

Abstract
Accumulation of misfolded α-synuclein is the pathological hallmark of Parkinson's disease (PD). Nevertheless, little is known about the mechanism contributing to α-synuclein aggregation and its further toxicity to dopaminergic neurons. Since oxidative stress can increase the expression and aggregation levels of α-synuclein, NADPH oxidases (Noxs), which are responsible for reactive oxygen species generation, could be major players in α-synucleinopathy. Previously, we demonstrated that Nox1 is expressed in dopaminergic neurons of the PD animal models as well as postmortem brain tissue of PD patients, and is responsible for oxidative stress and subsequent neuronal degeneration. Here, using paraquat (PQ)-based in vitro and in vivo PD models, we show that Nox1 has a crucial role in modulating the behavior of α-synuclein expression and aggregation in dopaminergic neurons.
We observed in differentiated human dopaminergic cells that Nox1 and α-synuclein expressions are increased under PQ exposure. Nox1 knockdown significantly reduced both α-synuclein expression and aggregation, supporting the role of Nox1 in this process. Furthermore, in rats exposed to PQ, the selective knockdown of Nox1 in the substantia nigra, using adeno-associated virus encoding Nox1-specific shRNA, largely attenuated the PQ-mediated increase of α-synuclein and ubiquitin expression levels as well as α-synuclein aggregates (proteinase K resistant) and A11 oligomers. Significant reductions in oxidative stress level and dopaminergic neuronal loss were also observed. Our data reveal a new mechanism by which α-synuclein becomes a neuropathologic protein through Nox1-mediated oxidative stress. This finding may be used to generate new therapeutic interventions that slower the rate of α-synuclein aggregation and the progression of PD pathogenesis.
Introduction
Despite there being numerous studies performed to decipher the pathogenesis of Parkinson disease (PD), the exact cause rendering this pathology remains unknown, indicating a multifactorial etiology behind the disease. To understand the mechanism of neuronal death occurring in PD, it is important to solve the enigma of plausible interaction between different factors, like oxidative stress and genetic factors, which may culminate in dopaminergic neurodegeneration.
α-Synuclein, the principal component of Lewy bodies, has been reported as a cause of PD (Beyer et al., 2009; Cookson, 2009). The encoding gene of α-synuclein, SNCA, is the first gene where a strong correlation between its functional mutations and familial form of PD was demonstrated (Polymeropoulos et al., 1997; Krüger et al., 1998; Zarranz et al., 2004). While the molecular mechanism underlying its toxic effects on the nigrostriatal system is largely unknown, the aberrant expression and aggregation of α-synuclein have been considered as potential causes involved in neuronal toxicity (Vekrellis et al., 2011).
Evidence suggested that paraquat (PQ) is a specific neurotoxin for dopaminergic neurodegeneration in the substantia nigra (SN) (Brown et al., 2006; Gatto et al., 2010), and the structure of PQ suggests that increased oxidative stress might be the reason of its toxicity. The SN of PD patients' postmortem brain tissues showed high oxidative stress with an increase in lipid peroxidation (Dexter et al., 1989), oxidative damages in DNA (Zhang et al., 1999) and protein (Alam et al., 1997), and decreased glutathione levels (Sofic et al., 1992). Evidence has demonstrated that the NADPH oxidase (Nox) complexes also play a role in generating reactive oxygen species (ROS) beside mitochondria, and are implicated in several pathologic conditions in CNS (Sorce and Krause, 2009). Our previous results showed that Nox1, an isoform of the Nox family, has a role in PQ-mediated dopaminergic neuronal cell death both in vivo and in cell cultures (Cristóvão et al., 2009). Recently, we have demonstrated that Nox1/Rac1 is activated in dopaminergic neurons following 6-hydroxydopamine (6-OHDA) treatment as well, causing oxidative stress and consequential neuronal death (Choi et al., 2012). Since oxidative stress is generally considered as a factor affecting α-synuclein aggregation (Krishnan et al., 2003), PQ-mediated oxidative stress was also shown to increase α-synuclein aggregation and expression levels in the SN of mice (Manning-Bog et al., 2002). Halting the expression levels of α-synuclein in a mouse model of PD was shown to be beneficial with reduced progression of neurodegeneration (Nuber et al., 2008). Understanding how α-synuclein expression and aggregation is regulated will provide us with targets that ultimately may be used to control and reduce the progression of certain aspects of the disease phenotype.
In the current study, we have investigated the effect of Nox1-derived ROS on the expression and aggregation of α-synuclein in the SN of rats exposed to PQ. We demonstrated that PQ-treated rats show noticeable α-synuclein increased expression and aggregation, which were clearly reduced in Nox1 knockdown.
Materials and Methods
Cell cultures
ReNcell VM culture method.
For the in vitro experiments on human dopaminergic neurons, we have used human mesencephalic neuronal progenitor cell line available from Millipore (catalog number SCC008). The specialty of these cells is that they are isolated from fetal human ventral mesencephalic region and subsequently immortalized by introduction of v-myc. The cells can readily differentiate into dopaminergic neurons upon withdrawal of growth factors (Millipore). We have followed the culture method as indicated by the company with little modifications. Briefly, the cells were allowed to grow on laminin-coated (20 μg/ml) dishes in DMEM/F12-containing medium with B27 supplement, glutamax, heparin (10 U/ml), and gentamicin (50 μg/ml). This medium is called maintenance medium. Cell division was allowed by addition of the two growth factors in the medium viz., basic fibroblast growth factor (bFGF) and epidermal growth factor (EGF), both at a concentration of 20 ng/ml. To induce differentiation, both growth factors were removed from the media and the cells were allowed to differentiate for 14 d with changing medium every alternate day before harvesting or any treatment. After differentiation, cells were treated with 800 μm or 1000 μm PQ for 8 and 24 h.
Immortalized rat mesencephalic dopaminergic cell (N27 cells) culture.
The N27 cells were grown in RPMI 1640 medium containing 10% fetal bovine serum (FBS), 100 U penicillin, and 50 μg/ml streptomycin, in a humidified atmosphere of 5% CO2 at 37°C. N27 cultures were prepared for experiments by plating the cells on polystyrene tissue culture dishes at a density of 0.5 × 105 cells/well in 24-well culture plates with glass coverslip and at 1.5 × 105 cells/well in 6-well culture plates.
Animals and treatment paradigm
The experiments were performed on rats, in accordance with the National Institutes for Health Guide for the Care and Use of Laboratory Animals. All procedures were approved by the local Animal Care and Use Committee. Male Wistar rats (8–10 weeks; Charles River) were maintained in a temperature/humidity-controlled environment under a 12 h light/dark cycle with ad libitum access to food and water. As depicted in Figure 7A, each animal received four intraperitoneal injections, separated by 1 d, of either vehicle (saline) or PQ (10 mg/kg body weight; b.w.), according to a previously published dose (Manning-Bog et al., 2002; Harraz et al., 2008; Cristóvão et al., 2009). All animals were weighed at day 1 and 12. In the studies using the paradigm depicted in Figure 7A, 4 weeks before starting PQ intraperitoneal injection, animals were stereotaxically injected with various viral constructs at the right SN using the following coordinates: mediolateral, +2.0; anteroposterior, −5.3; dorsoventral, −6.8. Animals were organized into four groups: vector + vehicle: stereotaxically injected with adeno-associated virus (AAV) particles containing a green fluorescent protein (GFP) vector (vector) and then intraperitoneally injected with saline (vehicle) (n = 10); vector + PQ: stereotaxically injected with vector and then intraperitoneally injected with PQ (n = 10); shNox1 + PQ: stereotaxically injected with AAV particles harboring Nox1 shRNA and then intraperitoneally injected with PQ (n = 10); shNox1 + vehicle: stereotaxically injected with Nox1 shRNA and then intraperitoneally injected with vehicle (n = 8). AAV containing GFP vector was used as a negative control. Five days after the last PQ intraperitoneal injection, animals were killed. For Western blot analysis, brains were collected and total protein lysates from SN were prepared. For immunohistochemical analysis, animals were intracardially perfused before collecting the brains.
Figure 7.

Selective Nox1 targeting by AAV-mediated Nox1 knockdown in the rat SN. A, AAV2 viral particles and PQ injection paradigm diagram. To knockdown Nox1 in the SN, AAV2 particles harboring Nox1 shRNA were stereotaxically injected into the SN. PQ intraperitoneal injections were performed 4 weeks after AAV2 delivery. Rats were divided into four groups. Group vector + vehicle: stereotaxic injection of AAV2 particles containing GFP vector and then vehicle (saline) intraperitoneal injection; group vector + PQ: stereotaxic injection of AAV2 particles containing GFP vector and then PQ intraperitoneal injection; group shNox1 + PQ: stereotaxic injection of AAV2 particles harboring Nox1 shRNA-GFP and then PQ intraperitoneal injection; and group shNox1 + vehicle: stereotaxic injection of AAV2 particles harboring Nox1 shRNA-GFP and then vehicle intraperitoneal injection. Animals were given a total of four intraperitoneal injections of either vehicle or PQ (10 mg/kg b.w.) every 2 d. All groups were killed 5 d post last injection. B, Representative immunoblot and quantitative analysis of Nox1 protein determined in total lysates of rat ipsilateral SN tissues. β-Actin was used as an internal control. Nox1 protein levels were quantified using Quantity One software and normalized against β-actin. The results are expressed as percentage of vector + vehicle. Data are shown as the mean ± SEM. Statistical analysis was performed using one-way ANOVA followed by Bonferroni's multiple-comparison test; *p < 0.05, **p < 0.01, and ***p < 0.001. (C) Representative photomicrographs of Nox1-immunoreactivity in the SN sections of the contralateral and ipsilateral sides of brain sections. Scale bars, 50 μm.
Construction of shRNA delivery vector U6-CMV-EGFP/pAAV (AAV-Nox1 shRNA) and preparation of rAAV2 containing Nox1 shRNA
U6 promoter-driven shRNA expression system was established in AAV serotype 2 (AAV2) vector. Enhanced GFP (EGFP) expression is separately controlled by a cytomegalovirus (CMV) promoter as a marker for the transduction efficiency. Rat Nox1 shRNA was designed based on the siRNA sequence, which efficiently knocked down Nox1 expression in N27 cells (Cristóvão et al., 2009).
The plasmid DNA vector only or AAV-Nox1 shRNA was cotransfected with plasmids pHelper and pAAV-RC to HEK293 AAV cells using a standard calcium phosphate method. After 72 h, the cells were harvested and crude rAAV vector solutions were obtained by repeated freeze/thaw cycles. The cleared crude lysate was then applied on a heparin column. After the total lysate pass through the column, the matrix was washed twice with 25 ml of PBS with low NaCl (pH7.4, 0.1 m NaCl). The virus was then eluted with 15 ml of PBS with high NaCl (PBS; pH 7.4, 0.4 m NaCl). The elute was concentrated to ∼1 ml with a Millipore Biomax-100K NMWL filter device (UFV2BHK40) by centrifugation 4000 rpm, 15–40 min. To adjust the NaCl concentration to physiological levels, the filter device was refilled with PBS, pH 7.4, and the virus was concentrated to 250–300 μl. After removal of the virus-containing solution, the membrane of the filter device was washed three times with PBS, pH 7.4, which was added to the main part of the recombinant AAV2. The fractions containing high-titer rAAV vectors were collected and used for injection into animals. The number of rAAV genome copies was semiquantified by PCR within the CMV promoter region using primers 5′-GACGTCAATAATGACGTATG-3′ and 5′-GGTAATAGCGATGACTAATACG-3′. The final titers were 6.4 × 1011 genomes/μl (rAAV2-vector) and 5.5 × 1011 genomes/μl (rAAV2-NOX-1 shRNA). Each animal received 16.5 × 1011genomes of the respective rAAV-vector.
Construction of shRNA delivery vector pLVX-shRNA2-zsGreen1/LVX (Lenti-Nox1 shRNA) and preparation of lentivirus containing Nox1 shRNA
The same Nox1 shRNA used for AAV2 construct was cloned into a pLVX-shRNA2 vector containing zsGreen1 (Clontech). To make lentiviral particles, ViraPower Lentiviral Expression System (Invitrogen) was used. The three packaging plasmids pLP1, pLP2, and pLP/VSVG, were individually purified from the mixture based on the pattern of restriction enzyme digestion. pLVX-shRNA2-rNox1 and the three packaging plasmids were cotransfected to Lenti-X 293T cells (Clontech) according to the Xfect transfection reagent protocol (Clontech). For 6-well plates, 8 μg of each plasmid was mixed in a 1:1:1:1 ratio in 100 μl of Xfect polymer buffer and then added to the cells. After 48 h, the viral-containing medium was harvested and centrifuged briefly at 500 × g for 10 min to remove cellular debris and supernatant recovered and kept at −80°C until used.
Transient transfection of α-synuclein tagged with FLAG (N terminal) and Myc (C terminal)
Human α-synuclein was cloned into the p3xFLAG-myc-CMV-23 expression vector (Sigma) for N- and C-terminal tagging with FLAG and Myc, respectively. For transient overexpression of tagged α-synuclein, N27 cells were plated onto 24-well plates with coverslips at 0.5 × 105 cells per well 1 d before transfection. The next day, cells were transiently transfected with FLAG-WTsyn-myc. Briefly, 1 μg of plasmid DNA was mixed with 6 μl of Lipofectamine 2000 (Invitrogen) in 100 μl of Opti-MEM for 20 min before addition in the culture. After 6 h of incubation, the culture medium was changed and 100 μl of Nox1 shRNA/LVX viral particle was added to each well. Cells were maintained for an additional 36 h before treatment with 800 μm or 1000 μm PQ for 8 and 24 h.
Western blot
For Western blot, brain tissues were lysed on ice in radioimmunoprecipitation assay buffer containing 50 mm Tris/HCl, pH 8.0, 150 mm NaCl, 2 mm sodium orthovanadate, 1% Nonidet-P40 (NP-40), 0.5% sodium deoxycholate, and 0.1% SDS, containing 1% of a protease inhibitor mixture (AEBSF, pepstatinA, E-64, bestatin, leupeptin, and aprotinin). The soluble fraction was obtained and equal amounts of cell lysate protein were loaded in each lane of a 12% SDS-PAGE or 4/10% to 20% polyacrylamide gel. After electrophoresis and transfer onto a polyvinylidene difluoride (PVDF) membrane, specific protein bands were detected using appropriate primary antibodies (rabbit anti-Nox1, rabbit anti-α-synuclein, mouse anti-Ubiquitin, mouse anti-tyrosine hydroxylase (TH), and mouse anti-β-actin) and secondary antibodies conjugated to alkaline phosphatase or hydrogen peroxidase (anti-rabbit or anti-mouse) followed by Enhanced Chemifluorescence (ECF) detection or Enhanced Chemiluminescence (ECL).
Dot blot analysis
For dot blot, brain tissues were homogenized in a buffer containing 0.32 m sucrose, 1 mm NaHCO3, 1 mm MgCl2, 0.5 mm CaCl2, and 1% of a protease inhibitor mixture (AEBSF, pepstatinA, E-64, bestatin, leupeptin, and aprotinin). The soluble fraction was obtained by centrifugation at 1000 × g and 5 μl of each sample, containing the same amount of protein, was spotted in a PVDF membrane. Membrane was air dried for 4 h and blocked overnight at 4°C in 5% nonfat dry milk TBST (10 mm Tris-HCl, pH 7.8, 100 mm NaCl, 0.05% Tween 20) solution. Protein spots were detected using the primary antibody, rabbit anti-A11oligomer (0.5 μg/ml) from Invitrogen, and secondary antibody conjugated to hydrogen peroxidase (anti-rabbit) followed by ECL detection.
Measurement of oxidized proteins
The levels of protein carbonyl were measured in protein extracts from the SN tissues, using the OxiSelect Protein Carbonyl Immunoblot Kit (Cell Biolabs), according to the manufacturer's instructions, with small modifications. Briefly, 5% nonfat dry milk/PBST was used as blocking solution and antibody buffer, and the membrane was blocked for 1 h and incubated with the primary antibody overnight. Detection was performed using ECL.
Immunocytochemistry
After each respective treatment, cells were fixed in 4% paraformaldehyde (PFA) for 20 min and permeabilized with 0.1% Triton X-100 in PBS for 10 min. Blocking was performed by incubation with 20% goat or donkey serum in PBS containing 0.1% Tween 20 for 90 min at room temperature. The cells were then incubated for 120 min at room temperature with the following primary antibodies, according to the aim of the experiment: goat anti-Nox1 (1:50), rabbit anti-TH (1:10,000), mouse anti-Flag (1:150), mouse anti-α-synuclein (1:150), and rabbit anti-A11 (2.5 μg/ml). After washing, cells were incubated for 120 min with the appropriate secondary antibodies conjugated to Alexa Fluor 647 or Alexa Fluor 488 (1:1000). For nuclear visualization coverslips were incubated with 2 μm Hoechst for 10 min. For quantification of Flag+ and α-synuclein+ cells, >30 different fields per coverslip were analyzed on a Nikon inverted fluorescent microscope under a 20× and 40× magnification.
Immunohistochemistry
Following perfusion with saline and 4% PFA in PBS, brains were removed, and forebrain and midbrain blocks were immersion fixed in 4% PFA and cryoprotected in sucrose. Serial coronal sections (40 μm) were cut on a cryostat, collected in cryopreservative solution, and stored at −20°C. For immunolabeling studies, sections were incubated at room temperature with blocking solution for 1 h (5% FBS and 0.3% Triton X-100 in PBS, pH 7.5) and then with primary antibodies overnight. Finally, sections were incubated with secondary antibodies in blocking solution at room temperature for 1 h. The primary antibodies used were mouse anti-TH (1:10,000), rabbit anti-Nox1 (1:500), goat anti-4-HNE (1:700), mouse anti-ubiquitin (1:250), rabbit anti-α-synuclein (1:150), and rabbit anti-A11 oligomers (2.5 μg/ml). The secondary antibodies were biotinylated anti-rabbit IgG, anti-goat IgG, or anti-rat IgG (1:200). The staining procedure was performed as indicated by the manufacturer of the Vectastain ABC kit and the reaction product visualized using 3,3′-diaminobenzidine (DAB) reagent in TBS containing 0.02% H2O2. The numbers of TH-immunoreactive cells in the SN were counted using an optical fractionator. Analysis was performed using a system consisting of a Nikon Eclipse E600 microscope (Morrell Instruments) equipped with a computer-controlled LEP BioPoint motorized stage (Ludl Electronic Products), a DEI-750 video camera (Meyer Instruments), a Dell Dimension 4300 computer (Dell), and the Stereo Investigator (v. 4.35) software program (Microbrightfield).
Proteinase K digestion of cells and tissues for α-synuclein aggregates detection
Immunocytochemistry for proteinase K (PK)-resistant α-synuclein was performed based on a methodology reported previously (Neumann et al., 2002), with some modifications. Briefly, fixed cells were permeabilized with 0.1% Tween 20 and then digested for 30 min at 37°C with PK (10 μg/ml). PK was inactivated with 3 m guanidine thiocyanate in 10 mm Tris-HCl solution for 10 min. Between each step, cells were washed gently three times with PBS. Cells were then incubated for 60 min with blocking solution containing 10% donkey serum followed by an overnight incubation with mouse anti-α-synuclein antibody (1:150). The day after, cells were incubated with secondary antibodies conjugated to Alexa Fluor 647 donkey anti-mouse for 60 min and with 2 μm Hoechst solution for 10 min. For PK-resistant α-synuclein evaluation in rat SN, 40 μm sections from 4% PFA fixed tissues were washed twice in distillated water with 0.1% Tween 20, and then incubated for 30 min in TBST. The tissues were incubated for 90 min at 55°C with 50 μg/ml PK in TBST and further washed three times in TBS. PK was denatured by incubating the tissues in a 3 m guanidine thiocyanate in 10 mm Tris-HCl solution for 10 min. Sections were incubated with blocking solution for 1 h (TBST with 0.2% casein) and then with rabbit anti-α-synuclein (1:150) at 4°C overnight. Finally, sections were incubated with biotinylated anti-rabbit IgG (1:200 blocking solution) at room temperature for 1 h. The staining procedure was performed as indicated by the manufacturer of the Vectastain ABC kit and the reaction product visualized using DAB reagent in TBS containing 0.02% H2O2.
Data analysis and statistics
Statistical analysis was performed with GraphPad Prism v.5 (GraphPad Software). Data are expressed as percentages of values obtained in control conditions, and are presented as mean ± SEM of at least four animals (in vivo studies). Statistical analyses were performed using the one-way ANOVA or two-way ANOVA followed by Bonferroni's multiple-comparison test, or using Student's t test. Values of p < 0.05 were considered significant.
Reagents
FBS and gentamycin were purchased from Invitrogen BRL. Phenylmethylsulfonyl fluoride, NP-40, SP600125, Brij35, and bupropion were purchased from Sigma Chemicals. Mouse anti-TH was obtained from Transduction Laboratories; rabbit anti-Nox1, rabbit anti-α-synuclein, and mouse anti-ubiquitin were obtained from Santa Cruz Biotechnology; and mouse anti-α-synuclein from BD Transduction Laboratories. Goat anti-4-HNE and EGF were purchased from Millipore Bioscience Research Reagents. Rabbit anti-A11, Alexa Fluor 488 or Alexa Fluor 647, Hoechst 33342, Lipofectamine, ViraPower Lentiviral Expression System, 10–20% SDS polyacrylamide gel and 10–20% tricine gel, laminin, glutamax, DMEM/F12, and B27 supplement were purchased from Invitrogen. ECF Western Blotting kit was obtained from GE Healthcare Bioscience. Vectastain ABC kit, biotinylated anti-rabbit, anti-mouse IgG, or anti-goat IgG were from Vector Laboratories. Taq polymerase was from Roche Applied Science. PQ, protease inhibitor mixture (AEBSF, aprotinin, bestatin hydrochloride, E-64-[N-(trans-epoxysuccinyl)-l-leucine 4-guanidinobutylamide], leupeptin, pepstatin A), heparin, PK, and guanidine thiocyanate were from Sigma-Aldrich. bFGF was purchased from Peprotech. Millipore Biomax-100K NMWL filter device (UFV2BHK40) was purchased from Millipore. CMV-IRES-hrGFP/AAV system was purchased from Stratagene and the p3xFLAG-myc-CMV-23 expression vector from Sigma. pLVX-shRNA2-zsGreen1, Lenti-X 293T cells, and Xfect transfection reagent were purchased from Clontech. OxiSelect Protein Carbonyl Immunoblot Kit was purchased from Cell Biolabs. All other chemicals of reagent grade were from Sigma Chemicals or Merck.
Results
α-Synuclein and Nox1 increases in human dopaminergic neurons after PQ treatment
To evaluate the effects of PQ treatment on α-synuclein and Nox1 expression in human dopaminergic neurons, human ventral mesencephalic neuronal progenitor cell line, ReNcell VM, was used (Donato et al., 2007). We first differentiated ReNcell VM into human midbrain neurons. After differentiation for 14 days, the obtained cultures were immunopositive for Tuj-1, a neuron-specific class III β-tubulin. Moreover, the majority (80%) of differentiated cells were TH positive, a specific marker of dopaminergic neuron, with an increased level of TH protein compared with undifferentiated cells (Fig. 1A). The effects of PQ on expression of α-synuclein and Nox1 were evaluated on these differentiated human dopaminergic cells. We observed that differentiated dopaminergic neurons express α-synuclein, which increases over time under PQ treatment (Fig. 1B). Similarly, low basal level of Nox1 was highly elevated by PQ treatment, as shown by immunocytochemistry and Western blot analyses (Fig. 1C).
Figure 1.
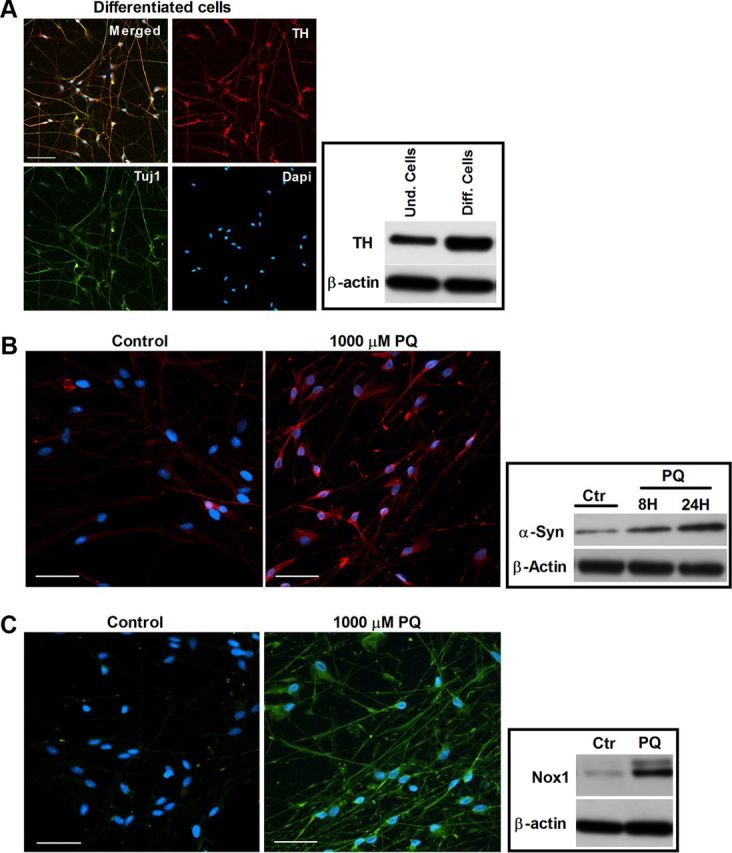
Increases in α-synuclein and Nox1 in human dopaminergic neurons exposed to PQ. A, Characterization of human ventral mesencephalic neuronal progenitor cell line, ReNcell VM, after differentiation (human dopaminergic neurons). Left, Depicts representative photomicrographs of TH, Tuj1, and DAPI immunostaining of ReNcell VM after 14 days differentiation. Right, Displays the expression of TH protein in ReNcell VM, before and after differentiation. B, α-Synuclein levels in differentiated human dopaminergic cells exposed to PQ. Left, Shows α-synuclein immunoreactivity (red). Right, Represents α-synuclein protein levels in immunoblot. C, Nox1 levels in differentiated human dopaminergic cells exposed 8 h to PQ. Left, Shows Nox1 immunoreactivity (green). Right, Illustrates Nox1 protein levels in immunoblot. β-Actin was used as an internal control. Und, undifferentiated; Diff, differentiated; Ctr, control; PQ, paraquat. Scale bars: 50 μm.
Overall, the results indicate that α-synuclein may be a key player in PQ-mediated dopaminergic neuronal toxicity. Moreover, the result also suggests that Nox1 may have an important role in the mechanism of human dopaminergic neurodegeneration as induced by PQ.
Nox1 knockdown significantly reduces PQ-induced α-synuclein expression and aggregation in dopaminergic cells
To further study the role of Nox1 in α-synucleinopathy caused by PQ in dopaminergic cells, the rat dopaminergic neuronal cell line, N27 cells, was investigated. PQ significantly increased the levels of α-synuclein expression. We observed 55 and 61% increases of α-synuclein protein levels in N27 cells exposed to 800 μm PQ for 8 and 24 h, respectively. When exposed to a 1000 μm dose of PQ, an increase of 60 and 27%, respectively, for 8 and 24 h incubation times was detected (Fig. 2A). Immunocytochemical evaluation showed that α-synuclein aggregation was also induced by PQ. As shown in Figure 2B, we could see increased immunoreactivity for α-synuclein in cultures treated with PQ, and, moreover, a pattern of aggregated α-synuclein was observed in treated cultures, which was not detected in the untreated ones (Fig. 2B, arrowheads). The quantification of aggregated α-synuclein-positive cells revealed an increase of α-synuclein aggregation in cultures exposed to PQ compared with the control. As shown in Figure 2D (open bars), a statistical increase of 62 and 64% in aggregation was found in cultures exposed for 24 h to 800 and 1000 μm PQ, respectively. To confirm the effect of PQ in α-synuclein aggregation, we have further evaluated the levels of α-synuclein resistant to PK digestion, since it was previously reported that α-synuclein aggregates are resistant to limited PK digestion (Neumann et al., 2002). As shown in Figure 3A, N27 cells exposed to PQ depict higher PK-resistant α-synuclein immunoreactivity, an indicator of higher α-synuclein aggregation. We have further evaluated α-synuclein aggregation by investigating the levels of A11 immunoreactivity in untreated and PQ-treated N27 cell, as the anti-A11 oligomer antibody was previously reported to efficiently detect α-synuclein aggregation (Winner et al., 2011). Figure 3B shows high A11 immunoreactivity in cultures exposed to PQ when compared with the controls, clearly indicating increased α-synuclein aggregation induced by PQ. As expected, this group of results infers that PQ induces increased levels of α-synuclein expression as well as aggregation.
Figure 2.
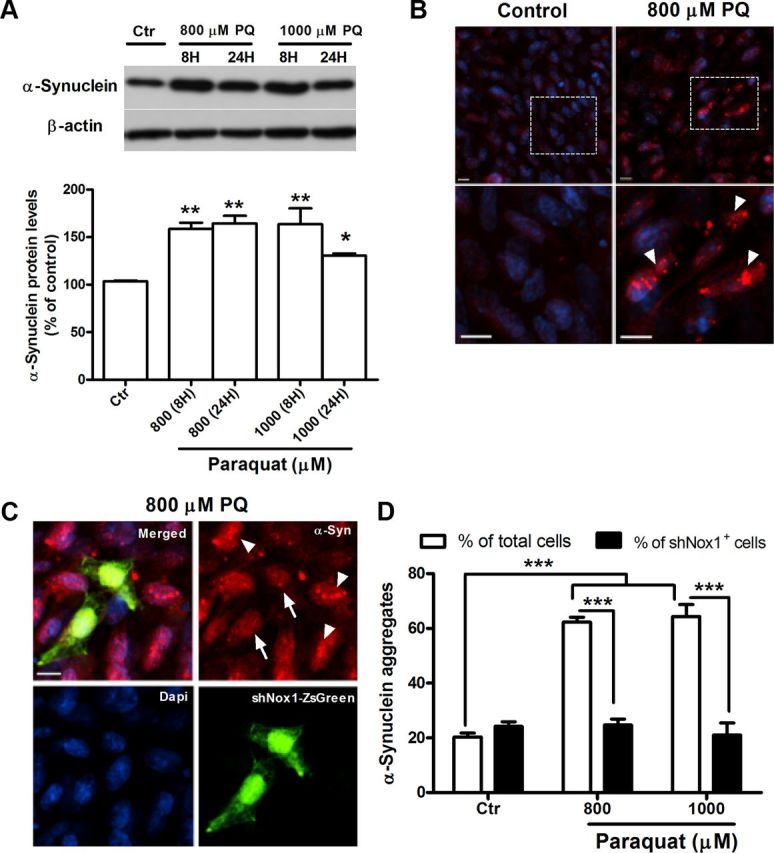
PQ induces increases of α-synuclein expression and aggregation in N27 cells, an event prevented by Nox1 knockdown. A, Representative immunoblot and quantitative analysis of α-synuclein protein levels. α-Synuclein protein was determined in total lysates of N27 cells exposed to PQ or control. β-Actin was used as an internal control. PQ significantly increased α-synuclein protein levels, which were quantified using Quantity One software and normalized against β-actin. B, Photomicrographs of aggregated α-synuclein immunoreactivity (red) in control and PQ-treated cells. The bottom shows higher magnification of respective boxed areas in the top. C, α-Synuclein fluorescence immunostaining of N27 cells incubated with Nox1 shRNA/LVX (shNox1-ZsGreen) viral particles for 36 h and then exposed to 800 μm PQ. shNox1-ZsGreen-infected cells were identified by green fluorescence (ZsGreen) in cells. D, Quantification of the cells depicting the bright, punctuated fluorescence, like the ones indicated with arrowheads in B and C. More than 30 assigned fields were analyzed in each independent experiment and in average the minimum number of total cells counted per condition was 700 cells. Data are shown as the mean ± SEM. Statistical analysis was performed using one-way ANOVA or two-way ANOVA, followed by Bonferroni's multiple-comparison test; *p < 0.05, **p < 0.01, and ***p < 0.001. Arrowheads specify cells with aggregated α-synuclein pattern, and the arrow indicates N27 cells showing double-staining for shNox1-ZsGreen and α-synuclein. Ctr, control; PQ, paraquat. Scale bars: 10 μm.
Figure 3.
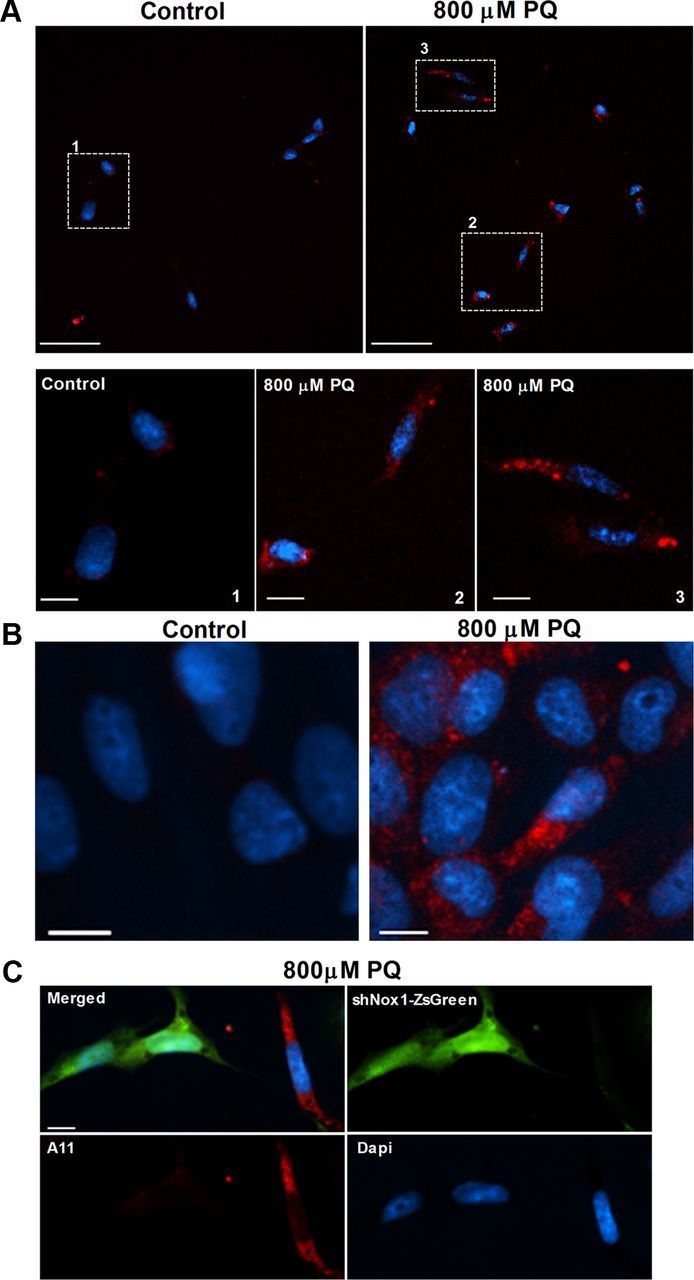
PQ increases levels of PK-resistant α-synuclein and A11 oligomer formation in N27, which is reversed by Nox1 knockdown. A, PK-resistant α-synuclein immunoreactivity in control and PQ-treated N27 cells. The bottom (scale bars: 10 μm) shows higher magnification of respective boxed areas shown in the top (scale bars: 50 μm). B, C, A11 immunoreactivity of control or PQ-treated N27 cells (B) and in N27 cells incubated with Nox1 shRNA/LVX (shNox1-ZsGreen) viral particles for 36 h exposed to 800 μm PQ (C). Scale bars: (for B, C), 10 μm.
To explore the contribution of Nox1 in the changes of α-synuclein induced by PQ, we knocked down Nox1, using lentivirus-mediated Nox1 shRNA overexpression (shNox1-ZsGreen), and exposed N27 cells to PQ. The results showed that PQ-induced α-synuclein aggregation was lower in cells overexpressing Nox1 shRNA (Fig. 2C). After quantifying the number of cells depicting both shNox1-ZsGreen and α-synuclein aggregates, no statistical differences were found between untreated and PQ-treated cultures as shown in Figure 2D (solid bars). The same result was found when analyzing A11 immunoreactivity in those cells. As shown in Figure 3C, shNox1-ZsGreen-positive cells clearly had lower immunoreactivity for A11 compared with shNox1-ZsGreen-negative cells.
Next, we sought to understand if Nox1 has a role only in the expression pathway of α-synuclein leading to protein increase and subsequent aggregation, or if it was also acting directly in its aggregation capability. Flag-tagged wild-type (WT) α-synuclein and shNox1-ZsGreen were overexpressed in N27 cells, and then cells were exposed to PQ. Strong cytoplasmic aggregation was induced by PQ treatment as detected by anti-flag immunostaining (Fig. 4A). A significant 2.5-fold increase of aggregation was observed in cells exposed to PQ when compared with control cells (Fig. 4C, open bars). Furthermore, when aggregation was analyzed in shNox1-ZsGreen-positive cells exposed to PQ, aggregation levels were not statistically different from control cells (Fig. 4C, solid bars). Aggregation was decreased by a 2.5-fold in shNox1-ZsGreen-positive cells (Fig. 4B, arrow) when compared with shNox1-ZsGreen-negative cells (Fig. 4B, arrowhead) for both concentrations of PQ (Fig. 4B,C).
Figure 4.

Nox1 knockdown inhibits aggregation of overexpressed WT α-synuclein in N27 cells induced by PQ. A, Representative pictures of flag-tagged WT α-synuclein immunoreactivity (red) in control and PQ-treated cells. Scale bars: (for top), 50 μm; (for bottom), 10 μm. B, Flag-tagged WT α-synuclein fluorescence immunostaining of N27 cells incubated with Nox1 shRNA/LVX (shNox1-ZsGreen) viral particles and exposed to 800 μm PQ. shNox1-ZsGreen-infected cells were identified by green fluorescence (ZsGreen) in cells. Scale bar, 10 μm. C, Quantification of the bright, punctuated fluorescent cells, indicated with arrowheads in A and B. More than 20 assigned fields were analyzed in each independent experiment and in average the minimum number of total cells counted per condition was 400 cells. Data are shown as the mean ± SEM. Statistical analysis was performed using one-way ANOVA or two-way ANOVA, followed by Bonferroni's multiple-comparison test; **p < 0.01 and ***p < 0.001. Arrowheads specify cells with aggregated α-synuclein pattern, and arrows indicate N27 cells depicting double-staining for shNox1-ZsGreen and α-synuclein-flag. Ctr, control; PQ, paraquat.
Altogether the above results are highly suggestive that Nox1 is an important intermediary in regulation of both expression and the aggregation process of α-synuclein in dopaminergic cells stressed with PQ.
PQ intraperitoneal injection causes increases in α-synuclein and Nox1 protein level as well as oxidative stress in the rat SN
To validate the significance of our in vitro results, we moved forward to in vivo studies, using a PQ-inducing rat model of PD. Our first observations showed that PQ injection in rats induced an increase of 50% in α-synuclein protein levels in the SN as determined by Western blot (Fig. 5A). α-Synuclein immunoreactivity in the SN was also increased after PQ administration, as shown in Figure 5B. The involvement of Nox1 in PQ-mediated dopaminergic cell death in mice was reported in a previous study of our group (Cristóvão et al., 2009). In the present study, PQ insult also induced increased Nox1 protein level in the rat SN. As shown in Figure 6A, animals exposed to PQ showed 58% higher levels of Nox1 protein than the group treated with vehicle. The upregulation of Nox1 in the SN of rats injected with PQ was further confirmed by immunohistochemistry as shown in Figure 6B.
Figure 5.
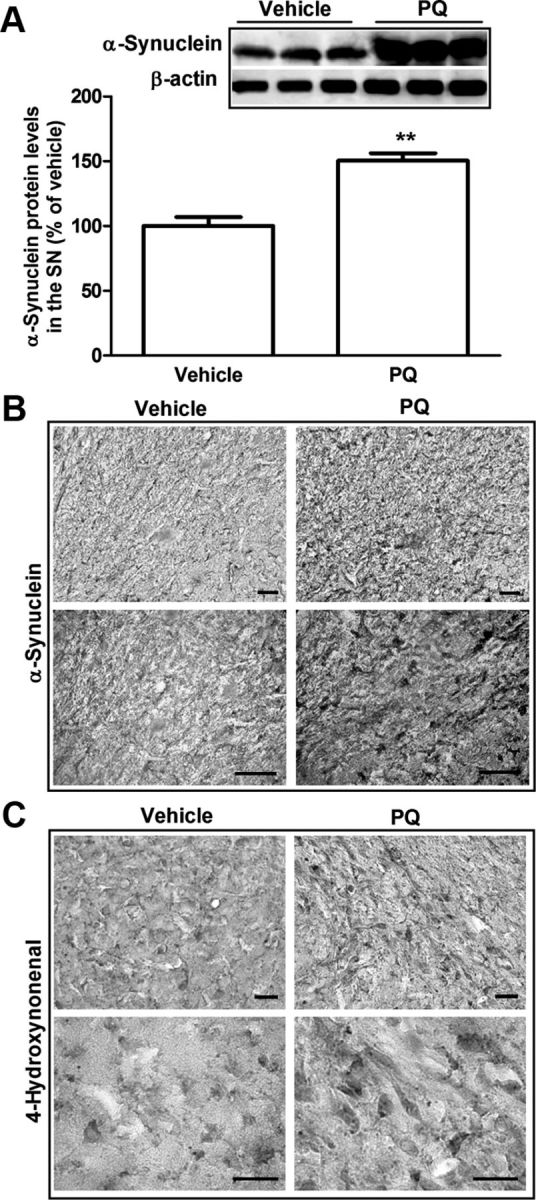
Increase in α-synuclein and lipid peroxidation in the SN of rats administered with PQ. A, Representative immunoblot and quantitative analysis of α-synuclein protein levels. α-Synuclein protein was determined in the total lysates of SN tissues of rats injected with vehicle or PQ by immunoblot analysis. β-Actin was used as an internal control. PQ significantly increased α-synuclein protein levels, which were quantified using Quantity One software and normalized against β-actin. B, C, Representative photomicrographs of α-synuclein (B) and 4HNE (C) immunostaining in the SN of rats treated with vehicle or PQ. Data are shown as the mean ± SEM. Statistical analysis was performed using the Student's t test; **p < 0.01. Scale bars: 50 μm.
Figure 6.
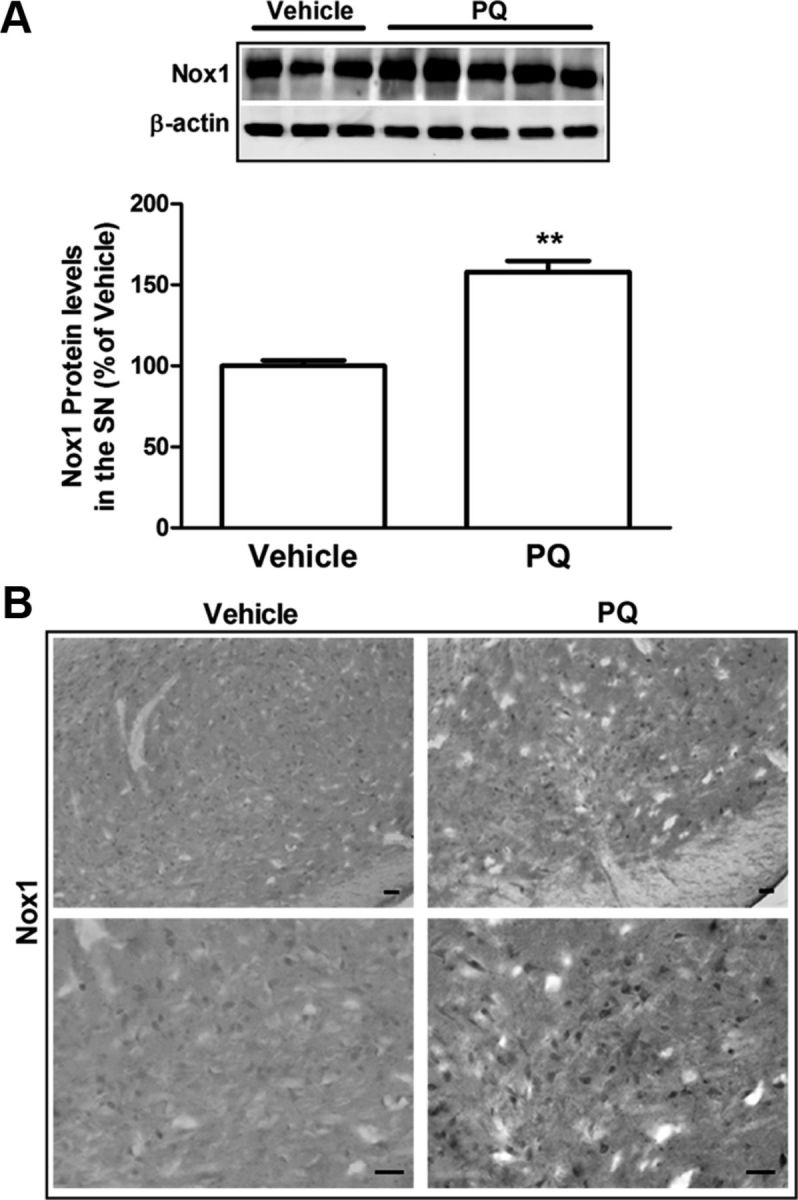
Increase in Nox1 protein levels in the SN of rats injected with PQ. A, Representative immunoblot and quantitative analysis of Nox1 protein levels. Nox1 protein was determined in the total lysates of SN tissues of rats injected with vehicle or PQ by immunoblot analysis. β-Actin was used as an internal control. PQ significantly increased Nox1 protein, which was quantified using Quantity One software and normalized against β-actin. B, Representative photomicrographs of Nox1-immunoreactivity in the SN sections of rats injected with vehicle or PQ. Nox1 immunoreactivity in the SN was increased in PQ-injected animals compared with vehicle. The result is expressed as percentage of vehicle. Data are shown as the mean ± SEM. Statistical analysis was performed using the Student's t test; **p < 0.01. Scale bars: 50 μm.
Nox enzymes are responsible for ROS production; we then evaluated if increases in Nox1 were also accompanied by increased oxidative stress markers in rat tissues exposed to PQ. As depicted in Figure 5C, an increase in 4-HNE immunoreactivity, a well established maker for lipid peroxidation, was found in the SN of rats treated with PQ compared with rats treated with vehicle. The above results are in accordance with the ones observed in vitro, and emphasize that under PQ insult, α-synuclein, Nox1, and oxidative stress may act as partners to enhance dopaminergic neurodegeneration.
The establishment of the rat model used in the present work was based on our previous reported results showing in mice the loss of dopaminergic neurons in the SN after PQ exposure (Cristóvão et al., 2009).
The specific knockdown of Nox1 reduced rat dopaminergic neuronal loss induced by PQ
To investigate the role of Nox1 in oxidative stress, dopaminergic neuronal death, and α-synuclein expression and aggregation changes induced by PQ in rats we achieved Nox1 knockdown in the SN by using AAV-mediated Nox1 shRNA overexpression. Nox1 knockdown was achieved by stereotaxic delivery of AAV2 particles into the rat SN. PQ intraperitoneal injections were performed 4 weeks after the AAV2 injection, as depicted in Figure 7A. AAV2 containing GFP vector was used as a negative control. To verify Nox1 knockdown efficiency, Nox1 levels in the SN of each group of animals were investigated by Western blot and immunohistochemistry analysis. Nox1 knockdown in the SN significantly reduced PQ-mediated Nox1 increases (Fig. 7B,C). As shown in Figure 7B, animals treated with vector + PQ showed statistically higher levels of Nox1 protein (70%) compared with animals treated with vector + vehicle. Compared with animals treated with vector + PQ, animals treated with shNox1 + PQ showed a 40% decrease in Nox1 protein levels in the SN (Fig. 7B). Nox1 levels in the SN of each group of animals were also investigated by immunohistochemistry, confirming the decrease in Nox1 immunoreactivity in the SN of animals exposed to Nox1shRNA + PQ compared with the ones exposed to vector + PQ (Fig. 7C). These results confirmed that AAV-mediated Nox1 knockdown in vivo significantly reduced PQ-mediated increase in Nox1 level, validating our knockdown method.
To investigate the contribution of Nox1 to the dopaminergic neurotoxicity induced by PQ in each group of animals, the levels of TH protein in the SN were investigated by Western blot and the numbers of TH-positive dopaminergic neurons in the substantia nigra pars compacta were obtained by stereological analysis. Administration of vector + PQ significantly reduced TH protein levels to 65% compared with the control group injected with vector + vehicle, while TH protein levels were recovered to 87% in the group in which Nox1 was knocked down before PQ exposure (Fig. 8B). The stereological count of TH-positive neurons showed that Nox1 knockdown significantly reduced PQ-elicited dopaminergic neuronal loss from 37% in the group treated with vector + PQ to 13% in the Nox1shRNA + PQ group (Fig. 8A). In addition, we found that Nox1 knockdown also reduced oxidative stress levels, as shown by the levels of lipid peroxidation and protein oxidation. Increased immunoreactivity of 4-HNE (Fig. 8C) and protein carbonyl (Fig. 8D,E) in animals treated with PQ was decreased in the Nox1shRNA + PQ group. Protein carbonyl levels were significantly increased by 95% after PQ exposure when compared with vector + PQ group, and reduced to 22% when Nox1 was knocked down (Fig. 8E). These results have shown that Nox1 knockdown reduced dopaminergic neuronal death and oxidative stress induced by PQ, which was in accordance with our two previous report observations showing that the Nox system plays an important role in PQ- and 6-OHDA-mediated dopaminergic neurotoxicity.
Figure 8.

Nox1 knockdown reduced SN dopaminergic neuronal death induced in rats administered with PQ. A, Representative photomicrographs of TH immunostaining and quantitative analysis of TH-positive dopaminergic neurons in the SN of rats after Nox1 knockdown. Representative photomicrographs of TH immunoreactivity in the SN of the contralateral and ipsilateral sides of brain sections of the four experimental groups. TH-positive neurons in the ipsilateral side were stereologically counted. B, Representative immunoblot and quantitative analysis of TH protein levels. TH protein was determined in total lysates of the rat's SN tissues in the ipsilateral side by immunoblot analysis. β-Actin was used as an internal control. TH protein levels were quantified using Quantity One software and normalized against β-actin. C, Representative photomicrographs of 4-HNE immunostaining in the SN of the contralateral and ipsilateral sides of brain sections of rats from shNox1 + PQ group. Scale bars, 50 μm. D, E, Immunoblot (D) and quantitative analysis (E) of protein carbonyl levels determined in total lysates of rats' ipsilateral SN tissues. β-Actin was used as an internal control. The results are expressed as percentage of vector + vehicle. Data are shown as the mean ± SEM. Statistical analysis was performed using one-way ANOVA followed by Bonferroni's multiple-comparison test; *p < 0.05, **p < 0.01, and ***p < 0.001.
Increased expression and aggregation of α-synuclein induced by PQ relies on Nox1 protein in the rat SN
Herein, we sought to evaluate the involvement of Nox1 in the effect of PQ on α-synuclein expression levels and aggregation in vivo. We investigated α-synuclein, protein aggregation, and ubiquitin levels in the SN of each group of animals by Western blot, dot blot, and immunohistochemical analyses. We found significant increases of 54, 68, and 43%, in α-synuclein, A11-positive oligomers and ubiquitin protein levels, respectively, in the vector + PQ-treated group when compared with the vector + vehicle group (Figs. 9A, 10A,C). Moreover, Nox1 knockdown reduced by 37, 50, and 43% the PQ-mediated α-synuclein, A11 oligomers, and ubiquitin levels, respectively, compared with the vector + PQ (Figs. 9A, 10A,C). To further evaluate in vivo the effect of Nox1 knockdown on PQ-induced α-synuclein aggregation we have also investigated the levels of α-synuclein resistant to PK digestion. We were able to observe an increase in PK-resistant α-synuclein immunoreactivity in the rat SN exposed to vector + PQ, but not in the vector + vehicle group. A significant reduction in PK-resistant α-synuclein staining was observed in the group in which Nox1 was knocked down before PQ injection (Nox1shRNA + PQ) (Fig. 9B). Immunohistochemistry evaluations revealed a significant increase in the immunoreactivity of A11 oligomers in the SN of rats exposed to PQ, which was decreased in the group exposed to PQ in which Nox1 was knocked down (Fig. 10B). The involvement of Nox1 in PQ-induced changes in ubiquitin in the SN was also evaluated and the significant increase in ubiquitin immunoreactivity observed in the rat SN exposed to vector + PQ was reversed by Nox1 knockdown (Nox1shRNA + PQ) (Fig. 10D). Together our results are highly suggestive of an active role of Nox1 in α-synucleinopathy induced by PQ at transcriptional levels as well as post-translational aggregation mechanism.
Figure 9.

Nox1 knockdown reduced PQ-mediated α-synuclein increase in the SN dopaminergic neurons. A, Representative immunoblot and quantitative analysis of α-synuclein protein levels. α-Synuclein protein was determined in total lysates of the rats SN tissues in the ipsilateral side by immunoblot analysis. β-Actin was used as an internal control. α-Synuclein protein levels were quantified using Quantity One software and normalized against β-actin. The results are expressed as percentage of vector + vehicle. Data are shown as the mean ± SEM. Statistical analysis was performed using one-way ANOVA followed by Bonferroni's multiple-comparison test; *p < 0.05 and **p < 0.01. B, Representative photomicrographs of PK-resistant α-synuclein immunoreactivity in the ipsilateral SN of brain sections of the four experimental groups. Increased PK-resistant α-synuclein immunostaining observed in the vector + PQ group was significantly decreased by Nox1 knockdown as observed in shNox1 + PQ group. Scale bars, 50 μm.
Figure 10.

Nox1 knockdown reduced PQ-mediated A11 oligomers and ubiquitin increase in the SN dopaminergic neurons. Representative immunoblot and quantitative analysis of A11 oligomers (A) and ubiquitin (C) levels determined in total lysates of the rats SN tissues in the ipsilateral side by immunoblot analysis. β-Actin was used as an internal control. A11 oligomer levels were quantified using Quantity One software and normalized against β-actin. The results are expressed as percentage of vector + vehicle. Data are shown as the mean ± SEM. Statistical analysis was performed using one-way ANOVA followed by Bonferroni's multiple-comparison test; *p < 0.05, **p < 0.01 Representative photomicrographs of A11 oligomers (B) and ubiquitin (D) immunoreactivity in the ipsilateral SN of brain sections of the three experimental groups. B, Bottom right shows higher magnification of the respective boxed area shown in the right top. Scale bars, 50 μm.
Discussion
In the present work, we perceive Nox1 as a crucial intermediary, between an environmental factor responsible for oxidative stress condition and α-synuclein transcriptional regulation and aggregation. We provide evidence that suggests, under oxidative stress, as induced by PQ, α-synuclein expression and aggregation levels are increased, which can be ameliorated to normal by Nox1 knockdown. And most important, dopaminergic loss in the SN of rats exposed to PQ can also be recovered by Nox1 knockdown, suggesting that Nox1-derived ROS play a crucial role in α-synuclein pathology as along with dopaminergic neuronal degeneration.
α-Synuclein is a distinctive genetic factor in PD pathogenesis, in which its alterations and mutations were linked to the development of the disease (Beyer et al., 2009; Cookson, 2009). Oxidative stress has also been largely mentioned as a strong contributor to the development of the disease, and it has also been involved in PQ-induced dopaminergic neurodegeneration (Dexter et al., 1994; Alam et al., 1997; Zhang et al., 1999). Physiologically, ROS are generated as a byproduct of several biological reactions from organelles like mitochondria, and Nox is the specialized system that produces ROS, but not as a byproduct (Sorce and Krause, 2009), and our recent studies demonstrated that Nox1 serves as a major contributing factor in dopaminergic neuronal degeneration in both 6-OHDA and PQ-mediated PD rodent models (Cristóvão et al., 2009; Choi et al., 2012). Our results using ReNcell VM cultures, which have been previously validated as an in vitro model of human dopaminergic neurons (Donato et al., 2007; Wood-Kaczmar et al., 2008), showed an increase in Nox1 level following PQ exposure (Fig. 1C). Nox1 and the oxidative stress marker were also found increased in rats exposed to PQ (Figs. 5, 6). The involvement of Nox1 in the mechanism of PQ-induced neurotoxicity was first demonstrated by our study, suggesting that Nox1 is involved in oxidative stress and consequent dopaminergic neuronal death (Cristóvão et al., 2009). More recently, we reported that Nox1-induced ROS also contributes to dopaminergic neurodegeneration induced by 6-OHDA (Choi et al., 2012), a well known toxin used to mimic PD pathogenesis in vitro and in vivo (Javoy et al., 1976; Terzioglu and Galter, 2008). We showed that the nuclear localization of Nox1 is responsible for nuclear DNA damage and degeneration of dopaminergic neurons after 6-OHDA treatment. Altogether this evidence emphasizes the importance of Nox1 as a crucial participant in dopaminergic neurodegeneration.
α-Synuclein point mutations, A30P, A53T, and E46K, were found in the familial forms of early onset PD and they are responsible for the changes in α-synuclein aggregation properties (Hardy et al., 2009). Interestingly, elevated expression of WT α-synuclein due to the multiplications of SNCA has also been identified in early onset familiar PD (Singleton et al., 2003; Chartier-Harlin et al., 2004), leading to the view that WT protein could cause PD in a dose-dependent manner. Although this fact fortifies the importance of the transcriptional regulation of α-synuclein, relatively few studies have focused on the role of oxidative stress in the expression level and transcriptional control of α-synuclein. This is at least partly due to the lack of proper in vitro and in vivo model systems that successfully demonstrate the increased endogenous α-synuclein level. Importantly, in the current work, both in vitro and in vivo models showed significant changes in α-synuclein expression under PQ exposure. The levels of α-synuclein in ReNcells and N27 cells cultures exposed to PQ were prominently increased with time (Figs. 1B, 2A), and significant increase in α-synuclein was also observed in the rat SN exposed to PQ as well (Fig. 5). Previous observations have shown that PQ increases α-synuclein expression levels and aggregation (Uversky et al., 2001; Manning-Bog et al., 2002), and that could be directly related to PQ-derived ROS generation (Krishnan et al., 2003). However, the molecular mechanism behind this effect is still elusive. Altogether these findings are suggestive of a possible relationship between increased ROS and the transcriptional regulation of α-synuclein, consistent with studies showing that toxic insults involving ROS production induce increased α-synuclein levels in the SN (McCormack et al., 2005). In PC12 cells as well as in primary cortical neurons from rat, it was demonstrated that α-synuclein expression in response to neurotrophins is regulated by the MAP/ERK and PI3-K pathways (Clough and Stefanis, 2007; Clough et al., 2011), which are also known to be activated under oxidative stress conditions (Miller et al., 2009). This suggests a possible relationship between increased ROS and transcriptional regulation of α-synuclein through these pathways. The above idea reinforces our hypothesis that Nox1-ROS generation might be a key regulator controlling α-synuclein expression. Nevertheless, the effects of ROS in transcriptional regulation are broad, including epigenetic alterations (Zawia et al., 2009), transcription factors binding regulation (Clough et al., 2009), or DNA damage (Turk et al., 1995). How Nox1-derived ROS regulates the transcription of α-synuclein remains to be investigated.
In addition to the effect of PQ in α-synuclein expression levels, we also found that PQ increased the aggregation of α-synuclein (Figs. 2–4) in N27 cells as well as in the rat SN, accompanied with increased levels of ubiquitin (Figs. 9, 10C,D). These findings are in agreement with previous reports showing increased α-synuclein aggregation in mice exposed to PQ (Manning-Bog et al., 2002). Based on this, we sought to enlarge our view of the disease paradigm and search for a potential relationship between Nox1 and α-synuclein aggregation. Nox1 knockdown achieved by viral delivery of shRNA against Nox1 significantly reduced α-synuclein aggregation in both in vitro and in vivo. A large number of reports have similarly shown that AAV-mediated shRNA delivery to the CNS for targeted knockdowns of specific genes can be achieved (Harper et al., 2005), including two of our recent works (Choi et al., 2011, 2012). On the other hand, the lentivirus system that delivers genes to cells showed higher infection efficiency then AAV2, but with less specificity, and was able to infect divided and nondivided cells. In that sense this system is more suitable for in vitro gene delivery using a cell culture system containing only one type of cells. Lentivirus-mediated Nox1 knockdown in N27 cells led to significant reduction in PQ-induced α-synuclein aggregation. Several methods were used for this evaluation and we found clear evidence that Nox1 is involved in the aggregation process of α-synuclein induced by PQ (Figs. 2C,D, 3C, 4B,C). Importantly, Nox1 knockdown also prevented aggregation of WT α-synuclein overexpressed, suggesting that along with its involvement in the transcriptional regulation, Nox1-induced ROS may also play a role in stabilizing the protein, leading to aggregation of α-synuclein.
AAV2-mediated Nox1 knockdown in the rat SN was shown to be effective not only in reducing Nox1 protein levels in the SN (Fig. 7B,C), but also in reducing oxidative stress (Fig. 8C) and dopaminergic neuronal death (Fig. 8A) induced by PQ. Nox1 knockdown induced a significant decrease in the total levels of α-synuclein expression (Fig. 9A) after PQ treatment, as well as a decrease in α-synuclein aggregation, as demonstrated by a decrease in PK-resistant α-synuclein (Fig. 9B). A11 oligomers (Fig. 10A,B) and ubiquitin (Fig. 10C,D) levels were also decreased, indicating that PQ-mediated α-synuclein aggregation is partially regulated by Nox1-derived ROS. These results are in agreement with other studies, which demonstrate that cytoplasmic α-synuclein aggregations can be induced by various ROS generators, such as hydroxyl radicals and peroxynitrite (Butterfield and Kanski, 2001; Matsuzaki et al., 2004). These metabolites are strong oxidants that can promote not only nitration but also oxidation of α-synuclein, favoring the stabilization of the protein polymer by forming stable cross-linked α-synuclein aggregates (Alvarez et al., 1999; Hashimoto et al., 1999; Souza et al., 2000).
In summary, our study provides strong evidence that Nox1 is involved in the mechanism responsible for generation of PQ-mediated oxidative stress conditions implicated in increased α-synuclein expression and aggregation, and dopaminergic neurodegeneration in the PQ-treated rat model of PD. This work also strengthens the possible relationship between oxidative stress and α-synuclein pathology in PD, introducing Nox1 as a key molecule that could serve as a good therapeutic target for PD and others α-synucleinopathies.
Footnotes
This work was supported by the National Institutes of Health Grant RO1 NS062827 (Y.-S.K.) and Michael J. Fox Foundation Grant Target Validation 2009 (Y.-S.K.). A.C.C. was supported by the Portuguese Foundation for Science and Technology (SFRH/BD/15889/2005 and SFRH/BPD/69643/2010). D.-H.C. was supported by Basic Science Research Program through the National Research Foundation of Korea funded by the Ministry of Education, Science, and Technology (2011-0017016).
References
- Alam ZI, Daniel SE, Lees AJ, Marsden DC, Jenner P, Halliwell B. A generalised increase in protein carbonyls in the brain in Parkinson's but not incidental Lewy body disease. J Neurochem. 1997;69:1326–1329. doi: 10.1046/j.1471-4159.1997.69031326.x. [DOI] [PubMed] [Google Scholar]
- Alvarez B, Ferrer-Sueta G, Freeman BA, Radi R. Kinetics of peroxynitrite reaction with amino acids and human serum albumin. J Biol Chem. 1999;274:842–848. doi: 10.1074/jbc.274.2.842. [DOI] [PubMed] [Google Scholar]
- Beyer K, Domingo-Sàbat M, Ariza A. Molecular pathology of lewy body diseases. Int J Mol Sci. 2009;10:724–745. doi: 10.3390/ijms10030724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown TP, Rumsby PC, Capleton AC, Rushton L, Levy LS. Pesticides and Parkinson's disease–is there a link? Environ Health Perspect. 2006;114:156–164. doi: 10.1289/ehp.8095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butterfield DA, Kanski J. Brain protein oxidation in age-related neurodegenerative disorders that are associated with aggregated proteins. Mech Ageing Dev. 2001;122:945–962. doi: 10.1016/s0047-6374(01)00249-4. [DOI] [PubMed] [Google Scholar]
- Chartier-Harlin MC, Kachergus J, Roumier C, Mouroux V, Douay X, Lincoln S, Levecque C, Larvor L, Andrieux J, Hulihan M, Waucquier N, Defebvre L, Amouyel P, Farrer M, Destée A. Alpha-synuclein locus duplication as a cause of familial Parkinson's disease. Lancet. 2004;364:1167–1169. doi: 10.1016/S0140-6736(04)17103-1. [DOI] [PubMed] [Google Scholar]
- Choi DH, Kim YJ, Kim YG, Joh TH, Beal MF, Kim YS. Role of matrix metalloproteinase 3-mediated alpha-synuclein cleavage in dopaminergic cell death. J Biol Chem. 2011;286:14168–14177. doi: 10.1074/jbc.M111.222430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi DH, Cristóvão AC, Guhathakurta S, Lee J, Joh TH, Beal MF, Kim YS. NADPH oxidase 1-mediated oxidative stress leads to dopamine neuron death in Parkinson's disease. Antioxid Redox Signal. 2012;16:1033–1045. doi: 10.1089/ars.2011.3960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clough RL, Stefanis L. A novel pathway for transcriptional regulation of alpha-synuclein. FASEB J. 2007;21:596–607. doi: 10.1096/fj.06-7111com. [DOI] [PubMed] [Google Scholar]
- Clough RL, Dermentzaki G, Stefanis L. Functional dissection of the alpha-synuclein promoter: transcriptional regulation by ZSCAN21 and ZNF219. J Neurochem. 2009;110:1479–1490. doi: 10.1111/j.1471-4159.2009.06250.x. [DOI] [PubMed] [Google Scholar]
- Clough RL, Dermentzaki G, Haritou M, Petsakou A, Stefanis L. Regulation of alpha-synuclein expression in cultured cortical neurons. J Neurochem. 2011;117:275–285. doi: 10.1111/j.1471-4159.2011.07199.x. [DOI] [PubMed] [Google Scholar]
- Cookson MR. alpha-Synuclein and neuronal cell death. Mol Neurodegener. 2009;4:9. doi: 10.1186/1750-1326-4-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cristóvão AC, Choi DH, Baltazar G, Beal MF, Kim YS. The role of NADPH oxidase 1-derived reactive oxygen species in paraquat-mediated dopaminergic cell death. Antioxid Redox Signal. 2009;11:2105–2118. doi: 10.1089/ars.2009.2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dexter DT, Carter CJ, Wells FR, Javoy-Agid F, Agid Y, Lees A, Jenner P, Marsden CD. Basal lipid peroxidation in substantia nigra is increased in Parkinson's disease. J Neurochem. 1989;52:381–389. doi: 10.1111/j.1471-4159.1989.tb09133.x. [DOI] [PubMed] [Google Scholar]
- Dexter DT, Holley AE, Flitter WD, Slater TF, Wells FR, Daniel SE, Lees AJ, Jenner P, Marsden CD. Increased levels of lipid hydroperoxides in the parkinsonian substantia nigra: an HPLC and ESR study. Mov Disord. 1994;9:92–97. doi: 10.1002/mds.870090115. [DOI] [PubMed] [Google Scholar]
- Donato R, Miljan EA, Hines SJ, Aouabdi S, Pollock K, Patel S, Edwards FA, Sinden JD. Differential development of neuronal physiological responsiveness in two human neural stem cell lines. BMC Neurosci. 2007;8:36. doi: 10.1186/1471-2202-8-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatto NM, Rhodes SL, Manthripragada AD, Bronstein J, Cockburn M, Farrer M, Ritz B. alpha-Synuclein gene may interact with environmental factors in increasing risk of Parkinson's disease. Neuroepidemiology. 2010;35:191–195. doi: 10.1159/000315157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy J, Lewis P, Revesz T, Lees A, Paisan-Ruiz C. The genetics of Parkinson's syndromes: a critical review. Curr Opin Genet Dev. 2009;19:254–265. doi: 10.1016/j.gde.2009.03.008. [DOI] [PubMed] [Google Scholar]
- Harper SQ, Staber PD, He X, Eliason SL, Martins IH, Mao Q, Yang L, Kotin RM, Paulson HL, Davidson BL. RNA interference improves motor and neuropathological abnormalities in a Huntington's disease mouse model. Proc Natl Acad Sci U S A. 2005;102:5820–5825. doi: 10.1073/pnas.0501507102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harraz MM, Marden JJ, Zhou W, Zhang Y, Williams A, Sharov VS, Nelson K, Luo M, Paulson H, Schöneich C, Engelhardt JF. SOD1 mutations disrupt redox-sensitive Rac regulation of NADPH oxidase in a familial ALS model. J Clin Invest. 2008;118:659–670. doi: 10.1172/JCI34060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto M, Hsu LJ, Xia Y, Takeda A, Sisk A, Sundsmo M, Masliah E. Oxidative stress induces amyloid-like aggregate formation of NACP/alpha-synuclein in vitro. Neuroreport. 1999;10:717–721. doi: 10.1097/00001756-199903170-00011. [DOI] [PubMed] [Google Scholar]
- Javoy F, Sotelo C, Herbet A, Agid Y. Specificity of dopaminergic neuronal degeneration induced by intracerebral injection of 6-hydroxydopamine in the nigrostriatal dopamine system. Brain Res. 1976;102:201–215. doi: 10.1016/0006-8993(76)90877-5. [DOI] [PubMed] [Google Scholar]
- Krishnan S, Chi EY, Wood SJ, Kendrick BS, Li C, Garzon-Rodriguez W, Wypych J, Randolph TW, Narhi LO, Biere AL, Citron M, Carpenter JF. Oxidative dimer formation is the critical rate-limiting step for Parkinson's disease alpha-synuclein fibrillogenesis. Biochemistry. 2003;42:829–837. doi: 10.1021/bi026528t. [DOI] [PubMed] [Google Scholar]
- Krüger R, Kuhn W, Müller T, Woitalla D, Graeber M, Kösel S, Przuntek H, Epplen JT, Schöls L, Riess O. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson's disease. Nat Genet. 1998;18:106–108. doi: 10.1038/ng0298-106. [DOI] [PubMed] [Google Scholar]
- Manning-Bog AB, McCormack AL, Li J, Uversky VN, Fink AL, Di Monte DA. The herbicide paraquat causes up-regulation and aggregation of alpha-synuclein in mice: paraquat and alpha-synuclein. J Biol Chem. 2002;277:1641–1644. doi: 10.1074/jbc.C100560200. [DOI] [PubMed] [Google Scholar]
- Matsuzaki M, Hasegawa T, Takeda A, Kikuchi A, Furukawa K, Kato Y, Itoyama Y. Histochemical features of stress-induced aggregates in alpha-synuclein overexpressing cells. Brain Res. 2004;1004:83–90. doi: 10.1016/j.brainres.2004.01.017. [DOI] [PubMed] [Google Scholar]
- McCormack AL, Atienza JG, Johnston LC, Andersen JK, Vu S, Di Monte DA. Role of oxidative stress in paraquat-induced dopaminergic cell degeneration. J Neurochem. 2005;93:1030–1037. doi: 10.1111/j.1471-4159.2005.03088.x. [DOI] [PubMed] [Google Scholar]
- Miller RL, James-Kracke M, Sun GY, Sun AY. Oxidative and inflammatory pathways in Parkinson's disease. Neurochem Res. 2009;34:55–65. doi: 10.1007/s11064-008-9656-2. [DOI] [PubMed] [Google Scholar]
- Neumann M, Kahle PJ, Giasson BI, Ozmen L, Borroni E, Spooren W, Müller V, Odoy S, Fujiwara H, Hasegawa M, Iwatsubo T, Trojanowski JQ, Kretzschmar HA, Haass C. Misfolded proteinase K-resistant hyperphosphorylated alpha-synuclein in aged transgenic mice with locomotor deterioration and in human alpha-synucleinopathies. J Clin Invest. 2002;110:1429–1439. doi: 10.1172/JCI15777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuber S, Petrasch-Parwez E, Winner B, Winkler J, von Hörsten S, Schmidt T, Boy J, Kuhn M, Nguyen HP, Teismann P, Schulz JB, Neumann M, Pichler BJ, Reischl G, Holzmann C, Schmitt I, Bornemann A, Kuhn W, Zimmermann F, Servadio A, et al. Neurodegeneration and motor dysfunction in a conditional model of Parkinson's disease. J Neurosci. 2008;28:2471–2484. doi: 10.1523/JNEUROSCI.3040-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL. Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science. 1997;276:2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, Lincoln S, Crawley A, Hanson M, Maraganore D, Adler C, Cookson MR, Muenter M, Baptista M, Miller D, Blancato J, et al. alpha-Synuclein locus triplication causes Parkinson's disease. Science. 2003;302:841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- Sofic E, Lange KW, Jellinger K, Riederer P. Reduced and oxidized glutathione in the substantia nigra of patients with Parkinson's disease. Neurosci Lett. 1992;142:128–130. doi: 10.1016/0304-3940(92)90355-b. [DOI] [PubMed] [Google Scholar]
- Sorce S, Krause KH. NOX enzymes in the central nervous system: from signaling to disease. Antioxid Redox Signal. 2009;11:2481–2504. doi: 10.1089/ars.2009.2578. [DOI] [PubMed] [Google Scholar]
- Souza JM, Giasson BI, Chen Q, Lee VM, Ischiropoulos H. Dityrosine cross-linking promotes formation of stable alpha-synuclein polymers. Implication of nitrative and oxidative stress in the pathogenesis of neurodegenerative synucleinopathies. J Biol Chem. 2000;275:18344–18349. doi: 10.1074/jbc.M000206200. [DOI] [PubMed] [Google Scholar]
- Terzioglu M, Galter D. Parkinson's disease: genetic versus toxin-induced rodent models. FEBS J. 2008;275:1384–1391. doi: 10.1111/j.1742-4658.2008.06302.x. [DOI] [PubMed] [Google Scholar]
- Turk PW, Laayoun A, Smith SS, Weitzman SA. DNA adduct 8-hydroxyl-2′-deoxyguanosine (8-hydroxyguanine) affects function of human DNA methyltransferase. Carcinogenesis. 1995;16:1253–1255. doi: 10.1093/carcin/16.5.1253. [DOI] [PubMed] [Google Scholar]
- Uversky VN, Li J, Fink AL. Pesticides directly accelerate the rate of alpha-synuclein fibril formation: a possible factor in Parkinson's disease. FEBS Lett. 2001;500:105–108. doi: 10.1016/s0014-5793(01)02597-2. [DOI] [PubMed] [Google Scholar]
- Vekrellis K, Xilouri M, Emmanouilidou E, Rideout HJ, Stefanis L. Pathological roles of alpha-synuclein in neurological disorders. Lancet Neurol. 2011;10:1015–1025. doi: 10.1016/S1474-4422(11)70213-7. [DOI] [PubMed] [Google Scholar]
- Winner B, Jappelli R, Maji SK, Desplats PA, Boyer L, Aigner S, Hetzer C, Loher T, Vilar M, Campioni S, Tzitzilonis C, Soragni A, Jessberger S, Mira H, Consiglio A, Pham E, Masliah E, Gage FH, Riek R. In vivo demonstration that alpha-synuclein oligomers are toxic. Proc Natl Acad Sci U S A. 2011;108:4194–4199. doi: 10.1073/pnas.1100976108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood-Kaczmar A, Gandhi S, Yao Z, Abramov AY, Miljan EA, Keen G, Stanyer L, Hargreaves I, Klupsch K, Deas E, Downward J, Mansfield L, Jat P, Taylor J, Heales S, Duchen MR, Latchman D, Tabrizi SJ, Wood NW. PINK1 is necessary for long term survival and mitochondrial function in human dopaminergic neurons. PloS one. 2008;3:e2455. doi: 10.1371/journal.pone.0002455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarranz JJ, Alegre J, Gómez-Esteban JC, Lezcano E, Ros R, Ampuero I, Vidal L, Hoenicka J, Rodriguez O, Atarés B, Llorens V, Gomez Tortosa E, del Ser T, Muñoz DG, de Yebenes JG. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann Neurol. 2004;55:164–173. doi: 10.1002/ana.10795. [DOI] [PubMed] [Google Scholar]
- Zawia NH, Lahiri DK, Cardozo-Pelaez F. Epigenetics, oxidative stress, and Alzheimer disease. Free Radic Biol Med. 2009;46:1241–1249. doi: 10.1016/j.freeradbiomed.2009.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Perry G, Smith MA, Robertson D, Olson SJ, Graham DG, Montine TJ. Parkinson's disease is associated with oxidative damage to cytoplasmic DNA and RNA in substantia nigra neurons. Am J Pathol. 1999;154:1423–1429. doi: 10.1016/S0002-9440(10)65396-5. [DOI] [PMC free article] [PubMed] [Google Scholar]