

Abstract
Infantile hemangiomas (IHs) are non-malignant, largely cutaneous vascular tumors affecting approximately 5–10% of children to varying degrees. During the first year of life, these tumors are strongly proliferative, reaching an average size ranging from 2 to 20 cm. These lesions subsequently stabilize, undergo a spontaneous slow involution and are fully regressed by 5 to 10 years of age. Systemic treatment of infants with the non-selective β-adrenergic receptor blocker, propranolol, has demonstrated remarkable efficacy in reducing the size and appearance of IHs. However, the mechanism by which this occurs is largely unknown. In this study, we sought to understand the molecular mechanisms underlying the effectiveness of β blocker treatment in IHs. Our data reveal that propranolol treatment of IH endothelial cells, as well as a panel of normal primary endothelial cells, blocks endothelial cell proliferation, migration, and formation of the actin cytoskeleton coincident with alterations in vascular endothelial growth factor receptor-2 (VEGFR-2), p38 and cofilin signaling. Moreover, propranolol induces major alterations in the protein levels of key cyclins and cyclin-dependent kinase inhibitors, and modulates global gene expression patterns with a particular affect on genes involved in lipid/sterol metabolism, cell cycle regulation, angiogenesis and ubiquitination. Interestingly, the effects of propranolol were endothelial cell-type independent, affecting the properties of IH endothelial cells at similar levels to that observed in neonatal dermal microvascular and coronary artery endothelial cells. This data suggests that while propranolol markedly inhibits hemangioma and normal endothelial cell function, its lack of endothelial cell specificity hints that the efficacy of this drug in the treatment of IHs may be more complex than simply blockage of endothelial function as previously believed.
Keywords: infantile hemangioma, propranolol, endothelial cells, angiogenesis
Introduction
Infantile hemangiomas (IHs) are the most common benign tumors in infancy affecting 5–10% of the population, and are largely composed of densely packed over-proliferating capillaries with high cellular density and the absence of an open lumen. These lesions are most prevalent in Caucasian children and are three times more common in female infants than male. The head and neck region is the most frequently involved area (60%), followed by the trunk (25%) and the extremities (15%), and these tumors exhibit a non-random distribution largely correlating with regions of embryological fusion (1). IHs have a predictable natural history, arising soon after birth to undergo a significant proliferative phase during the first year of life, followed by gradual involution over several years. Regression is complete in 50% of 5-year-old patients and 90% of 9-year-old patients. Because these tumors spontaneously regress and (for the majority of lesions) produce no long-term scarring, most children with IHs require no treatment. Despite their self-limiting course, in approximately 10% of cases depending on their anatomical site and/or size, there can be serious or life threatening complications requiring immediate intervention. The classical approaches for treating complicated IHs include the use of systemic or intralesional corticosteroids, chemotherapeutic agents such as vincristine, laser therapy, surgical resection or a combination of these treatments. The recent serendipitous discovery of β blockers as an effective therapy for IHs has revolutionized management of IHs to become the current gold standard (2).
Propranolol, which is administered systemically in pediatric patients with IHs, is a non-selective β-adrenergic receptor antagonist that blocks the action of epinephrine and norepinephrin. This drug has been shown to suppress angiogenesis via inhibition of proliferation, migration, barrier function, and induction of apoptosis in primary cultures of normal epithelial cells (3–6). The molecular mechanism of action for propranolol includes disrupting the epinephrine and norpinephrine regulation of cyclic AMP production, actin cytoskeletal dynamics and release of atherogenesis regulators (7–9). Only recently have investigations into the precise roles of propranolol in IHs revealed that this therapy blocks hemangioma endothelial growth and this effect may be through suppressing the production of nitric oxide and HIF1α regulation of vascular endothelial growth factor (VEGF) expression (10,11). Perplexingly, it is unknown how propranolol preferentially inhibits the growth of IHs, while spares the formation of new blood vessels necessary for growth and development of the infant. In this study, we sought to further evaluate the molecular mechanisms by which propranolol exerts its effects on human IH endothelial cells (HemECs). Furthermore, we compared the biological response of HemECs treated with propranolol to that of normal human endothelial cells treated with propranolol. Our data indicate that propranolol disrupts cell proliferation through modulation of key cell cycle regulators and blocks cell migration via alterations in the activation status of proteins essential for cytoskeletal dynamics. We further showed via microarray analysis that propranolol leads to large-scale changes in global gene expression, particularly in genes involved in lipid/sterol metabolism, cell cycle regulation, angiogenesis and post-translational modification. Interestingly, our data indicate that the effects of propranolol on HemECs are similar to that observed in normal endothelial cells, suggesting that this drug is not specific to HemECs.
Materials and methods
Cell lines and culture conditions
HemECs were previously isolated from proliferating-phase IH specimens collected from female infants (12). Primary cultures of neonatal human dermal microvascular endothelial cells (HDMVECs) and human coronary artery endothelial cells (HCAECs) were purchased from ATCC. These cell lines were cultured in vascular cell basal media (ATCC #PCS-100-030) and supplemented with 0.2% bovine brain extract, 5 ng/ml human epidermal growth factor, 10 mM L-glutamine, 0.75 U/ml heparin sulfate, 1 μg/ml hydrocortisone, 50 μg/ml ascorbic acid, 2% fetal bovine serum and penicillin/streptomycin. For all experiments cell lines were used at <10 passages.
RT-PCR
RNA was isolated from cells using the Ambion Purelink Mini kit according to the manufacturer’s directions. qRT-PCR was performed on an ABI7900HT RT-PCR system using TaqMan assays with predesigned primer sets for the genes of interest (Invitrogen). All RT-PCR experiments were performed in triplicate. Data shown are the average RQ value ± standard deviation of 4 replicates.
Western blot analysis
Cell lysates were collected after 48 h treatment and subjected to SDS-PAGE on gradient (4–15%) gels and subsequently transferred to PVDF for western blotting. p-vascular endothelial growth factor receptor-2 (VEGFR-2) (Cell Signaling #2478), VEGFR-2 (Cell Signaling #2479), p-p38 (Cell Signaling #4511), p-p44/42 (Cell Signaling #4370), p-SAPK/JNK (Cell Signaling #4668), p-ATF2 (Cell Signaling #5112), actin (Santa Cruz #SC47778), cyclin A1 (Abcam #ab13337), cyclin A2 (Abcam #7956), cyclin B2 (Abcam #18250), cyclin D1 (Cell Signaling #2978), cyclin D2 (Cell Signaling #3741), cyclin D3 (Cell Signaling #2936), cyclin E1 (Cell Signaling #4129), p15 (Cell Signaling #4822), p21 (Cell Signaling #2947), p27 (Cell Signaling #3698), cleaved caspase-9 (Cell Signaling #9509), cleaved caspase-3 (Cell Signaling #9664), p-FAK (Cell Signaling #3283), p-cofilin (Cell Signaling #3313), cofilin (Cell Signaling #3318), p-ERM (Cell Signaling #3149), ERM (Cell Signaling #3142), p-MYPT1 (Cell Signaling #4563) and MYPT1 (Cell Signaling #2634) antibodies were used at a 1:1000 dilution, followed by incubation with 1:1000 HRP-conjugated anti-mouse or anti-rabbit antibodies (as appropriate). Proteins were detected using Supersignal West Dura Extended Duration Substrate (Thermo Scientific) and digitally captured using a GE Image Quant LAS4000 imaging system.
Proliferation assay
Cells were plated at subconfluent density and subjected to the indicated treatments for 48 h. Images from 5 independent areas were collected at 1-h intervals using a Nikon Biostation CT time lapse imaging robot. Changes in cell density were calculated every 24 h by counting the number of cells in the selected field of vision. Data shown represent the average of 5 independent areas ± the standard deviation.
Flow cytometry
Cells were treated as indicated, trypsinized, and fixed in 70:30 ethanol:phosphate-buffered saline overnight. Cells were then stained with 200 μg/ml ethidium bromide plus 20 μg/ml RNase A and incubated overnight. DNA content was analyzed using an Accuri C6 flow cytometer. Data shown are representative of at least 3 independent experiments. Quantitative analysis of DNA content was performed using CFlow Plus software (Accuri) and is the average of triplicate data points.
Live/dead assay
Cells were treated as indicated, stained for 10 min with 5 μg/ml Hoechst and 5 μg/ml propidium iodide, and washed 3 times in PBS. A Nikon C2SI scanning laser confocal microscope was used to image the red and blue channels. Percent apoptosis (A) was calculated by the following formula: A = (number of red cells/number of blue cells) x 100. The data presented is the average of triplicates.
Migration assay
Confluent cultures were treated as indicated, scratch wounded, and the progress of ‘healing’ of the wound was monitored using a Nikon Biostation CT time lapse imaging robot. Migration speed was calculated by monitoring the movement of the ‘wound’ toward its center at each hour over a 12-h period.
Immunofluorescence and cytoskeletal organization calculations
Cells were grown on glass coverslips, treated as indicated and fixed in 4% paraformaldehyde. Then, the coverslips were blocked in 5% bovine serum albumin plus 0.5% Tween-20, incubated with 1:200 of the p-FAK antibody and detected with fluorescently conjugated secondary antibodies. Actin microfilaments were detected by staining with Rhodamine-conjugated phalloidin, and cell nuclei were detected with 4′,6-diamidino-2-phenylindole (DAPI). Immunofluorescent images were captured as z-stacks using a Nikon C2SI scanning laser confocal microscope. Image analysis of cytoskeletal organization included calculating the actin stress fiber correlation and binarizing this correlation image to determine fiber lengths using the FiberScore algorithm (13).
Microarray analysis
Total RNA was amplified and biotin-labeled using Illumina TotalPrep RNA Amplification kit (Ambion). A total amount of 750 ng of biotinylated aRNA was then briefly heat-denatured and loaded onto expression arrays to hybridize overnight. Following hybridization, arrays were labeled with Cy3-streptavidin and imaged on the Illumina ISCAN. Intensity values were transferred to Agilent GeneSpring GX microarray analysis software and data were filtered based on the quality of each call. Statistical relevance was determined using ANOVA with a Benjamini Hochberg FDR multiple testing correction (p<0.05). Data were then limited by fold change analysis to statistically relevant data points demonstrating a 2-fold or more change in expression.
Results
β-adrenergic receptor expression in HemECs and IHs
The presence of β-adrenergic receptors on normal human endothelial cells has been previously confirmed (14). However, despite the extensive use of systemic propranolol as an anti-IH agent, the expression of the three known β-adrenergic receptors in endothelial cells isolated from these benign tumors is unknown. Using RT-PCR, we evaluated the steady state mRNA expression of the three known β-adrenergic receptors (ADRB1, ADRB2 and ADRB3) in cultured HemECs. Our data revealed that ADRB1 and 2, although not ADRB3, were expressed in HemECs (Fig. 1). Moreover, similar results were observed for both HDMVECs and HCAECs, with equivalent levels of each receptor across the three endothelial cell lines.
Figure 1.
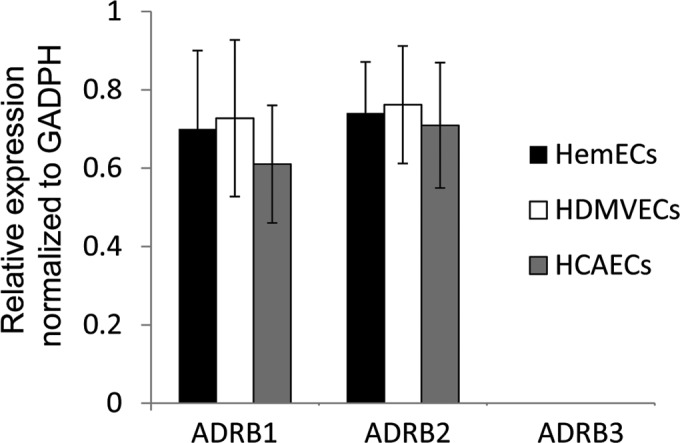
β-adrenergic receptor expression on infantile hemangioma (IH) and normal endothelial cells. RT-PCR expression assays measuring the steady state levels of ADRB1, ADRB2, and ADRB3 mRNA in primary cultures of human infantile hemangioma endothelial cells (HemECs), human dermal microvascular endothelial cells (HDMVECs) and human coronary artery endothelial cells (HCAECs). Expression data are represented as the relative abundance of each transcript normalized to the GAPDH levels.
Propranolol disrupts cell cycle progression
Despite the extensive use of propranolol, many of the mechanisms of action of this drug on IHs have been inferred from its effects on normal endothelial cells (7–9). Recent studies have suggested that propranolol may inhibit IHs by suppressing production of nitric oxide and HIF1α signaling (10,11). However, the signaling intermediates and many of the downstream effectors which modulate its action on IHs are largely unknown. To elucidate the molecular components at play following propranolol-induced inhibition of HemEC proliferation, we first examined the growth rates of HemECs, HDMVECs and HCAECs after treatment with a dose curve of propranolol. Our data indicate that the IC50 for propranolol-induced inhibition of proliferation was ∼50 μM for all three cell types (Fig. 2A and B), therefore for all subsequent experiments in this study we continued to use this concentration. Moreover, cell cycle analysis on the panel of endothelial cells using flow cytometry revealed that propranolol indiscriminately induced an increase in the proportion of cells in the G1 phase of the cell cycle, while reducing the proportion of cells in the S and G2/M phases (Fig. 2C, Table I). Equivalent results were observed in HDMVECs and HCAECs (Table I). To address the effects of propranolol on HIHEC proliferation, we analyzed the expression/activation status of a number of proteins involved in regulating proliferation. VEGFR-2 is a strong mitogenic regulator of endothelial cells that shows aberrant constitutive activation in HemECs (15), and its phosphorylation is reportedly blocked following propranolol treatment (5). Indeed, the 24-h treatment of HemECs with 50 mM propranolol resulted in sharply decreased VEGFR-2 phosphorylation (Fig. 2D). The mitogen activated protein kinases (MAPKs) are direct downstream effectors of VEGFR-2 regulating VEGF-induced endothelial proliferation. We tested the activation status of p38, p44, p42, SAPK, JNK, and the downstream effector ATF4 in HemECs experiencing 24 h of propranolol treatment. Of the proteins tested, the stress activated p38 (but not the stress activated SAPK or JNK) was the only one that exhibited a significant change in phosphorylation following propranolol treatment of HemECs (Fig. 2D). These data suggest that despite the inhibition of VEGFR-2 noted following the treatment, the major proliferative MAPKs such as p44 and p42 were not affected by propranolol treatment in HemECs. As flow cytometry analysis indicated that propranolol induced alterations in cell cycle progression, we performed western blot analysis on a panel of cell cycle regulators, discovering that this drug decreases the expression of key cyclin proteins (cyclins A1, A2, B2, D2 and D3) and increases the expression of important cell cycle inhibitors (p15, p21 and p27) (Fig. 2E). No change in the expression of Cdk2 or Cdk4 was observed following the treatment. These alterations in key cell cycle regulators likely account for propranolol-induced alterations in HemEC proliferation.
Figure 2.

Propranolol decreases the proliferation of human infantile hemangioma endothelial cells (HemECs). (A) HemECs, human dermal microvascular endothelial cells (HDMVECs), and human coronary artery endothelial cells (HCAECs) were treated with a dose curve of propranolol (0 to 100 μM) and cell proliferation was measured by counting changes in the number of cells/defined vision field over a 48-h period. (B) Time lapse microscopy image of sham and 50 μM propranolol treated HemECs over a 48-h period. (C) DNA content analysis of propidium iodide stained HemECs treated with sham or 50 μM propranolol for 48 h. (D) Western blot analysis detecting the levels of phosphorylated and total vascular endothelial growth factor receptor-2 (p-VEGFR-2 and VEGFR-2, respectively) and the phosphorylated forms of p38 (p-p38), p44 (p-p44), p42 (p-p42), stress activated protein kinase (p-SAPK), c-jun N-terminal kinase (p-JNK), and activating transcription factor 4 (p-ATF4) in HemECs treated for 24 h with sham or 50 μM propranolol. Actin levels were used as a loading control. (E) Western blot analysis detecting the levels of cyclins, cyclin dependent kinases, and cyclin dependent kinase inhibitors in HemECs treated for 24 h with sham or 50 μM propranolol. Actin levels were used as a loading control. Prop, propranolol.
Table I.
Percentage of endothelial cells in each cell cycle phase.
| Cells | Sham | Propranolol |
|---|---|---|
| HemECs | ||
| G1 | 68±2.3 | 74±2.2 |
| S | 8±0.6 | 5±0.3 |
| G2/M | 24±2.7 | 21±1.1 |
| HDMVECs | ||
| G1 | 69±3.0 | 75±1.6 |
| S | 8±4.1 | 4±0 |
| G2/M | 22±3.9 | 20±4.5 |
| HCAECs | ||
| G1 | 71±1.4 | 76±1.7 |
| S | 5±1.6 | 3±1.3 |
| G2/M | 23±3.3 | 20±2.5 |
HemECs, human infantile hemangioma endothelial cells; HDMVECs, human dermal microvascular endothelial cells; HCAECs, human coronary artery endothelial cells.
It is speculated that propranolol may increase apoptosis of IHs
To determine whether this drug affects apoptosis, we treated HemECs, HDMVECs and HCAECs with 50 μM propranolol for 3 days. As a control we treated HemECs with 5 μM cisplatin for an equivalent amount of time. Cells were co-stained with propidium iodide (which only stains the nuclei of dead cells) and Hoechst dye (which stains the nuclei of both live and dead cells). Calculation of the apoptotic index from each treatment revealed that 50 μM propranolol did not induce apoptosis of any of the cell lines tested, while 5 μM cisplatin resulted in almost 100% apoptosis (Fig. 3A and B). We did observe significant apoptosis in all of our cell lines at concentrations of propranolol higher than 150 μM (data not shown). To confirm our observations, we utilized western blotting to detect the cleavage products of the apoptotic initiator caspase-9 and the apoptotic effector caspase-3. Our data indicate that while 5 μM cisplatin strongly induced caspase cleavage, 50 μM propranolol failed to induce apoptotic signaling (Fig. 3C).
Figure 3.

Propranolol does not induce apoptosis in human infantile hemangioma endothelial cells (HemECs) at its effective inhibitory concentration. (A) Confocal imaging of HemECs treated for 72 h with sham or 50 μM propranolol and subsequently co-stained with propidium iodide (PI) and Hoechst dye (blue, Hoechst-positive nuclei; pink, Hoechst-positive/PI-positive nuclei). (B) Quantification of PI-positive nuclei in HemECs, human dermal microvascular endothelial cells (HDMVECs), and human coronary artery endothelial cells (HCAECs) treated for 72 h with sham or 50 μM propranolol. (C) Western blot analysis detecting the levels of cleaved caspase-9 and -3 (cl-caspase-9 and cl-caspase -3, respectively). Actin levels were used as a loading control.
Propranolol disrupts cell migration and actin cytoskeleton dynamics
Several reports have presented mixed results for the role of β-adrenergic receptor signaling in wound healing and cell migration, with evidence that inhibition of this class of receptors delays (16–19) or promotes (20–22) wound healing. Moreover, in cultured bovine aortic endothelial cells, β-adrenergic blockade with propranolol reportedly inhibits norepinephrine-induced induction of actin stress fibers (8), thus we sought to determine if similar effects on cell migration and cytoskeletal organization could be observed in HemECs treated with propranolol. HemECs, HDMVECs and HCAECs were grown to confluence, manually scratch wounded with a micropipette tip, and imaged using time lapse microscopy over a period of 12 h. As illustrated in Fig. 4A, treatment of HemECs with 50 μM propranolol resulted in a significant reduction in ‘wound’ closure compared to the control. Quantification of the time lapse images taken from the scratch migration assay revealed that propranolol dramatically reduced the migratory speed of HemECs (34% reduction), HMVECs (66% reduction) and HCAECs (67% reduction) (Fig. 4B), suggesting that propranolol is more effective at blocking proliferation of normal endothelial cells than HemECs. To identify signaling events that might shed light on how propranolol disrupts migration, we performed western blot analysis to detect the activation status of several known regulators of actin cytoskeletal dynamics including focal adhesion kinase (FAK), cofilin, ezrin/radixin/moesin (ERM), and myosin phosphatase (MYPT1), revealing that this drug effectively decreases the inhibitory phosphorylation of cofilin (an actin severing protein) at serine-3 (Fig. 4C). Given the increased activation of cofilin following propranolol treatment, we suspected that changes in the actin microfilament cytoskeleton would ensue. Indeed, immunofluorescent detection of actin stress fibers revealed that propranolol markedly inhibits actin polymerization in HemECs (Fig. 4D) and normal endothelial cells (data not shown) consistent with that expected from activation of cofilin. Computational analysis of actin stress fiber length using the FiberScore algorithm (Lichtenstein) demonstrated that propranolol-treated HemECs exhibit a greater than 1.5-fold reduction in average fiber length compared to the sham treatment (data not shown). Similar observations were observed for non-hemangioma endothelial cells (data not shown). In addition to altering actin stress fiber polymerization, propranolol shifted the subcellular localization of p-FAK from regions of colocalization with actin stress fibers in sham-treated cells to diffuse punctuate regions located throughout the cytoplasm in propranolol-treated cells (Fig. 4D). Despite alterations in p-FAK subcellular localization, no changes in the levels of p-FAK were observed in response to propranolol (Fig. 4C).
Figure 4.

Propranolol disrupts HIHEC migration and actin cytoskeleton dynamics. (A) Confluent monolayers of human infantile hemangioma endothelial cells (HemECs) were scratch wounded and treated with sham or 50 μM propranolol. Progress of migration was monitored using time lapse photography over a period of 12 h. (B) Quantification of the speed (μm/h) of HemECs, human dermal microvascular endothelial cells (HDMVECs), and human coronary artery endothelial cells (HCAECs) treated with sham or propranolol from the time lapse images of the scratch assay. (C) Western blot analysis detecting the levels of the total and phophorylated (p-) forms of focal adhesion kinase (FAK), cofilin, ezrin/radixin/moesin (ERM), and myosin phosphatase-targeting subunit 1 (MYPT1) in HemECs treated with sham or 50 μM propranolol for 48 h. Actin levels were used as a loading control. (D) Confocal immunofluorescent imaging of sham or 50 μM propranolol-treated HemECs co-stained with Rhodamine conjugated phalloidin (red), DAPI (blue), and antibodies against phospho-FAK. Prop, propranolol.
Propranolol disrupts global gene expression patterns in endothelial cells
Propranolol has been shown to affect the expression of cyclins across multiple cell types (5,23), gluconeogenic and glycolytic enzymes in the liver (24), epidermal growth factor 1 in cardiomyocytes (25) and pigment epithelial derived factor in the retina (26). However, these studies have focused on small subsets of genes and did not look at large-scale changes in genomic expression patterns. To evaluate the global gene expression changes in HemECs in response to propranolol and to examine how the identified changes compared with those observed in propranolol-treated normal human endothelial cells, we performed whole genome microarrays providing coverage for more than 47,000 transcripts and known splice variants across the human transcriptome. Our analysis identified 89 genes whose expression in HemECs was altered greater than 2-fold (p<0.05) in response to propranolol (32 genes significantly upregulated and 57 genes downregulated) (Table II). Several functional groupings of genes were identified included those involved in lipid and sterol metabolism (21% of total gene expression changes: HMGCS1, MSMO1, LDLR, DHCR7, SCD, ACAT2, LSS, FASN, MSMO1, SQLE, DHCR24, FDFT1, IDI1, FADS2, ACSS2, EBP, SC5DL, NSDHL and LPIN1), cell cycle regulation (14% of total gene expression changes: CDC2, CDCA8, CDKN3, PRC1, MCM4, CDC20, CDC45L, CCNA1, CCNB2, CCNA2, TOP2A and CDCA7), angiogenesis (5% of total gene expression changes: PGF, ANGPT2, ANGPTL4 and RGS4) and ubiquitin modifications (3% of total gene expression changes: UBE2T, UBE2C and UHRF1). Comparative analysis of genes whose expression was statistically altered by propranolol treatment in any of the three endothelial cell line tests revealed a strong correlation between cell lines (Fig. 5A). This data indicate that propranolol-induced gene expression changes are endothelial cell-type independent. Quantitative RT-PCR validation of ∼14% of the propranolol-responsive genes in HemECs revealed comparable alterations in gene expression similar to that revealed by microarray analysis, thus corroborating our microarray data through independent analysis (Fig. 5B).
Table II.
Alterations in gene expression (fold-change) induced by propranolol treatment.
| Gene symbol | Gene name | Accession no. | HIHEC | HDMVEC | HCAEC |
|---|---|---|---|---|---|
| HMGCS1 | 3-Hydroxy-3-methylglutaryl-CoA synthase 1 | NM_002130.6 | 4.6 | 5.8 | 6.2 |
| MSMO1 | Methylsterol monooxygenase 1, TV2 | NM_001017369.2 | 4.5 | 4.0 | 5.8 |
| INSIG1 | Insulin induced gene 1 | NM_198336.2 | 4.3 | 4.6 | 4.9 |
| LDLR | Low density lipoprotein receptor | NM_000527.4 | 3.6 | 3.8 | 3.9 |
| MVD | Mevalonate decarboxylase | NM_002461.1 | 3.6 | 5.7 | 3.4 |
| DHCR7 | 7-Dehydrocholesterol reductase | NM_001360.2 | 3.6 | 4.7 | 3.4 |
| SCD | Stearoyl-CoA desaturase | NM_005063.4 | 3.3 | 3.2 | 5.0 |
| ACAT2 | Acetyl-CoA acetyltransferase 2 | NM_005891.2 | 3.2 | 4.6 | 5.2 |
| LSS | Lanosterol synthase | NM_002340.5 | 3.2 | 4.5 | 5.1 |
| TM7SF2 | Transmembrane 7 superfamily member 2 | NM_003273.2 | 3.0 | 4.8 | 5.2 |
| HMGCR | 3-Hydroxy-3-methylglutaryl-CoA reductase | NM_000859.2 | 2.9 | 3.1 | 3.8 |
| FASN | Fatty acid synthase | NM_004104.4 | 2.8 | 4.6 | 4.3 |
| MSMO1 | Methylsterol monooxygenase 1, TV1 | NM_006745.4 | 2.8 | 2.0 | 3.0 |
| SQLE | Squalene epoxidase | NM_003129.3 | 2.7 | 2.6 | 3.6 |
| PSG4 | Pregnancy specific β-1-glycoprotein 4 | NM_002780.3 | 2.6 | 1.3 | 4.5 |
| DHCR24 | 24-Dehydrocholesterol reductase | NM_014762.3 | 2.5 | 2.4 | 3.3 |
| FDFT1 | Farnesyl-diphosphate farnesyltransferase 1 | NM_004462.3 | 2.5 | 2.2 | 3.2 |
| IDI1 | Isopentenyl-diphosphate δ isomerase 1 | NM_004508.2 | 2.4 | 2.3 | 3.4 |
| FADS2 | Fatty acid desaturase 2 | NM_004265.2 | 2.4 | 4.2 | 2.8 |
| NPC1 | Niemann-Pick disease, type C1 | NM_000271.4 | 2.3 | 2.3 | 3.8 |
| PFKFB4 | Fructose-2,6-biphosphatase 4 | NM_004567.2 | 2.2 | 2.8 | 2.3 |
| ACSS2 | Acyl-CoA synthetase family member 2, TV2 | NM_001076552.2 | 2.2 | 2.6 | 3.5 |
| ACSS2 | Acyl-CoA synthetase family member 2, TV1 | NM_018677.3 | 2.2 | 2.7 | 3.2 |
| EBP | Emopamil binding protein | NM_006579.2 | 2.1 | 2.4 | 2.3 |
| LOC100129668 | LOC100129669 | XM_001713607.1 | 2.1 | 2.2 | 2.5 |
| HMOX1 | Heme oxygenase (decycling) 1 | NM_002133.2 | 2.1 | 2.1 | 3.5 |
| SC5DL | Sterol-C5-desaturase-like | NM_006918.4 | 2.1 | 1.4 | 2.4 |
| NSDHL | NAD(P) dependent steroid dehydrogenase-like | NM_015922.2 | 2.1 | 2.3 | 2.2 |
| P2RX4 | Purinergic receptor P2X, 4 | NM_002560.2 | 2.1 | 1.7 | 1.8 |
| LPIN1 | Lipin 1 | NM_145693.1 | 2.0 | 1.6 | 2.9 |
| PGF | Placental growth factor | NM_002632.5 | 2.0 | 1.7 | 2.3 |
| ANGPT2 | Angiopoietin 2 | NM_001147.2 | 2.0 | 1.0 | 2.1 |
| LOC729009 | LOC729010 | XR_042330.1 | 2.0 | 1.6 | 2.6 |
| IL8 | Interleukin 8 | NM_000584.3 | −2.0 | −2.9 | −3.1 |
| CDK1 | Cyclin-dependent kinase 1 | NM_001786.4 | −2.0 | −1.8 | −2.1 |
| TUBB2C | Tubulin, β 4B Ivb | NM_006088.5 | −2.0 | −1.4 | −2.5 |
| PTTG1 | Pituitary tumor-transforming 1 | NM_004219.2 | −2.0 | −2.0 | −2.0 |
| OIP5 | Opa interacting protein 5 | NM_007280.1 | −2.0 | −2.3 | −2.1 |
| CDCA8 | Cell division cycle associated 8 | NM_018101.3 | 2.0 | −1.4 | −2.3 |
| TAGLN | Transgelin | NM_003186.3 | −2.0 | −1.7 | −2.9 |
| CDKN3 | Cyclin-dependent kinase inhibitor 3 | NM_005192.3 | −2.0 | −1.7 | −1.6 |
| ANLN | Anillin, actin binding protein | NM_018685.2 | −2.0 | −1.8 | −1.7 |
| HJURP | Holliday junction recognition protein | NM_018410.3 | −2.0 | −1.2 | −2.0 |
| PBK | PDZ binding kinase | NM_018492.2 | −2.0 | −1.6 | −2.5 |
| UBE2T | Ubiquitin-conjugating enzyme E2T (putative) | NM_014176.3 | −2.0 | −1.4 | −1.5 |
| STEAP1 | 6-Transmembrane epithelial antigen of the prostate 1 | NM_012449.2 | −2.0 | −1.8 | −1.8 |
| UBE2C | Ubiquitin-conjugating enzyme E2C, TV3 | NM_181800.1 | −2.0 | −1.9 | −2.8 |
| CKS1B | CDC28 protein kinase regulatory subunit 1B | NM_001826.2 | −2.0 | −1.6 | −1.9 |
| TACC3 | Transforming, acidic coiled-coil containing protein 3 | NM_006342.2 | −2.0 | −1.4 | −1.8 |
| NCAPG | Non-SMC condensin I complex, subunit G | NM_022346.3 | −2.0 | −1.7 | −1.6 |
| PCDH7 | Protocadherin 7 | NM_002589.2 | −2.0 | −1.2 | −2.2 |
| FAM64A | Family with sequence similarity 64, member A | NM_019013.2 | −2.1 | −1.2 | −2.0 |
| PRC1 | Protein regulator of cytokinesis 1 | NM_199413.1 | −2.1 | −1.5 | −2.3 |
| MELK | Maternal embryonic leucine zipper kinase | NM_014791.3 | −2.1 | −1.8 | −2.1 |
| TPX2 | TPX2, microtubule-associated | NM_012112.4 | −2.1 | −1.4 | −2.2 |
| MCM4 | Minichromosome maintenance complex CMPT 4, TV2 | NM_182746.2 | −2.1 | −1.6 | −1.9 |
| ZWINT | ZW10 interactor | NM_001005413.1 | −2.1 | −1.5 | −2.0 |
| KIFC1 | Kinesin family member C1 | NM_002263.2 | −2.1 | −1.4 | −2.2 |
| CDC20 | Cell division cycle 20 | NM_001255.2 | −2.2 | −2.0 | −4.0 |
| UBE2C | Ubiquitin-conjugating enzyme E2C, TV6 | NM_181803.1 | −2.2 | −1.8 | −2.4 |
| NCAPG2 | Non-SMC condensin II complex, subunit G2 | NM_017760.5 | −2.2 | −1.3 | −1.4 |
| SERPIND1 | Serpin peptidase inhibitor, clade D, member 1 | NM_000185.3 | −2.2 | −1.8 | −2.6 |
| CDC45L | Cell division cycle 45 | NM_003504.3 | −2.2 | −1.8 | −3.0 |
| LYAR | Ly1 antibody reactive | NM_017816.2 | −2.2 | −1.6 | −1.3 |
| CCNA1 | Cyclin A1 | NM_003914.3 | −2.2 | −2.2 | −1.9 |
| TRIP1 | Thyroid hormone receptor interactor 13 | NM_004237.3 | −2.2 | −1.9 | −2.6 |
| MPZL2 | Myelin protein zero-like 2, TV2 | NM_144765.2 | −2.2 | −1.3 | −2.4 |
| CEP55 | Centrosomal protein 55kDa | NM_018131.4 | −2.2 | −2.0 | −2.1 |
| CXCL1 | Chemokine (C-X-C motif) ligand 1 | NM_001511.3 | −2.2 | −2.5 | −2.8 |
| CCNB2 | Cyclin B2 | NM_004701.3 | −2.2 | −2.2 | −2.3 |
| KIF20A | Kinesin family member 20A | NM_005733.2 | −2.2 | −1.9 | −2.5 |
| RAD51AP1 | RAD51 associated protein 1 | NM_006479.4 | −2.2 | −1.6 | −2.2 |
| GINS2 | GINS complex subunit 2 | NM_016095.2 | −2.2 | −1.7 | −2.9 |
| FAM83D | Family with sequence similarity 83, member D | NM_030919.2 | −2.3 | −1.6 | −2.4 |
| KIAA0101 | KIAA0101 | NM_014736.4 | −2.3 | −2.0 | −2.3 |
| DLGAP5 | Discs, large (Drosophila) homolog-associated protein 5 | NM_014750.4 | −2.3 | −2.0 | −2.3 |
| CCNA2 | Cyclin A2 | NM_001237.3 | −2.3 | −2.2 | −2.8 |
| LOC399942 | LOC399943 | XM_934471.1 | −2.3 | −2.4 | −3.4 |
| MPZL2 | Myelin protein zero-like 2, TV1 | NM_005797.3 | −2.3 | −2.7 | −2.2 |
| TOP2A | Topoisomerase II α 170kDa | NM_001067.3 | −2.3 | −2.4 | −2.6 |
| RRM2 | Ribonucleotide reductase M2 | NM_001034.3 | −2.3 | −1.5 | −2.8 |
| FBXO5 | F-box protein 5 | NM_012177.3 | −2.4 | −1.9 | −2.6 |
| CDCA7 | Cell division cycle associated 7 | NM_031942.4 | −2.4 | −1.9 | −2.5 |
| MCM4 | Minichromosome maintenance complex CMPT 4, TV1 | NM_005914.3 | −2.4 | −1.5 | −2.0 |
| MAD2L1 | MAD2 mitotic arrest deficient-like 1 | NM_002358.3 | −2.4 | −2.3 | −2.1 |
| UHRF1 | Ubiquitin-like with PHD and ring finger domains 1 | NM_001048201.1 | −2.5 | −1.6 | −2.6 |
| ANGPTL4 | Angiopoietin-like 4 | NM_139314.1 | −2.8 | −2.8 | −2.9 |
| RGS4 | Regulator of G-protein signaling 4 | NM_005613.5 | −3.0 | −3.8 | −3.8 |
| IL1RL1 | Interleukin 1 receptor-like 1 | NM_003856.2 | −3.2 | 3.4 | −5.2 |
HDMVEC, human dermal microvascular endothelial cells; HCAEC, human coronary artery endothelial cells.
Figure 5.
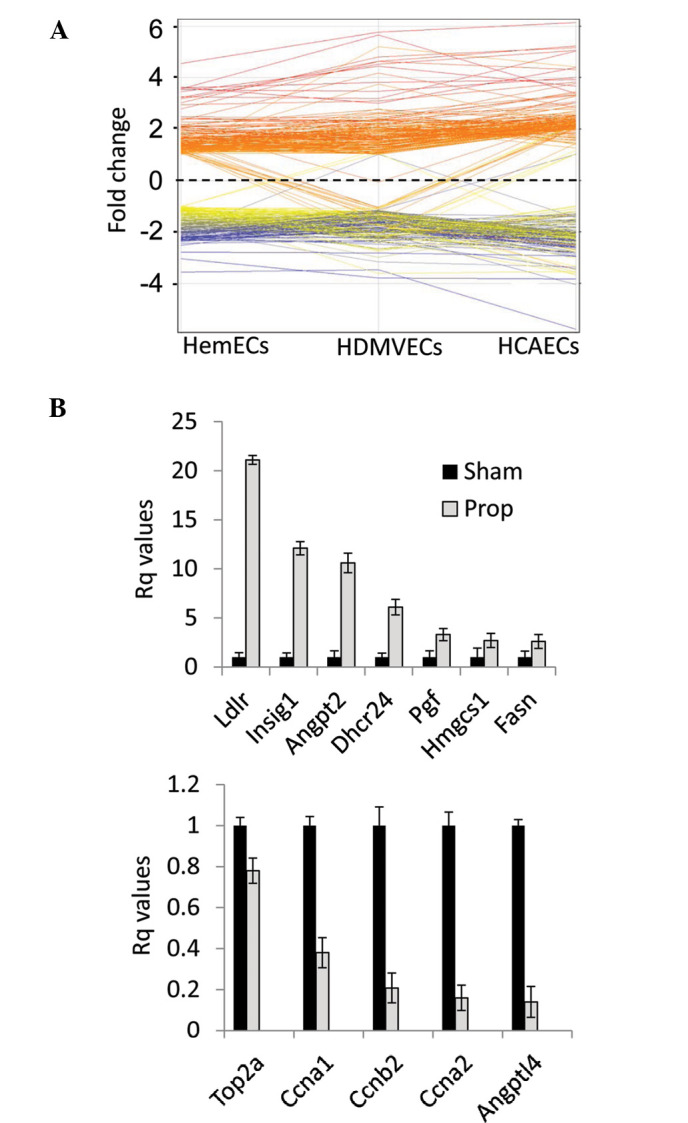
Propranolol induces significant alterations in global gene expression of human infantile hemangioma endothelial cells (HemECs). (A) Correlation map comparing the significant gene expression changes (>2 fold gene expression alteration, p<0.05) as determined by microarray analysis between HemECs, human dermal microvascular endothelial cells (HDMVECs), and human coronary artery endothelial cells (HCAECs) treated with sham or 50 μM propranolol for 24 h. (B) RT-PCR confirmation of a subset of genes in HemECs whose expression was statistically altered in the microarray.
Discussion
IHs as a whole are largely understudied considering the high prevalence of these lesions in children and the serious threat to health they pose in certain instances. To date, there remains a great deal of uncertainty as to the origin of these tumors, with evidence suggesting they may be caused by aberrant transplantation of placental endothelial cells (27), predisposing genetic factors (28,29) and/or tumor stem cell components (30). Despite the controversial origin of these tumors, proliferating IHs are characterized by an enhanced angiogenic capacity largely due to modulation of signaling pathways that regulate the VEGF signaling axis, while involuting IHs display a chronic inflammatory response and downregulation of angiogenesis regulators (31). The recent discovery that the β blocker propranolol is an effective treatment for IHs suggests that the sympathetic nervous system may play a key role in controlling IH growth. Epinephrine is a sympathomimetic amine that increases the activity of noradrenaline in post-synaptic cells, and is capable of enhancing vasodilation through activation of β-adrenergic receptors. β-adrenergic receptors have been shown to be expressed on normal capillary endothelial cells (32) and it is believed that β blockers, such as propranolol, inhibit epinephrine signaling through inducing vasoconstriction of endothelial cells and disruptions in key angiogenic processes (7–9,33). Moreover, β blockers have been shown to reduce the expression of VEGF in non-endothelial cells, thus leading to inhibition of angiogenic paracrine signaling (34). Despite the extensive use of propranolol in the treatment of IHs, very little has been done to determine the molecular mechanisms of propranolol in IH tumors. This drug is presumably believed to induce IH tumor regression through mechanisms similar to that noted in normal endothelial cells, and recent reports suggest that it may work in part through suppressing production of nitric oxide and HIF1α regulation of VEGF expression (10,11). In this study, we sought to investigate how propranolol disrupts HIHEC function and compare these effects to those seen in normal primary endothelial cell lines. Our data demonstrated that β-adrenergic receptors are expressed across a panel of HIHEC and normal endothelial cells. We further showed that propranolol disrupts HIHEC and normal endothelial cell cycle progression, migration, cytoskeletal dynamics, and gene expression, and we elucidated multiple downstream targets of propranolol including cell cycle progression regulators, cytoskeletal modulators and gene expression alterations.
The expression of β1- and β2-adrenergic receptors has been extensively studied in the cardiovascular system, with high expression occurring in cardiac myocytes and vascular smooth muscle cells (35). Although β3-knockout mice display increased hypotension in response to isoproternol (36), these receptors are suspected to play a lesser role in cardiovascular function compared to β1- and β2- receptors as they are expressed primarily in brown adipocytes, gallbladder and the colon (37). Our data indicate that both β1- and β2- (but not β3-) adrenergic receptors are expressed on primary cultures of HemECs. Interestingly, comparisons of the relative mRNA expression levels of these receptors on primary cultures of non-diseased endothelial cells revealed similar levels to that of HemECs, causing us to question if propranolol is selective for HemECs or if this drug demonstrates a similar level of inhibition for diseased and normal endothelial cells. Indeed, comparisons of the inhibitory effect of propranolol at its IC50 on cultures of HemECs, HDMVECs, and HCAECs revealed that this drug indiscriminately blocks proliferation, migration, and actin polymerization in an endothelial cell-type independent manner. This finding suggests that the mechanism of action for propranolol on IHs may extend beyond simply blocking endothelial cell function. For instance, IHs display a high pericyte density in the proliferating stage (38) and increased molecular markers of endothelial cell/pericyte interactions (31). Primary cultures of pericytes express functional adrenergic receptors and respond to autonomic vasoactive substances in vivo (39). As pericytes are responsible for a number of roles in the microvasculature including capillary maturation and stabilization, further studies should examine if propranolol inhibits IH growth through destabilization of endothelial cell/pericyte interactions. Another possibility that may account for the endothelial cell-type independent action of propranolol may have to do with the limitations of in vitro monolayer cell culture systems. Children undergoing systemic propranolol treatment for IHs often undergo side effects including bradycardia, hypotension and hypoglycemia (40), however significant disruptions of their existing vascular beds have not been reported. This suggests that propranolol may preferentially inhibit proliferating endothelial vasculature while sparing the quiescent established vasculature. However, this possibility is complicated by reports suggesting that propranolol improves wound healing - a process intimately dependent on neovascularization (20–22).
There is evidence that VEGFR-2 phosphorylation is controlled by β-adrenergic signaling (5,41,42), however the mechanisms underlying this effect remain to be determined. As HemECs are characterized by aberrant constitutive activation of VEGFR-2 signaling, we sought to determine whether propranolol is capable of attenuating this process. Similar to data reported in normal endothelial lines, propranolol effectively blocks VEGFR-2 phosphorylation on HemECs. We tested the effects of propranolol on known downstream targets of VEGF signaling revealing propranolol-induced alterations in p38 signaling, decreased cyclin expression and increased cyclin-dependent kinase inhibitor steady state mRNA levels. Moreover, propranolol treatment of the panel of endothelial cells resulted in reduced proliferation rates and increased percentages of cells in the G1 cell cycle phase. These alterations were coincident with significant changes in the levels of key cyclins and cell cycle inhibitors. Interestingly, of the panel of MAPK proteins that we tested, we saw increased phosphorylation only in p38, which is known to be responsive to stress stimuli such as cytokines, irradiation and shock. p38 plays a central role in inflammation and regulates the production of inflammatory mediators such as TNFα, IL1β, and COX2 (43), thus it is possible that increased p38 activation in propranolol-treated IHs may mediate IH regression through immune-mediated responses. A number of studies suggest that propranolol may induce apoptosis of HemECs. No caspase cleavage or apoptosis was observed at the IC50 (∼50 μM) for propranolol in HemECs or normal endothelial cells, although we did begin to see lethal effects of this treatment in all three endothelial cell lines at upwards of 150 μM. These findings do not rule out that propranolol-induced IH apoptosis plays a role in the efficacy of this treatment, but it does suggest that basic endothelial functions such as proliferation and migration display greater susceptibility to lower doses of propranolol than does apoptosis. We demonstrated that propranolol treatment of HemECs results in abolished stress fiber formation, and this effect may be due in part to decreased levels of phosphorylated cofilin following propranolol treatment. Cofilin is a cytoskeletal-binding protein critical for actin microfilament dynamics and reorganization by severing and depolymerizing actin filaments (44). Cofilin phosphorylation is an inhibitory event (45), thus the absence of stress fiber formation observed in propranolol-treated HemECs may be due in part to increased cofilin-mediated actin severing. This effect would certainly disrupt cell migration, and as the actin cytoskeleton is intimately tied to the regulation of cell cycle progression (46), may indirectly contribute to propranolol induced decreased cell proliferation.
As propranolol appears to work with great efficacy against IHs, similar inhibitory effects could potentially be observed in other vascular tumors such as angiosarcomas and Kaposi’s sarcomas. Indeed, propranolol has been tested in preclinical and clinical models of malignant tumors, demonstrating good efficacy in the treatment of melanoma (47), pancreatic (48), colorectal (49) and breast (50) cancer. Finally, given the cutaneous nature of IHs and the endothelial cell type-independent effects of propranolol observed in our study, topical delivery of β blockers (as opposed to systemic delivery) should be aggressively pursued. A controlled study of topical administration of the β blocker timolol on non-life threatening IHs revealed consistently good to moderate responses in 91.5% of infants (51), suggesting this therapy could specifically treat the lesion area while preventing potential collateral anti-vascular effects on the normal endothelium.
Acknowledgments
Support of this study was provided by a National Heart, Lung, and Blood Institute grant HL098931 and TTUHSC startup funds to BAB, a National Institute of Arthritis and Musculoskeletal and Skin Diseases grant (AR048564) to JB, a NASA EPSCoR award to NMSU, and internal support from NMSU to LB.
References
- 1.Waner M, North PE, Scherer KA, Frieden IJ, Waner A, Mihm MC., Jr The nonrandom distribution of facial hemangiomas. Arch Dermatol. 2003;139:869–875. doi: 10.1001/archderm.139.7.869. [DOI] [PubMed] [Google Scholar]
- 2.Léauté-Labrèze C, Dumas de la Roque E, Hubiche T, Boralevi F, Thambo JB, Taïeb A. Propranolol for severe hemangiomas of infancy. N Engl J Med. 2008;358:2649–2651. doi: 10.1056/NEJMc0708819. [DOI] [PubMed] [Google Scholar]
- 3.D’Angelo G, Lee H, Weiner RI. cAMP-dependent protein kinase inhibits the mitogenic action of vascular endothelial growth factor and fibroblast growth factor in capillary endothelial cells by blocking Raf activation. J Cell Biochem. 1997;67:353–366. [PubMed] [Google Scholar]
- 4.Sommers Smith SK, Smith DM. Beta blockade induces apoptosis in cultured capillary endothelial cells. In Vitro Cell Dev Biol Anim. 2002;38:298–304. doi: 10.1290/1071-2690(2002)038<0298:BBIAIC>2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 5.Lamy S, Lachambre MP, Lord-Dufour S, Béliveau R. Propranolol suppresses angiogenesis in vitro: inhibition of proliferation, migration, and differentiation of endothelial cells. Vascul Pharmacol. 2010;53:200–208. doi: 10.1016/j.vph.2010.08.002. [DOI] [PubMed] [Google Scholar]
- 6.Spindler V, Waschke J. Beta-adrenergic stimulation contributes to maintenance of endothelial barrier functions under baseline conditions. Microcirculation. 2011;18:118–127. doi: 10.1111/j.1549-8719.2010.00072.x. [DOI] [PubMed] [Google Scholar]
- 7.Makarski JS. Stimulation of cyclic AMP production by vasoactive agents in cultured bovine aortic and pulmonary artery endothelial cells. In Vitro. 1981;17:450–458. doi: 10.1007/BF02626746. [DOI] [PubMed] [Google Scholar]
- 8.Welles SL, Shepro D, Hechtman HB. Vasoactive amines modulate actin cables (stress fibers) and surface area in cultured bovine endothelium. J Cell Physiol. 1985;123:337–342. doi: 10.1002/jcp.1041230307. [DOI] [PubMed] [Google Scholar]
- 9.Brehm BR, Bertsch D, von Fallois J, Wolf SC. Beta-blockers of the third generation inhibit endothelin-1 liberation, mRNA production and proliferation of human coronary smooth muscle and endothelial cells. J Cardiovasc Pharmacol. 2000;36(Suppl 1):S401–S403. doi: 10.1097/00005344-200036051-00117. [DOI] [PubMed] [Google Scholar]
- 10.Dai Y, Hou F, Buckmiller L, Fan CY, Saad A, Suen J, Richter GT. Decreased eNOS protein expression in involuting and propranolol-treated hemangiomas. Arch Otolaryngol Head Neck Surg. 2012;138:177–182. doi: 10.1001/archoto.2011.1096. [DOI] [PubMed] [Google Scholar]
- 11.Chim H, Armijo BS, Miller E, Gliniak C, Serret MA, Gosain AK. Propranolol induces regression of hemangioma cells through HIF-1alpha-mediated inhibition of VEGF-A. Ann Surg. 2012;256:146–156. doi: 10.1097/SLA.0b013e318254ce7a. [DOI] [PubMed] [Google Scholar]
- 12.Boye E, Yu Y, Paranya G, Mulliken JB, Olsen BR, Bischoff J. Clonality and altered behavior of endothelial cells from hemangiomas. J Clin Invest. 2001;107:745–752. doi: 10.1172/JCI11432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lichtenstein N, Geiger B, Kam Z. Quantitative analysis of cytoskeletal organization by digital fluorescent microscopy. Cytometry A. 2003;54:8–18. doi: 10.1002/cyto.a.10053. [DOI] [PubMed] [Google Scholar]
- 14.Howell RE, Albelda SM, Daise ML, Levine EM. Characterization of beta-adrenergic receptors in cultured human and bovine endothelial cells. J Appl Physiol. 1988;65:1251–1257. doi: 10.1152/jappl.1988.65.3.1251. [DOI] [PubMed] [Google Scholar]
- 15.Jinnin M, Medici D, Park L, Limaye N, Liu Y, Boscolo E, Bischoff J, Vikkula M, Boye E, Olsen BR. Suppressed NFAT-dependent VEGFR1 expression and constitutive VEGFR2 signaling in infantile hemangioma. Nat Med. 2008;14:1236–1246. doi: 10.1038/nm.1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Souza BR, Santos JS, Costa AM. Blockade of beta1- and beta2-adrenoceptors delays wound contraction and re-epithelialization in rats. Clin Exp Pharmacol Physiol. 2006;33:421–430. doi: 10.1111/j.1440-1681.2006.04383.x. [DOI] [PubMed] [Google Scholar]
- 17.Romana-Souza B, Santos JS, Monte-Alto-Costa A. beta-1 and beta-2, but not alpha-1 and alpha-2, adrenoceptor blockade delays rat cutaneous wound healing. Wound Repair Regen. 2009;17:230–239. doi: 10.1111/j.1524-475X.2008.00453.x. [DOI] [PubMed] [Google Scholar]
- 18.Romana-Souza B, Porto LC, Monte-Alto-Costa A. Cutaneous wound healing of chronically stressed mice is improved through catecholamine blockade. Exp Dermatol. 2010;19:821–829. doi: 10.1111/j.1600-0625.2010.01113.x. [DOI] [PubMed] [Google Scholar]
- 19.Romana-Souza B, Otranto M, Vieira AM, Filgueiras CC, Fierro IM, Monte-Alto-Costa A. Rotational stress-induced increase in epinephrine levels delays cutaneous wound healing in mice. Brain Behav Immun. 2010;24:427–437. doi: 10.1016/j.bbi.2009.11.012. [DOI] [PubMed] [Google Scholar]
- 20.Romana-Souza B, Nascimento AP, Monte-Alto-Costa A. Propranolol improves cutaneous wound healing in streptozotocin-induced diabetic rats. Eur J Pharmacol. 2009;611:77–84. doi: 10.1016/j.ejphar.2009.03.053. [DOI] [PubMed] [Google Scholar]
- 21.Romana-Souza B, Nascimento AP, Monte-Alto-Costa A. Low-dose propranolol improves cutaneous wound healing of burn-injured rats. Plast Reconstr Surg. 2008;122:1690–1699. doi: 10.1097/PRS.0b013e31818cbf67. [DOI] [PubMed] [Google Scholar]
- 22.Mohammadi AA, Bakhshaeekia A, Alibeigi P, Hasheminasab MJ, Tolide-ei HR, Tavakkolian AR, Mohammadi MK. Efficacy of propranolol in wound healing for hospitalized burn patients. J Burn Care Res. 2009;30:1013–1017. doi: 10.1097/BCR.0b013e3181b48600. [DOI] [PubMed] [Google Scholar]
- 23.Musumeci M, Maccari S, Sestili P, Signore M, Molinari P, Ambrosio C, Stati T, Colledge WH, Grace AA, Catalano L, Marano G. Propranolol enhances cell cycle-related gene expression in pressure overloaded hearts. Br J Pharmacol. 2011;164:1917–1928. doi: 10.1111/j.1476-5381.2011.01504.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Erraji-Benchekroun L, Couton D, Postic C, Borde I, Gaston J, Guillet JG, André C. Overexpression of beta2-adrenergic receptors in mouse liver alters the expression of gluconeogenic and glycolytic enzymes. Am J Physiol Endocrinol Metab. 2005;288:E715–E722. doi: 10.1152/ajpendo.00113.2004. [DOI] [PubMed] [Google Scholar]
- 25.Patrizio M, Musumeci M, Stati T, Fecchi K, Mattei E, Catalano L, Marano G. Propranolol promotes Egr1 gene expression in cardiomyocytes via beta-adrenoceptors. Eur J Pharmacol. 2008;587:85–89. doi: 10.1016/j.ejphar.2008.04.017. [DOI] [PubMed] [Google Scholar]
- 26.Lashbrook BL, Steinle JJ. Beta-adrenergic receptor regulation of pigment epithelial-derived factor expression in rat retina. Auton Neurosci. 2005;121:33–39. doi: 10.1016/j.autneu.2005.05.006. [DOI] [PubMed] [Google Scholar]
- 27.Haggstrom AN, Beaumont JL, Lai JS, Adams DM, Drolet BA, Frieden IJ, Garzon MC, Holland KE, Horii KA, Lucky AW, et al. Measuring the severity of infantile hemangiomas: instrument development and reliability. Arch Dermatol. 2012;148:197–202. doi: 10.1001/archdermatol.2011.926. [DOI] [PubMed] [Google Scholar]
- 28.Boye E, Olsen BR. Signaling mechanisms in infantile hemangioma. Curr Opin Hematol. 2009;16:202–208. doi: 10.1097/MOH.0b013e32832a07ff. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Grimmer JF, Williams MS, Pimentel R, Mineau G, Wood GM, Bayrak-Toydemir P, Stevenson DA. Familial clustering of hemangiomas. Arch Otolaryngol Head Neck Surg. 2011;137:757–760. doi: 10.1001/archoto.2011.91. [DOI] [PubMed] [Google Scholar]
- 30.Khan ZA, Boscolo E, Picard A, Psutka S, Melero-Martin JM, Bartch TC, Mulliken JB, Bischoff J. Multipotential stem cells recapitulate human infantile hemangioma in immunodeficient mice. J Clin Invest. 2008;118:2592–2599. doi: 10.1172/JCI33493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Calicchio ML, Collins T, Kozakewich HP. Identification of signaling systems in proliferating and involuting phase infantile hemangiomas by genome-wide transcriptional profiling. Am J Pathol. 2009;174:1638–1649. doi: 10.2353/ajpath.2009.080517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Iaccarino G, Ciccarelli M, Sorriento D, Galasso G, Campanile A, Santulli G, Cipolletta E, Cerullo V, Cimini V, Altobelli GG, et al. Ischemic neoangiogenesis enhanced by beta2-adrenergic receptor overexpression: a novel role for the endothelial adrenergic system. Circ Res. 2005;97:1182–1189. doi: 10.1161/01.RES.0000191541.06788.bb. [DOI] [PubMed] [Google Scholar]
- 33.Storch CH, Hoeger PH. Propranolol for infantile haemangiomas: insights into the molecular mechanisms of action. Br J Dermatol. 2010;163:269–274. doi: 10.1111/j.1365-2133.2010.09848.x. [DOI] [PubMed] [Google Scholar]
- 34.Fredriksson JM, Lindquist JM, Bronnikov GE, Nedergaard J. Norepinephrine induces vascular endothelial growth factor gene expression in brown adipocytes through a beta-adrenoreceptor/cAMP/protein kinase A pathway involving Src but independently of Erk1/2. J Biol Chem. 2000;275:13802–13811. doi: 10.1074/jbc.275.18.13802. [DOI] [PubMed] [Google Scholar]
- 35.Taylor MR. Pharmacogenetics of the human beta-adrenergic receptors. Pharmacogenomics J. 2007;7:29–37. doi: 10.1038/sj.tpj.6500393. [DOI] [PubMed] [Google Scholar]
- 36.Rohrer DK, Chruscinski A, Schauble EH, Bernstein D, Kobilka BK. Cardiovascular and metabolic alterations in mice lacking both beta1- and beta2-adrenergic receptors. J Biol Chem. 1999;274:16701–16708. doi: 10.1074/jbc.274.24.16701. [DOI] [PubMed] [Google Scholar]
- 37.Krief S, Lönnqvist F, Raimbault S, Baude B, Van Spronsen A, Arner P, Strosberg AD, Ricquier D, Emorine LJ. Tissue distribution of beta 3-adrenergic receptor mRNA in man. J Clin Invest. 1993;91:344–349. doi: 10.1172/JCI116191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yuan SM, Jiang HQ, Ouyang TX, Xing X. The distribution and evolution of pericytes in infantile hemangioma. Zhonghua Zheng Xing Wai Ke Za Zhi. 2007;23:322–324. (In Chinese). [PubMed] [Google Scholar]
- 39.Ferrari-Dileo G, Davis EB, Anderson DR. Effects of cholinergic and adrenergic agonists on adenylate cyclase activity of retinal microvascular pericytes in culture. Invest Ophthalmol Vis Sci. 1992;33:42–47. [PubMed] [Google Scholar]
- 40.Siegfried EC, Keenan WJ, Al-Jureidini S. More on propranolol for hemangiomas of infancy. N Engl J Med. 359:2846. doi: 10.1056/NEJMc086443. author reply 2846–2847, 2008. [DOI] [PubMed] [Google Scholar]
- 41.Thaker PH, Han LY, Kamat AA, Arevalo JM, Takahashi R, Lu C, Jennings NB, Armaiz-Pena G, Bankson JA, Ravoori M, et al. Chronic stress promotes tumor growth and angiogenesis in a mouse model of ovarian carcinoma. Nat Med. 2006;12:939–944. doi: 10.1038/nm1447. [DOI] [PubMed] [Google Scholar]
- 42.Yang H, Xu Z, Iuvone PM, Grossniklaus HE. Angiostatin decreases cell migration and vascular endothelium growth factor (VEGF) to pigment epithelium derived factor (PEDF) RNA ratio in vitro and in a murine ocular melanoma model. Mol Vis. 2006;12:511–517. [PubMed] [Google Scholar]
- 43.Schieven GL. The biology of p38 kinase: a central role in inflammation. Curr Top Med Chem. 2005;5:921–928. doi: 10.2174/1568026054985902. [DOI] [PubMed] [Google Scholar]
- 44.Bamburg JR, Wiggan OP. ADF/cofilin and actin dynamics in disease. Trends Cell Biol. 2002;12:598–605. doi: 10.1016/s0962-8924(02)02404-2. [DOI] [PubMed] [Google Scholar]
- 45.Arber S, Barbayannis FA, Hanser H, Schneider C, Stanyon CA, Bernard O, Caroni P. Regulation of actin dynamics through phosphorylation of cofilin by LIM-kinase. Nature. 1998;393:805–809. doi: 10.1038/31729. [DOI] [PubMed] [Google Scholar]
- 46.Street CA, Bryan BA. Rho kinase proteins – pleiotropic modulators of cell survival and apoptosis. Anticancer Res. 2011;31:3645–3657. [PMC free article] [PubMed] [Google Scholar]
- 47.De Giorgi V, Gandini S, Grazzini M, Benemei S, Marchionni N, Geppetti P. Beta-blockers: a new and emerging treatment for melanoma. Recenti Prog Med. 2012;103:11–16. doi: 10.1701/1022.11152. (In Italian). [DOI] [PubMed] [Google Scholar]
- 48.Lin X, Luo K, Lv Z, Huang J. Beta-adrenoceptor action on pancreatic cancer cell proliferation and tumor growth in mice. Hepatogastroenterology. 2012;59:584–588. doi: 10.5754/hge11271. [DOI] [PubMed] [Google Scholar]
- 49.Jansen L, Below J, Chang-Claude J, Brenner H, Hoffmeister M. Beta blocker use and colorectal cancer risk: Population-based case-control study. Cancer. 2012 May 14; doi: 10.1002/cncr.26727. (Epub ahead of print). [DOI] [PubMed] [Google Scholar]
- 50.Pérez Piñero C, Bruzzone A, Sarappa MG, Castillo LF, Lüthy IA. Involvement of alpha2- and beta2-adrenoceptors on breast cancer cell proliferation and tumour growth regulation. Br J Pharmacol. 2012;166:721–736. doi: 10.1111/j.1476-5381.2011.01791.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chambers CB, Katowitz WR, Katowitz JA, Binenbaum G. A controlled study of topical 0.25% timolol maleate gel for the treatment of cutaneous infantile capillary hemangiomas. Ophthal Plast Reconstr Surg. 2012;28:103–106. doi: 10.1097/IOP.0b013e31823bfffb. [DOI] [PubMed] [Google Scholar]