

The G proteins Gα12/Gα13 and Gαq/Gα11 have opposing effects on vascular remodeling by differentially regulating vascular smooth muscle cell differentiation.
Abstract
Vascular smooth muscle (Sm) cells (VSMCs) are highly plastic. Their differentiation state can be regulated by serum response factor (SRF), which activates genes involved in Sm differentiation and proliferation by recruiting cofactors, such as members of the myocardin family and ternary complex factors (TCFs), respectively. However, the extracellular cues and upstream signaling mechanisms regulating SRF-dependent VSMC differentiation under in vivo conditions are poorly understood. In this study, we show that the procontractile signaling pathways mediated by the G proteins G12/G13 and Gq/G11 antagonistically regulate VSMC plasticity in different models of vascular remodeling. In mice lacking Gα12/Gα13 or their effector, the RhoGEF protein LARG, RhoA-dependent SRF-regulation was blocked and down-regulation of VSMC differentiation marker genes was enhanced. This was accompanied by an excessive vascular remodeling and exacerbation of atherosclerosis. In contrast, Sm-specific Gαq/Gα11 deficiency blocked activation of extracellular signal-regulated kinase 1/2 and the TCF Elk-1, resulting in a reduced VSMC dedifferentiation in response to flow cessation or vascular injury. These data show that the balanced activity of both G protein–mediated pathways in VSMCs is required for an appropriate vessel remodeling response in vascular diseases and suggest new approaches to modulate Sm differentiation in vascular pathologies.
Unlike skeletal or cardiac muscle cells, which are terminally differentiated, vascular smooth muscle (Sm) cells (SMCs [VSMCs]) retain a remarkable degree of plasticity throughout their lives. They can switch between a quiescent contractile state and phenotypes of increased proliferation, migration, and synthetic capacity (Owens, 1995). Dedifferentiation and redifferentiation of VSMCs are believed to be involved in vascular remodeling processes that physiologically enable vascular development and repair, as well as adaptation to chronically altered hemodynamics. However, dysregulation of VSMC plasticity also plays a role in the pathogenesis of vascular diseases such as atherosclerosis, restenosis after percutaneous interventions, and systemic as well as pulmonary hypertension (Owens et al., 2004).
The differentiation state of VSMCs is under the control of transcriptional regulators. Many genes involved in the regulation of SMC contractility are controlled by serum response factor (SRF), a widely expressed transcription factor which is believed to play a key role in the regulation of Sm differentiation (Miano et al., 2007; Owens, 2007). However, SRF can also induce the transcription of growth-related genes involved in Sm proliferation and dedifferentiation, and it is now well established that two families of transcriptional cofactors, the myocardin family (Pipes et al., 2006; Parmacek, 2007) and the ternary complex factor (TCF) family of Ets domain proteins (Treisman, 1994), differentially modulate the transcription of these distinct SRF target genes through their mutually exclusive binding to SRF (Wang et al., 2004). Whereas coactivators of the myocardin family, consisting of myocardin itself and myocardin-related transcription factors (MRTFs) A and B, promote VSMC differentiation, competitive binding of TCFs induces decreased expression of SMC-selective marker genes and VSMC proliferation (Mack, 2011). TCFs are phosphorylated and activated through the Ras/MAPK pathway (Posern and Treisman, 2006), whereas RhoA-mediated signaling has been shown to promote nuclear translocation of MRTFs and to induce Sm differentiation (Lu et al., 2001; Mack et al., 2001; Olson and Nordheim, 2010). However, the extracellular cues and upstream signaling mechanisms regulating SRF-dependent VSMC differentiation under in vivo conditions have remained poorly understood.
Most of the extracellular stimuli that regulate vascular Sm tone and increase contractility exert their effects via G protein–coupled receptors (GPCRs), which regulate two major pathways involving the heterotrimeric G proteins Gq/G11 and G12/G13. Whereas Gq/G11 mediates the activation of phospholipase C β-isoforms and subsequent Ca2+/calmodulin-dependent activation of myosin light chain kinase, the G12/G13 family of G proteins couples to Rho guanine nucleotide exchange factor (RhoGEF) proteins to activate RhoA and thereby induces a Ca2+-independent VSMC contraction via inhibition of myosin phosphatase (Gohla et al., 2000; Somlyo and Somlyo, 2003; Maguire and Davenport, 2005). Gq/G11-mediated signaling in VSMCs is required for basal vascular tone induced by vasoactive mediators, whereas both Gq/G11 and G12/G13 need to be activated for pathological increases in vascular tone like in hypertension (Wirth et al., 2008).
Here we report that the procontractile signaling pathways mediated by the G proteins G12/G13 and Gq/G11 antagonistically regulate SRF-dependent VSMC differentiation. Whereas G12/G13 promotes differentiation, Gq/G11 reduces SMC-selective marker gene expression and stimulates proliferation. Our data indicate that the balanced activities of both procontractile G protein–mediated signaling pathways control VSMC plasticity under basal conditions as well as after vascular injury or in response to changes in blood flow.
RESULTS
Reduced expression of Sm differentiation marker genes in Gα12/Gα13- but not in Gαq/Gα11-deficient vascular Sm
Using quantitative RT-PCR (qRT-PCR) analysis, we evaluated the effect of Sm-specific Gαq/Gα11 and Gα12/Gα13 deficiency on vascular gene expression in the media of arterial vessels using SMMHC-CreERT2;Gαqflox/flox;Gα11−/− mice (Sm-Gαq/Gα11-KO) and SMMHC-CreERT2;Gα12−/−;Gα13flox/flox mice (Sm-Gα12/Gα13-KO; Wirth et al., 2008). In different vessels from Sm-Gα12/Gα13-KO mice, we detected decreased mRNA levels of Sm differentiation marker genes such as Acta2 (α-Sm actin [α-SMA]), Myh11 (Sm myosin heavy chain [SMMHC]), Cnn1 (calponin-1), or Tagln (SM22), as well as of the Sm-enriched microRNAs 143 and 145, compared with WT and Sm-Gαq/Gα11-KO mice (Fig. 1 A and not depicted). Despite the down-regulation of genes encoding proteins of the contractile apparatus of SMCs, myometric experiments of agonist-induced contraction in carotid arteries did not reveal any functional defect in Sm-Gα12/Gα13-KO mice (Fig. 1 B). These results are in line with our previous finding that Sm-Gα12/Gα13-KO mice display a normal blood pressure profile (Wirth et al., 2008), suggesting that the observed subtle but significant changes in expression pattern are of no functional relevance.
Figure 1.

Differential effects of Gq/G11 and G12/G13 on neointima formation. (A) Relative levels of mRNAs encoding Sm differentiation markers normalized against 18S and of miR143/miR145 normalized against 4.5S RNA in the media of carotid arteries from WT, Sm-Gα12/Gα13-KO (Sm-12/13-KO), or Sm-Gαq/Gα11-KO (Sm-q/11-KO) mice (the data are representative for five to six males and three independent experiments per group). Levels in the media of WT mice were set as 100%. (B) Effect of increasing concentration of phenylephrine or KCl on the vascular tone of carotid arteries from WT or Sm-12/13-KO mice in percentage of maximal response (the data are representative for four males and two independent experiments per group). (C and D) Analysis of carotid artery remodeling after ligation. (C) Shown are sections of the carotid arteries from WT, Sm-12/13-KO, and Sm-q/11-KO mice at a distance of 250, 1,000, 2,000, and 3,000 µm from the ligation site as well as sections of the contralateral vessel 4 wk after ligation. (D) The neointima and media areas in sections at the indicated distances from the ligation site were determined (the data are representative for five to seven males and three independent experiments per group). (E) Carotid arteries from WT and Sm-12/13-KO animals 4 wk after ligation were stained for elastic fibers (left), proteoglycans (middle), and α-SMA (right). Shown are representative sections (the data are representative for four to six males and two independent experiments per group). (F and G) Carotid arteries from WT and Sm-12/13-KO mice were immunostained 4 wk after ligation for CD68 and α-SMA or CD3 and α-SMA. Nuclei were counterstained with DAPI. Shown are representative images of stained sections (F) as well as the proportions of CD3-, α-SMA–, or CD68-positive cells in the intima (int.), media (media.), or adventitia (adv.; G; the data are representative for four to five males and two independent experiments per group). Arrows in F indicate CD68 (left)- or CD3 (right)-positive cells. (H) Carotid arteries from WT, Sm-12/13-KO (12/13), or Sm-q/11-KO mice (q/11) were stained 3 d after ligation for α-SMA and Ki-67. Shown are representative images as well as a statistical analysis of the percentage of Ki-67–positive cells among α-SMA–positive cells (the data are representative for four males and two independent experiments per group). Shown are mean values ± SEM; *, P < 0.05; **, P < 0.01; ***, P < 0.001 (compared with WT). Bars: (C) 100 µm; (E, F, and H) 50 µm.
Gq/G11 and G12/G13 antagonistically regulate VSMC differentiation
As vascular remodeling processes such as neointima formation in response to vascular injury or flow cessation are often accompanied by decreased expression of SMC differentiation marker genes, we tested the response of Sm-specific Gαq/Gα11- and Gα12/Gα13-deficient mice to flow cessation in the carotid artery ligation model. In WT vessels, carotid artery ligation results in neointimal hyperplasia, adjacent to the ligation site. Histological evaluation 4 wk after ligation revealed a severely exaggerated response to flow cessation in Sm-Gα12/Gα13-KO mice, with excessive neointimal as well as medial hyperplasia (Fig. 1, C–E). By morphometric analysis, Sm-specific deletion of Gα12/Gα13 increased neointima and media thickness several fold compared with WT animals.
A closer analysis of the expanded media of ligated vessels from Sm-Gα12/Gα13-KO mice showed a disarray of elastic lamina and the presence of proteoglycans stained with Alcian blue (Fig. 1 E). Although the number of α-SMA–positive cells in the vessels of Sm-Gα12/Gα13-KO mice was increased compared with WT animals 4 wk after ligation, the proportion of CD68- and CD3-positive cells was indistinguishable between both genotypes (Fig. 1, F and G). VSMCs in the absence of Gα12/Gα13 showed an increased acute proliferative response to vascular injury as indicated by the strongly increased number of Ki-67 and α-SMA double-positive cells in the media 3 d after ligation (Fig. 1 H).
Surprisingly, an opposite phenotype was observed in Sm-Gαq/Gα11-KO mice, in which neointimal size was reduced compared with WT mice (Fig. 1, C and D). In most sections from Sm-Gαq/Gα11-KO mice, no neointima formation could be detected. This correlated with a reduced proliferative response to vascular injury compared with WT mice (Fig. 1 H). Because systemic blood pressure is reduced by 10–15% in Gαq/Gα11- deficient mice (Wirth et al., 2008), we tested whether the observed phenotype in Sm-Gαq/Gα11-KO animals was caused by hypotensive hemodynamics. In mice treated with the antihypertensives hydralazine or metoprolol in concentrations that reduced blood pressure to a level comparable with that in Sm-Gαq/Gα11-KO mice, the extent of neointima formation equaled that in untreated littermates (Fig. 2 A), suggesting that the reduced response of Sm-q/11-KO mice to flow cessation was unlikely to be a consequence of a reduced blood pressure.
Figure 2.

Effects of Gαq/Gα11 and Gα12/Gα13 deficiency on SMC differentiation marker gene expression. (A) Effect of hydralazine- and metoprolol-induced hypotension on neointima formation after carotid ligation. WT mice were treated without or with 500 mg/liter hydralazine or 2.5 g/liter metoprolol in the drinking water resulting in a 10–15% reduction of the mean arterial blood pressure in the treated group compared with untreated animals. Shown are the mean arterial blood pressure values during 2 d before treatment (before) and during days 3 and 4 after the start of treatment (after) and an evaluation of the neointima areas in sections from untreated and treated animals at the indicated distances from the ligation site (n = 3). (B) Relative levels of mRNAs encoding Sm differentiation markers as well as of miR143/miR145 in the media of carotid arteries from WT, Sm-12/13-KO, and Sm-q/11-KO mice 3 d after carotid artery ligation relative to the levels in the media of the sham-operated contralateral vessel (n = 5–6). (C) Carotid artery sections at a distance of 1,000 µm from the ligation site from WT, Sm-12/13-KO, and Sm-q/11-KO mice 7 d after sham operation (sham) or carotid artery ligation stained with an anti–α-SMA antibody. White boxes indicate enlarged areas at the bottom of each image. (D) 3 d after sham operation or ligation, carotid arteries of WT, Sm-Gα12/Gα13-KO (Sm-12/13-KO), or Sm-Gαq/Gα11-KO (Sm-q/11-KO) were prepared free of adventitia and intima, and lysates were analyzed by immunoblotting using antibodies against α-SMA, Sm22, and tubulin. (A–D) Shown is one representative of at least three experiments. Shown are mean values ± SEM; *, P < 0.05; **, P < 0.01; ***, P < 0.001 (compared with WT). Bars: (C, bottom) 40 µm; (C, top) 100 µm.
3 d after carotid artery ligation, RNA levels of Sm differentiation marker genes were significantly decreased in the media of operated WT vessels compared with contralateral control vessels (Fig. 2 B). In addition to the reduced basal expression of Sm differentiation marker genes, carotid artery ligation resulted in an exaggerated down-regulation of the genes in Sm-Gα12/Gα13-KO vessels (Fig. 2 B). In contrast, down-regulation of Sm differentiation marker genes was significantly reduced in Sm-Gαq/Gα11-KO compared with WT vessels. Consistent with the results from qRT-PCR, immunohistochemistry revealed a decreased α-SMA expression in response to carotid artery ligation in WT VSMCs. However, although α-SMA expression in Gα12/Gα13-deficient VSMCs was reduced after ligation to almost undetectable levels, no significant down-regulation was apparent in Gαq/Gα11-deficient VSMCs (Fig. 2 C). Similarly, immunoblotting of lysates of the media from sham-operated and ligated WT, Sm-Gα12/Gα13-KO, and Sm-Gαq/Gα11-KO vessels showed reduced levels of α-SMA and Sm22 in sham-operated Sm-Gα12/Gα13-KO media compared with WT (Fig. 2 D). 3 d after ligation, protein levels were more reduced in media from Sm-specific Gα12/Gα13-deficient vessels compared with WT media, whereas the reduction was less pronounced in media from ligated Sm-q/11-KO vessels when compared with WT (Fig. 2 D).
G12/G13 promotes VSMC differentiation through LARG-mediated activation of RhoA
As G12/G13 link GPCRs to activation of RhoA, we assessed whether RhoA is activated upon carotid artery ligation. Indeed, RhoA activity significantly increased upon carotid artery ligation in the media of WT and Sm-Gαq/Gα11-KO mice (Fig. 3 A). However, Sm-specific deletion of Gα12/Gα13 blocked activation of RhoA. G12/G13 activates RhoA through a subfamily of RhoGEFs consisting of p115-RhoGEF (Arhgef1), PDZ-RhoGEF (Arhgef11), and LARG (Arhgef12). Because LARG is the predominant RhoGEF effector of G12/G13 in VSMCs (Wirth et al., 2008), we analyzed mice with Sm-specific LARG deficiency (SMMHC-CreERT2;Arhgef12flox/flox [Sm-LARG-KO]). Sm-LARG-KO mice also lacked RhoA activation after carotid artery ligation (Fig. 3 A), and as in Gα12/Gα13-deficient mice, ligation resulted in excessive neointima and media hyperplasia, accompanied by a disarray of elastic lamina (Fig. 3, B and C) and enhanced down-regulation of α-SMA (Fig. 3 D). Thus, Sm-specific LARG-deficient mice phenocopy Sm-12/13-KO mice, indicating that the G12/G13-dependent differentiation of VSMCs in vivo involves LARG-dependent activation of RhoA.
Figure 3.

VSMC differentiation via RhoA/LARG. (A) Relative RhoA activity in the media of carotid arteries from WT, Sm-12/13-KO, Sm-LARG-KO, and Sm-q/11-KO mice 24 h after sham operation or carotid artery ligation (the data are representative for four to six males and three independent experiments per group). (B and C) The left common carotid artery of WT and Sm-LARG-KO mice was ligated and analyzed. Shown are sections of the vessel (B) as well as the determination of the neointima and media areas (C) at a distance of 250, 1,000, and 3,000 µm from the ligation site (the data are representative for six males and three independent experiments per group). (D) Carotid arteries from WT or Sm-LARG-KO mice were analyzed 7 d after sham operation or carotid artery ligation, and sections at ∼1,000 µm from the ligation site were stained with an anti–α-SMA antibody. White boxes indicate enlarged areas at the bottom of each panel. (E and F) VSMCs from carotid arteries of WT, Sm-12/13-KO, Sm-LARG-KO, or Sm-q/11-KO mice were isolated, starved for 48 h, and then incubated in the absence or presence of 20% FBS for 1 h. Cells were fixed and stained with an anti–MRTF-A antibody (E), and the fraction of cells with nuclear MRTF-A staining was determined (F; the data are representative for four to six males and three independent experiments per group). (G) Relative levels of mRNA encoding myocardin (myocd) in the media of carotid arteries in WT, Sm-12/13-KO, and Sm-q/11-KO mice 3 d after sham operation carotid artery ligation (the data are representative for three to six males and twp independent experiments per group). Shown are mean values ± SEM; *, P < 0.05; **, P < 0.01; ***, P < 0.001 (compared with WT, sham, or –serum conditions, respectively). Bars: (B and D [top]) 100 µm; (D, bottom) 40 µm; (E) 5 µm.
Cell-based studies have suggested that RhoA can induce SRF-dependent transcription of Sm marker genes through two mechanisms, up-regulation of myocardin expression (Wamhoff et al., 2004; Yoshida et al., 2004) and facilitation of MRTF nuclear translocation (Miralles et al., 2003; Lockman et al., 2004; Jeon et al., 2008). We therefore tested the effect of Sm-specific Gα12/Gα13 and LARG deficiency on nuclear translocation of MRTF-A in primary VSMCs. Serum-induced nuclear translocation of MRTF-A was absent in Gα12/Gα13- and LARG-deficient VSMCs (Fig. 3, E and F). In contrast, nuclear translocation of MRTF-A in Gαq/Gα11-deficient VSMCs was comparable with WT VSMCs (Fig. 3, E and F). We also investigated expression levels of myocardin and found reduced levels in the media of carotid arteries from sham-operated and operated Sm-Gα12/Gα13-KO mice compared with WT animals or animals with Sm-specific Gαq/Gα11 deficiency (Fig. 3 G).
Gq/G11 regulates MAPK-dependent induction of early response genes
To determine the mechanism that underlies the Sm-Gαq/Gα11-KO phenotype of reduced neointima formation, we analyzed the phosphorylation of the MAPK Erk1/2 as well as of the TCF Elk-1, which have been shown to mediate an inhibition of Sm differentiation (Mack, 2011). 24 h after ligation, we observed a significant increase of anti–phospho-Erk1/2 staining in the media of WT and Sm-Gα12/Gα13-KO mice but not in that of Sm-Gαq/Gα11-KO mice (Fig. 4, A and B). Similarly, the Erk1/2 substrate Elk-1 showed increased phosphorylation in WT and Sm-specific Gα12/Gα13-deficient mice compared with the media of Sm-Gαq/Gα11-KO animals 24 h after carotid artery ligation (Fig. 4, C and D). Finally, the ligation-induced and TCF-mediated up-regulation of early response genes such as Fos, Egr1, and Ets1 was blocked by Sm-specific Gαq/Gα11 deficiency, whereas their up-regulation was not affected in Sm-Gα12/Gα13-KO mice (Fig. 4, E–G).
Figure 4.

Gq/G11-mediated signaling in VSMCs after carotid artery ligation. (A and B) 24 h after carotid artery ligation or sham operation, carotid arteries of WT, Sm-12/13-KO, or Sm-q/11-KO mice were isolated and sectioned. Shown are sections at a distance of 250–500 µm from the ligation site stained with an anti–phospho-Erk1/2 antibody (A), and the relative pERK-positive area was determined (n = 3–4; B). (C and D) 24 h after carotid artery ligation or sham operation, carotid arteries from WT, Sm-12/13-KO, or Sm-q/11-KO mice were isolated and sectioned. Shown are sections at a distance of 250–500 µm from the ligation site stained with an anti–phospho-Elk1 (pElk1) antibody and counterstained with hematoxylin (n = 3–4; C). (D) The percentage of pElk1-positive nuclei is shown. (E–G) 3 d after carotid artery ligation or sham operation of WT, Sm-12/13-KO, or Sm-q/11-KO mice, the media of carotid arteries was isolated, and the levels of mRNAs encoding Fos, Ets1, or Egr1 were determined. (A–G) Shown are mean values ± SEM (n = 3–6); *, P < 0.05; **, P < 0.01; ***, P < 0.001 (compared with sham). Shown is one representative of at least three experiments. Bars, 20 µm.
G12/G13 and Gq/G11 antagonistically regulate the VSMC response to injury
Having uncovered an antagonistic role of G12/G13- and Gq/G11-mediated signaling in the regulation of SRF cofactors and neointima formation in the carotid artery ligation model, we wondered whether our findings apply analogously to other vascular remodeling processes. This prompted us to use a femoral artery injury model for restenosis. Our observations 4 wk after femoral artery injury paralleled those from the carotid artery ligation model. Sm-Gα12/Gα13-KO mice displayed excessive neointimal and medial hyperplasia, whereas Sm-specific deficiency of Gαq/Gα11 significantly attenuated the response to injury compared with WT animals (Fig. 5, A and B). Moreover, down-regulation of VSMC differentiation marker genes as determined by α-SMA staining was pronounced in Gα12/Gα13-deficient but not in Gαq/Gα11-deficient VSMCs (Fig. 5 C). This further indicates a critical role of Gq/G11 and G12/G13 signaling in the regulation of vascular Sm differentiation.
Figure 5.
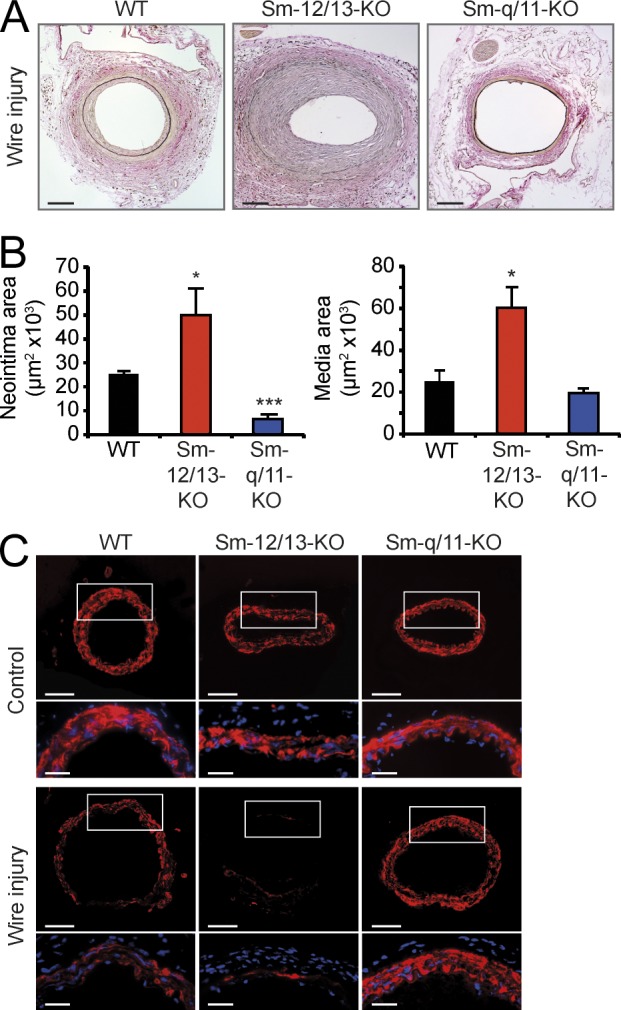
Effect of Sm-specific Gα deficiencies on vascular response to femoral artery injury. (A and B) 4 wk after femoral artery injury, femoral arteries from WT, Sm-12/13-KO, or Sm-q/11-KO mice were isolated and sectioned as described in Materials and methods. Shown are representative sections (A) and an evaluation of the neointima and media areas (n = 4; B). Shown are mean values ± SEM; *, P < 0.05; ***, P < 0.001 (compared with WT). (C) 7 d after femoral artery injury, femoral arteries from WT, Sm-12/13-KO, or Sm-q/11-KO mice were isolated. Shown are immunohistochemical analyses performed with an antibody against α-SMA. Boxes indicate magnified areas at the bottom of each panel. (A–C) Shown is one representative of at least three experiments. Bars: (A and C [top]) 100 µm; (C, bottom) 40 µm.
Sm-specific deficiency of Gα12/Gα13 promotes atherosclerotic plaque progression
As changes in vascular Sm differentiation may be involved in the pathogenesis of atherosclerosis (Dzau et al., 2002; Owens et al., 2004), we crossed Sm-Gα12/Gα13-KO mice with atherosclerosis-prone ApoE−/− mice. We did not analyze the effect of Sm-specific Gαq/Gα11 deficiency on atherosclerosis as any result would be confounded by the reduced vascular tone and blood pressure in Sm-Gαq/Gα11-KO mice (Wirth et al., 2008). After 12 wk of a high-fat diet, atherosclerotic lesions in ApoE-deficient Sm-Gα12/Gα13-KO mice compared with ApoE-deficient control mice were far more pronounced in terms of intimal plaque and media area (Fig. 6, A and B). In addition, we found total occlusions of the right common carotid artery in about a quarter of ApoE-deficient Sm-Gα12/Gα13-KO mice compared with none in the ApoE-deficient control mice. Consistent with the results obtained from carotid artery ligation and femoral artery injury, we observed a reduced α-SMA staining of the media in atherosclerotic vessels of Sm-12/13-KO mice (Fig. 6 C).
Figure 6.

Increased plaque size in Sm-specific Gα12/Gα13-deficient mice lacking ApoE. (A and B) After 12 wk of a high-fat diet, the innominate and right common carotid arteries of ApoE−/− or ApoE−/−;SMMHC-CreERT2;Gα12−/−;Gα13flox/flox (ApoE−/−;Sm-12/13-KO) mice were isolated and analyzed histologically. (A) Shown are representative sections. (B) The plaque and media areas in animals of both genotypes were determined in innominate arteries at a distance of 1,000 µm from the aortic arch (innom.), in the right common carotid artery at a distance of 1,000 µm from the bifurcation of the innominate artery (prox. RCCA), at a distance of 1,000 µm from the bifurcation of the common carotid artery (dist. RCCA), or in between (med. RCCA; the data are representative for nine males and two independent experiments per group). (C) The innominate artery or right common carotid artery (RCCA) of ApoE−/− or ApoE−/−;Sm-12/13-KO mice was isolated and stained with an anti–α-SMA antibody. (D) Atherosclerotic plaques from ApoE−/− or ApoE−/−;Sm-12/13-KO mice were immunostained with antibodies against α-SMA and Ki-67. Shown are representative images. The right image is a magnification of the area indicated by the white box, and arrows point to α-SMA and Ki-67 double-positive cells. The bar graphs show a statistical evaluation of the number of α-SMA–positive cells per cells in the plaque area and the percentage of Ki-67–positive cells per α-SMA–positive cells in plaques from ApoE−/− (WT) and ApoE−/−;Sm-12/13-KO mice (12/13; the data are representative for four males and two independent experiments per group). (E) Sections of atherosclerotic plaques from ApoE−/− or ApoE−/−;Sm-12/13-KO mice were immunostained with antibodies against CD68. Shown are individual images counterstained with DAPI (media and plaques [p] are marked) as well as a bar graph showing the statistical evaluation of the percentage of CD68-positive cells in plaques of both groups of animals (the data are representative for six males and two independent experiments per group). Shown are mean values ± SEM; *, P < 0.05; **, P < 0.01; ***, P < 0.001. Bars: (A and C) 100 µm; (D and E) 50 µm.
Immunostaining of atherosclerotic lesions of ApoE-deficient control mice and mice lacking Gα12/Gα13 in VSMCs with antibodies against α-SMA and against the proliferation marker Ki-67 revealed that the number of α-SMA–positive cells as well as the number of proliferating α-SMA–positive cells (Ki-67 and α-SMA double-positive cells) were significantly increased in lesions from Sm-specific Gα12/Gα13-deficient mice (Fig. 6, C and D). This suggests that lack of Gα12/Gα13 in VSMCs results in an increased propensity of α-SMA–positive cells in the atherosclerotic lesion to proliferate. Immunostaining with anti-CD68 antibodies revealed no significant differences in numbers of macrophages per plaque area in lesions from mice with Sm-specific Gα12/Gα13 deficiency compared with WT animals (Fig. 6 E).
DISCUSSION
VSMCs are highly plastic, and their dedifferentiation as well as their redifferentiation are thought to underlie many vascular remodeling processes. In this study, we show that the G proteins G12/G13 and Gq/G11 antagonistically regulate VSMC plasticity in different models of vascular remodeling. Our data from mice lacking Gα12/Gα13 or their effector, the RhoGEF protein LARG, specifically in SMCs reveal the dramatic consequences of an imbalance in the activity of the two G protein–mediated pathways. The predominance of Gq/G11- over G12/G13-LARG–mediated signaling promoted down-regulation of SMC differentiation marker genes and resulted in a highly abnormal phenotype of excessive vascular remodeling and exacerbation of atherosclerosis. In contrast, Sm-specific Gαq/Gα11 deficiency protected mice from neointimal hyperplasia in response to flow cessation or femoral artery injury.
Based on these findings, we conclude that G12/G13-mediated signaling is required to maintain VSMCs in a fully differentiated state and to prevent their unrestrained dedifferentiation in response to extracellular cues. Consistent with our observations, various vasoconstrictors acting through receptors coupled to both G12/G13 and Gq/G11 such as thrombin, lysophosphatidic acid, thromboxane A2, angiotensin-II, sphingosine-1-phosphate (S1P), or endothelin-1 have been reported to be capable of promoting the expression of Sm differentiation marker genes in vitro (Andrawis et al., 1996; Yoshida et al., 2004; Kim et al., 2009; Martin et al., 2009; Medlin et al., 2010). Our data from mice lacking Gα12/Gα13 or LARG specifically in SMCs suggest that Gα12/Gα13 promotes VSMC differentiation through LARG-mediated activation of RhoA. This is supported by in vitro data indicating that RhoA and LARG can mediate SMC differentiation (Lu et al., 2001; Mack et al., 2001; Gorenne et al., 2006; Medlin et al., 2010). In addition, evidence from cell-based studies shows that RhoA can induce SRF-dependent transcription of Sm differentiation marker genes through two distinct mechanisms, up-regulation of myocardin expression (Wamhoff et al., 2004; Yoshida et al., 2004; Martin et al., 2009) and facilitation of MRTF-A nuclear translocation (Miralles et al., 2003; Lockman et al., 2004; Jeon et al., 2008). Concordant with these data, we observed a down-regulation of myocardin expression in response to Gα12/Gα13 deletion in VSMCs in vivo. Moreover, our data show that G12/G13-LARG–mediated signaling is required for serum-induced nuclear translocation of MRTF-A. A precise analysis of the involvement of individual myocardin factors in the regulation of SMC differentiation has been difficult because of their overlapping expression and potential heterodimerization. Studies in mice lacking individual factors clearly indicate considerable functional redundancy in most types of VSMCs (Li et al., 2005; Oh et al., 2005; Pipes et al., 2005; Li et al., 2006; Sun et al., 2006). There are several effectors downstream of RhoA that can link RhoA to MRTFs such as Rho-kinase or mammalian homologue of diaphanous (mDia; Olson and Nordheim, 2010). A study using bone marrow chimeras has provided evidence that at least Rho-kinase subtype 1 in non-SMCs is involved in neointima formation (Noma et al., 2008); however, this study could not rule out a function of Rho-kinases in VSMCs. It is also possible that the RhoA-regulated actin nucleating protein mDia links G12/G13-RhoA to the regulation of SRF-mediated transcriptional events via MRTFs as it has been shown to promote actin polymerization and SMC-specific gene expression (Copeland and Treisman, 2002; Staus et al., 2007).
We found that the Gq/G11-mediated signaling pathway in VSMCs mediates the expression of early response genes and the suppression of SMC differentiation marker gene expression, as well as proliferation in response to flow cessation and vascular injury. In addition, our data indicate that this pathway involves activation of Erk1/2 as well as of its effector, the TCF Elk-1. This is consistent with earlier studies showing a central role for Erk in the regulation of TCFs (Treisman, 1994; Xi et al., 1997; Lockman et al., 2004). How Gq/G11 regulates Erk1/2 in VSMCs is not fully clear. In vitro studies and studies in other organ systems suggest that Gq/G11 can mediate Erk1/2 activation through transactivation of receptor tyrosine kinases (Hsieh et al., 2009) or by direct regulation of Erk1/2 through βγ-subunits released from Gq/G11 (Lorenz et al., 2009). In SMCs, several mechanisms have been described that link Gq/G11-mediated signaling to the activation of RhoA (Guilluy et al., 2010; Wuertz et al., 2010; Momotani et al., 2011). However, Gαq/Gα11 deletion in VSMCs did not affect RhoA activity in our in vivo experiments, indicating that RhoA activation via Gq/G11 is not critically involved in vascular remodeling.
Gq/G11- and G12/G13-mediated signaling pathways are able to increase myosin light chain phosphorylation and Sm tone through different mechanisms (Gohla et al., 2000; Somlyo and Somlyo, 2003; Maguire and Davenport, 2005; Wirth et al., 2008). In contrast to their synergistic function in vascular tone regulation, our data show that Gq/G11- and G12/G13-mediated signaling pathways antagonistically regulate the differentiation state of VSMCs at sites of vascular dysfunction or injury. Interestingly, both the Gq/G11-Erk1/2–mediated pathway mediating suppression of SMC-selective marker gene expression and proliferation as well as the G12/G13-Rho-mediated pathway, which promotes Sm differentiation, are activated in parallel in VSMCs shortly after induction of vascular injury. The net response of the vessel, which is also affected by other upstream signaling mechanisms involving integrins or receptor tyrosine kinases such as PDGFR-β (Ferns et al., 1991; Raines, 2000), is a dedifferentiation, which obviously requires intact Gq/G11-mediated signaling. The parallel activation of the G12/G13-mediated pathway, which promotes Sm differentiation, therefore appears counterintuitive. However, activation of the G12/G13-RhoA–mediated pathway prevented excessive loss of SMC-selective marker gene expression and proliferation of VSMCs in the course of vascular remodeling and may facilitate the redifferentiation of VSMCs, once the influence of proliferative stimuli declines. Thus, a well-balanced parallel activation of antagonistic pathways promoting differentiation and dedifferentiation of VSMCs appears to be required for the adaptive response to vascular injury or to changing hemodynamics. Alternatively or in addition, the balance of Gq/G11- and G12/G13-mediated signaling may be modulated in VSMCs during vascular remodeling by other factors such as regulators of G protein signaling, which have been shown to specifically affect G protein–mediated signaling in VSMCs (Heximer et al., 2003; Tang et al., 2003; Cho et al., 2008; Gunaje et al., 2011).
The receptors of most vasocontractile stimuli including angiotensin-II, S1P, thrombin, thromboxane A2, or endothelin-1 are dually coupled to Gq/G11 and G12/G13, albeit some receptors show a certain preference (Maguire and Davenport, 2005). The question therefore arises as to whether receptor ligands can be used to modulate Sm differentiation under pathological conditions. Pharmacological approaches indicate that individual S1P receptor subtypes play antagonistic roles in the regulation of neointima formation (Wamhoff et al., 2008), and global loss of the S1P-receptor S1P2, which efficiently couples to G12/G13 (Chun et al., 2010), increases susceptibility to injury-induced neointima formation (Shimizu et al., 2007). Thus, activation or blockade of receptor subtypes preferentially coupled to either of the two G protein families may promote VSMC differentiation and dedifferentiation, respectively. To efficiently reduce vascular remodeling by preferential inhibition of Gq/G11-mediated signaling, it may also be of interest to interfere directly with Gq/G11 or downstream signaling components. In addition, a preferential inhibition of Gq/G11- or activation of G12/G13-mediated signaling in VSMCs could be achieved by receptor ligands, which act as allosteric modulators or biased agonists (Reiter et al., 2012; Valant et al., 2012) on procontractile GPCRs.
Our data show that Gq/G11 and G12/G13, which both mediate effects of procontractile stimuli in VSMCs, antagonistically regulate VSMC differentiation by controlling the recruitment of transcriptional cofactors by SRF. Our in vivo experiments in different models of vascular disorders indicate that the balanced activity of both pathways in VSMCs controls the remodeling response of the vessel in vascular diseases. The opposite regulation of SMC differentiation marker gene expression by the two pathways may allow for modulation of VSMC differentiation under pathological conditions by biased GPCR ligands or by inhibitors of G protein–mediated signaling processes.
MATERIALS AND METHODS
Materials.
Phenylephrine, hydralazine, metoprolol, Alcian blue, and Accustain Elastic Stain were purchased from Sigma-Aldrich, DAPI was purchased from Invitrogen, and collagenases were purchased from Worthington Biochemical Corporation.
Genetic mouse models.
The generation of SMMHC-CreERT2 mice as well as genotyping procedures and induction of Cre activity by tamoxifen have been described previously (Wirth et al., 2008). The generation of floxed or null alleles of genes encoding G protein α-subunits Gαq (Gnaq), Gα11 (Gna11), Gα12 (Gna12), Gα13 (Gna13), or LARG (Arhgef12) as well as genotyping procedures have been described previously (Wettschureck et al., 2001; Moers et al., 2003; Herroeder et al., 2009). All animals were on a C57BL/6 background, and care and experimental procedures in this study were approved by the local authorities (Regierungspräsidia Darmstadt and Karlsruhe).
qRT-PCR.
Snap-frozen carotid media samples, isolated 3 d after ligation and sham operation, respectively, were disrupted and homogenized at 4°C using a mixer bead mill. RNA was then extracted using the RNeasy Micro kit (QIAGEN) for mRNA and the miRNeasy kit (QIAGEN) for microRNA according to the manufacturer’s instructions. 250 ng of total RNA was reverse transcribed using the Transcriptor High Fidelity kit (Roche). For microRNA, we used the NCode Vilo miRNA cDNA Synthesis kit (Invitrogen). Quantitative real-time PCR was performed with LightCycler 480 Probe Master (Roche) for mRNA analyses and LightCycler 480 SYBR Green Master (Roche) for microRNA analyses. Gene-specific RT-PCR primers were selected using the Roche ProbeFinder software. The relative amount of target mRNA normalized to 18S and of microRNA normalized to S4.5 RNA was calculated as previously described (Pfaffl et al., 2001). Primer sequences are as follows: 18s, forward 5′-GCAATTATTCCCCATGAACG-3′, reverse 5′-GGGACTTAATCAACGCAAGC-3′, probe #48; Myh11, forward 5′-TGGAGGCCAAGATTGCAC-3′, reverse 5′-GGCCGCCTGTTTCTCTCT-3′, probe #68; Acta2, forward 5′-CCAGCACCATGAAGATCAAG-3′, reverse 5′-TGGAAGGTAGACAGCGAAGC-3′, probe #58; SM22-α, forward 5′-GCAGTGTGGCCCTGATGTA-3′, reverse 5′-TCACCAATTTGCTCAGAATCA-3′, probe #5; Cnn1, forward 5′-GAAGGTCAATGAGTCAACTCAGAA-3′, reverse 5′-CCATACTTGGTAATGGCTTTGA-3′, probe #78; Myocd, forward 5′-AGGCTCCTCTGGACACAATC-3′, reverse 5′-TCCCAGGGTCTGACTGGTT-3′, probe #20; Elk1, forward 5′-GCTCCCCACACATACCTTGA-3′, reverse 5′-GGGTGCAATTGGACTCAGA-3′, probe #50; Fos, forward 5′-GAAGGGGCAAAGTAGAGCAG-3′, reverse 5′-CAGCTCCCTCCTCCGATT-3′, probe #46; Ets1, forward 5′-ACTGTGTGCCCTGGGTAAAG-3′, reverse 5′-CCTGATATGGTTTCACATCCTCT-3′, probe #50; Egr1, forward 5′-CCTATGAGCACCTGACCACA-3′, reverse 5′-TCGTTTGGCTGGGATAACTC-3′, probe #22; 4.5s, universal forward 5′-GTACTGCGCGTGGAGAGGAATT-3′, reverse 5′-GTCCAAACTCTTGGGAAGCAGA-3′; miR-143, universal forward 5′-GTACTGCGCGTGGAGAGGAATT-3′, reverse 5′-TGAGATGAAGCACTGTAGCTCA-3′; and miR-145, universal forward 5′-GTACTGCGCGTGGAGAGGAATT-3′, reverse 5′-CAGTTTTCCCAGGAATCCCTT-3′.
Western blotting.
Snap-frozen carotid media samples, isolated 3 d after ligation and sham operation, respectively, were homogenized on ice in lysis buffer, pH 8.0 containing 0.1 M Tris/HCl, 0.01 M EDTA, 10% SDS, 1× protease, and phosphatase inhibitor cocktail (Roche) using a handheld rotor–stator homogenizer (MM300; Retsch) and 3-mm stainless steel beads. The homogenates were centrifuged for 20 min at 10,000 g at 4°C to remove debris, and aliquots of the supernatants were assayed for total protein content by the BCA method (Thermo Fisher Scientific). Equal amounts of protein (10 µg per lane) were separated by 13% SDS-PAGE gels and transferred onto nitrocellulose membranes (Whatman; GE Healthcare). Immunoblotting was performed by using specific antibodies. In brief, polyclonal antisera against α-SM22 (Abcam), α-SMA (Abcam), and monoclonal antibody against α-tubulin (Sigma-Aldrich) were used. All primary antibodies were used at 1:200 dilutions, except for anti–α-tubulin, which was used at 1:3,000 dilutions. Secondary antibodies (1:3,000 dilutions) were from Cell Signaling Technology (goat anti–rabbit HRP and goat anti–mouse HRP). Immunoreactive proteins were visualized with corresponding HRP-conjugated secondary antibodies using a commercially available kit (Immobilon Western; Millipore) and developed through exposure to x-ray film.
Determination of RhoA activity.
Snap-frozen carotid media samples, isolated 24 h after ligation or sham operation, respectively, were disrupted and homogenized at 4°C using a mixer bead mill. Total protein concentrations where measured and equalized with lysis buffer. RhoA activity, determined by the amount of GTP-loaded RhoA, was then assessed using an absorbance-based ELISA-kit (G-LISA RhoA Activation Assay; Cytoskeleton) according to the manufacturer’s instructions. RhoA activity was expressed in relative units with the absorbance of sham-operated WT vessels serving as reference.
Carotid artery ligation.
Male mice at 12 wk of age were anesthetized, and left and right common carotid arteries were dissected after a midline incision of the neck. Distal ligation of the left common carotid artery was performed with a 7/0 silk thread (Serag-Wiessner).
Wire injury.
Male mice underwent femoral artery dilation at the age of 12 wk. After induction of anesthesia, a medial skin incision of the hind limb thigh was performed, and the femoral as well as the profunda femoris artery were dissected. Using 6/0 silk thread (Fine Science Tools) a loop was placed at the proximal femoral artery, and tension was applied to disrupt blood flow. A straight spring wire (diameter 0.38 mm; Cook) was introduced into the femoral artery via transverse arteriotomy of the profunda femoris artery. The wire was advanced ∼5 mm proximally and left in place for 1 min to dilate the femoral artery. After removal of the wire and ligation of the profunda femoris branch, blood flow was reconstituted.
Atherosclerosis.
To obtain Sm-specific Gα deficiency in ApoE−/− mice, ApoE−/− animals were crossed with tamoxifen-inducible Sm-Gα12/Gα13-KO mice. The offspring were then intercrossed. ApoE−/− mice without or with inducible Sm-specific Gα12/Gα13 deficiency were treated with tamoxifen at an age of 6 wk. At the age of 8 wk, a high-fat diet was started and continued for 12 wk. Thereafter, animals were sacrificed, and atherosclerotic lesions were analyzed as described under Histology and immunohistochemistry.
Telemetric blood pressure measurements and myography.
Telemetry experiments were performed as described previously (Wirth et al., 2008). A 10–15% decrease in mean arterial blood pressure was induced pharmacologically by application of 500 mg/liter hydralazine or 2.5 g/liter metoprolol in the drinking water. Treatment was started 7 d before and continued throughout the experiments. For isometric tension recordings, common carotid arteries were isolated after euthanization and PBS perfusion of mice. 3-mm-long carotid segments were prepared, mounted on a conventional myograph setup (610-M; Danish Myo Technology), and myography was performed as described previously (Wirth et al., 2008). Individual experiments were performed at least three times.
Histology and immunohistochemistry.
For histology, vessels were perfused and fixed in 4% PFA overnight. Vessels were then dehydrated using increasing concentrations of ethanol and embedded in paraffin. Paraffin-embedded arteries were cut in 5-µm serial cross sections, and predefined sections (see below) were mounted on slides and stained with Accustain Elastic Stain according to the manufacturer’s instructions. In ligated left common carotid arteries, cross sections from four predefined proximal distances from the ligation site (250, 1,000, 2,000, and 3,000 µm) were analyzed. In sham-operated right common carotid arteries, sections from within ∼1,000-µm distance to the bifurcation of internal and external carotid artery were analyzed. Femoral arteries were analyzed 4 wk after injury. Within the injured distal region (5,000 µm) of the left femoral artery, cross sections were analyzed in regular intervals of 1,000 µm to calculate a vessel mean for each parameter. Sections from the distal part of the contralateral right femoral artery (i.e., within ∼1,000 µm distance to the branching of the profunda femoris artery) were analyzed as an internal control. For the analysis of atherosclerotic vessels, cross sections of the innominate artery and right common carotid artery were analyzed. Photoshop CS5 extended software (Adobe) was used to measure circumferences of internal elastic lamina, external elastic lamina, and lumen, as well as medial, intimal, and luminal cross-sectional areas. Staining with Alcian blue was performed according to the manufacturer’s instructions.
For immunohistochemistry of paraffin sections, sections were dewaxed, rehydrated, and subsequently boiled for 18 min in 10 mM Tris/1 mM EDTA buffer, pH 9.0, for epitope retrieval. After 10-min incubation in 3% H2O2, sections were blocked in PBS supplemented with 5% normal goat serum for 2 h. After overnight incubation at 4°C with rabbit monoclonal anti–phospho-ERK1/2 (1:100; Cell Signaling Technology), rabbit polyclonal anti-phospho–Elk-1 (1:100; Cell Signaling Technology), or polyclonal rabbit anti–Sm α-actin (1:200; Abcam), primary antibodies were detected with biotinylated antibody to rabbit IgG followed by incubation with Avidin–Biotin complex reagent (Vectastain ABC System; Vector Laboratories) according to the manufacturer’s instructions. Staining was visualized using a diaminobenzidine (DAB) peroxidase substrate kit (Vector Laboratories) according to the manufacturer’s instructions, followed by counterstaining with hematoxylin for detection of nuclei where indicated. Immunostaining of phosphorylated Erk1/2 and phosphorylated Elk-1 was performed in carotid arteries that were harvested 24 h after carotid artery ligation. The fraction of positively stained medial area (phospho-Erk1/2) or positively stained nuclei (phospho–Elk-1) was quantified using Photoshop CS5 Extended software (Adobe). The threshold for positive staining was (pre)defined by an independent observer who was blinded to the treatment.
For cryoimmunohistochemistry, vessels were embedded in Tissue-Tek O.C.T. freezing medium, snap frozen in liquid nitrogen, and stored at −80°C until sectioning. Immunostaining of cryosections was performed using a rabbit anti-CD3 (clone SP7; 1:100; Abcam) or Alexa Fluor 488–conjugated rat anti-CD68 (clone FA-11; 1:100; AbD Serotec). After a 10-min fixation with 4% PFA and several washing steps with PBS, cryosections were incubated with primary antibodies at 4°C overnight followed by either CF555- or CF488-labeled anti–rabbit secondary antibody (1:300; Biotium) and counterstained with cy3- or FITC-conjugated anti–Sm α-actin antibody (1:300; Sigma-Aldrich) and DAPI. This protocol was adapted for Ki67 staining by a 20-min fixation with 4% PFA and an antigen retrieval in citrate buffer (20 min boiling). Overnight incubation of primary antibody anti–mouse Ki67 (clone TEC-3; 1:20; Dako) was followed by 1-h Cy3-conjugated anti–rat IgG (1:300; Millipore) incubation. Sections were viewed with a confocal microscope (SP5; Leica).
Primary VSMC culture and immunocytochemistry.
Primary VSMCs were isolated using a modified enzymatic digestion method based on Ray et al. (2001). 2 wk after tamoxifen induction, left and right common carotid arteries were isolated, perivascular tissue was removed, and vessels were cut into square pieces of ∼1-mm edge length, which were transferred into 5-ml tubes containing 1 mg/ml collagenase type II and 0.5 mg/ml elastase (both Worthington Biochemical Corporation) diluted in 500 µl serum-free DMEM. Tubes were incubated at 37°C, 5% CO2 for 3.5 h. To stop digestion, complete DMEM, i.e., supplemented with 10% FBS (Gibco), 2 mM l-glutamine, and 100 U/ml penicillin/streptomycin, was added. After centrifugation at 3,000 g for 5 min, cells were resuspended in 700 µl complete DMEM containing 2 µg/ml amphotericin B (Sigma-Aldrich) and plated on a single well of a 24-well plate. The following day, medium was changed to amphotericin-free medium. For immunocytochemistry, cells at passage 3 or 4 were plated on coverslips in 12-well plates and grown to confluency. After 48 h of starvation in serum-free medium, cells were stimulated for 1 h with complete DMEM containing 20% FBS. Cells were then rinsed with PBS, fixed with 4% PFA for 10 min, and blocked for 30 min at room temperature in 0.2% Triton/2 mg/ml bovine serum albumin. Cells were then incubated with rabbit polyclonal anti–MRTF-A antibody (1:200; Santa Cruz Biotechnology, Inc.). After extensive washing with PBS, cells were incubated with an Alexa Fluor 488–conjugated secondary antibody (1:200; anti-rabbit IgG; Invitrogen) for visualization, followed by 30-min incubation with DAPI (1:1,000; Invitrogen) for detection of nuclei.
Statistical analyses.
All values are expressed as mean ± SEM. Statistical comparison of two groups was performed using the unpaired Student’s t test with p-values < 0.05 considered statistically significant. If not stated otherwise, individual KOs (Sm-q/11-KO, LARG-KO, or Sm-12/13-KO) were only compared with WTs.
Acknowledgments
We wish to thank Svea Hümmer for excellent secretarial help.
This work was supported by the Transregional Collaborative Research Center 23 (SFB/TR23) of the German Research Foundation.
The authors declare no competing financial interests.
Footnotes
Abbreviations used:
- GPCR
- G protein–coupled receptor
- MRTF
- myocardin-related transcription factor
- qRT-PCR
- quantitative RT-PCR
- RhoGEF
- Rho guanine nucleotide exchange factor
- Sm
- smooth muscle
- SMC
- Sm cell
- SRF
- serum response factor
- TCF
- ternary complex factor
- VSMC
- vascular SMC
References
- Andrawis N.S., Wang E., Abernethy D.R. 1996. Endothelin-1 induces an increase in total protein synthesis and expression of the smooth muscle alpha-actin gene in vascular smooth muscle cells. Life Sci. 59:523–528 10.1016/0024-3205(96)00332-3 [DOI] [PubMed] [Google Scholar]
- Cho H., Park C., Hwang I.Y., Han S.B., Schimel D., Despres D., Kehrl J.H. 2008. Rgs5 targeting leads to chronic low blood pressure and a lean body habitus. Mol. Cell. Biol. 28:2590–2597 10.1128/MCB.01889-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun J., Hla T., Lynch K.R., Spiegel S., Moolenaar W.H. 2010. International Union of Basic and Clinical Pharmacology. LXXVIII. Lysophospholipid receptor nomenclature. Pharmacol. Rev. 62:579–587 10.1124/pr.110.003111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copeland J.W., Treisman R. 2002. The diaphanous-related formin mDia1 controls serum response factor activity through its effects on actin polymerization. Mol. Biol. Cell. 13:4088–4099 10.1091/mbc.02-06-0092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzau V.J., Braun-Dullaeus R.C., Sedding D.G. 2002. Vascular proliferation and atherosclerosis: new perspectives and therapeutic strategies. Nat. Med. 8:1249–1256 10.1038/nm1102-1249 [DOI] [PubMed] [Google Scholar]
- Ferns G.A., Raines E.W., Sprugel K.H., Motani A.S., Reidy M.A., Ross R. 1991. Inhibition of neointimal smooth muscle accumulation after angioplasty by an antibody to PDGF. Science. 253:1129–1132 10.1126/science.1653454 [DOI] [PubMed] [Google Scholar]
- Gohla A., Schultz G., Offermanns S. 2000. Role for G(12)/G(13) in agonist-induced vascular smooth muscle cell contraction. Circ. Res. 87:221–227 10.1161/01.RES.87.3.221 [DOI] [PubMed] [Google Scholar]
- Gorenne I., Jin L., Yoshida T., Sanders J.M., Sarembock I.J., Owens G.K., Somlyo A.P., Somlyo A.V. 2006. LPP expression during in vitro smooth muscle differentiation and stent-induced vascular injury. Circ. Res. 98:378–385 10.1161/01.RES.0000202802.34727.fd [DOI] [PubMed] [Google Scholar]
- Guilluy C., Brégeon J., Toumaniantz G., Rolli-Derkinderen M., Retailleau K., Loufrani L., Henrion D., Scalbert E., Bril A., Torres R.M., et al. 2010. The Rho exchange factor Arhgef1 mediates the effects of angiotensin II on vascular tone and blood pressure. Nat. Med. 16:183–190 10.1038/nm.2079 [DOI] [PubMed] [Google Scholar]
- Gunaje J.J., Bahrami A.J., Schwartz S.M., Daum G., Mahoney W.M., Jr 2011. PDGF-dependent regulation of regulator of G protein signaling-5 expression and vascular smooth muscle cell functionality. Am. J. Physiol. Cell Physiol. 301:C478–C489 10.1152/ajpcell.00348.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herroeder S., Reichardt P., Sassmann A., Zimmermann B., Jaeneke D., Hoeckner J., Hollmann M.W., Fischer K.D., Vogt S., Grosse R., et al. 2009. Guanine nucleotide-binding proteins of the G12 family shape immune functions by controlling CD4+ T cell adhesiveness and motility. Immunity. 30:708–720 10.1016/j.immuni.2009.02.010 [DOI] [PubMed] [Google Scholar]
- Heximer S.P., Knutsen R.H., Sun X., Kaltenbronn K.M., Rhee M.H., Peng N., Oliveira-dos-Santos A., Penninger J.M., Muslin A.J., Steinberg T.H., et al. 2003. Hypertension and prolonged vasoconstrictor signaling in RGS2-deficient mice. J. Clin. Invest. 111:1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh H.L., Tung W.H., Wu C.Y., Wang H.H., Lin C.C., Wang T.S., Yang C.M. 2009. Thrombin induces EGF receptor expression and cell proliferation via a PKC(delta)/c-Src-dependent pathway in vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 29:1594–1601 10.1161/ATVBAHA.109.185801 [DOI] [PubMed] [Google Scholar]
- Jeon E.S., Park W.S., Lee M.J., Kim Y.M., Han J., Kim J.H. 2008. A Rho kinase/myocardin-related transcription factor-A-dependent mechanism underlies the sphingosylphosphorylcholine-induced differentiation of mesenchymal stem cells into contractile smooth muscle cells. Circ. Res. 103:635–642 10.1161/CIRCRESAHA.108.180885 [DOI] [PubMed] [Google Scholar]
- Kim M.R., Jeon E.S., Kim Y.M., Lee J.S., Kim J.H. 2009. Thromboxane a(2) induces differentiation of human mesenchymal stem cells to smooth muscle-like cells. Stem Cells. 27:191–199 10.1634/stemcells.2008-0363 [DOI] [PubMed] [Google Scholar]
- Li J., Zhu X., Chen M., Cheng L., Zhou D., Lu M.M., Du K., Epstein J.A., Parmacek M.S. 2005. Myocardin-related transcription factor B is required in cardiac neural crest for smooth muscle differentiation and cardiovascular development. Proc. Natl. Acad. Sci. USA. 102:8916–8921 10.1073/pnas.0503741102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S., Chang S., Qi X., Richardson J.A., Olson E.N. 2006. Requirement of a myocardin-related transcription factor for development of mammary myoepithelial cells. Mol. Cell. Biol. 26:5797–5808 10.1128/MCB.00211-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockman K., Hinson J.S., Medlin M.D., Morris D., Taylor J.M., Mack C.P. 2004. Sphingosine 1-phosphate stimulates smooth muscle cell differentiation and proliferation by activating separate serum response factor co-factors. J. Biol. Chem. 279:42422–42430 10.1074/jbc.M405432200 [DOI] [PubMed] [Google Scholar]
- Lorenz K., Schmitt J.P., Schmitteckert E.M., Lohse M.J. 2009. A new type of ERK1/2 autophosphorylation causes cardiac hypertrophy. Nat. Med. 15:75–83 10.1038/nm.1893 [DOI] [PubMed] [Google Scholar]
- Lu J., Landerholm T.E., Wei J.S., Dong X.R., Wu S.P., Liu X., Nagata K., Inagaki M., Majesky M.W. 2001. Coronary smooth muscle differentiation from proepicardial cells requires rhoA-mediated actin reorganization and p160 rho-kinase activity. Dev. Biol. 240:404–418 10.1006/dbio.2001.0403 [DOI] [PubMed] [Google Scholar]
- Mack C.P. 2011. Signaling mechanisms that regulate smooth muscle cell differentiation. Arterioscler. Thromb. Vasc. Biol. 31:1495–1505 10.1161/ATVBAHA.110.221135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mack C.P., Somlyo A.V., Hautmann M., Somlyo A.P., Owens G.K. 2001. Smooth muscle differentiation marker gene expression is regulated by RhoA-mediated actin polymerization. J. Biol. Chem. 276:341–347 10.1074/jbc.M005505200 [DOI] [PubMed] [Google Scholar]
- Maguire J.J., Davenport A.P. 2005. Regulation of vascular reactivity by established and emerging GPCRs. Trends Pharmacol. Sci. 26:448–454 [DOI] [PubMed] [Google Scholar]
- Martin K., Weiss S., Metharom P., Schmeckpeper J., Hynes B., O’Sullivan J., Caplice N. 2009. Thrombin stimulates smooth muscle cell differentiation from peripheral blood mononuclear cells via protease-activated receptor-1, RhoA, and myocardin. Circ. Res. 105:214–218 10.1161/CIRCRESAHA.109.199984 [DOI] [PubMed] [Google Scholar]
- Medlin M.D., Staus D.P., Dubash A.D., Taylor J.M., Mack C.P. 2010. Sphingosine 1-phosphate receptor 2 signals through leukemia-associated RhoGEF (LARG), to promote smooth muscle cell differentiation. Arterioscler. Thromb. Vasc. Biol. 30:1779–1786 10.1161/ATVBAHA.110.209395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miano J.M., Long X., Fujiwara K. 2007. Serum response factor: master regulator of the actin cytoskeleton and contractile apparatus. Am. J. Physiol. Cell Physiol. 292:C70–C81 10.1152/ajpcell.00386.2006 [DOI] [PubMed] [Google Scholar]
- Miralles F., Posern G., Zaromytidou A.I., Treisman R. 2003. Actin dynamics control SRF activity by regulation of its coactivator MAL. Cell. 113:329–342 10.1016/S0092-8674(03)00278-2 [DOI] [PubMed] [Google Scholar]
- Moers A., Nieswandt B., Massberg S., Wettschureck N., Grüner S., Konrad I., Schulte V., Aktas B., Gratacap M.P., Simon M.I., et al. 2003. G13 is an essential mediator of platelet activation in hemostasis and thrombosis. Nat. Med. 9:1418–1422 10.1038/nm943 [DOI] [PubMed] [Google Scholar]
- Momotani K., Artamonov M.V., Utepbergenov D., Derewenda U., Derewenda Z.S., Somlyo A.V. 2011. p63RhoGEF couples Gα(q/11)-mediated signaling to Ca2+ sensitization of vascular smooth muscle contractility. Circ. Res. 109:993–1002 10.1161/CIRCRESAHA.111.248898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noma K., Rikitake Y., Oyama N., Yan G., Alcaide P., Liu P.Y., Wang H., Ahl D., Sawada N., Okamoto R., et al. 2008. ROCK1 mediates leukocyte recruitment and neointima formation following vascular injury. J. Clin. Invest. 118:1632–1644 10.1172/JCI29226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh J., Richardson J.A., Olson E.N. 2005. Requirement of myocardin-related transcription factor-B for remodeling of branchial arch arteries and smooth muscle differentiation. Proc. Natl. Acad. Sci. USA. 102:15122–15127 10.1073/pnas.0507346102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson E.N., Nordheim A. 2010. Linking actin dynamics and gene transcription to drive cellular motile functions. Nat. Rev. Mol. Cell Biol. 11:353–365 10.1038/nrm2890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owens G.K. 1995. Regulation of differentiation of vascular smooth muscle cells. Physiol. Rev. 75:487–517 [DOI] [PubMed] [Google Scholar]
- Owens G.K. 2007. Molecular control of vascular smooth muscle cell differentiation and phenotypic plasticity. Novartis Found. Symp. 283:174–191, discussion :191–193: 238–241 10.1002/9780470319413.ch14 [DOI] [PubMed] [Google Scholar]
- Owens G.K., Kumar M.S., Wamhoff B.R. 2004. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol. Rev. 84:767–801 10.1152/physrev.00041.2003 [DOI] [PubMed] [Google Scholar]
- Parmacek M.S. 2007. Myocardin-related transcription factors: critical coactivators regulating cardiovascular development and adaptation. Circ. Res. 100:633–644 10.1161/01.RES.0000259563.61091.e8 [DOI] [PubMed] [Google Scholar]
- Pfaffl M.W. 2001. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29:e45 10.1093/nar/29.9.e45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pipes G.C., Sinha S., Qi X., Zhu C.H., Gallardo T.D., Shelton J., Creemers E.E., Sutherland L., Richardson J.A., Garry D.J., et al. 2005. Stem cells and their derivatives can bypass the requirement of myocardin for smooth muscle gene expression. Dev. Biol. 288:502–513 10.1016/j.ydbio.2005.10.014 [DOI] [PubMed] [Google Scholar]
- Pipes G.C., Creemers E.E., Olson E.N. 2006. The myocardin family of transcriptional coactivators: versatile regulators of cell growth, migration, and myogenesis. Genes Dev. 20:1545–1556 10.1101/gad.1428006 [DOI] [PubMed] [Google Scholar]
- Posern G., Treisman R. 2006. Actin’ together: serum response factor, its cofactors and the link to signal transduction. Trends Cell Biol. 16:588–596 10.1016/j.tcb.2006.09.008 [DOI] [PubMed] [Google Scholar]
- Raines E.W. 2000. The extracellular matrix can regulate vascular cell migration, proliferation, and survival: relationships to vascular disease. Int. J. Exp. Pathol. 81:173–182 10.1046/j.1365-2613.2000.00155.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray J.L., Leach R., Herbert J.M., Benson M. 2001. Isolation of vascular smooth muscle cells from a single murine aorta. Methods Cell Sci. 23:185–188 10.1023/A:1016357510143 [DOI] [PubMed] [Google Scholar]
- Reiter E., Ahn S., Shukla A.K., Lefkowitz R.J. 2012. Molecular mechanism of β-arrestin-biased agonism at seven-transmembrane receptors. Annu. Rev. Pharmacol. Toxicol. 52:179–197 10.1146/annurev.pharmtox.010909.105800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu T., Nakazawa T., Cho A., Dastvan F., Shilling D., Daum G., Reidy M.A. 2007. Sphingosine 1-phosphate receptor 2 negatively regulates neointimal formation in mouse arteries. Circ. Res. 101:995–1000 10.1161/CIRCRESAHA.107.159228 [DOI] [PubMed] [Google Scholar]
- Somlyo A.P., Somlyo A.V. 2003. Ca2+ sensitivity of smooth muscle and nonmuscle myosin II: modulated by G proteins, kinases, and myosin phosphatase. Physiol. Rev. 83:1325–1358 [DOI] [PubMed] [Google Scholar]
- Staus D.P., Blaker A.L., Taylor J.M., Mack C.P. 2007. Diaphanous 1 and 2 regulate smooth muscle cell differentiation by activating the myocardin-related transcription factors. Arterioscler. Thromb. Vasc. Biol. 27:478–486 10.1161/01.ATV.0000255559.77687.c1 [DOI] [PubMed] [Google Scholar]
- Sun Y., Boyd K., Xu W., Ma J., Jackson C.W., Fu A., Shillingford J.M., Robinson G.W., Hennighausen L., Hitzler J.K., et al. 2006. Acute myeloid leukemia-associated Mkl1 (Mrtf-a) is a key regulator of mammary gland function. Mol. Cell. Biol. 26:5809–5826 10.1128/MCB.00024-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang K.M., Wang G.R., Lu P., Karas R.H., Aronovitz M., Heximer S.P., Kaltenbronn K.M., Blumer K.J., Siderovski D.P., Zhu Y., Mendelsohn M.E. 2003. Regulator of G-protein signaling-2 mediates vascular smooth muscle relaxation and blood pressure. Nat. Med. 9:1506–1512 (published erratum appears in Nat. Med. 2004. 10:105) 10.1038/nm958 [DOI] [PubMed] [Google Scholar]
- Treisman R. 1994. Ternary complex factors: growth factor regulated transcriptional activators. Curr. Opin. Genet. Dev. 4:96–101 10.1016/0959-437X(94)90097-3 [DOI] [PubMed] [Google Scholar]
- Valant C., Robert Lane J., Sexton P.M., Christopoulos A. 2012. The best of both worlds? Bitopic orthosteric/allosteric ligands of g protein-coupled receptors. Annu. Rev. Pharmacol. Toxicol. 52:153–178 10.1146/annurev-pharmtox-010611-134514 [DOI] [PubMed] [Google Scholar]
- Wamhoff B.R., Bowles D.K., McDonald O.G., Sinha S., Somlyo A.P., Somlyo A.V., Owens G.K. 2004. L-type voltage-gated Ca2+ channels modulate expression of smooth muscle differentiation marker genes via a rho kinase/myocardin/SRF-dependent mechanism. Circ. Res. 95:406–414 10.1161/01.RES.0000138582.36921.9e [DOI] [PubMed] [Google Scholar]
- Wamhoff B.R., Lynch K.R., Macdonald T.L., Owens G.K. 2008. Sphingosine-1-phosphate receptor subtypes differentially regulate smooth muscle cell phenotype. Arterioscler. Thromb. Vasc. Biol. 28:1454–1461 10.1161/ATVBAHA.107.159392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z., Wang D.Z., Hockemeyer D., McAnally J., Nordheim A., Olson E.N. 2004. Myocardin and ternary complex factors compete for SRF to control smooth muscle gene expression. Nature. 428:185–189 10.1038/nature02382 [DOI] [PubMed] [Google Scholar]
- Wettschureck N., Rütten H., Zywietz A., Gehring D., Wilkie T.M., Chen J., Chien K.R., Offermanns S. 2001. Absence of pressure overload induced myocardial hypertrophy after conditional inactivation of Galphaq/Galpha11 in cardiomyocytes. Nat. Med. 7:1236–1240 10.1038/nm1101-1236 [DOI] [PubMed] [Google Scholar]
- Wirth A., Benyó Z., Lukasova M., Leutgeb B., Wettschureck N., Gorbey S., Orsy P., Horváth B., Maser-Gluth C., Greiner E., et al. 2008. G12-G13-LARG-mediated signaling in vascular smooth muscle is required for salt-induced hypertension. Nat. Med. 14:64–68 10.1038/nm1666 [DOI] [PubMed] [Google Scholar]
- Wuertz C.M., Lorincz A., Vettel C., Thomas M.A., Wieland T., Lutz S. 2010. p63RhoGEF—a key mediator of angiotensin II-dependent signaling and processes in vascular smooth muscle cells. FASEB J. 24:4865–4876PubMed 10.1096/fj.10-155499 [DOI] [PubMed] [Google Scholar]
- Xi X.P., Graf K., Goetze S., Hsueh W.A., Law R.E. 1997. Inhibition of MAP kinase blocks insulin-mediated DNA synthesis and transcriptional activation of c-fos by Elk-1 in vascular smooth muscle cells. FEBS Lett. 417:283–286 10.1016/S0014-5793(97)01303-3 [DOI] [PubMed] [Google Scholar]
- Yoshida T., Hoofnagle M.H., Owens G.K. 2004. Myocardin and Prx1 contribute to angiotensin II-induced expression of smooth muscle alpha-actin. Circ. Res. 94:1075–1082 10.1161/01.RES.0000125622.46280.95 [DOI] [PubMed] [Google Scholar]