

Abstract
Although high HDL-cholesterol levels are associated with decreased cardiovascular risk in epidemiological studies, recent genetic and pharmacological findings have raised doubts about the beneficial effects of HDL. Raising HDL levels in animal models by infusion or over expression of apolipoprotein A-I has shown clear vascular improvements, such as delayed atherosclerotic lesion progression and accelerated lesion regression, along with increased reverse cholesterol transport. Inflammation and other factors, such as myeloperoxidase mediated oxidation, can impair HDL production and HDL function, in regard to its reverse cholesterol transport, antioxidant, and anti-inflammatory activities. Thus, tests of HDL function, which have not yet been developed as routine diagnostic assays, may prove useful and be a better predictor of cardiovascular risk than HDL-cholesterol levels.
Human high density lipoprotein (HDL) is a heterogeneous collection of lipoprotein particles with a density between 1.063 and 1.21 g/ml. When human HDL is run on a size exclusion column or non-denaturing gradient gels, it is evident that HDL is polydisperse with several discrete particle sizes evident. Ultracentrifugation can separate two major density sub factions, HDL2 (density between 1.063 and 1.125 g/ml) and HDL3 (density between 1.125 and 121 g/ml). The proteomics of HDL is very complex1, but the overwhelming majority of HDL particles contain apolipoprotein A-I (apoAI), which is the most abundant apolipoprotein in normal human plasma. Many HDL particles also contain apoAII, the second most abundant protein in HDL, and those that do carry apoAII can be separated by immunoisolation. Many of the less abundant proteins associated with HDL are found on only a small fraction of HDL particles, increasing the diversity of HDL particles. A useful way to get a snapshot of the diversity of apoAI-containing particles is through 2D nondenaturing gel electrophoresis, followed by blotting and staining with an antibody against human apoAI, which yields a complex pattern of pre-beta, alpha, and pre-alpha particles of various sizes. Generally speaking, the pre-beta migrating particles represent small lipid-free and lipid-poor apoAI, while the alpha1, 2, and 3 particles represent spherical HDL of decreasing sizes2.
The metabolism of HDL initiates with apoAI synthesis in the liver and intestine, but in order to form HDL, apoAI must interact with cells expressing ABCA1, the gene defective in Tangier disease. Mouse models of tissue specific ABCA1-deficiency demonstrate that hepatic ABCA1 plays the largest role in generating HDL, but that non-hepatic tissues also play significant roles in HDL formation3. Nascent HDL released by ABCA1 expressing cells contains cellular phospholipids (PL) and free cholesterol (FC), and this particle is the substrate for lethicin:cholesterol acyltransferase (LCAT) which esterifies FC into cholesteryl ester (CE), building up the hydrophobic core necessary to generate spherical alpha HDL particles. Further HDL remodeling by plasma and cell surface enzymes is complex and includes processes mediated by ABCG1, hepatic lipase, endothelial lipase, cholesterol ester transfer protein (CETP), and phospholipid transfer protein. In humans, HDL-cholesterol (HDL-C) can be returned to the liver via two pathways: direct hepatic uptake by scavenger receptor B1 (SR-B1); or through CETP exchange of HDL-CE for TG in apoB-containing lipoproteins, followed by hepatic uptake of these apoB-containing particles by the LDL receptor.
HDL-C is commonly known as the “good” cholesterol as high levels of HDL-C are associated with reduced levels of cardiovascular disease (CVD), and low levels of HDL-C are associated with increased CVD, in multiple epidemiological studies. The concept that HDL-C is protective against incident coronary heart disease for subjects in different strata of low density lipoprotein-cholesterol (LDL-C) levels was first demonstrated in the Framingham study in the 1970s and 1980s4, 5. In addition, the incidence of low HDL-C (<35 mg/dl) was ~3-fold higher among men with premature (<60 years of age) coronary heart disease than in age matched controls6. However, recent human studies have cast some doubt on the “good cholesterol” HDL hypothesis. A genetic method called mendelian randomization found that a score derived from 14 common genetic variants that is associated with HDL-C levels, and not other lipoprotein traits, is not associated with myocardial infarction7. Furthermore, two recent drug trials, one of the CETP inhibitor torcetrapib, and the other of extended-release niacin, did not show beneficial cardiovascular outcomes despite increased HDL-C levels8, 9. However, torcetrapib had off target effects raising blood pressure, and the niacin trial had many design flaws. Since there is abundant evidence in mouse models (described below) that increased HDL is associated with decreased atherosclerosis progression and accelerated atherosclerosis regression, a new theory has surfaced, that the levels of HDL-C may not be an ideal indicator of coronary heart disease risk, rather HDL function may be a better indicator. This was also made clear in the mouse model of SR-B1 deficiency, in which high levels of plasma HDL-C accumulate, but these mice developed more severe atherosclerosis when bred onto the hyperlipidemic apoE-deficient background10. In this review, we will discuss one of the major functions of HDL, its role in promoting reverse cholesterol transport, along with several mechanisms by which HDL can become dysfunctional, and new clinical evidence linking HDL functionality to cardiovascular outcomes.
Reverse Cholesterol Transport and Assays for HDL/apoAI Function
HDL has been ascribed with many atheroprotective activities, such as its antioxidant, anti-inflammatory, endothelial cell maintenance functions, and its activity in mediating reverse cholesterol transport (RCT). While the relative role of the above activities in mediating HDL’s protective effect have not been directly compared, it is our opinion that RCT likely plays an important role in its atheroprotective effect, making this topic the focus of our review. The RCT hypothesis, first put forth by Glomset, proposes that HDL acts to accept cholesterol from the periphery, such as arterial wall cells, and deliver it to the liver, where it can be directly excreted into the bile or be metabolized into bile salts before excretion11.
HDL function can be measured in several in vitro assays. Fogelman and colleagues pioneered cell-based and cell-free assays to measure the anti-inflammatory and antioxidant activities of HDL12, 13. LDL added to endothelial cells co-cultured with smooth muscle cells undergoes oxidation and induces expression of monocyte chemotactic factors and increases monocyte binding and transmigration. The addition of HDL can impair this response, demonstrating HDL’s antioxidant and anti-inflammatory activities. However, HDL from patients undergoing an acute phase reaction did not possess antioxidant activity and did not inhibit monocyte chemotaxis but actually increased it, demonstrating pro-inflammatory activity14. Through these and similar studies, Fogelman’s group concluded that HDL can become dysfunctional. Another example of apoAI’s anti-inflammatory activity is its ability to reduce the macrophage response to endotoxin, suppressing the type I interferon response15. Several of the HDL accessory proteins such as paraoxonase and apoL-I are associated with apoAI’s antioxidant activity16.
HDL’s activities relevant to endothelial cell maintenance and inflammation can be assayed by measuring: i) the ability of HDL to promote NO production by cultured endothelial cell; ii) the protection of cultured endothelial cells from apoptotic stimuli such as exposure to ultraviolet light; and iii) the ability of HDL to reduce endothelial cell expression of the pro inflammatory adhesion protein VCAM-1 after treatment with an inflammatory cytokine17, 18. HDL isolated from type 2 diabetic patients has reduced levels of these endothelial protective activities indicative of diabetes inducing HDL dysfunction17.
The first step in RCT is the efflux of cellular cholesterol. ApoAI, other exchangeable apolipoproteins, and mimetic peptides that share the amphipathic helical structure of these apolipoproteins, can all accept cellular FC and PL in an ABCA1 dependent fashion19, 20. Plasma derived HDL can also accept cholesterol via ABCG1 or SR-B1. However, depending upon its method of preparation, plasma HDL may also contain some ABCA1-dependent lipid acceptor activity. There is some evidence to suggest that this may be due to lipoprotein remodeling releasing lipid-free apoAI, since HDL2 or reconstituted HDL made in vitro from apoAI and phosphatidylcholine, is virtually devoid of this activity21, 22. It is relatively simple to measure the activity of HDL, apoAI, or plasma/serum fractions for their function in RCT via their ability to act as acceptors of cholesterol from cholesterol loaded macrophages or cells with regulated expression of ABCA1, ABCG1, or SR-B1. One prepares [3H or 14C]cholesterol labeled cells, chases these cells with the specific acceptor, and calculates cholesterol efflux as the % of the cellular [3H or 14C]cholesterol that appears in the media. HDL recovered from sepsis patients is reported to have decreased cholesterol accepting activity, indicating that sepsis is associated with dysfunctional HDL in regard to its role in RCT23.
Cholesterol efflux assays are quite useful for examining the first steps in the RCT pathway; however, it had been a long standing challenge to prove the RCT hypothesis in vivo. Dan Rader and colleagues developed a simple in vivo RCT assay in mice in which cholesterol labeled macrophages are injected i.p, and the cholesterol radioactivity can be followed into the plasma, liver, and feces; and, this study showed that RCT is modulated by apoAI expression levels24. RCT is calculated as the % of the injected radioactivity found in the plasma, liver, and feces. This assay can be modified using different types of donor cells and different sites of donor cell injection25; and, a recent review has cataloged all studies through 2011 using this assay26. Although this assay has been widely adapted, what is really measured is not net efflux of cholesterol mass from the donor cells, but unidirectional movement of the radiolabeled cholesterol tracer from the donor cells to the plasma and onwards, and this movement may occur by exchange with endogenous cholesterol pools rather than by net efflux. In order to get around this caveat, Weibel et al. developed a method in which macrophages were encapsulated in hollow fibers and then implanted into recipient mice. Following a one day incubation in vivo, the cells are recovered and the cholesterol mass of the cells is compared pre and post in vivo incubation27. While there is a reduction in foam cholesterol mass after implantation into wild type mice (indicative of foam cell regression), there is an increase after implantation into hyperlipidemic LDL receptor-deficient mice (indicative of foam cell formation). Although numerous HDL turnover studies have been performed in humans, it would be attractive to have a good method to assess the entire RCT pathway from macrophages to feces in humans. Although this methodology has not been developed yet, recently a [13C]cholesterol infusion study in humans demonstrated the ability to monitor cholesterol in different pools and use kinetic modeling to estimate the reverse cholesterol transport pathway28. However, this method does not specifically assess the contribution of foam cells, and thus, whether this design is germane to coronary atherosclerosis is not known.
Several studies demonstrate changes in HDL structure and function associated with inflammation. For example, de Beer and colleagues have shown marked HDL remodeling after treatment of mice with endotoxin, or in humans after surgery, producing acute phase HDL, in which serum amyloid A (SAA) and group IIa secretory phospholipase A2 are induced while apoAI levels are repressed29. Since SAA has cholesterol acceptor activity, the consequences of inflammation on RCT must be determined empirically. Two studies employed LPS injections in mice to induce inflammation and the acute phase response, and both reported impaired RCT, with the block occurring primarily in cholesterol mobilization from the liver to the intestine, consistent with large decreases in hepatic Abcg5, Abcg8, and Cyp7a1 gene expression23, 30. A third study used zymosan to induce inflammation and the acute phase response, but zymosan is also effective in leading to the release of myeloperoxidase (MPO), an enzyme known to impair apoAI and HDL function (discussed below). This study also found impaired RCT, but at the first step where cholesterol is mobilized from the injected foam cells to the plasma compartment. Furthermore, diluted plasma from the zymosan treated mice had decreased ABCA1-dependent cholesterol acceptor activity31. In addition, direct injection of MPO and hydrogen peroxide into mice also leads to impaired RCT to the plasma, liver, and fecal compartments23.
HDL Effects on Atherosclerosis Progression and Regression
Given the many studies establishing HDL particles as cholesterol acceptors in vitro, the extrapolation of its effects to the in vivo situation predicted protection from atherosclerosis. The simple reasoning was that by its promotion of cholesterol efflux from macrophages in plaques, increasing HDL particles would either retard the formation of foam cells from macrophages (in the setting of progression) or unload excess cholesterol from these cells after they formed (in the setting of regression). As additional “pleiotropic” effects of HDL have been uncovered (discussed above), the expectation that HDL would be beneficial in reducing coronary artery disease (CAD) risk has further increased. This optimism is sustained by many investigators in the face of recent reports that higher levels of plasma HDL-C in genetic or pharmacologic studies were not associated with decreased CAD risk7, 32, most likely because there is scant evidence that HDL-C is a reliable biomarker of HDL function; in fact, there is accumulating evidence to the contrary, reviewed below. HDL proponents point to the direct evidence that raising the level of functional HDL particles by either increasing their hepatic production or by HDL infusion results in atheroprotective effects (decreased progression or increased regression of plaques). Though these data have largely come from pre-clinical studies, there are also reports also from the clinical literature. Both types of studies will now be briefly summarized, starting with pre-clincial investigations.
Progression
In the standard mouse models of atherosclerosis (LDLr−/−, apoE−/−), increased production of HDL particles has been achieved by either transgenic or adenoviral means. Only a few examples can be given in this brief review, but in all cases, the content of macrophages and macrophage-derived foam cells, the central cellular components of the atherosclerotic plaque, were decreased as a result. Two groups made apoE−/− mice transgenic for human apoAI (“hAI/EKO” mice) and showed that atherosclerosis progression was suppressed by >80% even after 8 months on chow diets, on which the mice sustained non-HDL-C levels of > 400 mg/dL 33, 34. Both apoAI levels (a rough indicator of HDL particle number) and HDL-C were increased. These results were extended to show atheroprotection when the mice were fed a western-type diet (WD), which further elevated non-HDL-C to over 1000 mg/dL 35. Examples of the viral approach are the reports in which either apoE−/− 36 or LDLr−/− mice 37–39 fed chow (apoE−/−) or WD (LDLr−/−) were infected with an adenovirus or an AAV containing DNA encoding human apoAI. Again, plasma levels of apoAI were increased (though not always HDL-C) 38, and the early progression of atherosclerosis was suppressed. The infusion approach was also successful in suppressing atherosclerosis progression in rabbits 40.
There are a number of clinical studies in which plasma levels of HDL-C have been raised, with inconsistent outcomes on CAD risk observed (some showed benefit, others did not). It is not possible to summarize this complex area here, but it has been discussed in a number of recent articles, including one by two of us 41. Simply stated, a major difficulty in interpreting the available clinical studies is that in none of them has it been established whether an increase in functional HDL particles was achieved.
Regression
Though retarding the progression of atherosclerosis is highly desirable, perhaps regression is more relevant to the typical clinical scenario, in that by the time risk factor reduction is medically undertaken, many people will already have significant plaque burden, and in secondary prevention situations, will already have documented CAD. As in the progression studies above, there are strong supporting data from pre-clinical studies that by increasing the number of functional HDL particles, pre-existing plaques can undergo remarkable remodeling, particularly in the content and inflammatory phenotype of plaque macrophages and macrophage-foam cells. There are also clinical studies, albeit limited, consistent with these findings.
Considering first the pre-clinical models, the raising of plasma levels of apoAI by viral or infusion means after plaques formed resulted in significant reductions of the macrophage and macrophage-foam cell content in LDLr−/− 42 or apoE−/− 43 mice. These data agreed with earlier work in rabbits, in which human HDL infusions also regressed atherosclerosis 44. One of the limitations of both the viral and infusion approaches to study regression of atherosclerosis has been the relative short term nature of the treatments, either because of the cumbersome logistics of repeated injections/infusions or the transient expression of viral vectors. To overcome these limitations, we adopted an aortic transplant approach by using as recipients hAI/EKO mice 45, 46. By transferring an atherosclerotic aortic arch from a donor mouse into a recipient with a different plasma lipoprotein profile, the environment that the plaque cells are exposed to is changed quickly, and is spontaneously sustained for long periods of time. To study the effects of increasing HDL-C levels on plaques, aortic arches from apoE−/− mice (low HDL-C, high non-HDL-C) were transplanted into recipient hAI/EKO mice (normal HDL-C, high non-HDL-C). With regard to the above mentioned distinction between HDL-C and HDL particles, it is important to note that there is good correlation between plasma levels of apoAI and HDL-C in hAI/EKO mice.
Remarkably, despite persistent elevated non-HDL-C in hAI/EKO recipients, the plaque content of CD68+ cells (macrophages and macrophage-derived foam cells) decreased by >50% one week after transplantation. Interestingly, the decreased content of plaque CD68+ cells was associated with their emigration from the plaques and induction of their chemokine receptor CCR7, a factor we have previously shown to be required for regression in the transplant model 47. Based on a recent study of another mouse model of atherosclerosis regression 48, it is also possible that some reduction of plaque macrophage content was due to decreased monocyte recruitment, but this remains to be determined. The induction of CCR7 is likely related to changes in the sterol content of foam cells when they are placed in a regression environment, given that its promoter has a putative sterol regulatory element (SRE) that appears to be active in vivo 49. In support of this mechanism are the data from our study showing that there were reductions in cholesteryl ester content of the plaques in the hAI/EKO recipients, compared to the donor mice, accompanied by the induction of the classic SRE-regulated gene, HMGCoA-reductase in CD68+ cells captured from plaques 46.
Another important result was the change in the inflammatory state of plaque CD68+ cells. There was decreased expression of inflammatory factors and enrichment of markers of the M2 (“anti-inflammatory“) macrophage state in the hAI/EKO recipients, compared to the donor mice 46. Macrophage heterogeneity in human atherosclerotic plaques is widely recognized, with both M1 (activated) and M2 markers being detectable in lesions 50, 51, but little is known about the factors that regulate M2 marker expression in plaques in vivo. How raising HDL-C accomplished this in our model is under investigation, but in this regard, it is interesting to note that an apoAI-mimetic peptide inhibited M1 and promoted M2 changes in macrophages in vitro 52, 53.
Another approach to raise the level of functioning HDL particles has come from microRNA research. MiR-33, an intronic miRNA located within the gene encoding sterol-regulatory element binding protein-2, inhibits hepatic expression of both ABCA1 and ABCG1, reducing HDL-C concentrations, as well as ABCA1 expression in macrophages, thus resulting in decreased cholesterol efflux 54. In LDLr−/− mice treated with an inhibitor of miR-33, HDL-C levels rose concomitant with enrichment of M2 markers in plaque CD68+ cells 55. The treated mice also exhibited plaque regression with fewer macrophages and macrophage-derived foam cells. The therapeutic potential of miR-33 inhibitors to cause similar benefits in people was suggested by plasma levels of HDL-C and apoAI being raised in treated non-human primates 56. Thus, antagonism of miR-33 may represent a novel approach to enhancing macrophage cholesterol efflux and raising HDL-C levels in the future.
Clinical Studies of Regression by ApoAI/HDL Therapies
Turning to the clinical investigations, there are a limited number of human studies in which HDL levels have been manipulated by infusion, and the effects on plaques assessed. In the first 57, patients at high risk for cardiovascular disease were infused with either an artificial form of HDL (apoAI milano/phospholipid complexes) or saline (placebo) once a week for 5 weeks. By intravascular ultrasound (IVUS), there was a significant reduction in atheroma volume (−4.2%) in the combined (high and low dose) treatment group, though no dose response was observed of a higher vs. lower dose of the artificial HDL. There was no significant difference in atheroma volume compared to the placebo group, but the study was not powered for a direct comparison. In the second infusion study, high-risk patients received 4 weekly infusions with reconstituted HDL (rHDL; containing wild type apoAI) or saline (placebo) 58. Similar to the previous study, there was a significant decrease in atheroma volume (−3.4%) (as assessed by IVUS) after treatment with rHDL compared to baseline, but not compared to placebo (which the study was not powered for). However, the rHDL group had statistically significant improvements in a plaque characterization index and in a coronary stenosis score on quantitative coronary angiography compared to the placebo group. In the third infusion trial 59, a single dose of reconstituted human HDL was infused into patients undergoing femoral atherectomies, with the procedure performed 5–7 days later. Compared to the control group (receiving saline solution), in the excised plaque samples in the HDL infusion group, macrophage activation state (e.g.,. VCAM-1 expression) as well as cell size (due to diminished lipid content) were reduced.
Based on the evidence reviewed above, as well as on other reports in the literature, HDL has the potential to retard the progression of atherosclerosis or promote its regression by modulating a number of steps, including the oxidation of LDL, the activation of endothelium, the recruitment of circulating monocytes and their conversion to foam cells, the activation and inflammatory state of macrophages, and their retention or emigration. From the small amount of clinical work consistent with plaque-protective benefits of functional HDL, it is tempting to speculate that at least some of these effects will be operative in people; however, one major limitation that necessarily limits enthusiasm is that there are no outcome studies to show that any of the clinical endpoints measured to date (e.g., plaque volume, inflammatory state of macrophages) are correlated with decreased events.
Myeloperoxidase modification of apoAI/HDL
Posttranslational modification of apoAI can directly lead to HDL dysfunction. For example, copper oxidation, malondialdehyde, or lipid peroxide treatment of HDL alters apoAI structure and function (reviewed in 60), HDL from diabetic subjects can have glycated apoAI with altered lipid binding activity, and incubation of HDL with glucose impaired its anti-inflammatory and antioxidant activities 61, 62. One of the best studied modifications of apoAI is mediated by MPO, a leukocyte derived heme protein abundant in neutrophils, monocytes and a subset of tissue macrophages. Part of the innate immune host defense system, MPO uses hydrogen peroxide to generate an array of reactive oxidant and free radical species that are antimicrobial, such as hypochlorous acid (HOCl). These same species can also foster spurious oxidative injury to normal tissues as well, such as within atherosclerotic plaque, where MPO has been shown to promote both protein modifications and initiate lipid peroxidation. Once released from activated leukocytes, in the circulation and within lesions, MPO has been shown to bind to HDL. This tight binding, which has been mapped to helix 8 region of apoAI63, likely accounts for the selective oxidative targeting of apoAI within HDL for modification by MPO generated oxidants63. These and other recent observations support a role for MPO serving as an enzymatic catalyst for site-specific modification of apoAI and HDL leading to functional impairment within the artery wall. Indeed, HDL isolated from human atherosclerotic plaque has been shown to co-immunoprecipitate with MPO. Moreover, mass spectrometry analyses of apoAI within HDL recovered from human atheroma is markedly enriched in protein-bound 3-chlorotyrosine and 3-nitrotyrosine, post translational modifications of protein tyrosine residues indicative of protein exposure to MPO-generated reactive chlorinating and nitrating oxidants63, 64. In several clinical studies, circulating apoAI recovered from CAD subjects demonstrates dramatic increases in chlorotyrosine, a specific molecular fingerprint of MPO-catalyzed oxidation, than apoAI isolated from plasma from healthy controls63–66. The 100–500 fold enrichment in levels of MPO-specific oxidation products within apoAI recovered from either plasma or human atherosclerotic plaque serves as strong evidence for MPO selectively targeting apoAI for oxidative modification in vivo63. In one clinical study, subjects with an elevated apoAI chlorotyrosine or nitrotyrosine content were shown to have a 16-fold or 6-fold, respectively, greater likelihood of having cardiovascular disease63. Consistent with the notion that MPO selectively modifies apoAI in the artery wall, histological studies demonstrate colocalization of apoAI along with both MPO, and other MPO-generated oxidation products within human plaque67, 68. Additional studies have shown that the extent to which apoAI harbors MPO-generated oxidative modifications such as chlorotyrosine is strongly associated with further impairment in the cholesterol efflux function of apoAI, particularly via the ABCA1 dependent cholesterol efflux pathway63–66.
MPO generated reactive chlorinating oxidants are known to favor modification of Cys, Met, Lys, His, Tyr, and Trp residues on proteins. The sites of oxidative modification to apoAI recovered from human plasma and atherosclerotic plaque have been mapped by mass spectrometry63,64. Initial in vitro studies demonstrated MPO-catalyzed oxidative modification occurs preferentially at residues on helix 8 (e.g. Tyr 192), in close spatial proximity with the site on apoAI where MPO has been shown to bind63. Additional residues in close spatial proximity to this region have been shown to serve as preferred sites of oxidative modification, such as Trp 72, and Tyr 166 63–66. Critical to these studies is the recognition that oxidized apoAI binds to lipid less effectively, and is often not HDL associated69, so traditional buoyant density lipoprotein isolation methods prior to mass spectrometry analyses can substantially underestimate the degree of oxidative modification, and lead to spurious conclusions70. Although there is general consensus that MPO, or its reactive product HOCl, inactivates apoAI’s cholesterol acceptor activity, the actual site of apoAI modification responsible for this dysfunction is controversial. The creation of Trp-free apoAI isoforms has been informative in this regard. Replacing all four Trp residues with Leu (the 4WL isoform) led to a dysfunctional apoAI; however, replacing the Trp residues with Phe (the 4WF isoform) led to a fully functional apoAI, but one that is resistant to becoming dysfunctional after MPO or HOCl treatment71. Thus apoAI’s Trp residues appear to be the Achilles heel in leading to its loss of cholesterol acceptor activity from MPO-generated HOCl. It is possible to speculate that the oxidant resistant 4WF apoAI isoform may be a better therapeutic reagent to promote the regression of atherosclerotic plaques, making “good cholesterol” even better, since it would be anticipated to have a prolonged biological half-life within the pro-oxidant MPO-rich vulnerable atherosclerotic plaque.
Subsequent studies have revealed that MPO-induced modification of apoAI/HDL inhibits additional HDL functions. For example, oxidative modification of apoAI Tyr166 through MPO-catalyzed nitrating and chlorinating pathways is linked to functional impairment of HDL binding to LCAT, and LCAT activation and activity72. Similarly, oxidation of apoAI Met148 also impairs LCAT activation73. HDL exposure to MPO or HOCl results in loss of HDL’s anti-apototic and anti-inflammatory activities, and specifically the loss of SR-B1 binding activity, while increasing its pro-inflammatory activities, such as endothelial cell NF-kB activation and VCAM-1 expression18. Some of the effects of MPO on generating dysfunctional apoAI and HDL are summarized in Figure 1. HDL exposure to MPO generated oxidants can serve as a potent inhibitor of platelet activation and aggregation induced by physiologic agonists, suggesting that not all oxidative modifications of HDL result in adverse cardiovascular phenotypes74.
Figure 1.
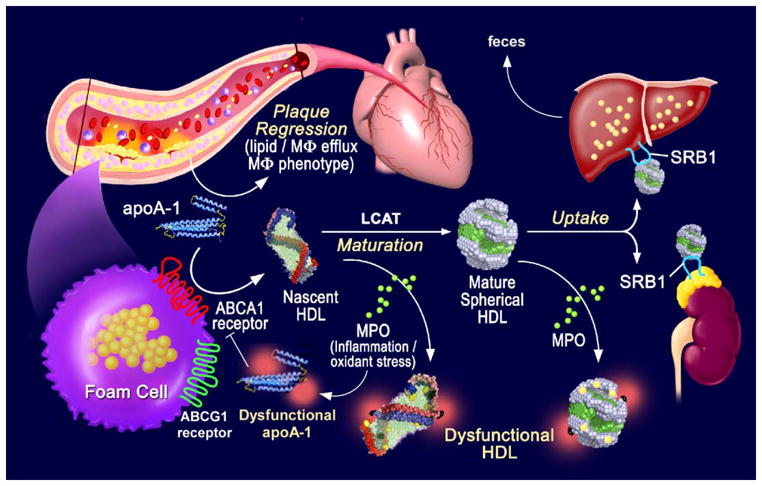
HDL dysfunction impairs reverse cholesterol transport. Reverse cholesterol transport is initiated from arterial macrophages by the interaction of lipid free apoAI with cellular ABCA1 to generate nascent HDL. ABCG1 adds additional cholesterol to HDL and LCAT esterifies HDL cholesterol to generate mature spherical HDL particles. HDL-C uptake by steroidogenic tissues and the liver is mediated by SR-B1. The liver excretes sterols and bile acids into the intestine for output in the feces. MPO generated oxidants modify apoAI, blocking its lipidation by cellular ABCA1 and leading to decreased nascent HDL biogenesis. MPO also impairs LCAT mediated particle maturation and generates dysfunctional and pro-inflammatory HDL.
HDL Function as a Diagnostic Indicator
Tests for HDL function or bioma associated with dysfunctional HDL may be useful for identifying subjects at risk for CAD. For example, plasma MPO levels are positively associated with CAD and the risk of a subsequent major adverse cardiac event75, 76. Similarly, the level of apoAI chlorotyrosine, detected by mass spectrometry, is also a predictor of cardiovascular disease63. Although this assay may be for a potential means of quantifying dysfunctional HDL levels, it is not suitable for routine clinical use. The identification of specific modified amino acid residues linked to loss of function in apoAI may facilitate the development of clinical immunoassays specific for oxidized apoAI/dysfunctional HDL. Several tests of HDL function were described above, and one high impact study used the cholesterol acceptor activity of human apoB depleted serum in cultured macrophages as a surrogate indicator of HDL function. Khera et al. measured this activity in three large patient cohorts and found lower acceptor activity in CAD vs. control subjects32. While cholesterol acceptor activity was correlated with HDL-C levels, variation in HDL-C was associated with only 26% of the variation in cholesterol acceptor activity. After partitioning of the subjects into quartiles based on their cholesterol acceptor activity, those in the highest quartile were associated with an odds ratio for CAD of 0.28 vs. those in the lowest quartile; and this effect remained significant after adjustment for traditional risk factors including apoAI and HDL-C. In a logistic regression model containing traditional risk factors, efflux capacity was significantly associated with decreased CAD risk, although this measure did not appreciably add to the sensitivity vs. specificity compared to the model using just HDL-C. As an add on to their study, Khera et al. measured the change in cholesterol acceptor activity in a small set of subjects after treatment with the PPARγ agonist Pioglitazone or with a statin. Although both treatments led to small increases in HDL-C, only the Pioglitazone treated group had a small (11%), but significant increase in cholesterol acceptor activity32.
In the future, we anticipate additional large clinical studies using cholesterol acceptor activity and other measures of HDL function and apoAI modification. Since the Khera et al. study was a retrospective study, it will be of interest whether these measures can also predict prospectively who will go on to have subsequent events. If these studies are successful, measures of HDL function/dysfunction may be useful as a criterion to select patients for treatment to prevent CAD, as well as to select the specific drug therapy used.
Acknowledgments
Sources of Funding
This work was supported by the NIH grant PO1 HL098055 to EAF, SLH, and JDS. BH was supported by a fellowship from the German Research Foundation (DFG: HE 6092/1-1).
Footnotes
Disclosures
Dr. Hazen reports being listed as co-inventor on pending and issued patents held by the Cleveland Clinic relating to cardiovascular diagnostics and therapeutics. Dr. Hazen reports having been paid as a consultant or speaker for the following companies: Abbott Diagnostics, Cleveland Heart Lab, Esperion, Lilly, Liposcience Inc., Merck & Co., Inc., and Pfizer Inc. Dr. Hazen reports receiving research funds from Abbott, Cleveland Heart Lab, Liposcience Inc., and Pfizer Inc. Dr. Hazen reports having the right to receive royalty payments for inventions or discoveries related to cardiovascular diagnostics or therapeutics from the companies shown below: Abbott Laboratories, Inc., Cleveland Heart Lab., Esperion, Frantz Biomarkers, LLC, Liposcience Inc., and Siemens. Dr. Smith reports being listed as co-inventor on pending and issued patents held by the Cleveland Clinic relating to cardiovascular diagnostics and therapeutics. Dr. Smith reports having been paid as a consultant for Esperion, and receiving research funds from Esperion. Dr. Smith reports having the right to receive royalty payments for inventions or discoveries related to cardiovascular diagnostics or therapeutics from Cleveland Heart Lab. and Esperion.
References
- 1.Gordon SM, Deng J, Lu LJ, Davidson WS. Proteomic Characterization of Human Plasma High Density Lipoprotein Fractionated by Gel Filtration Chromatography. Journal of Proteome Research. 2010;9:5239–5249. doi: 10.1021/pr100520x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Asztalos BF, Tani M, Schaefer EJ. Metabolic and functional relevance of HDL subspecies. Current Opinion in Lipidology. 2011;22:176–185. doi: 10.1097/MOL.0b013e3283468061. [DOI] [PubMed] [Google Scholar]
- 3.Singaraja RR, Van Eck M, Bissada N, Zimetti F, Collins HL, Hildebrand RB, Hayden A, Brunham LR, Kang MH, Fruchart J-C, Van Berkel TJC, Parks JS, Staels B, Rothblat GH, Fievet C, Hayden MR. Both Hepatic and Extrahepatic ABCA1 Have Discrete and Essential Functions in the Maintenance of Plasma High-Density Lipoprotein Cholesterol Levels In Vivo. Circulation. 2006;114:1301–1309. doi: 10.1161/CIRCULATIONAHA.106.621433. [DOI] [PubMed] [Google Scholar]
- 4.Castelli W, Garrison R, Wilson P, Abbott R, Kalousdian S, Kannel W. Incidence of coronary heart disease and lipoprotein cholesterol levels: The framingham study. JAMA: The Journal of the American Medical Association. 1986;256:2835–2838. [PubMed] [Google Scholar]
- 5.Gordon T, Castelli W, Hjortland M, Kannel W, Dawber T. High density lipoprotein as a protective factor against coronary heart disease. The Framingham Study. Am J Med. 1977;62:707–714. doi: 10.1016/0002-9343(77)90874-9. [DOI] [PubMed] [Google Scholar]
- 6.Genest JJ, McNamara JR, Salem DN, Schaefer EJ. Prevalence of risk factors in men with premature coronary artery disease. The American Journal of Cardiology. 1991;67:1185–1189. doi: 10.1016/0002-9149(91)90924-a. [DOI] [PubMed] [Google Scholar]
- 7.Voight BF, Peloso GM, Orho-Melander M, Frikke-Schmidt R, Barbalic M, Jensen MK, Hindy G, Holm H, Ding EL, Johnson T, et al. Plasma HDL cholesterol and risk of myocardial infarction: a mendelian randomisation study. The Lancet. 2012;380:572–580. doi: 10.1016/S0140-6736(12)60312-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barter PJ, Caulfield M, Eriksson M, Grundy SM, Kastelein JJP, Komajda M, Lopez-Sendon J, Mosca L, Tardif J-C, Waters DD, Shear CL, Revkin JH, Buhr KA, Fisher MR, Tall AR, Brewer B. Effects of Torcetrapib in Patients at High Risk for Coronary Events. New England Journal of Medicine. 2007;357:2109–2122. doi: 10.1056/NEJMoa0706628. [DOI] [PubMed] [Google Scholar]
- 9.Investigators A-H. Niacin in Patients with Low HDL Cholesterol Levels Receiving Intensive Statin Therapy. New England Journal of Medicine. 2011;365:2255–2267. doi: 10.1056/NEJMoa1107579. [DOI] [PubMed] [Google Scholar]
- 10.Braun A, Trigatti BL, Post MJ, Sato K, Simons M, Edelberg JM, Rosenberg RD, Schrenzel M, Krieger M. Loss of SR-BI Expression Leads to the Early Onset of Occlusive Atherosclerotic Coronary Artery Disease, Spontaneous Myocardial Infarctions, Severe Cardiac Dysfunction, and Premature Death in Apolipoprotein E-Deficient Mice. Circulation Research. 2002;90:270–276. doi: 10.1161/hh0302.104462. [DOI] [PubMed] [Google Scholar]
- 11.Glomset JA. The plasma lecithin:cholesterol acyltransferase reaction. Journal of Lipid Research. 1968;9:155–167. [PubMed] [Google Scholar]
- 12.Navab M, Hama SY, Hough GP, Subbanagounder G, Reddy ST, Fogelman AM. A cell-free assay for detecting HDL that is dysfunctional in preventing the formation of or inactivating oxidized phospholipids. Journal of Lipid Research. 2001;42:1308–1317. [PubMed] [Google Scholar]
- 13.Navab M, Imes SS, Hama SY, Hough GP, Ross LA, Bork RW, Valente AJ, Berliner JA, Drinkwater DC, Laks H. Monocyte transmigration induced by modification of low density lipoprotein in cocultures of human aortic wall cells is due to induction of monocyte chemotactic protein 1 synthesis and is abolished by high density lipoprotein. The Journal of Clinical Investigation. 1991;88:2039–2046. doi: 10.1172/JCI115532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Van Lenten BJ, Hama SY, de Beer FC, Stafforini DM, McIntyre TM, Prescott SM, La Du BN, Fogelman AM, Navab M. Anti-inflammatory HDL becomes pro-inflammatory during the acute phase response. Loss of protective effect of HDL against LDL oxidation in aortic wall cell cocultures. The Journal of Clinical Investigation. 1995;96:2758–2767. doi: 10.1172/JCI118345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Suzuki M, Pritchard DK, Becker L, Hoofnagle AN, Tanimura N, Bammler TK, Beyer RP, Bumgarner R, Vaisar T, de Beer MC, de Beer FC, Miyake K, Oram JF, Heinecke JW. High-density lipoprotein suppresses the type I interferon response, a family of potent antiviral immunoregulators, in macrophages challenged with lipopolysaccharide. Circulation. 2010;122:1919–1927. doi: 10.1161/CIRCULATIONAHA.110.961193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Davidson WS, Silva RA, Chantepie S, Lagor WR, Chapman MJ, Kontush A. Proteomic analysis of defined HDL subpopulations reveals particle-specific protein clusters: relevance to antioxidative function. Arterioscler Thromb Vasc Biol. 2009;29:870–876. doi: 10.1161/ATVBAHA.109.186031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sorrentino SA, Besler C, Rohrer L, Meyer M, Heinrich K, Bahlmann FH, Mueller M, Horvath T, Doerries C, Heinemann M, Flemmer S, Markowski A, Manes C, Bahr MJ, Haller H, von Eckardstein A, Drexler H, Landmesser U. Endothelial-Vasoprotective Effects of High-Density Lipoprotein Are Impaired in Patients With Type 2 Diabetes Mellitus but Are Improved After Extended-Release Niacin Therapy. Circulation. 2010;121:110–122. doi: 10.1161/CIRCULATIONAHA.108.836346. [DOI] [PubMed] [Google Scholar]
- 18.Undurti A, Huang Y, Lupica JA, Smith JD, DiDonato JA, Hazen SL. Modification of High Density Lipoprotein by Myeloperoxidase Generates a Pro-inflammatory Particle. Journal of Biological Chemistry. 2009;284:30825–30835. doi: 10.1074/jbc.M109.047605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Remaley AT, Schumacher UK, Stonik JA, Farsi BD, Nazih H, Brewer HB. Decreased Reverse Cholesterol Transport from Tangier Disease Fibroblasts. Arteriosclerosis, Thrombosis, and Vascular Biology. 1997;17:1813–1821. doi: 10.1161/01.atv.17.9.1813. [DOI] [PubMed] [Google Scholar]
- 20.Remaley AT, Thomas F, Stonik JA, Demosky SJ, Bark SE, Neufeld EB, Bocharov AV, Vishnyakova TG, Patterson AP, Eggerman TL, Santamarina-Fojo S, Brewer HB. Synthetic amphipathic helical peptides promote lipid efflux from cells by an ABCA1-dependent and an ABCA1-independent pathway. Journal of Lipid Research. 2003;44:828–836. doi: 10.1194/jlr.M200475-JLR200. [DOI] [PubMed] [Google Scholar]
- 21.Cuchel M, Lund-Katz S, de la Llera-Moya M, Millar JS, Chang D, Fuki I, Rothblat GH, Phillips MC, Rader DJ. Pathways by Which Reconstituted High-Density Lipoprotein Mobilizes Free Cholesterol From Whole Body and From Macrophages. Arteriosclerosis, Thrombosis, and Vascular Biology. 2010;30:526–532. doi: 10.1161/ATVBAHA.109.196105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang N, Lan D, Chen W, Matsuura F, Tall AR. ATP-binding cassette transporters G1 and G4 mediate cellular cholesterol efflux to high-density lipoproteins. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:9774–9779. doi: 10.1073/pnas.0403506101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Annema W, Nijstad N, Tolle M, de Boer JF, Buijs RVC, Heeringa P, van der Giet M, Tietge UJF. Myeloperoxidase and serum amyloid A contribute to impaired in vivo reverse cholesterol transport during the acute phase response but not group IIA secretory phospholipase A2. Journal of Lipid Research. 2010;51:743–754. doi: 10.1194/jlr.M000323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang Y, Zanotti I, Reilly MP, Glick JM, Rothblat GH, Rader DJ. Overexpression of Apolipoprotein A-I Promotes Reverse Transport of Cholesterol From Macrophages to Feces In Vivo. Circulation. 2003;108:661–663. doi: 10.1161/01.CIR.0000086981.09834.E0. [DOI] [PubMed] [Google Scholar]
- 25.Wang M-D, Franklin V, Marcel YL. In Vivo Reverse Cholesterol Transport From Macrophages Lacking ABCA1 Expression Is Impaired. Arteriosclerosis, Thrombosis, and Vascular Biology. 2007;27:1837–1842. doi: 10.1161/ATVBAHA.107.146068. [DOI] [PubMed] [Google Scholar]
- 26.Annema W, Tietge U. Regulation of reverse cholesterol transport - a comprehensive appraisal of available animal studies. Nutrition & Metabolism. 2012;9:25. doi: 10.1186/1743-7075-9-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Weibel GL, Hayes S, Wilson A, Phillips MC, Billheimer J, Rader DJ, Rothblat GH. Novel In Vivo Method for Measuring Cholesterol Mass Flux in Peripheral Macrophages. Arteriosclerosis, Thrombosis, and Vascular Biology. 2011;31:2865–2871. doi: 10.1161/ATVBAHA.111.236406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Turner S, Voogt J, Davidson M, Glass A, Killion S, Decaris J, Mohammed H, Minehira K, Boban D, Murphy E, Luchoomun J, Awada M, Neese R, Hellerstein M. Measurement of Reverse Cholesterol Transport Pathways in Humans: In Vivo Rates of Free Cholesterol Efflux, Esterification, and Excretion. Journal of the American Heart Association. 2012:1. doi: 10.1161/JAHA.112.001826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.van der Westhuyzen DR, de Beer FC, Webb NR. HDL cholesterol transport during inflammation. Current Opinion in Lipidology. 2007;18:147–151. doi: 10.1097/MOL.0b013e328051b4fe. [DOI] [PubMed] [Google Scholar]
- 30.McGillicuddy FC, de la Llera Moya M, Hinkle CC, Joshi MR, Chiquoine EH, Billheimer JT, Rothblat GH, Reilly MP. Inflammation Impairs Reverse Cholesterol Transport In Vivo. Circulation. 2009;119:1135–1145. doi: 10.1161/CIRCULATIONAHA.108.810721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Malik P, Berisha SZ, Santore J, Agatisa-Boyle C, Brubaker G, Smith JD. Zymosan-mediated inflammation impairs in vivo reverse cholesterol transport. Journal of Lipid Research. 2011;52:951–957. doi: 10.1194/jlr.M011122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Khera AV, Cuchel M, de la Llera-Moya M, Rodrigues A, Burke MF, Jafri K, French BC, Phillips JA, Mucksavage ML, Wilensky RL, Mohler ER, Rothblat GH, Rader DJ. Cholesterol Efflux Capacity, High-Density Lipoprotein Function, and Atherosclerosis. New England Journal of Medicine. 2011;364:127–135. doi: 10.1056/NEJMoa1001689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Plump AS, Scott CJ, Breslow JL. Human apolipoprotein A-I gene expression increases high density lipoprotein and suppresses atherosclerosis in the apolipoprotein E-deficient mouse. Proc Natl Acad Sci U S A. 1994;91:9607–9611. doi: 10.1073/pnas.91.20.9607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Paszty C, Maeda N, Verstuyft J, Rubin EM. Apolipoprotein AI transgene corrects apolipoprotein E deficiency-induced atherosclerosis in mice. J Clin Invest. 1994;94:899–903. doi: 10.1172/JCI117412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Choudhury RP, Rong JX, Trogan E, Elmalem VI, Dansky HM, Breslow JL, Witztum JL, Fallon JT, Fisher EA. High-density lipoproteins retard the progression of atherosclerosis and favorably remodel lesions without suppressing indices of inflammation or oxidation. Arterioscler Thromb Vasc Biol. 2004;24:1904–1909. doi: 10.1161/01.ATV.0000142808.34602.25. [DOI] [PubMed] [Google Scholar]
- 36.Benoit P, Emmanuel F, Caillaud JM, Bassinet L, Castro G, Gallix P, Fruchart JC, Branellec D, Denefle P, Duverger N. Somatic gene transfer of human ApoA-I inhibits atherosclerosis progression in mouse models. Circulation. 1999;99:105–110. doi: 10.1161/01.cir.99.1.105. [DOI] [PubMed] [Google Scholar]
- 37.Belalcazar LM, Merched A, Carr B, Oka K, Chen KH, Pastore L, Beaudet A, Chan L. Long-term stable expression of human apolipoprotein A-I mediated by helper-dependent adenovirus gene transfer inhibits atherosclerosis progression and remodels atherosclerotic plaques in a mouse model of familial hypercholesterolemia. Circulation. 2003;107:2726–2732. doi: 10.1161/01.CIR.0000066913.69844.B2. [DOI] [PubMed] [Google Scholar]
- 38.Lebherz C, Sanmiguel J, Wilson JM, Rader DJ. Gene transfer of wild-type apoA-I and apoA-I Milano reduce atherosclerosis to a similar extent. Cardiovasc Diabetol. 2007;6:15. doi: 10.1186/1475-2840-6-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Van Craeyveld E, Gordts SC, Nefyodova E, Jacobs F, De Geest B. Regression and stabilization of advanced murine atherosclerotic lesions: a comparison of LDL lowering and HDL raising gene transfer strategies. J Mol Med. 2011;89:555–567. doi: 10.1007/s00109-011-0722-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Miyazaki A, Sakuma S, Morikawa W, Takiue T, Miake F, Terano T, Sakai M, Hakamata H, Sakamoto Y, Natio M, et al. Intravenous injection of rabbit apolipoprotein A-I inhibits the progression of atherosclerosis in cholesterol-fed rabbits. Arterioscler Thromb Vasc Biol. 1995;15:1882–1888. doi: 10.1161/01.atv.15.11.1882. [DOI] [PubMed] [Google Scholar]
- 41.Hewing B, Moore KJ, Fisher EA. HDL and cardiovascular risk: time to call the plumber? Circulation Research. 2012 doi: 10.1161/CIRCRESAHA.112.280958. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tangirala RK, Tsukamoto K, Chun SH, Usher D, Pure E, Rader DJ. Regression of atherosclerosis induced by liver-directed gene transfer of apolipoprotein A-I in mice. Circulation. 1999;100:1816–1822. doi: 10.1161/01.cir.100.17.1816. [DOI] [PubMed] [Google Scholar]
- 43.Shah PK, Yano J, Reyes O, Chyu KY, Kaul S, Bisgaier CL, Drake S, Cercek B. High-dose recombinant apolipoprotein A-I(milano) mobilizes tissue cholesterol and rapidly reduces plaque lipid and macrophage content in apolipoprotein e-deficient mice. Potential implications for acute plaque stabilization. Circulation. 2001;103:3047–3050. doi: 10.1161/hc2501.092494. [DOI] [PubMed] [Google Scholar]
- 44.Badimon JJ, Badimon L, Fuster V. Regression of atherosclerotic lesions by high density lipoprotein plasma fraction in the cholesterol-fed rabbit. J Clin Invest. 1990;85:1234–1241. doi: 10.1172/JCI114558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rong JX, Li J, Reis ED, Choudhury RP, Dansky HM, Elmalem VI, Fallon JT, Breslow JL, Fisher EA. Elevating high-density lipoprotein cholesterol in apolipoprotein E-deficient mice remodels advanced atherosclerotic lesions by decreasing macrophage and increasing smooth muscle cell content. Circulation. 2001;104:2447–2452. doi: 10.1161/hc4501.098952. [DOI] [PubMed] [Google Scholar]
- 46.Feig JE, Rong JX, Shamir R, Sanson M, Vengrenyuk Y, Liu J, Rayner K, Moore K, Garabedian M, Fisher EA. HDL promotes rapid atherosclerosis regression in mice and alters inflammatory properties of plaque monocyte-derived cells. Proc Natl Acad Sci U S A. 2011;108:7166–7171. doi: 10.1073/pnas.1016086108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Feig JE, Ma Y, Randolph GJ, Torra IP, Garabedian MJ, Fisher EA. CCR7 is functionally required for atherosclerosis regression and is activated by LXR. Arterioscler Thromb Vasc Biol. 2006;26:e-50. [Google Scholar]
- 48.Potteaux S, Gautier EL, Hutchison SB, van Rooijen N, Rader DJ, Thomas MJ, Sorci-Thomas MG, Randolph GJ. Suppressed monocyte recruitment drives macrophage removal from atherosclerotic plaques of Apoe−/− mice during disease regression. J Clin Invest. 2011;121:2025–2036. doi: 10.1172/JCI43802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Feig JE, Shang Y, Rotllan N, Vengrenyuk Y, Wu C, Shamir R, Torra IP, Fernandez-Hernando C, Fisher EA, Garabedian MJ. Statins Promote the Regression of Atherosclerosis via Activation of the CCR7-Dependent Emigration Pathway in Macrophages. PLoS One. 2011;6:e28534. doi: 10.1371/journal.pone.0028534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bouhlel MA, Derudas B, Rigamonti E, Dievart R, Brozek J, Haulon S, Zawadzki C, Jude B, Torpier G, Marx N, Staels B, Chinetti-Gbaguidi G. PPARgamma activation primes human monocytes into alternative M2 macrophages with anti-inflammatory properties. Cell Metab. 2007;6:137–143. doi: 10.1016/j.cmet.2007.06.010. [DOI] [PubMed] [Google Scholar]
- 51.Chinetti-Gbaguidi G, Baron M, Bouhlel MA, Vanhoutte J, Copin C, Sebti Y, Derudas B, Mayi T, Bories G, Tailleux A, Haulon S, Zawadzki C, Jude B, Staels B. Human atherosclerotic plaque alternative macrophages display low cholesterol handling but high phagocytosis because of distinct activities of the PPARgamma and LXRalpha pathways. Circ Res. 2011;108:985–995. doi: 10.1161/CIRCRESAHA.110.233775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gupta H, Dai L, Datta G, Garber DW, Grenett H, Li Y, Mishra V, Palgunachari MN, Handattu S, Gianturco SH, Bradley WA, Anantharamaiah GM, White CR. Inhibition of lipopolysaccharide-induced inflammatory responses by an apolipoprotein AI mimetic peptide. Circ Res. 2005;97:236–243. doi: 10.1161/01.RES.0000176530.66400.48. [DOI] [PubMed] [Google Scholar]
- 53.Smythies LE, White CR, Maheshwari A, Palgunachari MN, Anantharamaiah GM, Chaddha M, Kurundkar AR, Datta G. Apolipoprotein A-I mimetic 4F alters the function of human monocyte-derived macrophages. Am J Physiol Cell Physiol. 2010;298:C1538–1548. doi: 10.1152/ajpcell.00467.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rayner KJ, Suarez Y, Davalos A, Parathath S, Fitzgerald ML, Tamehiro N, Fisher EA, Moore KJ, Fernandez-Hernando C. MiR-33 contributes to the regulation of cholesterol homeostasis. Science. 2010;328:1570–1573. doi: 10.1126/science.1189862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rayner KJ, Sheedy FJ, Esau CC, Hussain FN, Temel RE, Parathath S, van Gils JM, Rayner AJ, Chang AN, Suarez Y, Fernandez-Hernando C, Fisher EA, Moore KJ. Antagonism of miR-33 in mice promotes reverse cholesterol transport and regression of atherosclerosis. J Clin Invest. 2011;121:2921–2931. doi: 10.1172/JCI57275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rayner KJ, Esau CC, Hussain FN, McDaniel AL, Marshall SM, van Gils JM, Ray TD, Sheedy FJ, Goedeke L, Liu X, Khatsenko OG, Kaimal V, Lees CJ, Fernandez-Hernando C, Fisher EA, Temel RE, Moore KJ. Inhibition of miR-33a/b in non-human primates raises plasma HDL and lowers VLDL triglycerides. Nature. 2011;478:404–407. doi: 10.1038/nature10486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nissen SE, Tsunoda T, Tuzcu EM, Schoenhagen P, Cooper CJ, Yasin M, Eaton GM, Lauer MA, Sheldon WS, Grines CL, Halpern S, Crowe T, Blankenship JC, Kerensky R. Effect of recombinant ApoA-I Milano on coronary atherosclerosis in patients with acute coronary syndromes: a randomized controlled trial. Jama. 2003;290:2292–2300. doi: 10.1001/jama.290.17.2292. [DOI] [PubMed] [Google Scholar]
- 58.Tardif JC, Gregoire J, L’Allier PL, Ibrahim R, Lesperance J, Heinonen TM, Kouz S, Berry C, Basser R, Lavoie MA, Guertin MC, Rodes-Cabau J. Effects of reconstituted high-density lipoprotein infusions on coronary atherosclerosis: a randomized controlled trial. Jama. 2007;297:1675–1682. doi: 10.1001/jama.297.15.jpc70004. [DOI] [PubMed] [Google Scholar]
- 59.Shaw JA, Bobik A, Murphy A, Kanellakis P, Blombery P, Mukhamedova N, Woollard K, Lyon S, Sviridov D, Dart AM. Infusion of reconstituted high-density lipoprotein leads to acute changes in human atherosclerotic plaque. Circ Res. 2008;103:1084–1091. doi: 10.1161/CIRCRESAHA.108.182063. [DOI] [PubMed] [Google Scholar]
- 60.Smith JD. Dysfunctional HDL as a diagnostic and therapeutic target. Arterioscler Thromb Vasc Biol. 2010;30:151–155. doi: 10.1161/ATVBAHA.108.179226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hedrick CC, Thorpe SR, Fu MX, Harper CM, Yoo J, Kim SM, Wong H, Peters AL. Glycation impairs high-density lipoprotein function. Diabetologia. 2000;43:312–320. doi: 10.1007/s001250050049. [DOI] [PubMed] [Google Scholar]
- 62.Calvo C, Talussot C, Ponsin G, Berthézène F. Non enzymatic glycation of apolipoprotein A-I. Effects on its self-association and lipid binding properties. Biochem Biophys Res Commun. 1988;153:1060–1067. doi: 10.1016/s0006-291x(88)81336-6. [DOI] [PubMed] [Google Scholar]
- 63.Zheng L, Nukuna B, Brennan M-L, Sun M, Goormastic M, Settle M, Schmitt D, Fu X, Thomson L, Fox PL, Ischiropoulos H, Smith JD, Kinter M, Hazen SL. Apolipoprotein A-I is a selective target for myeloperoxidase-catalyzed oxidation and functional impairment in subjects with cardiovascular disease. The Journal of Clinical Investigation. 2004;114:529–541. doi: 10.1172/JCI21109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zheng L, Settle M, Brubaker G, Schmitt D, Hazen SL, Smith JD, Kinter M. Localization of Nitration and Chlorination Sites on Apolipoprotein A-I Catalyzed by Myeloperoxidase in Human Atheroma and Associated Oxidative Impairment in ABCA1-dependent Cholesterol Efflux from Macrophages. Journal of Biological Chemistry. 2005;280:38–47. doi: 10.1074/jbc.M407019200. [DOI] [PubMed] [Google Scholar]
- 65.Bergt C, Pennathur S, Fu X, Byun J, O’Brien K, McDonald TO, Singh P, Anantharamaiah GM, Chait A, Brunzell J, Geary RL, Oram JF, Heinecke JW. The myeloperoxidase product hypochlorous acid oxidizes HDL in the human artery wall and impairs ABCA1-dependent cholesterol transport. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:13032–13037. doi: 10.1073/pnas.0405292101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pennathur S, Bergt C, Shao B, Byun J, Kassim SY, Singh P, Green PS, McDonald TO, Brunzell J, Chait A, Oram JF, O’Brien K, Geary RL, Heinecke JW. Human Atherosclerotic Intima and Blood of Patients with Established Coronary Artery Disease Contain High Density Lipoprotein Damaged by Reactive Nitrogen Species. Journal of Biological Chemistry. 2004;279:42977–42983. doi: 10.1074/jbc.M406762200. [DOI] [PubMed] [Google Scholar]
- 67.Hazell LJ, Arnold L, Flowers D, Waeg G, Malle E, Stocker R. Presence of hypochlorite-modified proteins in human atherosclerotic lesions. The Journal of Clinical Investigation. 1996;97:1535–1544. doi: 10.1172/JCI118576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Malle E, Wag G, Thiery J, Sattler W, Grone H-J. Hypochlorite-Modified (Lipo)Proteins Are Present in Rabbit Lesions in Response to Dietary Cholesterol. Biochemical and Biophysical Research Communications. 2001;289:894–900. doi: 10.1006/bbrc.2001.6074. [DOI] [PubMed] [Google Scholar]
- 69.Peng D-Q, Wu Z, Brubaker G, Zheng L, Settle M, Gross E, Kinter M, Hazen SL, Smith JD. Tyrosine Modification Is Not Required for Myeloperoxidase-induced Loss of Apolipoprotein A-I Functional Activities. Journal of Biological Chemistry. 2005;280:33775–33784. doi: 10.1074/jbc.M504092200. [DOI] [PubMed] [Google Scholar]
- 70.Shao B, Pennathur S, Heinecke JW. Myeloperoxidase Targets Apolipoprotein A-I, the Major High Density Lipoprotein Protein, for Site-Specific Oxidation in Human Atherosclerotic Lesions. Journal of Biological Chemistry. 2010;287:6375–6386. doi: 10.1074/jbc.M111.337345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Peng D-Q, Brubaker G, Wu Z, Zheng L, Willard B, Kinter M, Hazen SL, Smith JD. Apolipoprotein A-I Tryptophan Substitution Leads to Resistance to Myeloperoxidase-Mediated Loss of Function. Arteriosclerosis, Thrombosis, and Vascular Biology. 2008;28:2063–2070. doi: 10.1161/ATVBAHA.108.173815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wu Z, Wagner MA, Zheng L, Parks JS, Shy JM, Smith JD, Gogonea V, Hazen SL. The refined structure of nascent HDL reveals a key functional domain for particle maturation and dysfunction. Nat Struct Mol Biol. 2007;14:861–868. doi: 10.1038/nsmb1284. [DOI] [PubMed] [Google Scholar]
- 73.Shao B, Cavigiolio G, Brot N, Oda MN, Heinecke JW. Methionine oxidation impairs reverse cholesterol transport by apolipoprotein A-I. Proc Natl Acad Sci U S A. 2008;105:12224–12229. doi: 10.1073/pnas.0802025105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Valiyaveettil M, Kar N, Ashraf MZ, Byzova TV, Febbraio M, Podrez EA. Oxidized high-density lipoprotein inhibits platelet activation and aggregation via scavenger receptor BI. Blood. 2008;111:1962–1971. doi: 10.1182/blood-2007-08-107813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Brennan M-L, Penn MS, Van Lente F, Nambi V, Shishehbor MH, Aviles RJ, Goormastic M, Pepoy ML, McErlean ES, Topol EJ, Nissen SE, Hazen SL. Prognostic Value of Myeloperoxidase in Patients with Chest Pain. New England Journal of Medicine. 2003;349:1595–1604. doi: 10.1056/NEJMoa035003. [DOI] [PubMed] [Google Scholar]
- 76.Zhang R, Brennan M-L, Fu X, Aviles RJ, Pearce GL, Penn MS, Topol EJ, Sprecher DL, Hazen SL. Association between myeloperoxidase levels and risk of coronary artery disease. JAMA: The Journal of the American Medical Association. 2001;286:2136–2142. doi: 10.1001/jama.286.17.2136. [DOI] [PubMed] [Google Scholar]