

Abstract
Objective
Blood-brain barrier (BBB) dysfunction caused by activation of matrix metalloproteinases (MMPs) is a pathological feature in vascular/neurological disease. Here, we describe the mechanisms of BBB dysfunction and neuroinflammation as a result of MMP-3/9 activation and disruption of VEGF-A/VEGFR-2 interaction, impairing effective angiogenesis.
Methods and Results
We investigate the hypothesis in human brain endothelial cells and animal model of chronic alcohol ingestion. Proteome array analysis, zymography, immunofluorescence and Western blotting techniques detected the activation, expression and levels of MMP-3 and MMP-9. We found that degradation of VEGFR-2 and BBB proteins viz, occludin, claudin-5, and ZO-1 by MMP-3/9 causes rupture of capillary endothelium and BBB leakiness. Impairment of BBB integrity was demonstrated by increased permeability of dye tracers and Fluo-3/calcein-AM labeled monocytes adhesion or infiltration, and decrease in trans-endothelial electrical resistance. Alcohol-induced degradation of endothelial VEGFR-2 by MMP-3/9 led to a subsequent elevation of cellular/serum VEGF-A level. The decrease in VEGFR-2 with subsequent increase in VEGF-A level led to apoptosis and neuroinflammation via the activation of caspase-1 and IL-1β release. The use of MMPs, VEGFR-2 and caspase-1 inhibitors helped to dissect the underlying mechanisms.
Conclusions
Alcohol-induced MMPs activation is a key mechanism for dysfunction of BBB via degradation of VEGFR-2 protein and activation of caspase-1 or IL-1β release. Targeting VEGF-induced MMP-3/9 activation can be a novel preventive approach to vascular inflammatory disease in alcohol abuse.
Keywords: Alcohol, blood-brain barrier, vascular inflammation, VEGF/VEGFR-2, matrix metalloproteinase
INTRODUCTION
The blood–brain barrier (BBB) is consisted of highly specialized intercellular tight junction (TJ) proteins that regulate the trafficking of ions and molecules into the brain from blood circulation 1. The TJ proteins such as occludin, claudins and zonula occludens (ZO-1-3) connect the brain endothelial cells to form a tight monolayer for ensuring structural integrity and selective permeability across the BBB 2. Occludin and Claudins act as sieve proteins that are linked to actin cytoskeleton via the intracellular anchoring protein ZOs 3. Disruption of BBB function/structure integrity is commonly observed in neurological diseases such as stroke 4, Alzheimer’s disease 5, HIV-1 encephalitis 6 and multiple sclerosis 7, 8. The cellular and molecular mechanisms of BBB damage and neurovascular inflammation are not well known in these neurological diseases.
One common mechanism of BBB leakiness is regulated by activation of matrix metalloproteinases (MMPs, zinc-dependent endopeptidases) during oxidative stress 9. Activation of MMP-1, MMP-2, and MMP-9 has been shown to degrade BBB basement membrane and TJ proteins that lead to increased permeability and immune cell infiltration into the brain 9, 10. Such an increase in MMPs activities and BBB damage are also demonstrated in animal model of ischemic stroke 11, 12. Significantly, there is a strong association between activation of MMPs and degradation of BBB basement membrane proteins in human after ischemic and hemorrhagic stroke 13, 14. Surprisingly, there is very little information as to why the inherent angiogenesis fails to repair the damaged basement membrane and TJ proteins of these capillaries.
Vascular endothelial growth factor (VEGF-A), an agonist to VEGFR-2 is a key regulator of angiogenic response and endothelium wound healing 15. The binding of VEGF-A to VEGFR-2 induces receptor dimerization and autophosphorylation, which promotes angiogenesis and repair of the damaged existing vasculature 16, 17. Tyrosine kinase receptors such as VEGFR-1, VEGFR-2 and VEGFR-3 are expressed exclusively in endothelial cells 18. Out of this, the intracellular VEGFR-2 (also known as KDR/Flk-1) plays a central role in the proliferation and vascular endothelium function 19, 20. Thus, lack of VEGFR-2 can terminate the embryonic stage due to the absence of endothelium development 16. But, over expression of VEGF in brain endothelium has been shown to diminish TJ proteins and BBB function 21, 22. VEGF seems to exert protective 23 as well as destructive 24 effects on cerebral vascular function (BBB integrity). These biphasic effects appear to depend on the physiological concentrations of VEGF. The mechanism by how VEGF signaling disrupts the BBB function in the context of VEGFR-2 and MMPs activation remains elusive in brain vascular biology.
Recently, we reported that selective phosphorylation of VEGFR, insulin receptor and non-receptor Src kinase was associated with MMP-1, MMP-2 and MMP-9 activation and BBB dysfunction during acute ethanol exposure and oxidative stress 9. Here, we examined the effects of 50 mM ethanol (0.2 % v/v) as acute exposure in cell culture and 5% (v/v) ethanol liquid intake for 9 weeks as animal model of chronic alcohol intake on VEGF/VEGFR-2 regulation and MMPs activation. The concentrations of ethanol use in this study is within the physiologically detectable range because in human up to 0.31 % (68 mM) blood alcohol levels are detected in moderate to severe intoxicated alcohol drinkers 25, 26. The purpose of the study is to dissect out the molecular mechanisms of BBB disruption by ethanol exposure in acute condition and then validate the findings in animal model of moderately high alcohol ingestion in chronic condition. We propose the idea that physiological level of VEGF in acute alcohol intake triggers the activation of MMP-3/9 in brain endothelium via the autophosphorylation of VEGFR-2. The vasculature damage (BBB damage) caused by MMPs as a result of chewing up the TJ proteins and basement membrane components is likely to be repaired by VEGF-mediated VEGFR-2 phosphorylation in acute condition. However, in chronic alcohol intake, we propose that a sustained MMPs activation causes the degradation of TJ and VEGFR-2 proteins. The inability of VEGFR-2 to actively regulate the vascular repair process leads to chronic rupture of capillaries and leakiness of BBB, thereby, initiating the neurovascular inflammation. We propose that this decrease in VEGFR-2 levels enhances VEGF-A in endothelium and in the blood serum, which appears to be responsible for the sustained activation of MMP-3/9 via the caspase-1 and IL-1β release pathway. We conclude that excess VEGF production in chronic alcoholism may be a detrimental risk factor for neurological disorders such as hypertension and stroke.
METHODS
Reagents and Cell culture
The source of antibodies, chemicals, primary human brain endothelial cells (hBECs), and cell culture techniques are described in detail in Supplemental material. Briefly, primary hBECs were cultured in type 1 rat-tail collagen and fibronectin pre-coated plates as described previously 27.
Animal studies
Five-week old weight-match male rats were pair fed the Lieber–DeCarli control or 29% calorie intake (5% v/v) ethanol liquid-diets for 9 weeks. Animals were sacrificed at 10th week on different days depending on the nature of experiments with/without cell infusion. Particularly, experiments that involve infusion of fluo-3 labeled cells into the common carotid artery as previously described28 require a few days to complete the whole experimental conditions. The levels of alcohol, VEGF-A and IL-1β were determined in blood samples, and blood alcohol level was determined to be 8.8 – 24 mM (0.05–0.11%) in alcohol diet ingested animals (determined by EtOH assay kit, Diagnostic Chemicals Ltd, Charlottetown, PEI, Canada). This huge variation in blood alcohol levels is commonly in animal studies due to nocturnal feeding habit of rats, variation in ethanol metabolic and clearance rate. The detectable levels of blood alcohol can be increased and the variable range can be narrowed in this animal model. This can be achieved by starving the animal overnight follow by feeding them at about 5-6 am, and then collect the blood samples at about 7-8 am. This was not done in the present study because we knew the outcome from our past experience.
Immunofluorescence and microscopy
Changes in cell adhesion/migration, expression of TJ proteins, MMPs, caspase-1, VEGFR2 were analyzed in intact external cerebral capillary vessels and brain tissue sections containing the internal capillaries, or hBECs cultured on cover slips by immunohistochemical stainings and fluorescent microscopy. For detail protocols and dilution of respective antibody, please see supplemental material.
Western blotting
Changes in protein levels of MMP-3, MMP-9, occludin, claudin-5, ZO-1, VEGFR-2, p-VEGFR-2tyr1054, p-VEGFR-2tyr1175, caspase-1 and IL-1β in intact capillary vessels, brain tissue sections and hBECs culture were analyzed by Western blotting. See detail experimental protocol in the supplemental material.
Dot blot MMPs array analysis
Human matrix metalloproteinases antibody array (Ray Biotech, Norcross, GA) was used to analyze the regulation of MMPs in control or EtOH treated samples as per manufacturer’s instructions.
Zymography of MMP Activity
We determined the MMPs activities in hBECs culture, brain tissue and microvessel homogenates by zymography as per well-established method 29. Gelatinolytic (for MMP9) or stromelysin (for MMP3) activities were determined by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) using a 10% polyacrylamide gel containing 0.1% gelatin or a 12% gel containing 0.1% casein at 125 V for 90 minutes at 4°C and stained with 0.5% coomassie brilliant blue R-250. See detail experimental protocol in the supplemental material.
Enzyme-Linked Immunosorbent Assay (ELISA)
Using commercial enzyme-linked immunosorbent assay kits (ELISA), the levels of VEGF-A (R&D Systems, Minneapolis, MN) and IL-1β (BD Biosciences, San Jose, CA) were analyzed in cell culture media and blood serum as per manufacturer’s instructions.
TUNEL and PARP analyses
Using the terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL, Roche diagnostics, IN) assay kit, cell apoptosis was determined in tissue sections and cell culture as per manufacture’s instructions. To confirm the TUNEL assay, Western blotting determined the break down of 113 kDa poly-ADP-Ribose-Polymerase (PARP) to 89 kDa and 24 kDa PARP fragments using rabbit anti-PARP primary antibody (BD Biosciences). Detection of 89 kDa PARP fragment is an early marker for apoptosis, which is mediated by caspase-1/3 signaling pathway.
Transendothelial electrical Resistance
To determine the integrity of BBB function, a highly sensitive 1600R ECIS system (Applied Biophysics, Troy, NY) determined the changes in trans-endothelial electrical resistance (TEER) across the BBB as previously described 9. See detail protocol in the supplemental material.
Cell adhesion and migration across BBB
Immune cells adhesion on the endothelium and migration across the BBB in an in vivo and in vitro were analyzed by fluorescence-labeled cells as described previously 28,30. See detail protocol in the supplemental material.
Data analysis
All the results are expressed as the mean ±SEM. Statistical analysis of the data was performed using Graphpad Prism V5 (Sorrento Valley, CA). Two-way ANOVA with Dunnett’s post-hoc tests determined the differences between control and experimental conditions, and a p-value of less than 0.05 was considered significant.
RESULTS
Activation of MMP-3/9 causes BBB leakiness
Activation of MMPs diminishes tight junction (TJ) protein expression in brain endothelium and microvessels
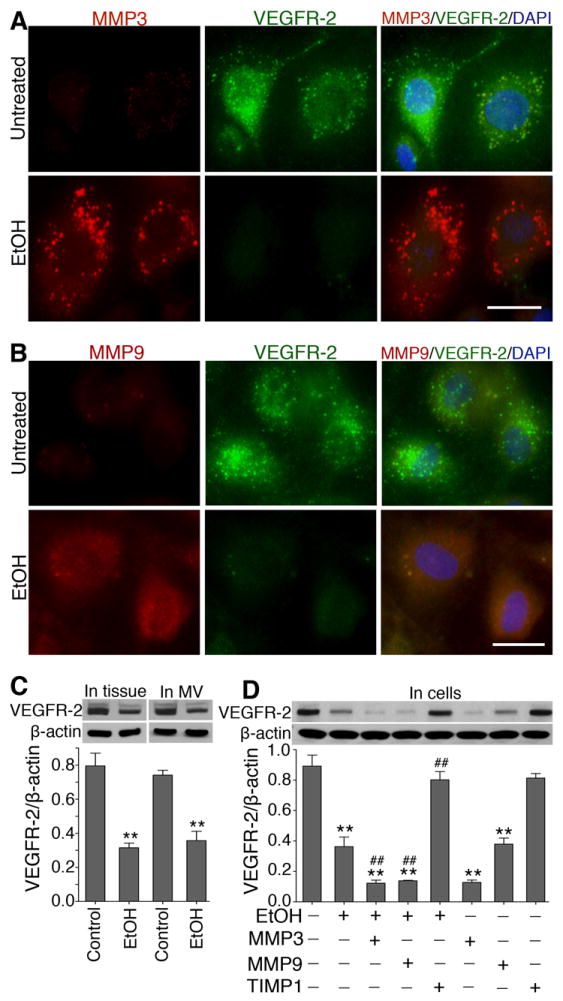
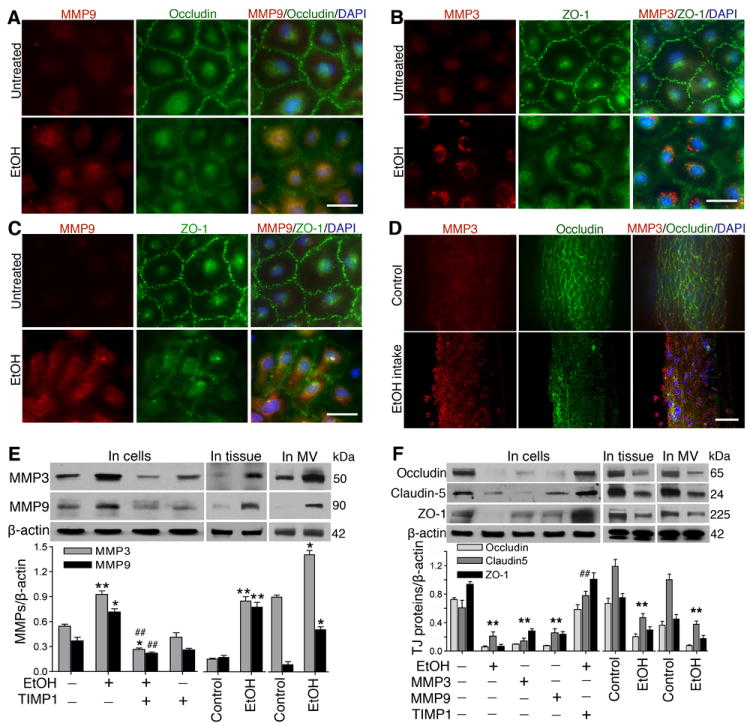
Degradation of extracellular matrix and basement membrane proteins is the key function of matrix metalloproteinases (MMPs). We have shown that VEGFR is involved in activation of MMPs and BBB damage in acute alcohol exposure 9. To identify the specific types of MMPs, we first examined the changes in the expression of ten different types of MMPs by proteome array in primary hBECs culture following exposure to 50 mM ethanol for 24 hr. Out of the 10 MMPs, ethanol exposure significantly elevated the levels of MMP-3 and MMP-9 compared with untreated hBECs in culture (Supplemental Figure IA through IC), and these observations were confirmed by immunofluorescence staining and Western blotting. Further, detection of MMPs activity by zymography validated the changes in expression and protein levels of MMPs in brain endothelial cells and rat brain tissues and microvessels. Thus, we found that alcohol increased the gelatinolytic (for MMP9) or stromelysin (for MMP3) activity with/without VEGF treatment in cell culture or in brain tissue and microvessels (Supplemental Figure II). Thus, in this study, MMP-3 and MMP-9 were targeted as the causative factors for disruption and leakiness of neurovascular function (the BBB). As expected, co-localization of MMPs and TJ proteins revealed that increase in the expression of MMP-3 and MMP-9 reciprocally diminished the expression of occludin and ZO-1 by alcohol treatment compared with untreated control cells (Figure 1A through 1C; Supplemental Figure III). In parallel, we also analyzed the co-localization of MMP-3 or MMP-9 and occludin in intact rat brain microvessels from control and chronic alcohol intake. In agreement with the findings in primary hBEC culture, we observed that increase in MMP-3 or MMP-9 expression correlated with a decrease in occludin expression in intact capillary vessels (Figure 1D) and in microvessels of brain tissue section (Supplemental Figure IV) from alcohol ingested animal compared with controls.
Figure 1. Activation of MMPs by ethanol degrade TJ proteins.
A-D, Immunofluorescent staining of (A) MMP-9 (red), occludin (green), and DAPI (blue), (B) MMP-3 (red), ZO-1 (green), and DAPI (blue), (C) MMP-9 (red), ZO-1 (green), and DAPI (blue) in hBECs, and (D) MMP-3 (red), occludin (green), DAPI (blue) in intact brain microvessels of rat. E-F, Western blot analyses of MMP-3, MMP-9, and actin; Occludin, Claudin-5, ZO-1 and actin in hBEC lysate protein, whole brain tissue homogenates, and in protein extract from isolated microvessels. For all hBECs culture work, cells were treated with or without EtOH (50 mM) in the presence or absence of MMP-3 or MMP-9 (100 ng/mL each) or TIMP1 (100 ng/mL) for 24 hr. Bar graphs show the results that are expressed as ratio of MMP-3/9, occludin, claudin-5 or ZO-1 to that of β-actin bands. Values are mean ±SEM; n=4. *p<0.05; **p<0.01 vs control in E and F; ##p<0.01 vs EtOH in E or MMP-3/9 in F. Scale bar = 40 μm in A, B and C and 5 μm in D.
Reduction in TJ proteins by MMPs disrupts the integrity of BBB interface
Changes in the expression of MMP-3, MMP-9, and TJ proteins were validated by Western blot analyses in lysates protein from hBEC culture, intact brain microvessels, and brain tissue homogenates. Similar to microscopy imaging data, Western blot analyses confirmed that alcohol in deed enhanced the levels of MMP-3 and MMP-9 proteins (Figure 1E) as well as decreased the levels of occludin, claudin-5 and ZO-1 proteins (Figure 1F) in hBEC culture, intact capillary vessels, and in brain tissue. Degradation of occludin, claudin-5 and ZO-1 by addition of exogenous MMP-3 or MMP-9 proved the point that BBB is targeted by MMPs activation. The attenuating effect of TIMP1 on TJ proteins in the absence or presence of ethanol further validated the notion that activation of MMPs lead to disruption and leakiness of the BBB integrity.
Loss of TJ integrity enhances permeability and leakiness of BBB
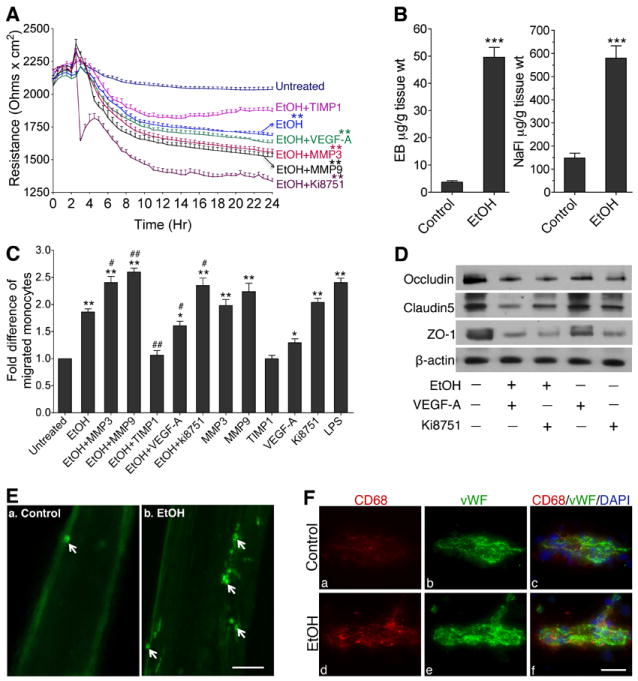
As functional assays, we assessed the integrity of BBB by trans-endothelial electrical resistance (TEER), permeability of BBB by sodium fluorescein/Evan Blue (NaFl/EB) tracers, and infiltration of immune cells across the BBB and into the brain by calcein-AM or Fluo-3 labeled monocytes. Assessing the changes in real-time TEER by a highly sensitive ECIS system showed a significant reduction in BBB electrical resistance by EtOH or in combination with MMP-3 or MMP-9, which exacerbated the effect of EtOH in a time-dependent manner (Figure 2A). The fact that TIMP1 prevented the effect of EtOH on TEER reduction reinforced the direct role MMPs on BBB dysfunction. The role of VEGFR-2 in the integrity of BBB was come from the treatment of Ki8751 (VEGFR-2 kinase inhibitor), where TEER value was significantly reduced and with EtOH the effect was exacerbated (Figure 2A). Disruption of this BBB integrity was further supported by increased permeability of small molecular weight tracer NaFl (MW=376) and large molecular weight tracer EB (MW=961) into the brain of chronic alcohol intake rats compared with respective controls (Figure 2B).
Figure 2. Activation of MMP-3/9 causes BBB leakiness.
A, Kinetic profile of the trans-endothelial electrical resistance (TEER) across the BBB following EtOH, MMP-3/9, TIMP1, VEGF-A or Ki8751 treatment. After maintaining a stable resistance for 2 hr, TEER was monitored at 400 Hz with 10 min intervals. Reproducible results were obtained in 3 individual experiments. B, In vivo permeability of Evans Blue (EB, 5 μM) and Sodium fluorescein (Na-Fl, 5 μM) across the BBB (n=5). C, Migration of monocytes across the in vitro model of BBB after treatment of various test compounds as shown in figure. Data are expressed as fold difference from untreated cells. D, Effects of EtOH (50 mM), VEGF-A (100 ng/mL), Ki8751 (10 μM), EtOH+VEGF-A or EtOH+Ki8751 on occludin, claudin-5 and ZO-1 levels in hBECs after 24 hr exposure (Western blotting). E, Fluo-3 labeled macrophage adhesion/migration in brain capillary following infusion of cells into the common carotid artery. F, Immunofluorescent staining of CD68 (green) and von Willibrand factor (green) merged with DAPI (blue) in microvessel of brain tissue section. Scale bar indicates 5 μm in all panels. *p<0.05; **p<0.01 vs untreated or control in A, B and C; #p<0.05; ##p<0.01 vs EtOH (second bar) in C.
Leakiness of BBB enhances immune cell infiltration into the brain
Migration of calcein-AM labeled monocytes across the endothelium showed a significant 2-2.5 fold increments of immune cell migration in EtOH or EtOH in combination with MMP-3 or MMP-9 compared with controls (Figure 2C). The effect of EtOH on cell migration was comparable to that of lipopolysaccharide (LPS), which was used here as positive control. As expected, TIMP1, an inhibitor of MMPs attenuated the effect of EtOH on monocyte migration. VEGF-A alone did not cause robust cell migration. However, inhibition of VEGFR-2 by Ki8751 exacerbated the effect of EtOH on cell adhesion and migration across the BBB. Thus, we further investigated the involvement of VEGFR-2 on BBB integrity in hBEC culture. Western blot analyses revealed that treatment of hBECs with Ki8751 selectively down-regulated the levels of occludin, claudin-5 and ZO-1 proteins either in the presence or absence of alcohol exposure, suggesting the synergistic association between VEGFR-2 and brain vascular function (Figure 2D). Moreover, addition of VEGF-A (100 ng/ml) exogenously decreased the level of occludin, claudin-5 and ZO-1 proteins (Figure 2D), suggesting that antagonizing the VEGFR-2 function by a high concentration of VEGF-A can impair the integrity of BBB.
Finally, utilization of our animal model of chronic alcohol intake allowed us to validate the notion that neuroinflammatory process was initiated by BBB damage. We found that infusion of bone marrow derived rat monocytes and macrophages labeled with Fluo-3 into the right common carotid artery demonstrated an increase in cell adhesion and infiltration at the damage sites of the brain capillary alcohol intake animal compared with pair-fed controls (Figure 2E). Further, infiltration of immune cells into brain was analyzed by staining with antibody to CD68 in brain tissue sections from rats that were not subjected to cell infusion. Our data confirmed that there was more number of infiltrated immune cells in the brain of alcohol intake than the pair-fed controls (Figure 2F). Taken together, these results indicate that activation of MMPs and diminished repair function of VEGFR-2 may lead to vascular leakiness, and enhanced cell adhesion/infiltration for initiation of cerebral inflammatory disease in alcohol abuse.
MMPs degrade VEGFR-2 protein
VEGFR-2 is a key regulator of angiogenesis and vascular wound healing, which is mediated by the physiological concentration of VEGF-A. We reported a correlation between MMPs activation and VEGF/VEGFR signaling responsible for the disruption of BBB function during alcohol exposure 9. Here, we evaluate the underlying mechanisms of MMP-VEGFR-2/VEGF interplay for causing vascular leakiness in rat model of alcohol intake and in hBEC culture. Our results show that ethanol increased the expression of MMP-3 and MMP-9, and decreased the expression of VEGFR-2 in hBECs (Figure 3A and 3B). Alcohol-induced reduction in VEGFR-2 protein was validated by Western blot in protein samples from brain tissue and microvessel of rats (Figure 3C). To prove that degradation of VEGFR-2 protein by MMPs was a possible mechanism for endothelium dysfunction, we treated hBECs with exogenous MMP-3 and MMP-9 in the presence or absence of EtOH or TIMP1. Addition of MMP-3 or MMP-9 significantly reduced the level of VEGFR-2 protein either in the presence or absence of EtOH compared with untreated control (Figure 3D). Importantly, TIMP1 mitigated the MMP-mediated degradation of VEGFR-2 protein, supporting the hypothesis that MMP-3/9 activation and VEGFR-2 reduction was involved in alcohol-induced leakiness of the BBB function.
Figure 3. Alcohol-induced activation of MMPs degrades VEGFR-2 protein.
A-B, Immunofluorescent staining of (A) MMP-3 (red), VEGFR-2 (green), and DAPI (blue); (B) MMP-9 (red), VEGFR-2 (green), and DAPI (blue) in hBECs with or without exposure to 50 mM EtOH for 24 hr. Scale bar indicates 20 μm in all panels. C, Western blot analysis of VEGFR-2 protein in cortical brain tissue homogenates and brain microvessel protein. D, Western blot analysis of VEGFR-2 protein in hBEC lysate proteins following exposure to respective test compounds for 24 hr. Bar graphs show the results that are expressed as ratio of VEGFR-2 to that of β-actin bands, and values are mean ±SEM; n=4. **p<0.01 vs untreated or control in C and D; ##p<0.01 vs EtOH (second bar) in D.
Degradation of VEGFR-2 elevates cellular and serum VEGF-A levels
The decrease in VEGFR-2 protein that we observed here is expected to elevate the cellular and circulatory VEGF-A levels, because the platelets and endothelial cells will continue to secrete VEGF-A while VEGFR-2/VEGFR-1 is unable to bind to VEGF-A inefficiently. Thus, we quantified the levels of VEGF-A in blood serum samples and supernatants of hBECs cultured media by ELISA. Our results show that alcohol elevated the levels of VEGF-A in serum and cellular culture media compared with respective controls (Figure 4A and 4B). Interestingly, MMP-3 and MMP-9 also increased the levels of VEGF-A either in the presence or absence of alcohol, while TIMP1 (inhibitor of MMPs) decreased the effect of alcohol on VEGF-A regulation. These data suggest that MMP-3/9 was involved in the malfunctioning of VEGFR-2 causing increase extracellular VEGF-A levels. To address the idea that this excess level of VEGF-A in turn triggers the activation of MMP-3/9, we then determined the changes in protein levels of MMP-3, MMP-9 and VEGFR-2 after treatment of VEGF-A with hBECs in culture. The kinetic profile data showed that an increase in MMP-3 or MMP-9 protein level was inversely proportional to a decrease in VEGFR-2 protein (Figure 4C) following VEGF-A treatment. The subtle difference between MMP-3, MMP-9 and VEGFR-2 alterations appeared to occur at 12-24 hr after VEGF-A exposure. Thus we treated hBEC culture with VEGF-A for 24 hr with or without EtOH and analyzed the alterations of MMP-3, MMP-9 and VEGFR-2 proteins level by Western blotting. In deed, we confirmed that activation of MMP-3 or MMP-9 by physiological excess VEGF-A level led to the degradation of VEGFR-2 protein, which was further aggravated by the presence of alcohol (Figure 4D). We then examined the phosphorylation of VEGFR-2 at two sites of phospho-VEGFR-2 tyr1054 and phospho-VEGFR-2 tyr1175 in cell culture and animal model of chronic alcohol intake. Using specific antibody to phospho-VEGFR-2tyr1054 and phospho-VEGFR-2tyr1175, we detected the phosphorylation of VEGFR-2 by Western blotting. EtOH or VEGF significantly increased the phosphorylation of VEGFR-2 at these two tyrosine residues in hBECs culture similar to alcohol-induced increase in whole brain or microvessel tissues compared with respective controls (Figure 4E). As expected, TIMP1 or Ki8751 significantly inhibited the phosphorylation of VEGFR-2.
Figure 4. Degradation of VEGFR-2 causes elevation of VEGF-A levels.
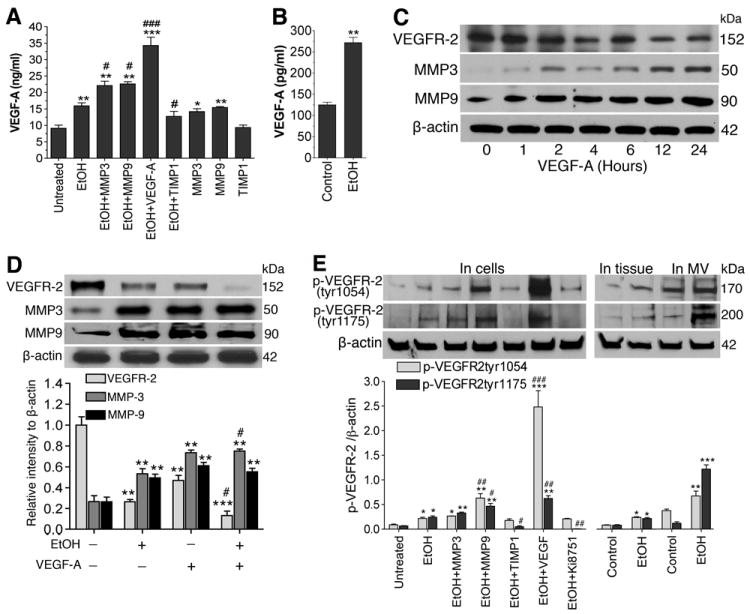
A-B, ELISA shows the levels of VEGF-A in: (A) hBEC cell culture supernatants in different treatment conditions, (B) blood serum. C, Western blot analysis of alterations in VEGFR-2, MMP-3 and MMP-9 protein levels after treatment of hBECs with VEGF-A (100 ng/mL) at different time points. D, Changes in protein levels of VEGFR-2, MMP-3 and MMP-9 after treatment of hBECs with EtOH (50 mM), or VEGF-A, or in combination for 24 hr. E, Western blot analysis for the changes in protein levels of phosphorylated VEGFR-2 (at tyr1054 and tyr1175) after treatment of hBECs in different treatment conditions for 24 hour as shown and in EtOH treated rat brain tissue and microvessels lysates. Results are expressed as ratio of VEGFR-2 or MMP-3/9 to that of β-actin in D and p-VEGFR-2tyr1054 or p-VEGFR-2tyr1175 to that of β-actin bands in E. Values are mean ±SEM, n=3 to 5 in all experiments. *p<0.05; **p<0.01 vs untreated or control in A and B; ##p<0.05; ##p<0.01; ###p<0.001 vs EtOH (second bar) in A and E.
These findings suggest that reduction of VEGFR-2 by MMPs may be further exacerbated by VEGF signaling loop in alcohol-induced neurovascular disease. These data suggest that alcohol-induced degradation of VEGFR-2 causes increase in phosphorylation of VEGFR-2 protein as result of VEGF elevation and translocation of VEGFR-2 to the nuclear membrane, which has been observed in endothelial cells 31, 32.
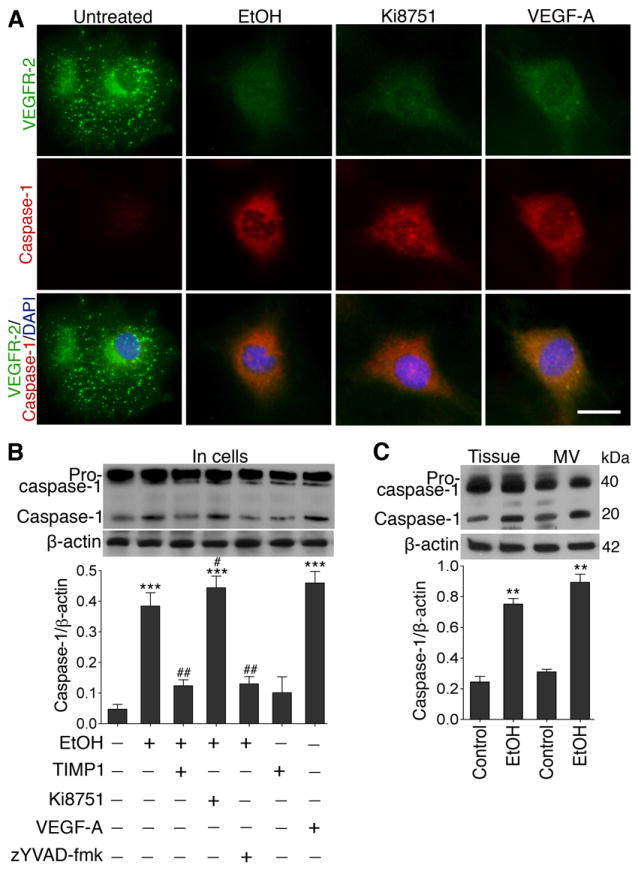
Caspase-1 activation controls the VEGF signaling loop
To test the hypothesis whether alcohol-induced increase in VEGF-A (due to VEGFR-2 reduction) could activate caspase-1, we analyzed the expression of caspase-1 in hBECs exposed to EtOH, Ki8751, VEGF-A, z-YVAD-fmk (caspase-1 inhibitor), and TIMP1. It was apparent that EtOH, VEGF-A or inhibition of VEGFR-2 respectively increased the expression of caspase-1 accompanied by an abrupt decrease in VEGFR-2 protein expression (Figure 5A). These changes in caspase-1 protein expression were validated by the changes in protein levels in total protein extracts from hBECs, brain tissue and microvessel homogenates by Western blot analyses (Figure 5B). EtOH, VEGF-A, or Ki8751 respectively increased caspase-1 protein without significantly affecting the levels of the pro-caspase-1. As expected, z-YVAD-fmk inhibited the maturation of pro-caspase-1 to caspase-1. Interestingly, TIMP1 was able to inhibit pro-caspase-1 maturation, suggesting that sparing the VEGFR-2 degradation by MMPs can reinforce the VEGF-VEGFR-2 interaction for vascular repair process. The effects of EtOH on caspase-1 activation in brain tissue and microvessel of chronic alcohol intake animal and pair-fed control are further demonstrated with a co-localization of brain endothelial marker protein vWF (Supplemental Figure V).
Figure 5. Elevation of VEGF-A by alcohol activates caspase-1.
A, Changes in the expression of VEGFR-2 (green), caspase-1 (red), DAPI (blue) in hBECs after exposure to EtOH (50 mM), Ki8751 (10 μM) and VEGF-A (100 ng/mL) for 24 hr. Scale bar =20 μm. B-C, Immunoblot analysis to show the alterations in caspase-1 protein levels in: (B) hBECs culture following treatment with respective test compounds, and (C) brain cortical tissue and microvessel homogenates protein. Bar graphs show the results, which are expressed as ratio of caspase-1 to that of β-actin bands. Values are mean ±SEM; n=4 to 5. **p<0.01; ***p<0.001 vs untreated or control in B and C; #p<0.05; ##p<0.01 vs EtOH (second bar) in B.
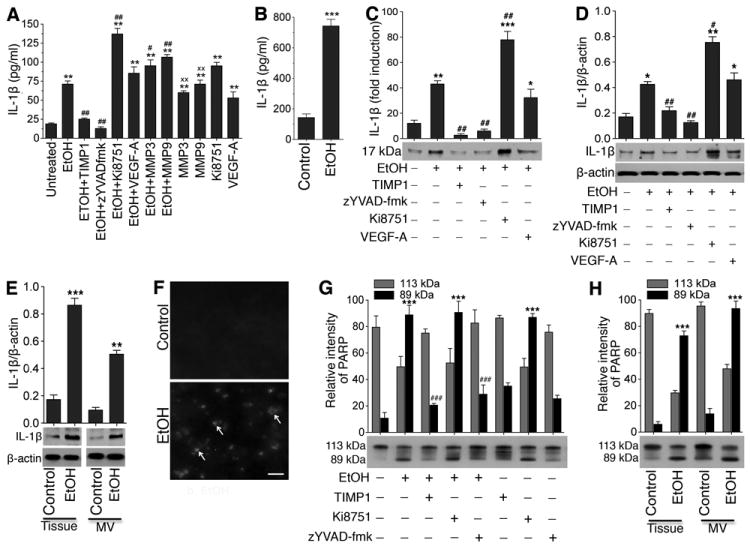
Activation of caspase-1 matures IL-1β
Since activation of caspase-1 leads to production of interleukin 1 beta (IL-1β), we then analyzed the level of IL-1β in cell-cultured media and in blood serum from chronic animal studies by ELISA. We observed that treatment of hBECs with exogenous MMP-3, MMP-9, VEGF-A, or Ki8751 significantly increased the levels of IL-1β in the presence or absence of EtOH, however TIMP1 and z-YVAD-fmk prevented the effects of EtOH on IL-1β elevation (Figure 6A). These in vitro studies findings were further validated by our in vivo studies, where the levels of IL-1β in the blood serum of EtOH-diet consumed animal had about 7-fold higher than the pair-fed control (Figure 6B). Note that the levels of IL-1β in the blood serum were much higher than that present in cell cultured supernatants. Further, to validate the results of these extracellular and circulating IL-1β levels, we also analyzed the actual protein contents of IL-1β in cell-cultured supernatants, cell lysates, and whole brain tissue/microvessel homogenates by Western blotting. Similar to ELISA assayed results, inclusion of MMP-3, MMP-9, VEGF-A, or Ki8751 in alcohol condition significantly elevated the levels of IL-1β in cell-cultured supernatants (Figure 6C), in cell lysates protein (Figure 6D), and in whole brain tissue or microvessel homogenates (Figure 6E). Taken together, these data indicate that MMP-mediated loss of VEGFR-2 function and subsequent elevation of VEGF-A levels in alcohol intake lead to activation of caspase-1 and release of proinflammatory IL-1β. Thus, release of this cytokine via the VEGF signaling pathway may be the reversible loop mechanism for the activation of MMP-3/9 by VEGF-A, which was observed in data presented in Figure 4A and 4B.
Figure 6. Activation of caspase-1 matures IL-1β and causes neuroinflammation and cell apoptosis.
A-B, ELISA assay of IL-1β levels in: (A) hBEC culture supernatants of different treatment conditions, and (B) blood serum (100 μL of serum was used per well) of control and chronic alcohol intake rats. C-E, Western blot analyses of IL-1β protein levels in: (C) hBEC culture supernatants expressed as fold induction of IL-1β protein levels, (D) hBEC lysate protein, and (E) whole brain tissue and microvessel homogenates protein Bar graphs show the fold induction of expression of IL-1β in different treatments in C; ratio of expression of IL-1β to that of β-actin bands in D and E. F, TUNEL staining in rat brain cortical tissue section. The arrow indicates the TUNEL-positive cells. Scale bar =40 μm. G-H, Changes in PARP protein levels by Western blotting using anti-PARP antibody in: (G) hBEC lysate and (H) whole brain cortical tissue or microvessel tissue homogenates. Bar graphs data show the changes in arbitrary relative intensity of 113 kDa and 89 kDa fragments of PARP in cell culture and brain tissue samples. Values are mean ±SEM; n=3 to 4. **p<0.01; ***p<0.001 vs untreated or control in C, D and E; ##p<0.01 vs EtOH (second bar) in C and D. ***p<0.001 vs respective fragment in G and H.
Caspase-1 and IL-1β causes cell apoptosis
As a functional read out assay for caspase-1 activation and IL-1β release, we next evaluated the extent of cell apoptosis by TUNEL and Poly-ADP-Ribose-Polymerase (PARP) induction Western blot in hBECs culture and in animal studies. It was obvious that there was much higher positive staining of TUNEL in brain tissue from alcohol-diet ingested animal than the pair-fed control brain tissue section (Figure 6F). These findings were further substantiated by the induction of 89 kDa PARP in hBECs (Figure 6G) and in rat brain tissue or microvessel homogenates (Figure 6H) following alcohol exposure. Similar to IL-1β release data, Ki8751 with or without EtOH up-regulated the level of 89 kDa PARP, while TIMP1 or z-YVAD-fmk decreased the levels of 89 kDa PARP induction. These results suggest that down-regulation of VEGFR-2 mediated caspase-1 activation and IL-1β release causes cell apoptosis.
DISCUSSION
It is now widely accepted that MMP-induced leakiness of the BBB is a key event for the development of vascular and neurological disease. Here, we demonstrated that activation of MMP-3/9 by ethanol impaired the TJ proteins of the BBB in hBECs culture and brain capillary of animal compared with respective controls. Alcohol-induced decrease in real-time trans-endothelial electrical resistance supported the loss of BBB integrity, which was validated by enhanced permeability of Na-Fl/EB tracers across the capillary and into the brain. These findings suggested that leakiness of BBB might be susceptible for initiation of inflammatory neurological disease such as atherosclerosis and stroke. As a proof-of-concept to this inflammatory process, our in vitro and in vivo study findings indicated an increase immune cell (calcein-AM/Fluo3 labeled monocytes) adhesion and infiltration into the brain. Interestingly, TIMP significantly but not completely reversed the effect of EtOH on TEER unlike that of monocyte infiltration. This may be attributed to the fact that TIMP is an inhibitor of MMPs but not an inhibitor of ethanol metabolism. Thus, TIMP is unable to prevent the effect of acetaldehyde and oxidative damage of the BBB. Degradation of the BBB basement by MMPs plays a significant role for migration of immune cells into the brain, thus, inhibition of MMPs by TIMP was effective in reversing the effect of ethanol for infiltration of monocytes. Since activation of MMPs is involved in BBB leakiness 9, 33, inhibition of MMPs has been proposed as beneficial approach to alleviate vascular inflammatory diseases 34. It is now evident that disruption of BBB and neurovascular components correlates with inflammatory disease like multiple sclerosis 7, 35 and ischemic stroke 36.
Our idea is that a sustained activation of MMPs by chronic alcohol alcohol intake leads to degradation of VEGFR-2 protein, thereby, unable to repair the leakiness of the capillary. Our data supported this argument because exposure of hBECs to exogenous MMP-3 or MMP-9 led to the reduction of VEGFR-2 protein, which was further validated by the fact that activation of MMP-3 or MMP-9 in brain microvessel correlated with a down-regulation of VEGFR-2 protein. We have not examined the cleavage site(s) of VEGFR-2 by MMPs, however it has been shown that cleavage of VEGFR-2 by MMPs occur at multiple positions of the extracellular domain of the endothelium VEGFR-2 37, 38. There seems to be a strong association between VEGFR-2 signaling and activation of MMPs in vascular biology, because it has been shown that inhibition of VEGFR-2 phosphorylation by propranolol blocked the secretion of MMP-2 in endothelial cells 39. Conversely, interaction of VEGF and VEGFR-2 (i.e. phosphorylation of VEGFR-2) appears to down-regulate MMP-9 through a STAT1 activation pathway 40. In this study, the authors suggest that tyrosine phosphorylation of STAT1 by VEGF may responsible for down-regulation of MMP-9. Thus, Ito et al. (2009) demonstrated that degradation of VEGFR-1 by MMP-7 exploited the VEGF function to deleterious health effects 41. Here we propose that acute exposure of alcohol activates MMP-3/9 via the phosphorylation of VEGFR-2, while chronic exposure of alcohol activates MMPs by a sustained VEGF-A loop pathway at the expense of TJ/VEGFR-2 proteins degradation by MMPs.
In the present study, we demonstrated that high concentration of VEGF initially increased the expression of VEGFR-2 protein, but decreased the VEGFR-2 protein time-dependently with a gradual increase in MMP-3 or MMP-9. These findings indicate that binding of VEGF to VEGFR-2 initially caused the dimerization of VEGFR-2 and increased the expression of receptor protein. Since the binding sites of VEGFR-2 get saturated, the excess VEGF level began to activate MMP-3/9 via the caspase-1 activation loop. In turn, MMPs activation degraded VEGFR-2 protein and further elevated the extra- and intra- cellular VEGF levels. Thus, activation of MMPs by VEGF not only degrade VEGFR-2 but also shown to decrease the BBB tight junction proteins occludin and claudin-5 in autoimmune disease 21, 42 similar to our present findings. In physiological condition, platelets and endothelial cells constantly produce VEGF-A. As such impairment of VEGFR-2/VEGFR-1 is expected to increase the levels of VEGF-A in the blood circulation. The present studies showed that impairment of VEGFR-2 was accompanied by high levels of VEGF-A (270 pg/ml) in the serum of alcohol intake animal compared with pair-fed controls (125 pg/ml). However, the levels of VEGF-A detected in hBEC culture with or without exposure to EtOH or MMP was in the range of 9-22 ng/ml, which was much higher than the VEGF-A levels detected in the serum. This high level of VEGF-A in cell culture supernatants could be contributed by the presence of exogenous VEGF-A in the culture media in addition to EtOH-stimulated secretion by the brain endothelial cells. Similar to these findings, others have reported the elevation of serum VEGF levels in human alcoholics 43 and alcohol exposed endothelial cells 44. Interestingly, the serum level of VEGF-A detected in the present studies was comparable to the serum VEGF levels of diabetic patients (257-365 pg/ml), where the mean VEGF-A level in serum of healthy subjects was 216 pg/ml 45. Similarly, the level of mean VEGF-A in serum of cancer patients was 291-380 pg/ml compared with the mean VEGF level of 219 pg/ml in healthy subjects 46. All these findings clearly indicate that detection of abnormal VEGF levels in blood samples can be a standard diagnostic marker for neurovascular disease, diabetes and cancers. It must be noted here that normal physiological level of VEGF is an essential growth factor required for endothelial cell growth and vasculogenesis. Interruption of this physiological homeostasis such as by impairing the VEGF receptors can lead to elevation of VEGF, which in turn may promote activation of MMPs and growth of cancer cells. Here we describe for the first time that alcohol-induced decrease in VEGFR-2 raises the VEGF above the physiological level, and this sustained increase in VEGF level in turn activates the MMP-3/9 through caspase-1 activation and IL-1β release. Activation of caspase-1 and release of IL-1β release were validated by TUNEL cell apoptosis and induction of PARP in hBECs and in animal studies.
In summary, the alteration of the normal VEGF/VEGFR-2 interaction appears to dictate the outcome of this vascular disease, because VEGF is a promoter of angiogenesis (BBB repair process) at physiological level and an activator of MMPs at high level. Here, we discuss the mechanism of switch by VEGF for causing BBB leakiness in the context of MMPs activation and VEGFR-2 reduction in alcohol abuse. We found that activation of MMP-3/9 by alcohol not only altered the TJ proteins and basement membrane components, but also degraded the VEGFR-2 protein. While disruption of TJ proteins and basement membrane led to leakiness of BBB and vascular inflammation, the impairment of VEGFR-2 function led to inefficient repair process of the leaky BBB as well as elevation of VEGF levels in the circulation. This sustained elevation of VEGF-A above the physiological level triggered the activation of MMP-3/9 through a caspase-1 and IL-1β release to cause inflammation and brain cell death. Supplemental Figure VI depicts the schematic presentation of our findings.
Supplementary Material
Acknowledgments
This work was supported by NIH/NIAAA grants: R21 AA020370-01A1 and RO1 AA017398. We thank Dr. Yuri Persidsky for providing us the primary human endothelial cells.
SOURCE OF FUNDING
This study was funded by NIH/NIAAA grants: R21 AA020370-01A1 and RO1 AA017398.
Footnotes
DISCLOSURES
None
References
- 1.Rubin LL, Staddon JM. The cell biology of the blood-brain barrier. Annu Rev Neurosci. 1999;22:11–28. doi: 10.1146/annurev.neuro.22.1.11. [DOI] [PubMed] [Google Scholar]
- 2.Hawkins BT, Davis TP. The blood-brain barrier/neurovascular unit in health and disease. Pharmacol Rev. 2005;57:173–185. doi: 10.1124/pr.57.2.4. [DOI] [PubMed] [Google Scholar]
- 3.Fanning AS, Jameson BJ, Jesaitis LA, Anderson JM. The tight junction protein ZO-1 establishes a link between the transmembrane protein occludin and the actin cytoskeleton. J Biol Chem. 1998;273:29745–29753. doi: 10.1074/jbc.273.45.29745. [DOI] [PubMed] [Google Scholar]
- 4.Wardlaw JM, Doubal F, Armitage P, Chappell F, Carpenter T, Munoz Maniega S, Farrall A, Sudlow C, Dennis M, Dhillon B. Lacunar stroke is associated with diffuse blood-brain barrier dysfunction. Ann Neurol. 2009;65:194–202. doi: 10.1002/ana.21549. [DOI] [PubMed] [Google Scholar]
- 5.Fiala M, Liu QN, Sayre J, Pop V, Brahmandam V, Graves MC, Vinters HV. Cyclooxygenase-2-positive macrophages infiltrate the Alzheimer’s disease brain and damage the blood-brain barrier. Eur J Clin Invest. 2002;32:360–371. doi: 10.1046/j.1365-2362.2002.00994.x. [DOI] [PubMed] [Google Scholar]
- 6.Persidsky Y, Heilman D, Haorah J, Zelivyanskaya M, Persidsky R, Weber GA, Shimokawa H, Kaibuchi K, Ikezu T. Rho-mediated regulation of tight junctions during monocyte migration across the blood-brain barrier in HIV-1 encephalitis (HIVE) Blood. 2006;107:4770–4780. doi: 10.1182/blood-2005-11-4721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McCandless EE, Piccio L, Woerner BM, Schmidt RE, Rubin JB, Cross AH, Klein RS. Pathological expression of CXCL12 at the blood-brain barrier correlates with severity of multiple sclerosis. Am J Pathol. 2008;172:799–808. doi: 10.2353/ajpath.2008.070918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gaitan MI, Shea CD, Evangelou IE, Stone RD, Fenton KM, Bielekova B, Massacesi L, Reich DS. Evolution of the blood-brain barrier in newly forming multiple sclerosis lesions. Ann Neurol. 2011;70:22–29. doi: 10.1002/ana.22472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Haorah J, Schall K, Ramirez SH, Persidsky Y. Activation of protein tyrosine kinases and matrix metalloproteinases causes blood-brain barrier injury: Novel mechanism for neurodegeneration associated with alcohol abuse. Glia. 2008;56:78–88. doi: 10.1002/glia.20596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gursoy-Ozdemir Y, Qiu J, Matsuoka N, Bolay H, Bermpohl D, Jin H, Wang X, Rosenberg GA, Lo EH, Moskowitz MA. Cortical spreading depression activates and upregulates MMP-9. J Clin Invest. 2004;113:1447–1455. doi: 10.1172/JCI21227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim GW, Gasche Y, Grzeschik S, Copin JC, Maier CM, Chan PH. Neurodegeneration in striatum induced by the mitochondrial toxin 3-nitropropionic acid: role of matrix metalloproteinase-9 in early blood-brain barrier disruption? J Neurosci. 2003;23:8733–8742. doi: 10.1523/JNEUROSCI.23-25-08733.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Machado LS, Kozak A, Ergul A, Hess DC, Borlongan CV, Fagan SC. Delayed minocycline inhibits ischemia-activated matrix metalloproteinases 2 and 9 after experimental stroke. BMC Neurosci. 2006;7:56. doi: 10.1186/1471-2202-7-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rosell A, Alvarez-Sabin J, Arenillas JF, Rovira A, Delgado P, Fernandez-Cadenas I, Penalba A, Molina CA, Montaner J. A matrix metalloproteinase protein array reveals a strong relation between MMP-9 and MMP-13 with diffusion-weighted image lesion increase in human stroke. Stroke. 2005;36:1415–1420. doi: 10.1161/01.STR.0000170641.01047.cc. [DOI] [PubMed] [Google Scholar]
- 14.Rosell A, Ortega-Aznar A, Alvarez-Sabin J, Fernandez-Cadenas I, Ribo M, Molina CA, Lo EH, Montaner J. Increased brain expression of matrix metalloproteinase-9 after ischemic and hemorrhagic human stroke. Stroke. 2006;37:1399–1406. doi: 10.1161/01.STR.0000223001.06264.af. [DOI] [PubMed] [Google Scholar]
- 15.Carmeliet P. Angiogenesis in life, disease and medicine. Nature. 2005;438:932–936. doi: 10.1038/nature04478. [DOI] [PubMed] [Google Scholar]
- 16.Shalaby F, Rossant J, Yamaguchi TP, Gertsenstein M, Wu XF, Breitman ML, Schuh AC. Failure of blood-island formation and vasculogenesis in Flk-1-deficient mice. Nature. 1995;376:62–66. doi: 10.1038/376062a0. [DOI] [PubMed] [Google Scholar]
- 17.Millauer B, Wizigmann-Voos S, Schnurch H, Martinez R, Moller NP, Risau W, Ullrich A. High affinity VEGF binding and developmental expression suggest Flk-1 as a major regulator of vasculogenesis and angiogenesis. Cell. 1993;72:835–846. doi: 10.1016/0092-8674(93)90573-9. [DOI] [PubMed] [Google Scholar]
- 18.Neufeld G, Cohen T, Gengrinovitch S, Poltorak Z. Vascular endothelial growth factor (VEGF) and its receptors. FASEB J. 1999;13:9–22. [PubMed] [Google Scholar]
- 19.Baffert F, Le T, Sennino B, Thurston G, Kuo CJ, Hu-Lowe D, McDonald DM. Cellular changes in normal blood capillaries undergoing regression after inhibition of VEGF signaling. Am J Physiol Heart Circ Physiol. 2006;290:H547–H559. doi: 10.1152/ajpheart.00616.2005. [DOI] [PubMed] [Google Scholar]
- 20.Gerber HP, Hillan KJ, Ryan AM, Kowalski J, Keller GA, Rangell L, Wright BD, Radtke F, Aguet M, Ferrara N. VEGF is required for growth and survival in neonatal mice. Development. 1999;126:1149–1159. doi: 10.1242/dev.126.6.1149. [DOI] [PubMed] [Google Scholar]
- 21.Argaw AT, Gurfein BT, Zhang Y, Zameer A, John GR. VEGF-mediated disruption of endothelial CLN-5 promotes blood-brain barrier breakdown. Proc Natl Acad Sci U S A. 2009;106:1977–1982. doi: 10.1073/pnas.0808698106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Morin-Brureau M, Lebrun A, Rousset MC, Fagni L, Bockaert J, de Bock F, Lerner-Natoli M. Epileptiform activity induces vascular remodeling and zonula occludens 1 downregulation in organotypic hippocampal cultures: role of VEGF signaling pathways. J Neurosci. 2011;31:10677–10688. doi: 10.1523/JNEUROSCI.5692-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Young PP, Fantz CR, Sands MS. VEGF disrupts the neonatal blood-brain barrier and increases life span after non-ablative BMT in a murine model of congenital neurodegeneration caused by a lysosomal enzyme deficiency. Exp Neurol. 2004;188:104–114. doi: 10.1016/j.expneurol.2004.03.007. [DOI] [PubMed] [Google Scholar]
- 24.Chi OZ, Hunter C, Liu X, Tan T, Weiss HR. Effects of VEGF on the blood-brain barrier disruption caused by hyperosmolarity. Pharmacology. 2008;82:187–192. doi: 10.1159/000151433. [DOI] [PubMed] [Google Scholar]
- 25.Zuba D, Piekoszewski W, Pach J, Winnik L, Parczewski A. Concentration of ethanol and other volatile compounds in the blood of acutely poisoned alcoholics. Alcohol. 2002;26:17–22. doi: 10.1016/s0741-8329(01)00186-0. [DOI] [PubMed] [Google Scholar]
- 26.Deutch SR, Christian C, Hoyer S, Christensen EF, Dragsholt C, Hansen AC, Kristensen IB, Hougaard K. Drug and alcohol use among patients admitted to a Danish trauma centre: a prospective study from a regional trauma centre in Scandinavia. Eur J Emerg Med. 2004;11:318–322. doi: 10.1097/00063110-200412000-00004. [DOI] [PubMed] [Google Scholar]
- 27.Haorah J, Knipe B, Gorantla S, Zheng J, Persidsky Y. Alcohol-induced blood-brain barrier dysfunction is mediated via inositol 1,4,5-triphosphate receptor (IP3R)-gated intracellular calcium release. J Neurochem. 2007;100:324–336. doi: 10.1111/j.1471-4159.2006.04245.x. [DOI] [PubMed] [Google Scholar]
- 28.Alikunju S, Abdul Muneer PM, Zhang Y, Szlachetka AM, Haorah J. The inflammatory footprints of alcohol-induced oxidative damage in neurovascular components. Brain Behav Immun. 2011;25(Suppl 1):S129–136. doi: 10.1016/j.bbi.2011.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li DQ, Meller D, Liu Y, Tseng SC. Overexpression of MMP-1 and MMP-3 by cultured conjunctivochalasis fibroblasts. Invest Ophthalmol Vis Sci. 2000;41:404–410. [PubMed] [Google Scholar]
- 30.Ramirez SH, Heilman D, Morsey B, Potula R, Haorah J, Persidsky Y. Activation of peroxisome proliferator-activated receptor gamma (PPARgamma) suppresses Rho GTPases in human brain microvascular endothelial cells and inhibits adhesion and transendothelial migration of HIV-1 infected monocytes. J Immunol. 2008;180:1854–1865. doi: 10.4049/jimmunol.180.3.1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Domingues I, Rino J, Demmers JA, de Lanerolle P, Santos SC. VEGFR2 translocates to the nucleus to regulate its own transcription. PLoS One. 2011;6:e25668. doi: 10.1371/journal.pone.0025668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lampugnani MG, Orsenigo F, Gagliani MC, Tacchetti C, Dejana E. Vascular endothelial cadherin controls VEGFR-2 internalization and signaling from intracellular compartments. J Cell Biol. 2006;174:593–604. doi: 10.1083/jcb.200602080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Reijerkerk A, Kooij G, van der Pol SM, Khazen S, Dijkstra CD, de Vries HE. Diapedesis of monocytes is associated with MMP-mediated occludin disappearance in brain endothelial cells. FASEB J. 2006;20:2550–2552. doi: 10.1096/fj.06-6099fje. [DOI] [PubMed] [Google Scholar]
- 34.Raffetto JD, Khalil RA. Matrix metalloproteinases and their inhibitors in vascular remodeling and vascular disease. Biochem Pharmacol. 2008;75:346–359. doi: 10.1016/j.bcp.2007.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bennett J, Basivireddy J, Kollar A, Biron KE, Reickmann P, Jefferies WA, McQuaid S. Blood-brain barrier disruption and enhanced vascular permeability in the multiple sclerosis model EAE. J Neuroimmunol. 2010;229:180–191. doi: 10.1016/j.jneuroim.2010.08.011. [DOI] [PubMed] [Google Scholar]
- 36.Tsuji K, Aoki T, Tejima E, Arai K, Lee SR, Atochin DN, Huang PL, Wang X, Montaner J, Lo EH. Tissue plasminogen activator promotes matrix metalloproteinase-9 upregulation after focal cerebral ischemia. Stroke. 2005;36:1954–1959. doi: 10.1161/01.STR.0000177517.01203.eb. [DOI] [PubMed] [Google Scholar]
- 37.Turk BE, Huang LL, Piro ET, Cantley LC. Determination of protease cleavage site motifs using mixture-based oriented peptide libraries. Nat Biotechnol. 2001;19:661–667. doi: 10.1038/90273. [DOI] [PubMed] [Google Scholar]
- 38.Tran ED, DeLano FA, Schmid-Schonbein GW. Enhanced matrix metalloproteinase activity in the spontaneously hypertensive rat: VEGFR-2 cleavage, endothelial apoptosis, and capillary rarefaction. J Vasc Res. 2010;47:423–431. doi: 10.1159/000281582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lamy S, Lachambre MP, Lord-Dufour S, Beliveau R. Propranolol suppresses angiogenesis in vitro: inhibition of proliferation, migration, and differentiation of endothelial cells. Vascul Pharmacol. 2010;53:200–208. doi: 10.1016/j.vph.2010.08.002. [DOI] [PubMed] [Google Scholar]
- 40.Ugarte-Berzal E, Redondo-Munoz J, Eroles P, Del Cerro MH, Garcia-Marco JA, Terol MJ, Garcia-Pardo A. VEGF/VEGFR2 interaction down-regulates matrix metalloproteinase-9 via STAT1 activation and inhibits B chronic lymphocytic leukemia cell migration. Blood. 2010;115:846–849. doi: 10.1182/blood-2009-08-239426. [DOI] [PubMed] [Google Scholar]
- 41.Ito TK, Ishii G, Saito S, Yano K, Hoshino A, Suzuki T, Ochiai A. Degradation of soluble VEGF receptor-1 by MMP-7 allows VEGF access to endothelial cells. Blood. 2009;113:2363–2369. doi: 10.1182/blood-2008-08-172742. [DOI] [PubMed] [Google Scholar]
- 42.Kanazawa M, Igarashi H, Kawamura K, Takahashi T, Kakita A, Takahashi H, Nakada T, Nishizawa M, Shimohata T. Inhibition of VEGF signaling pathway attenuates hemorrhage after tPA treatment. J Cereb Blood Flow Metab. 2011;31:1461–1474. doi: 10.1038/jcbfm.2011.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Heberlein A, Muschler M, Lenz B, Frieling H, Buchl C, Groschl M, Riera R, Kornhuber J, Bleich S, Hillemacher T. Serum levels of vascular endothelial growth factor A increase during alcohol withdrawal. Addict Biol. 2010;15:362–364. doi: 10.1111/j.1369-1600.2010.00215.x. [DOI] [PubMed] [Google Scholar]
- 44.Radek KA, Kovacs EJ, Gallo RL, DiPietro LA. Acute ethanol exposure disrupts VEGF receptor cell signaling in endothelial cells. Am J Physiol Heart Circ Physiol. 2008;295:H174–H184. doi: 10.1152/ajpheart.00699.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Deguchi T, Hashiguchi T, Horinouchi S, Uto T, Oku H, Kimura K, Makisumi K, Arimura K. Serum VEGF increases in diabetic polyneuropathy, particularly in the neurologically active symptomatic stage. Diabet Med. 2009;26:247–252. doi: 10.1111/j.1464-5491.2009.02680.x. [DOI] [PubMed] [Google Scholar]
- 46.Obermair A, Tempfer C, Hefler L, Preyer O, Kaider A, Zeillinger R, Leodolter S, Kainz C. Concentration of vascular endothelial growth factor (VEGF) in the serum of patients with suspected ovarian cancer. Br J Cancer. 1998;77:1870–1874. doi: 10.1038/bjc.1998.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.