitrophenyl phosphate) decreased the extent of hepatic necrosis caused by AcINH but not that caused by AcHz, which suggested that the hepatotoxic species came from INH being acetylated and then hydrolyzed to AcHz.2,5 Together, these studies provided evidence that the hepatotoxic effects of INH are mainly due to the bioactivation of AcHz and not the parent drug.4,5 However, the hypothesis that AcHz is responsible for INH-induced hepatotoxicity in humans was driven by these studies in rats in which the characteristics of the toxicity are very different from the toxicity in humans. Later, in an acute rabbit model, hydrazine (Hz) was proposed to be the metabolite that leads to hepatotoxicity.6,7 The chemistry of the oxidation of INH is very similar to that of AcHz. We had previously shown that INH is oxidized by myeloperoxidase or activated neutrophils, and this oxidation appeared to involve a reactive intermediate.8 Therefore, the greater oxidation of AcHz than INH in the rat presumably reflects the enzyme specificity in this species, which could be different in humans. More recently, we found preliminary evidence that a reactive metabolite of the parent drug binds to the liver of mice.9 In this study, we used an antibody to study the covalent binding of INH in rats, mice, and human liver microsomes (HLM) in detail.
Materials and Methods
Trapping of the Reactive Metabolite
INH (Sigma; Oakville, ON) and N-α-acetyl-l-lysine (NAL) or glutathione (both from Fisher Scientific; Ottawa, ON) were dissolved in water and incubated with 1 mg/mL of human liver microsomes (HLM, pooled from 50 donors, BD Biosciences; Mississauga, ON) in phosphate buffer (50 mM, pH 7.4) at concentrations of 500 μM for INH and 1 mM for NAL and glutathione. The reactions were initiated by the addition of a NADPH-regenerating system (solutions A and B; BD Biosciences) and incubated at 37 °C for 30 min. To quench the reaction, 3 volumes of ice-cold methanol were added, and the mixture was allowed to sit at −20 °C for 30 min. After centrifugation (11,000g for 10 min), supernatants were dried under a stream of N2. The samples were reconstituted with the initial mobile phase, and 10 μL samples were analyzed using a 150 × 3 mm Luna 3 μm C18(2) 100A column (Phenomenex; Torrance, CA) with a methanol/10 mM aqueous ammonium acetate (pH 4.0) gradient at a flow of 0.2 mL/min. Initially, methanol was 10% for 2 min with a linear gradient to 95% methanol over 8 min.
INA Activated Ester, INH Dimer, and Isonicotinic Acid-N-α-acetyl-l-lysine (INA-NAL) Adduct
The INH dimer (INA-INH) was synthesized as previously described;10 similarly, the activated ester of isonicotinic acid (INA) was synthesized by the reaction of isonicotinoyl chloride hydrochloride (Fisher Scientific) with N-hydroxysuccinimide (NHS, Sigma) to form INA-NHS as previously shown.11
For the synthesis of INA-NAL, to a stirred solution of NAL (0.2 g; 0.001 mol) in anhydrous methanol (50 mL) was added INA-NHS (0.22 g; 0.001 mol). The mixture was refluxed for 2 h and concentrated in vacuo. The residue was taken up in methanol and purified by column chromatography (silica gel, 230 × 33 mm, mesh 230–400, Sigma) using a gradient starting with 4% methanol in CHCl3 and increasing up to 10% methanol. The structure was confirmed by 1H NMR, 13C NMR, and mass spectrometry. Yield: 0.21 g (30%). 1H NMR (400 MHz; d-MeOH) δ: 8.7 (d; 2H); 7.8 (d; 2H); 4.4 (m; 1H); 3.4 (m; 2H); 1.95 (s; 3H); 1.9 (m; 2H); 1.6 (m; 2H); 1.5 (m; 2H). 13C NMR (400 MHz; d-MeOH) δ: 175, 172, 166, 149, 143, 122, 53, 39, 32, 29, 23, 21. MS (ESI): m/z: 294 [M + H]+.
Conjugation of INH to BSA/Blue Carrier Protein and Antibody Production
INH was coupled to bovine serum albumin (BSA, Sigma) by adding a 10-fold molar excess (based on 58 lysine residues/BSA) of the activated ester (INA-NHS, 0.2 g) to a stirred solution of BSA (0.1 g) in buffer (see Table S1, Supporting Information). INH was coupled to Blue Carrier Protein (Fisher Scientific) in a similar manner with a 10-fold molar excess (based on a maximum of 699 lysine residues/Protein Blue protein) of INA-NHS (3.3 mg) to 20 mg of protein. The mixtures of INA-NHS with protein were stirred for 1–2 h at room temperature, and low mass products removed with a 10,000 MW cut off filter. INA coupling to BSA was confirmed by mass spectrometry, while a trinitrobenzene sulfonic acid assay (Sigma) was used to measure free amino groups and calculate coupling to Blue Carrier Protein because of its high mass. All protein concentrations were measured using the bicinchoninic acid (BCA) kit (Fisher Scientific). Production of polyclonal antibodies in rabbits against INA was carried out by the Division of Comparative Medicine at the University of Toronto using INA coupled to Blue Carrier Protein coupled as the antigen. The antibody production schedule involved a primary immunization of rabbits with 500 μg of antigen dissolved in phosphate-buffered saline (pH 7.4) followed by two subsequent immunizations of 250 and 100 μg of antigen. For primary immunizations, Freund’s complete adjuvant was used to induce an immune response, and subsequent immunizations involved Freund’s incomplete adjuvant.
Animal Treatment
Mice (BalbcAnNCrl or C57BL/6NCrl) and rats, Brown Norway/Crl (BN) or Wistar Crl:WI, 6–8 weeks of age, were purchased from Charles River Laboratories (Montreal, QC) and were allowed to acclimatize for one week before treatment. INH was thoroughly mixed with food and given to rodents at a dose of 0.2% of INH by weight in food or dissolved in saline and given by gavage as specified. Food was given in small jars ad libitum, and the amount consumed was measured; this resulted in an INH dose of about 300–450 mg/kg/day for mice and up to 100 mg/kg/day for rats. Blood was collected from the saphenous vein in mice using heparin-coated tubes and from the tail vein in rats. As biomarkers of liver injury, the activity of alanine aminotransferase (ALT, Thermo Scientific, Middletown, VA) and sorbitol dehydrogenase (SDH, Catachem; Oxford, CT) were measured as described by the manufacturer. Activity of glutamate dehydrogenase (GLDH, Randox; Crumlin, UK) was slightly modified where reagent 1 was premixed with reagent 2, and to this mixture was added to serum and the absorbance monitored for at least 5 min. All animal experiments were approved by the University of Toronto Animal Care Committee.
INH, AcHz, and Hz Blood Level Measurements
The procedure for the quantitation of serum INH and its metabolites was adapted from Sarich et al.7 To 25 μL of serum was added 375 μL of ice cold methanol containing 4-dimethylaminoantipyrene (Sigma) as the internal standard, the mixture was placed at −20 °C for 30 min, and then it was centrifuged at 11,000g for 5 min. To 200 μL of supernatant was added 200 μL of 1 M formic acid in water. To this mixture was added 100 μL of derivatizing agent (3-methoxybenzaldehyde, Sigma) made up 1:10 in 50% 2-propanol in methanol and incubated at room temperature in the dark with rocking for 2 h. After 2 h, the samples were diluted, and the metabolite levels were analyzed using an LC-MS system with a 30 × 2 mm Gemini 5 μm C18 100A column (Phenomenex) and a mobile phase consisting of methanol/10 mM aqueous ammonium acetate (pH 4.0) gradient at a flow of 0.2 mL/min. Initial % of methanol was 0 for 2 min with a linear gradient to 95% methanol over 5 min. Optimizations for multireaction monitoring were performed using the synthetic standards (Sigma) for N′-(3-methoxybenzylidene)isonicotinohydrazide (Q1/Q3: 255.95/121.2), 3-methoxybenzaldehyde[(3-methoxyphenyl)methylene]hydrazone (Q1/Q3: 268.81/136.0), and N′-(3-methoxybenzylidene)acetohydrazide (Q1/Q3: 192.82/151.2).
Western Blotting
Mouse liver microsomes (MLM) and rat liver microsomes (RLM) were prepared from male C57BL/6 mice or male BN rats. Briefly, the liver was homogenized in phosphate-buffered saline (pH 7.4) and centrifuged at 9,000g for 10 min at 4 °C, and the supernatant from this centrifugation (S9) was recovered and centrifuged again at 100,000g for 50 min at 4 °C. The pellet from this last centrifugation contained the microsomes and was resuspended in 20% glycerol and 0.4% KCl in phosphate-buffered saline (pH 7.4). In vitro incubations of microsomes with INH utilized a microsome concentration of 0.5 mg/mL, and 10 μg of protein/lane was loaded on the gel for Western blotting. For in vivo studies, the S9 fraction was prepared in the presence of protease inhibitors (Sigma), and 20 μg of protein/lane was loaded on the gel. The protein was separated by electrophoresis (8% SDS–PAGE) and transferred onto a nitrocellulose membrane (Bio-Rad, Mississauga, ON). Each Western blot was repeated at least twice, and each time, the concentration of protein loaded was measured using the BCA kit. Rabbit anti-INH antibody was used as the primary antibody, and goat antirabbit IgG-peroxidase (Sigma) was used as the secondary antibody. Bound peroxidase was detected using Supersignal West Pico Chemiluminescent Substrate (Fisher Scientific). Mouse monoclonal anti-GAPDH (Sigma) was used as the loading control and detected by goat antimouse IgG-peroxidase (Jackson ImmunoResearch; West Grove, PA). Super signal enhanced molecular weight markers were used (Fisher Scientific).
Histopathology and Immunohistochemistry
Formalin-fixed, paraffin-embedded liver sections were stained with hematoxylin and eosin (H&E) by the department of pathology at the University for Sick Children (Toronto, ON). For immunohistochemical analysis, rabbit anti-INH antibodies were used as the primary antibody, and goat antirabbit IgG-peroxidase (Sigma) was used as the secondary antibody. Each experiment was repeated at least twice, and the signal was developed using NovaRed (Vector; Burlington, ON) with Mayers hematoxylin (Sigma) as the counter stain.
Statistical Analysis
Statistical analyses were performed using GraphPad prism (GraphPad Software, San Diego, CA). Data was analyzed using two-way ANOVA or the Mann–Whitney U test.
Results
The reactive metabolite of INH was trapped with NAL in an incubation of HLM with a NADPH-generating system. Two products were observed (m/z 292 and 243), which corresponded to INA-NAL with a retention time of 9 min and an INH dimer (INA-INH) with a retention time of 9.9 min (Table 1 and Figure 1). The NAL adduct and INH dimer were identical on LC-MS/MS to the synthetic products. The fragmentation pattern of the protonated molecular ion of INA-INH (m/z 243) in the positive ion mode was m/z 243 (0%), 137.0 (11%), 124.4 (31%), 121.3 (57%), 107 (13%), 105.1 (23%), 93.1 (47%), 79.1 (100%), 66.2 (27%), and that for the dimer, INA-NAL, in the negative ion mode was m/z 292 (0%), 121.1 (11%), 77.9 (100%), 57.9 (7%). No glutathione adduct was detected when the NAL was replaced by glutathione. Given the structure of the product, it is likely that this reactive metabolite is a diazohydroxide that reacts with hard nucleophiles such as primary amines to form an amide.
Table 1. INH Adduct Formation in Human Liver Microsomesa.
| HLM | NADPH | INH | NAL | adducts observed | tr (min) |
|---|---|---|---|---|---|
| + | – | – | – | no peak | – |
| + | + | – | – | no peak | – |
| + | + | + | – | INA-INH | 9.9 |
| + | + | + | + | INA-NAL | 9.0 |
INA-INH = the INH dimer and INA-NAL = the N-α-acetyl-l-lysine adduct.
Figure 1.

INH adduct formation in human liver microsomes. INH = isoniazid, NAL = N-α-acetyl-l-lysine, HLM = human liver microsomes, INA-INH = INH dimer, and INA-NAL = N-α-acetyl-l-lysine adduct.
An antibody was produced by immunization of rabbits with a protein modified by reaction with an activated ester of INA that should mimic the covalent binding of this diazohydroxide reactive metabolite of INH. This antibody was tested for specificity against INH and cross-reactivity with binding of AcHz by both ELISA and Western blotting (Figure 2). The antibody detected INH binding to many hepatic proteins, and the binding was specific, i.e., no binding was observed to hepatic proteins from untreated controls, and binding was blocked by preincubation of the serum with INH. We found that after chronic treatment of mice with INH, a large amount of INH was bound to the livers of male C57BL/6 mice (Figure 3A). This was also true for female C57BL/6 and Balb C mice (Figure S1, Supporting Information). No major differences were observed between the degree of covalent binding in male vs female C57BL/6 mice (Figure 3B). There may be slightly more INH binding in female C57BL/6 than female Balb C mice, but the duration of treatment was also longer (Figure 3C), and there was a similar difference in binding between male and female Balb C mice (Figure S1, Supporting Information). The amount of covalent binding increased with each dose of INH with day 7 having much greater covalent binding than days 1 and 3, and the amount of binding was greatest when the drug was given in food for three weeks (Figure 3D). Presumably, this reflects the half-lives of the proteins modified. Immunohistochemical analysis showed that the binding was mostly centrilobular (Figure 3E,F). This was true for both mice and rats (data not shown). Covalent binding also occurred in rats, both BN and Wistar rats (Figure 4A,B), with Wistar rats having a slightly greater amount of covalent binding than BN rats even though Wistar rats were treated for four weeks instead of five (Figure 4C). The densest bands were observed at about 37 and 50 kDa. In BN rats, there was not much difference in the amount of covalent binding when the drug was given by gavage rather than in food (Figure 4A). In comparison to mice, the covalent binding of INH to hepatic proteins in rats was less (Figure 4D). Because of this, the sensitivity had to be increased, which led to noticeable artifact bands in proteins from untreated control rats; however, the difference between treated and control animals was clear (Figure 4A,B)
Figure 2.

Specificity of the anti-INH antibody. (A) Antibody specificity was tested by ELISA. The plate was either coated with BSA modified with INA (BSA-INA) or BSA alone. Preimmune serum (SPre) or serum after immunization with Blue Carrier Protein modified with INA (SAft) diluted at 1:100,000 was used as the primary antibody. In the third and fifth column, the primary antibody was preincubated with INH or NAL, respectively, at a concentration of 200 μM for 30 min at room temperature. (B) Antibody was tested for cross-reactivity with binding due to AcHz. Female C57BL/6 mice were treated with either INH or AcHz (Fisher Scientific) by gavage for 7 days at 50 mg/kg/day (n = 2 for each group). There was no binding to hepatic proteins from untreated control and AcHz-treated mice, whereas hepatic proteins from INH-treated mice showed a large number of bands modified with INH. (C) Antiserum was tested for specificity on Western blots by preincubation with INH at 200 μM or 2 mM for 1 h at 4 °C, which prevented binding to the INH-modified hepatic proteins. (D) Binding of the anti-INH serum to INH-modified liver proteins on Western blots was compared to that of the preimmune serum from the same animal.
Figure 3.

Covalent binding of INH to hepatic proteins in mice. (A) Male C57BL/6 (n = 4) untreated controls or treated with INH (0.2% of INH by weight in food) for 5 weeks. (B) Male vs female C57BL/6 (n = 4) treated with INH (0.2% of INH by weight in food) for 5 weeks. (C) Female C57BL/6 mice (n = 3) treated for 5 weeks vs Balb C mice (n = 3) treated for 3 weeks, both with 0.2% INH by weight in food. (D) Female Balb C mice treated with INH; either by gavage at 100 mg/kg/day (n = 2) for a period of 1, 3, or 7 days or with 0.2% INH by weight in food for 3 weeks. (E,F) Immunohistochemical staining in the livers of female C57BL/6 mice in untreated control vs those treated with 0.2% INH by weight in food for 5 weeks. Magnification: 5×.
Figure 4.

Covalent binding of INH to hepatic proteins in rats. (A) BN rats (n = 3) in untreated controls or treated with INH either by gavage at a dose of 150 mg/kg/day or with 0.2% INH by weight in food for 5 weeks. (B) Wistar rats (n = 4) in untreated controls or treated with INH by gavage at 150 mg/kg/day for 4 weeks. (C) Comparison of covalent binding between male Wistar rats and male BN rats (n = 4) treated with INH by gavage at a dose of 150 mg/kg/day for 4 and 5 weeks, respectively. (D) Comparison of covalent binding between male C57BL/6 mice (n = 4) treated with 0.2% INH by weight in food for 5 weeks vs male Wistar rats (n = 4) treated with INH by gavage at a dose of 150 mg/kg/day for 4 weeks.
In Vitro Binding of INH to Human, Mouse, and Rat Liver Microsomes
To relate our in vivo animal studies to humans, we investigated the oxidation of INH by HLM to a reactive metabolite. INH binding to HLM was greater at 100 μM INH than 10 μM (Figure 5A). There was a small amount of INH binding to HLM in the absence of a NADPH-generating system (Figure 5A). This could be due to a small amount of endogenous peroxidase activity or to nucleophilic attack on proteins by the hydrazine group of INH. Binding of INH to MLM was also apparent and is consistent with the in vivo studies (Figure 5B). The higher concentrations are above therapeutic serum concentrations, but such concentrations may occur in the liver clinically during first pass through the liver. Covalent binding of INH to HLM was less than that to MLM, and it was time and concentration dependent (Figure 5C–F). The difference between HLM and MLM was more apparent with the higher concentrations of INH. A comparison among rat, mouse, and human liver microsomes revealed that the amount of covalent binding is greatest in mouse followed by rat and human liver microsomes (Figure 6).
Figure 5.

In vitro covalent binding of INH to hepatic microsomes (A) to HLM with and without an NADPH-generating system; (B) to MLM with and without a NADPH-generating system; (C) to HLM as a function of time and INH concentration; (D) to MLM as a function of time and INH concentration; (E,F) direct comparison of binding between HLM and MLM at INH concentrations of 10 and 100 μM, respectively.
Figure 6.

Comparison of INH covalent binding to rat, mouse, and human liver microsomes at an INH concentration of 50 μM in the presence or absence of a NADPH generating system.
Treatment of Wistar and BN Rats with INH
In other studies, we have found BN rats to be more sensitive to immune-mediated reactions than other strains;12 therefore, we treated male BN rats with INH using a variety of protocols to try to develop an animal model. A dose of 200 mg/kg/day caused rats to appear ill after 7 days, but again histology only showed signs of steatosis (data not shown). Lower doses such as 150 mg/kg/day and 50 mg/kg twice daily or INH given in food for up to 5 weeks also did not lead to an increase in SDH with the exception of when INH was given by gavage, and then after the first week, the SDH returned to normal despite continued treatment (Figure 7A). An experiment in which INH was administered by gavage twice daily had to be discontinued at the end of the first week because the animals lost significant weight mainly due to reduced food intake (Table 2). The group that was gavaged at an INH dose of 150 mg/kg/day also lost weight after one week; however, at the end of the first week, powdered food was mixed with water to make the food more palatable, and this resulted in the rats eating food again, and they could be treated for up to 5 weeks (Figure 7B). At the end point, ALT and GLDH were also measured in addition to SDH in all groups, but no change in SDH or GLDH was observed, but rather a decrease in ALT was seen when the drug was given in food and by gavage (Table S2, Supporting Information). The decrease in ALT levels in treated animals is consistent with the fact that the ALT assay is inhibited by drugs such as INH that can react with pyridoxal-5′-phosphate.13 This is because INH forms a Schiff base with pyridoxal-5′-phosphate, a cofactor for the ALT assay. We recently demonstrated the inhibition of the ALT assay where BN rats were dosed with INH (400 mg/kg/day) for up to 7 days, which produced systemic toxicity. As predicted, ALT levels were lower in treated rats, but the SDH levels were elevated.14 The H&E slides from BN rats dosed with 400 mg/kg/day showed signs of steatosis but no other evidence of liver injury (Figure S2, Supporting Information). Treatment of Wistar rats for up to 4 weeks with 150 mg/kg/day of INH by gavage produced no obvious signs of liver injury and no steatosis as seen by H&E staining.
Figure 7.

Serum SDH activities and body weights in INH-treated BN rats. (A) SDH activity in BN rats treated with INH by gavage at 150 mg/kg/day, 50 mg/kg twice daily, or given at 0.2% INH by weight in food. (B) Body weight of BN rats given INH as in panel A. Values represent the mean ± SE. Analyzed for statistical significance by two-way ANOVA. Significantly different from the control group (*p < 0.05; **p < 0.01; ***p < 0.001).
Table 2. Food Consumption in Mice/Rats and Daily INH Dosea.
| food consumption (g/day) | avg animal weight range (g) | avg INH intake in mg/kg/day | ||
|---|---|---|---|---|
| BN rats | control | 15.6 ± 1.1 | 240–300 | |
| gavaged at 150 mg/kg/day | 4.7 ± 1.0*** | 220–240 | ||
| gavaged at 50 mg/kg twice daily | 3.5 ± 2.0*** | 180–240 | ||
| INH given in food | 9.1 ± 0.6*** | 210–240 | 79–87 | |
| mice | male C57BL/6 | |||
| control | 5.0 ± 0.3 | 22–26 | ||
| INH | 4.6 ± 0.3 | 22–24 | 386–420 | |
| female C57BL/6 | ||||
| control | 5.1 ± 0.3 | 16–20 | ||
| INH | 4.2 ± 0.3** | 16–20 | 420–525 | |
| male Balb C | ||||
| control | 4.6 ± 0.3 | 22–23 | ||
| INH | 2.9 ± 0.2*** | 18–20 | 290–322 | |
| female Balb C | ||||
| control | 4.6 ± 0.1 | 17–19 | ||
| INH | 3.1 ± 0.3*** | 15–17 | 367–416 | |
INH was given to BN rats (n = 4) by gavage as specified or at 0.2% INH by weight in food for 5 weeks maximum. C57BL/6 (n = 4) mice were treated with INH at 0.2% by weight in food for 5 weeks, and Balb C (n = 4) were treated at the same dose for 3 weeks. Animal body weight range was estimated from Figures 5 and 6, and the amount of food consumed was calculated on the basis of weight range and by assuming homogenous mixing of drug with food. Values represent the mean ± standard error of the mean (SE) with 4 animals per group. The data were analyzed for statistical significance by the Mann-Whitney U test. Significantly different from control group (*p < 0.05; **p < 0.01; ***p < 0.001).
In none of the groups were we able to find evidence of liver injury that represented an animal model with hepatotoxicity similar to that which occurs in humans. The biochemical findings were consistent with the H&E staining that showed no necrosis or inflammatory cell infiltrate, although some steatosis was observed when BN rats were gavaged with INH at a dose of 200 or 400 mg/kg/day for 7 days (Figure S2, Supporting Information), but it resolved despite continued treatment when INH was given for up to 5 weeks either by gavage or in food.
Treatment of C57BL/6 and Balb C Mice with INH
Because INH failed to cause significant liver injury in rats and we found greater covalent binding of INH in mice, we also investigated INH-induced liver injury in mice. Treatment of C57BL/6 mice with INH for up to 5 weeks did not result in an increase in GLDH (Figure 8A,B). In contrast, GLDH levels were elevated in Balb C mice, with male Balb C mice having a greater increase at week 1 (Figure 8E) and female Balb C mice at week 3 (Figure 8F). At the 3 week end point, SDH was only elevated in female Balb C mice, while ALT was generally decreased in all the treated groups (Table S3, Supporting Information). Liver histology was normal in Balb C (data not shown) and C57BL/6 mice (Figure S2, Supporting Information). Food intake was decreased in Balb C mice (Table 2) resulting in a significant decrease in body weight (Figure 8G,H). In general, C57BL/6 mice ate more food that was mixed with the drug than Balb C mice, and this resulted in a higher body weight and higher INH dose; C57BL/6 mice could be treated for longer (Table 2). We gave the drug in food because INH has a short half-life, and this provided a more consistent blood level than once-a-day oral gavage. Given the amount of food that mice and rats consume, and based on their average body weight, the mice received a greater dose of INH (about 400 mg/kg/day) than rats (about 90 mg/kg/day, Table 2). This method of drug administration produced blood levels in mice that were comparable to the Cmax in humans.15 To compare INH and its metabolite blood levels in mice vs rats, we treated male C57BL/6 and male BN rats with INH in food for one week. Mice had INH blood levels of 4.5 ± 0.9 μg/mL, which is comparable to the Cmax in humans,15 but rats had about 3-fold lower INH concentrations (1.5 ± 0.1 μg/mL; Figure 9A). In contrast, rats had more than double the concentration of AcHz compared to that of mice (rat/mouse AcHz ratio of 2.5), while the concentration of Hz was only slightly higher in rats compared to that in mice (Rat/Mouse Hz ratio of 1.5). This resulted in a higher INH to AcHz ratio in mice (Figure 9B) indicating that the relative exposure of mice and rats to INH and AcHz is quite different.
Figure 8.

GLDH activities and body weights in mice. (A–D) Male and female C57BL/6 mice were treated at 0.2% INH by weight in food for 5 weeks. (E–H) Male and female Balb C mice were treated at 0.2% INH by weight in food for 3 weeks. Values represent the mean ± SE from 4 animals per group. The data were analyzed for statistical significance by two-way ANOVA. Significantly different from the control group (*p < 0.05; **p < 0.01; ***p < 0.001).
Figure 9.

Serum concentrations of INH and INH/AcHz ratio in mice and rats. INH was given at a dose of 0.2% by weight in food for up to one week. Values represent the mean ± SE for mice (n = 5) and for rats (n = 4). The data were analyzed for statistical significance by the Mann–Whitney U test. Significantly different groups (*p < 0.05).
Discussion
On the basis of acute toxicity studies in rats from several decades ago, it is generally accepted that the hepatotoxic effects of INH are due to the bioactivation of AcHz.2−5 However, the present study demonstrates that INH itself can be oxidized to a reactive metabolite that binds to hepatic proteins. The ability to trap this reactive metabolite with NAL and the structure of the adduct suggest that the metabolite responsible for the binding is a diazohydroxide (see the graphic in the abstract section). The alternative carbocation formed by the loss of nitrogen would have an extraordinarily short half-life16 making it difficult to trap with a nucleophile and unlikely to bind in vivo to any proteins other than the enzyme that formed it; however, it is conceivable that the diazene is susceptible to nucleophilic attack. This is also not the chemistry that would be expected of a free radical intermediate. The chemistry of INH and AcHz oxidation is similar; therefore, their relative contribution to covalent binding would be determined by the relative rates of acetylation, hydrolysis, the affinities of P450 isozymes for the two hydrazides, and the relative contributions of other clearance pathways such as pyruvate conjugation. These parameters are likely to be species dependent, and although we do not have quantitative data for all of these parameters, the relative concentration of AcHz was greater in rats than mice, while the covalent binding and blood levels of INH were greater in mice (Figure 4D and Figure 9A). The contribution of INH bioactivation relative to that of AcHz to covalent binding is presumably greater in mice than in rats; therefore, human slow acetylators,15 who are at increased risk of hepatotoxicity, are more comparable to mice. This makes conclusions based on studies in rats suspect.
With this information about the bioactivation of INH, we tried to develop an animal model of INH-induced hepatotoxicity with characteristics similar to those of INH-induced liver injury in humans. The typical features of INH-induced hepatotoxicity in humans include a delay in onset, and in more severe cases of liver injury, the histopathology is associated with centrilobular necrosis with a mild lymphocytic infiltrate, often with eosinophils.17,18 We found that ALT was not a reliable assay to measure INH liver injury because INH interferes with the assay as previously reported.13 Even though this has been reported, it is important to note because this problem is not generally known, and ALT is commonly used in studies of INH-induced liver injury, both in animals and humans. This may be less of an issue in humans because it is commonly recognized that INH depletes pyridoxal phosphate leading to peripheral neuropathies; therefore, patients are usually, but not always, given vitamin B6 to prevent this problem. In subsequent studies, we used SDH or GLDH rather than trying to modify the ALT assay.
Another difficulty in developing an animal model of INH-induced hepatotoxicity is the route of administration. Because of its short half-life, it was found that smaller, more frequent doses of INH lead to greater hepatotoxicity than one large dose.2 Wistar rats that were treated with INH did not display any signs of hepatotoxicity as seen by H&E staining. We found that in BN rats, high doses of INH (200 and 400 mg/kg/day) led to signs of central nervous system (CNS) toxicity. A smaller dose of 150 mg/kg/day decreased food intake and weight gain (Figure 7F); therefore, animals could be maintained for longer, but even 50 mg/kg given twice daily led to signs of CNS toxicity. When INH was administered in food, it produced INH blood levels in mice that were comparable to therapeutic levels in humans, but the blood levels in rats at the same dose were less than half those in mice (Figure 9A). Thus administration of INH to rats in food led to fewer signs of CNS toxicity, and animals could be maintained for five weeks. Because of the short half-life of INH, this method of administration has the advantage of providing more constant blood levels. However, despite trying different species and strains of animals and experimentation with dose and mode of administration, we were not able to develop an animal model of INH-induced liver injury similar to what occurs in humans. We did see evidence of steatosis that resolved despite continued treatment, which suggests that INH can cause mitochondrial injury (Figure S2, Supporting Information). This is consistent with other published studies where cotreatment of INH with rifampicin caused steatosis and mitochondria oxidative stress in mice and rats.19,20 The amount of covalent binding was not the only predictor of hepatotoxicity because INH binding was similar to both C57BL/6 and Balb C (Figure 3C), but Balb C mice had a greater increase in GLDH and SDH levels (Figure 8 and Table S3, Supporting Information). Although there was liver injury with some treatments, in no case did it appear to be a good model of the idiosyncratic liver injury observed in humans. Unfortunately, this inability to reproduce idiosyncratic drug reactions in animals is typical of such studies.
How do these data relate to INH-induced liver injury in humans? Most studies indicate that slow acetylators are at increased risk of INH-induced liver injury.21 The fact that slow acetylators appear to be at increased risk has been explained on the basis that they are exposed to more AcHz. This is because, although slow acetylators form less AcHz, clearance of AcHz by a second acetylation is also slower, and the net result is a small increase in AcHz exposure.22,23 However, the difference in blood levels of INH between fast and slow acetylators is even greater than that of AcHz;15,23 therefore, the increased risk associated with the slow acetylator phenotype in humans is more easily explained if a reactive metabolite of INH is responsible for the liver injury.
Although INH-induced liver injury has been classed as metabolic idiosyncrasy, implying that it is not immune-mediated, there are cases that have clear evidence of an immune mechanism with immediate fever and increase in ALT on rechallenge.9,17 An immune mechanism is also the easiest way to explain the idiosyncratic nature of INH-induced liver injury, and the absence of features of a hypersensitivity reaction is not good evidence against an immune mechanism. Furthermore, we have found that patients treated with INH who have an increase in ALT also have an increase in Th17 cells, which provides additional evidence for the involvement of the immune system (unpublished observations). In addition, there is direct evidence for an immune mechanism involving covalent binding of INH. Specifically, Warrington et al. found that lymphocytes from patients with mild INH-induced liver injury proliferated when incubated with INH- or INA-modified protein but not to INH itself.24,25 If the injury was more severe, the lymphocytes also proliferated when incubated with INH itself. This positive lymphocyte transformation test provides strong evidence that INH-induced liver injury is mediated by the adaptive immune system, and furthermore, the recognition of INH-modified proteins suggests that the immune response is against INH-modified proteins, which fits nicely with our observation that INH binds to human hepatic proteins. In more severe cases, the immune response spreads so that the parent drug is also recognized. In addition, although it has yet to be replicated, the finding of an increased risk of INH-induced liver injury in patients who have the HLA-DQB1 gene *0201 provides additional evidence for an immune mechanism.26 It also seems that modification of protein by INH would be more likely to induce an immune response than would acetylation (the result of AcHz reactive metabolite binding) because its structure is more “foreign”. However, cell damage caused by bioactivation of AcHz could also contribute to the induction of an immune response; therefore, both reactive metabolites may play a role in INH-induced liver injury. In addition, INH and AcHz could also be oxidized to free radicals that cause cell injury that would not be detected by covalent binding assays but could contribute to cell damage and the induction of an immune response.
In conclusion, in contrast to previous studies in rats, we found that INH is oxidized to a reactive metabolite that covalently binds to mouse and human hepatic proteins. This is consistent with previous studies that found that lymphocytes from patients with INH induced liver injury responded to INH or INH modified proteins. Also, in contrast to previous assertions that INH-induced liver injury is not immune-mediated, the lymphocyte transformation test data along with other data such as clinical cases that have features of an immune response and an association with a specific HLA genotype provide evidence that INH induced liver injury is immune-mediated.
Glossary
Abbreviations
- AcHz
acetylhydrazine
- AcINH
acetylisoniazid
- ALT
alanine aminotransferase
- BSA
bovine serum albumin
- BN
Brown Norway
- CNS
central nervous system
- GLDH
glutamate dehydrogenase
- H&E
hematoxylin and eosin
- HLM
human liver microsomes
- Hz
hydrazine
- INA
isonicotinic acid
- INH
isoniazid
- INA-BSA
isonicotinic acid coupled to bovine serum albumin
- INA-INH
isonicotinic acid coupled to isoniazid
- INA-NAL
isonicotinic acid coupled to N-α-acetyl-l-lysine
- INA-NHS
reactive N-hydroxysuccinimide ester of isonicotinic acid
- LC-MS
liquid chromatography coupled to mass spectrometry
- MLM
mouse liver microsomes
- NAL
N-α-acetyl-l-lysine
- NHS
N-hydroxysuccinimide
- RLM
rat liver microsomes
- SDH
sorbitol dehydrogenase
Supporting Information Available
Covalent binding of INH to hepatic proteins in mice; liver histology of mice and rats treated with INH; percentage of BSA and blue carrier protein lysine residues modified by INA-NHS; GLDH and ALT activities after 5 weeks of treatment of BN rats with INH; and SDH and ALT activities in Balb C and C57BL/6 mice. This material is available free of charge via the Internet at http://pubs.acs.org.
This work was supported by the Canadian Institute of Health Research (CIHR).
I.G.M. is a current trainee of the “Drug Safety and Effectiveness Cross-Disciplinary Training” program which is funded by CIHR. J.U. holds the Canada’s Research Chair in Adverse Drug Reactions.
The authors declare no competing financial interest.
Supplementary Material
References
- (2010) Severe isoniazid-associated liver injuries among persons being treated for latent tuberculosis infection: United States, 2004–2008. MMWR 59, 224–229. [PubMed] [Google Scholar]
- Mitchell J. R.; Zimmerman H. J.; Ishak K. G.; Thorgeirsson U. P.; Timbrell J. A.; Snodgrass W. R.; Nelson S. D. (1976) Isoniazid liver injury: clinical spectrum, pathology, and probable pathogenesis. Ann. Intern. Med. 84, 181–192. [DOI] [PubMed] [Google Scholar]
- Snodgrass W.; Potter W. Z.; T. J.; Jollow D. J.; Mitchell J. R. (1974) Possible mechanism of isoniazid-related hepatic injury. Clin. Res. 22, 323A. [Google Scholar]
- Timbrell J. A.; Mitchell J. R.; Snodgrass W. R.; Nelson S. D. (1980) Isoniazid hepatoxicity: the relationship between covalent binding and metabolism in vivo. J. Pharmacol. Exp. Ther. 213, 364–369. [PubMed] [Google Scholar]
- Nelson S. D.; Mitchell J. R.; Timbrell J. A.; Snodgrass W. R.; Corcoran G. B. (1976) Isoniazid and iproniazid: activation of metabolites to toxic intermediates in man and rat. Science 193, 901–903. [DOI] [PubMed] [Google Scholar]
- Noda A.; Hsu K. Y.; Noda H.; Yamamoto Y.; Kurozumi T. (1983) Is isoniazid-hepatotoxicity induced by the metabolite, hydrazine?. J. UOEH 5, 183–190. [DOI] [PubMed] [Google Scholar]
- Sarich T. C.; Youssefi M.; Zhou T.; Adams S. P.; Wall R. A.; Wright J. M. (1996) Role of hydrazine in the mechanism of isoniazid hepatotoxicity in rabbits. Arch. Toxicol. 70, 835–840. [DOI] [PubMed] [Google Scholar]
- Hofstra A. H.; Li-Muller S. M.; Uetrecht J. P. (1992) Metabolism of isoniazid by activated leukocytes. Possible role in drug-induced lupus. Drug. Metab. Dispos. 20, 205–210. [PubMed] [Google Scholar]
- Metushi I. G.; Cai P.; Zhu X.; Nakagawa T.; Uetrecht J. P. (2011) A fresh look at the mechanism of isoniazid-induced hepatotoxicity. Clin. Pharmacol. Ther. 89, 911–914. [DOI] [PubMed] [Google Scholar]
- Bernhardt P. V.; Chin P.; Richardson D. R. (2001) Unprecedented oxidation of a biologically active aroylhydrazone chelator catalysed by iron(III): serendipitous identification of diacylhydrazine ligands with high iron chelation efficacy. J. Biol. Inorg. Chem. 6, 801–809. [DOI] [PubMed] [Google Scholar]
- Christensen J. B. (2001) A simple method for synthesis of active esters of isonicotinic and picolinic acids. Molecules 6, 47–51. [Google Scholar]
- Zhu X.; Li J.; Liu F.; Uetrecht J. P. (2011) Involvement of T helper 17 cells in D-penicillamine-induced autoimmune disease in Brown Norway rats. J. Toxicol. Sci. 120, 331–338. [DOI] [PubMed] [Google Scholar]
- O’Brien P. J.; Slaughter M. R.; Polley S. R.; Kramer K. (2002) Advantages of glutamate dehydrogenase as a blood biomarker of acute hepatic injury in rats. Lab. Anim. 36, 313–321. [DOI] [PubMed] [Google Scholar]
- Ng W.; Lobach A. R.; Zhu X.; Chen X.; Liu F.; Metushi I. G.; Sharma A.; Li J.; Cai P.; Ip J.; Novalen M.; Popovic M.; Zhang X.; Tanino T.; Nakagawa T.; Li Y.; Uetrecht J. (2012) Animal models of idiosyncratic drug reactions. Adv. Pharmacol. 63, 81–135. [DOI] [PubMed] [Google Scholar]
- Fukino K.; Sasaki Y.; Hirai S.; Nakamura T.; Hashimoto M.; Yamagishi F.; Ueno K. (2008) Effects of N-acetyltransferase 2 (NAT2), CYP2E1 and glutathione-S-transferase (GST) genotypes on the serum concentrations of isoniazid and metabolites in tuberculosis patients. J. Toxicol. Sci. 33, 187–195. [DOI] [PubMed] [Google Scholar]
- Jencks B. D. S. a. W. P. (1989) Mechanism of solvolysis of substituted benzoyl halides. J. Am. Chem. Soc. 8470–8479. [Google Scholar]
- Maddrey W. C.; Boitnott J. K. (1973) Isoniazid hepatitis. Ann. Intern. Med. 79, 1–12. [DOI] [PubMed] [Google Scholar]
- Black M.; Mitchell J. R.; Zimmerman H. J.; Ishak K. G.; Epler G. R. (1975) Isoniazid-associated hepatitis in 114 patients. Gastroenterology 69, 289–302. [PubMed] [Google Scholar]
- Chowdhury A.; Santra A.; Bhattacharjee K.; Ghatak S.; Saha D. R.; Dhali G. K. (2006) Mitochondrial oxidative stress and permeability transition in isoniazid and rifampicin induced liver injury in mice. J. Hepatol. 45, 117–126. [DOI] [PubMed] [Google Scholar]
- Sodhi C. P.; Rana S. V.; Mehta S. K.; Vaiphei K.; Attari S.; Mehta S. (1997) Study of oxidative-stress in isoniazid-rifampicin induced hepatic injury in young rats. Drug. Chem. Toxicol. 20, 255–269. [DOI] [PubMed] [Google Scholar]
- Huang Y. S.; Chern H. D.; Su W. J.; Wu J. C.; Lai S. L.; Yang S. Y.; Chang F. Y.; Lee S. D. (2002) Polymorphism of the N-acetyltransferase 2 gene as a susceptibility risk factor for antituberculosis drug-induced hepatitis. Hepatology 35, 883–889. [DOI] [PubMed] [Google Scholar]
- Peretti E.; Karlaganis G.; Lauterburg B. H. (1987) Increased urinary excretion of toxic hydrazino metabolites of isoniazid by slow acetylators. Effect of a slow-release preparation of isoniazid. Eur. J. Clin. Pharmacol. 33, 283–286. [DOI] [PubMed] [Google Scholar]
- Lauterburg B. H.; Smith C. V.; Todd E. L.; Mitchell J. R. (1985) Pharmacokinetics of the toxic hydrazino metabolites formed from isoniazid in humans. J. Pharmacol. Exp. Ther. 235, 566–570. [PubMed] [Google Scholar]
- Warrington R. J.; Tse K. S.; Gorski B. A.; Schwenk R.; Sehon A. H. (1978) Evaluation of isoniazid-associated hepatitis by immunological tests. Clin. Exp. Immunol. 32, 97–104. [PMC free article] [PubMed] [Google Scholar]
- Warrington R. J.; McPhilips-Feener S.; Rutherford W. J. (1982) The predictive value of the lymphocyte transformation test in isoniazid-associated hepatitis. Clin. Allergy 12, 217–222. [DOI] [PubMed] [Google Scholar]
- Sharma S. K.; Balamurugan A.; Saha P. K.; Pandey R. M.; Mehra N. K. (2002) Evaluation of clinical and immunogenetic risk factors for the development of hepatotoxicity during antituberculosis treatment. Am. J. Respir. Crit. Care Med. 166, 916–919. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.