

Abstract
Prevailing approaches to manage autoimmune thrombotic disorders, such as heparin-induced thrombocytopenia, antiphospholipid syndrome and thrombotic thrombocytopenic purpura, include immunosuppression and systemic anticoagulation, though neither provides optimal outcome for many patients. A different approach is suggested by the concurrence of autoantibodies and their antigenic targets in the absence of clinical disease, such as platelet factor 4 in heparin-induced thrombocytopenia and β2-glycoprotein-I (β2GPI) in antiphospholipid syndrome. The presence of autoantibodies in the absence of disease suggests that conformational changes or other alterations in endogenous protein autoantigens are required for recognition by pathogenic autoantibodies. In thrombotic thrombocytopenic purpura, the clinical impact of ADAMTS13 deficiency caused by autoantibodies likely depends on the balance between residual antigen, that is, enzyme activity, and demand imposed by local genesis of ultralarge multimers of von Willebrand factor. A corollary of these concepts is that disrupting platelet factor 4 and β2GPI conformation (or ultralarge multimer of von Willebrand factor oligomerization or function) might provide a disease-targeted approach to prevent thrombosis without systemic anticoagulation or immunosuppression. Validation of this approach requires a deeper understanding of how seemingly normal host proteins become antigenic or undergo changes that increase antibody avidity, and how they can be altered to retain adaptive functions while shedding epitopes prone to elicit harmful autoimmunity.
Introduction
Autoantibody-mediated hematologic disorders are common and diverse, targeting many cell types and coagulation proteins and leading to complications that can include thrombosis, bleeding, infection, or vasculitis. There is expanding understanding of host defects that permit autoantibodies to emerge, characteristics of autoantibodies that promote disease development, and interventions that impede autoantibody production. Although immunosuppression would, in theory, provide the most rational way to address the autoimmune response, such an approach, even if targeted to errant disease-specific B-cell clones, would not often provide therapeutic effects efficacy on a time scale necessary to alleviate acute and potentially life-threatening complications, such as thrombosis.
For the 3 thrombotic disorders considered here, heparin-induced thrombocytopenia (HIT), antiphospholipid syndrome (APS), and thrombotic thrombocytopenic purpura (TTP), there is compelling evidence that autoantibodies are pathogenic, but these same autoantibodies may also circulate in the absence of clinical disease. In each disorder, thrombosis occurs episodically and shows regional predilections not entirely explained by circulating antibody titer or antigen specificity. This suggests that, although the autoantibodies are necessary, they are not sufficient for disease expression and additional factors modify the structure of endogenous proteins to induce or enhance recognition by autoantibody in the case of HIT and APS or that modulate effects downstream of autoantibody engagement with its cognate autoantigen in TTP.
Moreover, these disorders share clinical features, including acute onset of thrombosis not readily amenable to immunosuppression and variable response to anticoagulation. Therefore, although this model adds an additional layer of complexity to our understanding of pathogenesis, identifying and characterizing these processes may provide new therapeutic opportunities. Agents that antagonize the structural reorganization of endogenous protein autoantigens in HIT and APS or act on the substrate of the autoantigen in TTP, ultralarge multimers of von Willebrand factor (ULVWF), processes we refer to as “antigen” or “substrate withdrawal,” respectively, might lead to new, highly targeted approaches to prevent or treat these 3 acute-onset autoimmune thrombotic disorders and might mitigate current reliance on nonspecific immunosuppression or systemic anticoagulation.
Antigen withdrawal: is there precedence?
Before addressing the 3 thrombotic disorders HIT, APS, and TTP, we first asked whether there is precedence for the hypothesis that the onset and duration of certain autoantibody-mediated disorders are regulated at the level of antigen expression and organization. We found that the most straightforward support for this concept comes from the effect of antigen withdrawal in several disorders characterized by antibody-mediated thrombocytopenia.
Immune thrombocytopenia induced by drugs
The concept of antigen withdrawal is most clearly exemplified by the various ways in which self-reactive anti-platelet antibodies are induced by drugs or their metabolites.1 In the case of abciximab, pathogenic antibodies are directed to the murine component of the chimeric Fab fragment. Drugs such as penicillin elicit self-reactive antibodies by binding to host cell proteins, whereas others (eg, glycoprotein IIb/IIIa antagonists) might expose immunogenic epitopes in native proteins to preexisting antibodies or induce new epitopes causing delayed onset of disease. It is theorized that some drug-induced antibodies (eg, quinine, quinidine) arise from a preexisting population of low-affinity antibodies that recognize both the drug and its target but only bind with high affinity when both are present. The drug contains elements that mirror both the antigenic site and the complementarity determining regions of the Fab, serving as a bridge that improves “fit.” The relevance of these findings is that drug-induced thrombocytopenia typically resolves soon after the drug is discontinued, although the antibodies may persist for years and cause rapid onset of disease on reexposure to antigen (eg, quinine). Less frequently, the autoantibodies bind to platelets in a drug-independent manner (eg, alemtuzimab2) but still only persist for weeks to months.
Immune thrombocytopenia induced by vaccines and microbes
A second example of antigen withdrawal leading to resolution of clinical disease comes from the experience with thrombocytopenia after vaccination or infection. Acute thrombocytopenia develops within 42 days after mumps-measles-rubella vaccination in children,3 with an estimated incidence of one case per 40 000 doses. Antiplatelet glycoprotein IIb/IIIa antibodies have been documented and patients respond to treatment used for primary immune thrombocytopenia (ITP). Importantly, thrombocytopenia is self-limiting in 80%-90% of cases and typically resolves within 6 weeks. “ITP” may also develop after infection with Helicobacter pylori, HIV, hepatitis C, and probably others (eg, in children).4 Antimicrobial antibodies that cross-react with glycoprotein IIb/IIIa or uncharacterized platelet antigens have been identified. Most patients with HIV-related ITP respond to reduction in viral load. In contrast, the response rate to eradication of H pylori vary regionally from ∼ 50% in Japan and Italy to ∼ 5% in native-born United States patients, for unknown reasons that may relate to the generation of microbe-independent antibodies, analogous to the evolution of MALT lymphoma.5 The responsiveness of ITP to eradication of hepatitis C or other infections is less well characterized.6 We posit that, upon “antigen withdrawal,” whether by discontinuation of the drug, dissipation of the antigen in the case of vaccination, or successful antimicrobial therapy, the impetus to generate platelet-directed memory B cells wanes and the normal host immune system resets to eliminate residual self-reactive antibody-producing cells.
We next asked whether this insight into the pathogenesis of antibody-mediated thrombocytopenia is also applicable to disorders characterized by antibody-mediated thrombosis in which a seemingly normal constitutive host protein in an otherwise seemingly immunologically normal host becomes a persistent autoantigen.
HIT: autoantigen oligomerization
HIT is an autoantibody-mediated thrombotic disorder that occurs in ∼ 1% of patients receiving at least 5 days of unfractionated heparin (UFH) in therapeutic doses, a lower proportion of those treated exclusively with low molecular weight heparin, and rarely after exposure to the anticoagulant pentasaccharide fondaparinux. HIT is caused by antibodies that recognize platelet factor 4 (PF4), a member of the CXC chemokine family, in a complex with heparin (H), other polysaccharides, or cellular glycosaminoglyans (GAGs). The reader is referred elsewhere for detailed reviews on the pathogenesis of thrombosis, diagnosis, and management of HIT.7,8
Here, we address the question of why the prevalence of antibodies to PF4/heparin (H) or GAGs after intense platelet activation and release of PF4, such as those undergoing coronary bypass or other major surgery, greatly exceeds the incidence of HIT.9 Antibody titer and IgG isotype contribute to clinical risk,10,11 but many exceptions have been reported and host response factors (eg, trauma, vascular function) may contribute as well. HIT antibodies bind to PF4/H or PF4/GAG complexes over a narrow molar range of reactants at which they activate platelets.12 Although there is emerging literature that epitope specificity contributes to risk,13 the focus here is on the antigen and how changes in its composition and recognition by “HIT antibodies” may contribute to pathogenesis without a change in the antibody itself.
PF4 is a 32-amino acid protein synthesized predominantly by megakaryocytes and stored in the α-granules of platelets bound to chondroitin sulfate side chains on serglycin. PF4 monomers exist in equilibrium with dimers and tetramers, with the latter likely to predominate within the granules based on estimated concentration. Polymorphisms and posttranslational modifications of PF4 have not been reported. It has been proposed that, when platelets are activated, they release PF4/chondroitin sulfate complexes. A proportion of these complexes bind to the cell surface through the chondroitin sulfate moiety. PF4 also binds extensively to endothelium and other hematopoietic cells that express GAGs with higher affinity for PF4, such as heparan sulfate.14
How does this help to explain the discrepancy between the incidence of seropositivity and clinical disease? Insight into this issue begins with the finding that PF4 and heparin form complexes that vary in size depending on the molar ratio of reactants.15 Binding of HIT antibody to PF4/H occurs over a relatively narrow range of molar ratios of reactants (∼ 2 molecules of PF4 to 1 molecule of UFH, assuming an average molecular weight of 15 kDa). The requirements for antigen induction have been analyzed systematically by incubating plasma containing HIT antibodies with PF4 and diverse, characterized polysaccharides. The results showed that the polysaccharide plays a “permissive role” in antigen formation: a gradation of antigenicity is imparted to PF4 by the polysaccharides that depends on chain length, chemical composition, and extent of branching.16 At the optimal antigenic ratio of reactants, PF4 and UFH form ultra-large complexes (ULCs) in solution (molecular weight > 670 kDa).17 The concentration of heparin required to form ULCs and the percent of total complexes they represent vary inversely with the length of the “heparin.” Fondaparinux does not form ULCs, which correlates with its low risk of causing HIT. ULCs bind multiple anti–PF4/H HIT-like antibodies per complex and are more potent inducers of platelet activation than are smaller complexes that contain an estimated 2 PF4 and 2 UFH molecules. ULCs are stable over 24 hours but are readily dissociated by small changes in heparin or PF4 concentration.17 The in vivo relevance of these findings is supported by the relationship between the concentration of exogenous PF4 and binding of anti-PF4/H antibodies to platelets and monocytes (wherein antibody binding increases with addition of PF4 until an optimal concentration is reached above which binding falls) and the expression level of endogenous PF4 and the development of thrombocytopenia and thrombosis in an animal model.12,14 The finding that activation of platelets and monocytes increases PF4 binding capacity may provide insight into why the risk of thrombosis is greater in settings, such as bypass surgery and trauma, among others, that are characterized by intense PF4 release, inflammation, and vascular injury.
The immunogenicity of PF4-heparin complexes in vivo correlates with their physical properties detected by measurements of dynamic light scattering and ζ-potential. Complexes enlarge through colloidal electrostatic interactions. Particle size peaks (> 2000 nm) at PF4/UFH ratios that minimize ζ-potential.18 However, immunogenicity is enhanced by a net cationic surface charge (higher PF4/UFH ratios) that may permit complexes to bind more efficiently to GAG-containing antigen-presenting cells.18 Analysis using atomic force microscopy shows that heparin approximates and aligns PF4 tetramers, which may enhance antibody binding.19 Chemical oligomerization of PF4 also increases the binding of a pathogenic HIT-like monoclonal antibody, KKO, but not an isotype-matched nonpathogenic antibody, RTO, that binds to PF4/H with the same Bmax by ELISA.13 This suggests that pathogenic antibodies may recognize an epitope clustered by heparin, which then increases their avidity and thereby helps offset their dissociation and dissipation by blood flow in vivo (Figure 1).
Figure 1.

Oligomerization of the HIT antigen. (A) PF4 and ULC formation and dissociation. PF4 tetramers exist in a dynamic equilibrium with dimers and monomers. Under normal circumstances, tetramerization is favored when PF4 is bound to cellular GAGs (right pointing arrow on left side of figure), leading to a high surface density of PF4 and a propensity to form oligomers that are capable of binding multiple HIT antibodies. Heparin promotes the formation of PF4 ULCs in solution and on cell surface GAGs, which clusters antibody as well. The addition of 2-O, 3-O desulfated heparin (ODSH) disrupts ULCs into smaller complexes that bind fewer antibodies (right side of figure). Similarly, PF4 antagonists impede PF4 tetramerization (upward pointing arrows on left side of figure), leading to a lower surface density of PF4 tetramers, less propensity to form ULCs, and fewer sites for antibody binding. (B) Schematic of pathogenic versus nonpathogenic antibody binding. Simplest model showing distinction between effects of heparin on binding of pathogenic (KKO) and nonpathogenic (RTO) anti-PF4 antibodies. Heparin (orange) binds to a circumferential band of cationic residues on the surface of each PF4 tetramer (blue); the interrupted line represents binding to the distal side of the tetramer. Heparin neutralizes cationic charge repulsion among PF4 tetramers forming oligomeric complexes (shown here as a dimer for simplicity), which approximates the binding sites for KKO (panel B 1A,1B). Epitope approximation increases the avidity of KKO through increased proximity to more than 1 binding site on PF4 (1B). Some KKO antibodies may bind to epitopes on neighboring tetramers stabilizing ULCs induced by heparin (1A). In contrast, heparin has no such effect or may partially inhibit exposure of the epitope recognized by RTO (2). (C) Disruption of PF4 tetramerization. Two PF4 dimers are shown as ribbon diagrams based on the published crystal structure. A PF4 antagonist (gray) is bound to the lower dimer (red/blue) preventing association with the upper dimer (purple/cyan).
Clustering of PF4/GAG/IgG on cell surfaces may sustain engagement of FcRγIIA on platelets,20 signal through FcRγI on monocytes,21 and inhibit PF4-mediated activation of protein C22 and possibly stimulate prothrombotic pathways involving endothelium, monocytes, and other cell types.23 These properties may differ from antibodies associated with other autoimmune, alloimmune, or drug-induced thrombocytopenias that lead predominantly to platelet opsonization followed by accelerated clearance.
A corollary to these findings is the prediction that disrupting PF4 oligomerization will reduce antibody avidity and prothrombotic sequelae by impeding or disrupting formation of pathogenic ULCs. Several lines of evidence support this concept. First, increasing concentrations of UFH in vitro and in vivo12,14,17 or the addition of undersulfated, nonanticoagulant heparin dissociate H/PF4 complexes, reduce antigenicity, and displace PF4 from cells or cell-like surfaces.24,25 Second, a PF4 variant (PF4K50E) with a mutation at the dimer-dimer interface binds heparin comparably to wild-type PF4 but does not tetramerize spontaneously, forms few ULCs in the presence of heparin, and is poorly recognized by HIT antibodies over a wide range of UFH concentrations.17 Third, the crystal structure of the AB/CD dimer interface has been used to design small molecule inhibitors of tetramer formation. These PF4 antagonists prevent and reverse oligomerization of wild-type PF4 by heparin, inhibit HIT antibody binding, and block activation of platelets and a PF4/heparin/FcγIIA-dependent cell line by HIT antibodies26 (Figure 1).
APS: antigen conformation
The APS is characterized by arterial and/or venous thrombosis, unexplained recurrent abortions or fetal loss, and persistent positive tests for a lupus anticoagulant or anticardiolipin or anti–β2-glycoprotein I (β2GPI) antibodies.27 Treatment is predicated on systemic anticoagulation and secondary thromboprophylaxis.28 Immunosuppression is of unproven benefit, suggesting a need for novel, targeted intervention strategies. The clinical presentation, diagnosis, management, and diversity of antigenic targets have been reviewed elsewhere.29–32
Here we consider how antiphospholipid antibodies (APLAs) and their cognate antigens are found in plasma both in the presence and in the absence of disease. Although some APLAs recognize prothrombin or other proteins alone or in complex with phospholipid, we will focus on the phospholipid binding protein β2GPI, the most prevalent target of APLAs31 and the one best documented to be associated with thrombosis.33–35 We propose that enhanced exposure of antigenic regions in β2GPI underlies recognition by pathogenic anti-β2GPI antibodies and that preventing these changes might provide an unexplored approach to intervention in APS.
β2GPI is an abundant plasma protein (0.2 mg/mL), consisting of 326 amino acids within 4 typical and one atypical short consensus repeat modules. The atypical fifth short consensus repeat is positively charged, containing 18% lysine residues that mediate binding to phospholipid and anionic surfaces, such as DNA and heparin, through the 281-CKNKEKKC-288 sequence.36 β2GPI also binds to diverse cellular proteins, although the structural basis for binding has not been well defined.37–39 Crystallization of β2GPI reveals that the 5 modules assume a fishhook-shaped structure with individual short consensus repeats arranged as beads on a string.40,41 The 4 N-terminal modules form a slight right-handed spiral that joins the atypical fifth domain to form the tip of the hook. β2GPI contains 3 potential N-linked glycosylation sites in domain 3 (Asn143, Asn164, Asn174) and a fourth in domain 4, which contributes to the microheterogeneity of plasma β2GPI (Figure 2).
Figure 2.

Exposure of the APS antigen. The APLA epitope in β2GPI domain 1, composed at least in part by Arg39-Arg43, may become exposed and recognized by anti-β2GPI antibodies. Circulating β2GPI to be largely in a circular conformation (top left), in which this epitope (red dot) is not exposed. Shielding of this epitope may result from interactions between domain 1 (DI) and domain 5 (DV), or possibly by steric effects of carbohydrate residues originating from domains 3 (DIII) and 4 (DIV) (represented by the blue triangle). (A) Depicts “unfolding” of circular β2GPI to a fishhook-like shape after binding to anionic phospholipid. Binding to this surface is mediated by domain 5 and results in exposure of the domain 1 epitope. Binding of bivalent anti-β2GPI antibodies to the exposed epitope may then promote functional β2GPI dimerization. However, to stimulate unactivated cells, this complex would need to dissociate from phospholipid and subsequently bind cellular receptors. (B) Depicts the proposed effect of direct binding of β2GPI to putative cellular receptors (annexin A2, apoER2, GPIb; green semicircles). Subsequent β2GPI unfolding and cross-linking by anti-β2GPI antibodies may activate cells directly through receptor oligomerization. (C) Shows how binding of β2GPI to cellular receptors may lead to unfolding and subsequent cross-linking by PF4 tetramers depicted in blue. Cross-linking via PF4 might directly activate cells or facilitate the ability of anti-β2GPI antibodies to cross-link β2GPI. (D) Shows how partial epitope exposure may be induced by β2GPI deglycosylation, oxidation, or interactions with proteins and/or proteases derived from infectious agents. Subsequent binding of anti-β2GPI anti-bodies may then occur coincident with binding of β2GPI to cellular receptors. Antibody binding may stabilize and promote the unfolded conformation of β2GPI.
Antigenic sites in β2GPI have been mapped using human anti-β2GPI antibodies from persons with or without clinical manifestations of APS. Although antibodies may bind to each of the 5 domains, site-directed mutagenesis reveals that those found in patients with APS react primarily with amino acids 40-43 within domain 1.42,43 This site lies within a more complex, nonlinear epitope that involves R39-R43, D8, and D9 in domain 1 and the domain I-domain II interlinker region.44 Antibodies reactive with domain I correlate more closely with the presence of a lupus anticoagulant and a history of thrombosis than do those that bind exclusively to other parts of the molecule (odds ratio = 18.9; 95% CI, 6.8-53.2 vs 1.1; 95% CI, 0.4-2.8, respectively).34 The finding that recombinant β2GPI domain 1 blocked enhancement of femoral vein thrombosis in mice given APS IgG45 supports the pathophysiologic relevance of this region and provides a template to develop inhibitors of antibody binding.
In view of these findings, why are circulating antigen-antibody complexes not detected in all persons with anti-β2GPI antibodies,46 and why are most patients asymptomatic at most times and at most sites although the antigen and antibody are both abundant in plasma? One potential explanation is that the relevant epitopes are normally encrypted and that exposure requires a conformational or biochemical change in β2GPI in vivo before it is recognized by antibody. An alternative explanation is that antibodies only bind with clinically relevant avidity when β2GPI is deposited on an appropriate biologic surface (cell surface phospholipid or protein receptor) at sufficiently high density to engage both of its Fab arms.47 Three nonmutually exclusive mechanisms by which cryptic epitopes on β2GPI might be induced or exposed have been proposed.
De-encryption may involve a dynamic global conformational change in β2GPI structure.48 For example, plasma β2GPI is found predominantly in a circular or “closed” form, which it is thought is maintained by interactions between domains 1 and 548 (Figure 2). This circular conformation can be converted to the fishhook-like “open” structure, similar to that identified by crystallography. APS antibodies recognize only the open conformation of purified β2GPI, although both conformations are recognized on incubation with cardiolipin-coated surfaces.48 It has been proposed that binding of β2GPI to anionic phospholipids or receptors exposed on the surface of activated or apoptotic cells converts the circular form found in plasma to the open conformation, although this has not been demonstrated directly. It has also been proposed that β2GPI undergoes a conformational change when it binds to Streptococcus pyogenes protein H,49 leading to the development of APLA. Other antibodies may arise from molecular mimicry. The Val247/Val247 β2GPI genotype, which is more prevalent in patients with APS,50 may alter the tertiary structure of β2GPI either by impeding this interaction between domains IV and V or another mechanism that reduces the stability of the closed conformation.
De-encryption may also result from removing a protective shield provided by carbohydrates in domain 1.51 A subset of human anti-β2GPI antibodies that correlate with a clinical history of thrombosis bind to plasma β2GPI more avidly after it has been immobilized on a hydrophilic than on a hydrophobic surface, which may shift the orientation of carbohydrate residues. This interpretation is supported by the observation that this same subset of antibodies binds deglycosylated plasma β2GPI in a manner identical to recombinant β2GPI, which lacks carbohydrate.51 However, the role of carbohydrates in epitope masking remains unproven and may not fully explain either the circular conformation of β2GPI found in plasma or the difference in the pattern of proteolytic digestion products between the open and closed conformations of the molecule.48
Third, β2GPI may be altered by oxidation. Some APLAs recognize phospholipids that have undergone oxidation (eg, oxidized LDL).52 β2GPI can also be oxidized directly.53 Approximately 80% of circulating plasma β2GPI is maintained in the reduced form by platelet and endothelial cell oxidoreductases,54,55 which may protect cells from hydrogen peroxide–induced apoptosis.56 Free cysteines in reduced β2GPI, particularly Cys288 and Cys326, are susceptible to oxidation and nitrosylation as is the Cys32-Cys60 disulfide bond in domain 1, which spans the primary antigenic region.56 Oxidation increases the affinity of polyclonal rabbit and murine monoclonal antibodies for β2GPI,53 but its effect on patient-derived antibodies requires additional study.
As mentioned, APS is associated with oxidative stress.57,58 The clinical relevance of oxidation as a therapeutic target is supported by the observation that the total amount and proportion of β2GPI that is oxidized are increased in patients with APS and a history of thrombosis compared with healthy controls or persons with thrombosis who do not have APS.54 In vitro, some statin drugs that possess antioxidant activity inhibit endothelial cell activation by APLA,59 and preincubation of monocytes with coenzyme Q10 decreases APLA-induced oxidative stress and mitochondrial dysfunction.58 In a mouse model of ischemia-reperfusion injury to mesenteric vessels associated with oxidative stress, deposition of β2GPI and anti-β2GPI antibodies at the ischemic site exacerbated tissue injury. Tissue damage was attenuated by peptides corresponding to the lysine-rich lipid-binding regions in β2GPI domain 5 that may block its binding to tissues.60 Moreover, the antioxidant hydroxycholoroquine inhibits β2GPI–anti-β2GPI immune complexes from disrupting the annexin V shield formed over exposed phospholipids,61 although its effect on β2GPI conformation or epitope exposure is uncertain.
Fourth, the conformation of β2GPI may be altered by interaction with proteins released in response to cell injury. Artificially dimerized β2GPI stimulates platelet adhesion.62 Although dimers or oligomers have yet to be detected in plasma, they might form on cell surfaces under specific conditions. For example, β2GPI dimerizes when it binds to PF4,63 which is released when platelets are activated. Modeling strategies predict that one β2GPI molecule binds through anionic patches within the “fishhook” region to the A and C chains of one PF4 tetramer, whereas a second β2GPI binds to the B and D chains of the tetramer63 (Figures 1C and 2). PF4, which binds primarily to cellular GAGs, might function as a pathophysiologic inducer of β2GPI dimerization, enhancing its avidity for cellular receptors or exposed phospholipid at sites of inflammation or vascular injury.63 It would be of interest to determine whether inhibitors of PF4 tetramerization13,24,25 or its contact sites on β2GPI might reduce anti-β2GPI binding. Annexin A2,37 apoER2,62 GPIb,39 and other cell surface receptors for β2GPI might provide additional potential targets for intervention. It is unclear which, if any, of these molecular partners induces conformational changes in β2GPI that promote binding of anti-β2GPI antibody and initiate intracellular signaling.
Lastly, cell activation or damage by anti-β2GPI antibodies in some experiments is amplified by complement.64 Incorporation of complement components into β2GPI-containing immune complexes may also stabilize their binding to cell surfaces, which may in turn recruit additional antibodies and provide a strong stimulatory signal to B cells. Complement activation products are elevated in patients with APLAs,64 although the correlation with thrombosis is not firmly established. Successful treatment of catastrophic APS with the anti-C5 antibody, eculizimab, has been reported.65 The emergence of low molecular weight inhibitors of C3 may offer alternative approaches to treatment.
TTP: autoantigen function
TTP is a potentially fatal syndrome characterized by profound thrombocytopenia, microangiopathic hemolytic anemia, and varying degrees of organ dysfunction.66 Although a few cases are associated with inherited mutations of the ADAMTS13 gene,67 most are caused by acquired autoantibodies that inhibit plasma ADAMTS13 activity.68,69 This compromises proteolytic processing of VWF and leads to persistence of ULVWF, exaggerated platelet aggregation, and thrombus formation in small arteries and capillaries. Treatment of TTP is predicated on removing antibody by plasma exchange, creating antigen excess or interfering with antibody production to prevent relapse. Here we explore the possibility of interfering with antibody binding by changing the structure of ADAMTS13 or by inhibiting the interaction of uncleaved ULVWF with platelets, analogous to inhibiting thrombin generated by autoantibodies in HIT and APS.
Similar to HIT and APS, in which autoantibodies appear to be necessary but not sufficient for disease expression, anti-ADAMTS13 inhibitory autoantibodies may be present during disease remission. This suggests that the clinical impact of decreased ADAMTS13 depends on additional factors that affect the balance between antigen, reflected in enzyme activity, and the amount of substrate that needs to be processed. In addition, ADAMTS13 inhibitory autoantibodies are routinely assayed using recombinant ADAMTS13 or ADAMTS13 derived from the plasma of healthy persons. Neo-autoepitope formation, as postulated for HIT and APS, has not been shown to play a role in TTP, although the possibility that binding of ADAMTS13 to ULVWF enhances autoantibody affinity has not been excluded.
VWF is a glycoprotein produced in endothelial cells and megakaryocytes that consists of a 2-40 subunit polypeptide linked by disulfide bridges. Each 250 kDa subunit is capable of simultaneous or sequential binding to collagen, platelet receptors GPIb and GPIIb/IIIa, and factor VIII through distinct sites. Upon stimulation, endothelial cells release ULVWF, which forms “string-like” polymers that remain anchored on cell membranes via an unknown mechanism.70–72 Plasma ADAMTS13 rapidly and efficiently cleaves cell-bound ULVWF and ULVWF/VWF after Tyr1605 in the central A2 domain in the presence71 or absence of flow.73 Released ULVWF multimers undergo further degradation by plasma ADAMTS13 in the microvasculature under high shear74 (Figure 3A). Cleavage is accelerated when VWF binds to physiologic cofactors, such as factor VIII and platelets.75,76
Figure 3.
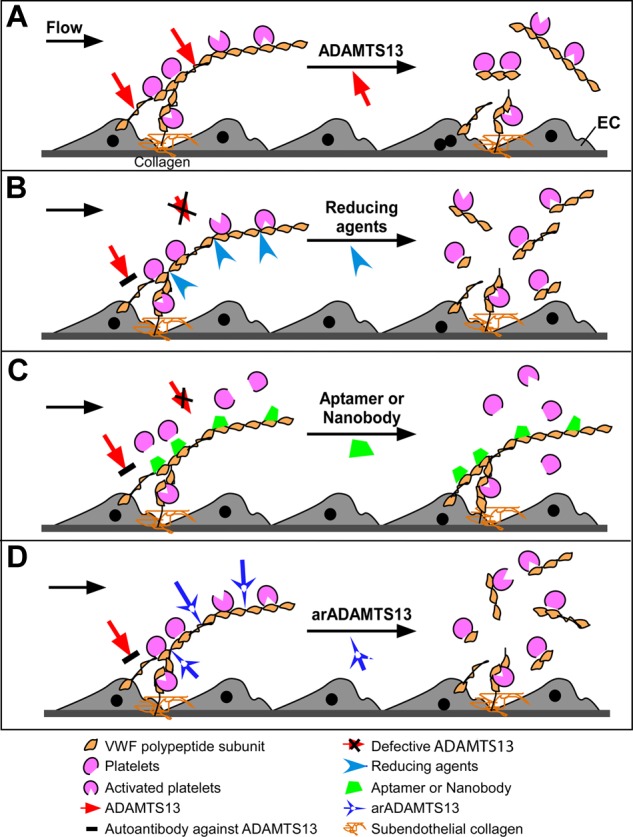
Nonimmunosuppression strategies to treat autoantibody-mediated TTP. VWF is secreted from activated endothelial cells as ULVWF multimers, which recruit flowing platelets and form thrombi. (A) In the presence of flow and/or platelet binding, VWF is stretched to expose its A2 domain that is cleaved by ADAMTS13, resulting in dissociation of platelet aggregates. (B) In the absence of ADAMTS13 activity because of hereditary mutations of ADAMTS13 or acquired autoantibodies that block ADAMTS13 function, a disulfide bond reducing agent, such as N-acetylcysteine, or the C-terminal TSP1 repeats of ADAMTS1399,100 may be able to reduce the disulfide bridges linking VWF polypeptide subunits disassembling the VWF multimers. (C) In the setting of insufficient ADAMTS13 activity as described in panel B, aptamer or nanobody may bind to the VWF-A1 domain, inhibit the interaction between ULVWF and platelet receptor GPIb, and eliminate thrombus formation under flow. (D) Anti-ADAMTS13 inhibitory autoantibodies may be bypassed through the use of novel recombinant ADAMTS13 preparations that have been engineered to retain VWF cleaving activity while deleting autoantibody-binding epitopes.
The imbalance between plasma levels of ADAMTS13 and ULVWF may contribute to the pathogenesis of arterial thrombosis. Inflammatory cytokines may enhance antibody production by promoting the addition of N-linked oligosaccharides, which may accelerate endocytosis of ADAMTS13 through mannose receptors on dendritic cells and thereby enhance antigen presentation. Inflammation may also contribute to this imbalance, independent of an increase in ADAMTS13 inhibitory antibodies. For example, reactive oxygen species released from activated neutrophils can modify Met1606 (or less commonly Tyr1605) in VWF, inhibiting its cleavage by ADAMTS13.77 Inflammatory cytokines also trigger the release of ULVWF from the endothelium while concomitantly suppressing the synthesis of ADAMTS13 in the liver and endothelial cells.78
It has been proposed that the balance between ADAMTS13 activity and VWF can be restored, even in the presence of ADAMTS13 autoantibodies, using N-acetylcysteine, which reduces the VWF intra-A1 and intersubunit disulfide bonds, thereby altering the mean size and platelet adhesive function of VWF multimers in vitro and in vivo79 (Figure 3B). Alternatively, acute episodes of TTP might be attenuated by pharmacologic inhibition of the interaction of platelet GPIb with the A1 domain of VWF using an anti-VWF aptamer (eg, ARC1779)80,81 or nanobody (eg, ALX-0081)82–84 (Figure 3C). A continuous infusion of ARC1779 suppressed VWF activity and led to a rise in platelet counts in patients with hereditary TTP.84,85 ARC1779 and nanobody ALX-0081 appear to be well tolerated. However, their clinical efficacy in animal models or in humans with autoimmune TTP has not yet been reported.
Another potential approach to treat acute episodes of autoantibody-mediated TTP makes use of novel recombinant ADAMTS13 preparations that retain VWF cleavage activity but are unaffected by autoantibodies that inhibit native wild-type ADAMTS13 (Figure 3D). Development of such enzymes has been made possible by first characterizing the binding of autoantibodies to ADAMTS13 on a molecular level. Nearly all adults with acquired TTP harbor autoantibodies that recognize the spacer domain of ADAMTS13, particularly exosite 3 (Tyr659-Tyr665) and its surrounding residues (Arg568 and Phe592),86,87 even when antibodies to other domains are present.69 The surface/charged amino acid residues in exosite 3 may be essential for binding and proteolytic cleavage of VWF88,89 and are required to attenuate arterial thrombosis in vivo.86,89,90 Substitution of these residues with Ala dramatically reduces ADAMTS13 proteolytic activity and inhibition by autoantibodies.89 When a more subtle change of these residues in the exosite 3 and surrounding areas is introduced, the resulting ADAMTS13 variants exhibit normal or enhanced proteolytic activity. Importantly, some gain-of-function variants also acquire resistance to in vitro inhibition by anti-ADAMTS13 autoantibodies.86 We are currently evaluating the efficacy of these reengineered ADAMTS13 variants in a murine model of acquired TTP to assess their potential as short-term therapy.
Antigen modulation in other autoimmune disorders
The concurrence of autoantibodies and their autoantigenic targets in the apparent absence of disease is not unique to autoimmune thrombotic disorders. Patients treated for autoimmune hemolytic anemia may have residual red cells with a positive direct antiglobulin test and high titers of antiglobulin-reacting IgG in their plasma yet show no evidence of hemolysis. Similarly, antiplatelet autoantibodies may be found in patients with a history of idiopathic ITP during clinical remission. The lack of red cell or platelet destruction is not readily explained by waxing and waning of autoantigen expression or autoantibody titer or avidity of autoantibody but may be related to antibody modifications, such as galactosylation or fucosylation, which can influence effector function.91
Evidence for altered self-antigen structure is also not unique to the hematologic disorders. One of the most striking accounts of altered self-antigens involves the autoantibody reactivity to citrullinated peptides, which are a sensitive and specific serologic marker for rheumatoid arthritis.92 Conversion of arginine residues to citrulline by deimination is catalyzed by peptidylarginine deiminase type 4.93 The peptidylarginine deiminase type 4 enzyme acts on histones and, intriguingly, is essential for the release of neutrophil extracellular chromatin traps that capture pathogens.94 Patients with Felty syndrome, a form of rheumatoid arthritis that includes the additional features of splenomegaly and neutropenia, produce autoantibodies that preferentially bind to deiminated histones.94 Other examples of autoantigen modifications include oxidation (discussed above), phosphorylation (eg, B-cell epitopes in the La/SSB autoantigen),95 and epigenetic changes (eg, the presence of unmethylated CpG motifs enhancing the immunogenicity of DNA).96 Another mechanism by which autoantigens may exhibit altered antigenicity is through formation of immune complexes. For example, rheumatoid factor antibodies bind other antibodies, generating antiantibody complexes to a wide array of antigens. Similarly, anti-DNA antibodies, by binding to DNA, can be complexed with proteins that also bind to DNA. Formation of these intermolecular complexes can increase the avidity of self-antigen/antibody interactions while also potentially promoting epitope spreading by recruiting a diverse pool of T cells. Finally, the cellular context of autoantigen expression is also important. Several autoantigens that are associated with systemic lupus erythematosus can be expressed within apoptotic blebs,97 and circulating microparticles containing nuclear and cytosolic antigens may stimulate the immune system by simultaneously triggering several different classes of receptors within a highly localized space.98
In conclusion, we suggest that the variability in the clinical expression of HIT and APS can be explained in part by structural changes in endogenous PF4 and β2GPI that induce antigenic epitopes or cause the proteins to cluster. In patients with TTP, disease expression correlates with increased “demand” for limited residual autoantigen, ADAMTS13, by stress-induced increases in ULVWF. We posit that modifying the antigenic target in the settings of HIT and APLAs or modulating the downstream effects of autoantigen engagement in the context of TTP provides new therapeutic strategies to alleviate the acute, potentially life-threatening, consequences of systemic thrombosis while obviating the need for nonspecific immunosuppression or anticoagulation.
Note added in proof: The text states that the anti-thrombotic efficacy of targeting VWF in models of thrombosis had not been reported. Such findings are reported in the 120:13 issue of Blood by Navarette et al.101
Acknowledgments
This work was supported in part by grants from the National Institutes of Health (NIH HL110860, D.B.C. and BSS; grant P50HL081011, K.R.M.) and the American Heart Association (Established Investigator Award 0940100N), and grants from the NIH (HL074124, X.L.Z.; grant R56-AI090842, E.L.P.; and grant HLP50-HL81012, D.L.S.).
Authorship
Contribution: D.B.C., K.R.M., X.L.Z., R.S.S., E.L.P., and D.L.S. conceptualized and wrote the text and developed the figures and final manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Douglas B. Cines, Department of Pathology and Laboratory Medicine, Perelman School of Medicine, University of Pennsylvania, 513 A Stellar-Chance, 422 Curie Blvd, Philadelphia, PA 19104-4274; e-mail: dcines@mail.med.upenn.edu.
References
- 1.Aster RH, Bougie DW. Drug-induced immune thrombocytopenia. N Engl J Med. 2007;357(6):580–587. doi: 10.1056/NEJMra066469. [DOI] [PubMed] [Google Scholar]
- 2.Cuker A, Coles AJ, Sullivan H, et al. A distinctive form of immune thrombocytopenia in a phase 2 study of alemtuzumab for the treatment of relapsing-remitting multiple sclerosis. Blood. 2011;118(24):6299–6305. doi: 10.1182/blood-2011-08-371138. [DOI] [PubMed] [Google Scholar]
- 3.Mantadakis E, Farmaki E, Buchanan GR. Thrombocytopenic purpura after measles-mumps-rubella vaccination: a systematic review of the literature and guidance for management. J Pediatr. 2010;156(4):623–628. doi: 10.1016/j.jpeds.2009.10.015. [DOI] [PubMed] [Google Scholar]
- 4.Cines DB, Bussel JB, Liebman HA, Luning Prak ET. The ITP syndrome: pathogenic and clinical diversity. Blood. 2009;113(26):6511–6521. doi: 10.1182/blood-2009-01-129155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cines DB. ITP: time to bug off? Blood. 2007;110:3818–3819. [Google Scholar]
- 6.Cines DB, Liebman H, Stasi R. Pathobiology of secondary immune thrombocytopenia. Semin Hematol. 2009;46(1 Suppl 2):S2–S14. doi: 10.1053/j.seminhematol.2008.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arepally GM, Ortel TL. Heparin-induced thrombocytopenia. Annu Rev Med. 2010;61:77–90. doi: 10.1146/annurev.med.042808.171814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cuker A, Cines DB. How I treat heparin-induced thrombocytopenia. Blood. 2012;119(10):2209–2218. doi: 10.1182/blood-2011-11-376293. [DOI] [PubMed] [Google Scholar]
- 9.Bauer TL, Arepally G, Konkle BA, et al. Prevalence of heparin-associated antibodies without thrombosis in patients undergoing cardiopulmonary bypass surgery. Circulation. 1997;95(5):1242–1246. doi: 10.1161/01.cir.95.5.1242. [DOI] [PubMed] [Google Scholar]
- 10.Greinacher A, Ittermann T, Bagemuhl J, et al. Heparin-induced thrombocytopenia: towards standardization of platelet factor 4/heparin antigen tests. J Thromb Haemost. 2010;8(9):2025–2031. doi: 10.1111/j.1538-7836.2010.03974.x. [DOI] [PubMed] [Google Scholar]
- 11.Bakchoul T, Giptner A, Najaoui A, Bein G, Santoso S, Sachs UJ. Prospective evaluation of PF4/heparin immunoassays for the diagnosis of heparin-induced thrombocytopenia. J Thromb Haemost. 2009;7(8):1260–1265. doi: 10.1111/j.1538-7836.2009.03465.x. [DOI] [PubMed] [Google Scholar]
- 12.Rauova L, Zhai L, Kowalska MA, Arepally GM, Cines DB, Poncz M. Role of platelet surface PF4 antigenic complexes in heparin-induced thrombocytopenia pathogenesis: diagnostic and therapeutic implications. Blood. 2006;107:2346–2353. doi: 10.1182/blood-2005-08-3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sachais BS, Litvinov RI, Yarovoi SV, et al. Dynamic antibody binding properties in the pathogenesis of HIT. Blood. 2012;120(5):1137–1142. doi: 10.1182/blood-2012-01-407262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rauova L, Hirsch JD, Greene TK, et al. Monocyte-bound PF4 in the pathogenesis of heparin-induced thrombocytopenia. Blood. 2010;116(23):5021–5031. doi: 10.1182/blood-2010-03-276964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bock PE, Luscombe M, Marshall SE, Pepper DS, Holbrook JJ. The multiple complexes formed by the interaction of platelet factor 4 with heparin. Biochem J. 1980;191:769–776. doi: 10.1042/bj1910769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Greinacher A, Alban S, Dummel V, Franz G, Mueller-Eckhardt C. Characterization of the structural requirements for a carbohydrate based anticoagulant with a reduced risk of inducing the immunologic type of heparin-associated thrombocytopenia. Thromb Haemost. 1995;74:886–892. [PubMed] [Google Scholar]
- 17.Rauova L, Poncz M, McKenzie SE, et al. Ultralarge complexes of PF4 and heparin are central to the pathogenesis of heparin-induced thrombocytopenia. Blood. 2005;105(1):131–138. doi: 10.1182/blood-2004-04-1544. [DOI] [PubMed] [Google Scholar]
- 18.Suvarna S, Espinasse B, Qi R, et al. Determinants of PF4/heparin immunogenicity. Blood. 2007;110(13):4253–4260. doi: 10.1182/blood-2007-08-105098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Greinacher A, Gopinadhan M, Gunther JU, et al. Close approximation of two platelet factor 4 tetramers by charge neutralization forms the antigens recognized by HIT antibodies. Arterioscler Thromb Vasc Biol. 2006;26(10):2386–2393. doi: 10.1161/01.ATV.0000238350.89477.88. [DOI] [PubMed] [Google Scholar]
- 20.Kelton JG, Sheridan D, Santos A, et al. Heparin-induced thrombocytopenia: laboratory studies. Blood. 1988;72(3):925–930. [PubMed] [Google Scholar]
- 21.Kasthuri RS, Glover SL, Jonas W, et al. PF4/heparin-antibody complex induces monocyte tissue factor expression and release of tissue factor positive microparticles by activation of FcgammaRI. Blood. 2012;119(22):5285–5293. doi: 10.1182/blood-2011-06-359430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kowalska MA, Krishnaswamy S, Rauova L, et al. Antibodies associated with heparin-induced thrombocytopenia (HIT) inhibit activated protein C generation: new insights into the prothrombotic nature of HIT. Blood. 2011;118(10):2882–2888. doi: 10.1182/blood-2011-02-335208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cines DB, Tomaski A, Tannenbaum S. Immune endothelial cell injury in heparin-associated thrombocytopenia. N Engl J Med. 1987;316:581–589. doi: 10.1056/NEJM198703053161004. [DOI] [PubMed] [Google Scholar]
- 24.Krauel K, Hackbarth C, Furll B, Greinacher A. Heparin-induced thrombocytopenia: in vitro studies on the interaction of dabigatran, rivaroxaban, and low-sulfated heparin, with platelet factor 4 and anti-PF4/heparin antibodies. Blood. 2012;119(5):1248–1255. doi: 10.1182/blood-2011-05-353391. [DOI] [PubMed] [Google Scholar]
- 25.Joglekar MV, Quintana Diez PM, Marcus S, et al. Disruption of PF4/H multimolecular complex formation with a minimally anticoagulant heparin (ODSH). Thromb Haemost. 2012;107(4):717–725. doi: 10.1160/TH11-11-0795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sachais BS, Rux AH, Cines DB, et al. Rational design and characterization of platelet factor 4 antagonists for the study of heparin-induced thrombocytopenia. Blood. 2012;119(25):5955–5962. doi: 10.1182/blood-2012-01-406801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Miyakis S, Lockshin MD, Atsumi T, et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost. 2006;4(2):295–306. doi: 10.1111/j.1538-7836.2006.01753.x. [DOI] [PubMed] [Google Scholar]
- 28.Giannakopoulos B, Krilis SA. How I treat the antiphospholipid syndrome. Blood. 2009;114(10):2020–2030. doi: 10.1182/blood-2009-05-220756. [DOI] [PubMed] [Google Scholar]
- 29.Palomo I, Segovia F, Ortega C, Pierangeli S. Antiphospholipid syndrome: a comprehensive review of a complex and multisystemic disease. Clin Exp Rheumatol. 2009;27(4):668–677. [PubMed] [Google Scholar]
- 30.Giannakopoulos B, Passam F, Rahgozar S, Krilis SA. Current concepts on the pathogenesis of the antiphospholipid syndrome. Blood. 2007;109(2):422–430. doi: 10.1182/blood-2006-04-001206. [DOI] [PubMed] [Google Scholar]
- 31.de Groot PG, Urbanus RT. The significance of autoantibodies against beta2-glycoprotein I. Blood. 2012;120(2):266–274. doi: 10.1182/blood-2012-03-378646. [DOI] [PubMed] [Google Scholar]
- 32.Ruiz-Irastorza G, Crowther M, Branch W, Khamashta MA. Antiphospholipid syndrome. Lancet. 2010;376(9751):1498–1509. doi: 10.1016/S0140-6736(10)60709-X. [DOI] [PubMed] [Google Scholar]
- 33.Urbanus RT, Derksen RH, de Groot PG. Current insight into diagnostics and pathophysiology of the antiphospholipid syndrome. Blood Rev. 2008;22(2):93–105. doi: 10.1016/j.blre.2007.09.001. [DOI] [PubMed] [Google Scholar]
- 34.de Laat B, Pengo V, Pabinger I, et al. The association between circulating antibodies against domain I of beta2-glycoprotein I and thrombosis: an international multicenter study. J Thromb Haemost. 2009;7(11):1767–1773. doi: 10.1111/j.1538-7836.2009.03588.x. [DOI] [PubMed] [Google Scholar]
- 35.De Laat B, Derksen RH, Reber G, et al. An international multicentre-laboratory evaluation of a new assay to detect specifically lupus anticoagulants dependent on the presence of anti-beta2-glycoprotein autoantibodies. J Thromb Haemost. 2011;9(1):149–153. doi: 10.1111/j.1538-7836.2010.04068.x. [DOI] [PubMed] [Google Scholar]
- 36.Lozier J, Takahashi N, Putnam FW. Complete amino acid sequence of human plasma beta 2-glycoprotein I. Proc Natl Acad Sci U S A. 1984;81(12):3640–3644. doi: 10.1073/pnas.81.12.3640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ma K, Simantov R, Zhang JC, Silverstein R, Hajjar KA, McCrae KR. High affinity binding of beta 2-glycoprotein I to human endothelial cells is mediated by annexin II. J Biol Chem. 2000;275(20):15541–15548. doi: 10.1074/jbc.275.20.15541. [DOI] [PubMed] [Google Scholar]
- 38.Pennings MT, van Lummel M, Derksen RH, et al. Interaction of beta2-glycoprotein I with members of the low density lipoprotein receptor family. J Thromb Haemost. 2006;4(8):1680–1690. doi: 10.1111/j.1538-7836.2006.02036.x. [DOI] [PubMed] [Google Scholar]
- 39.Shi T, Giannakopoulos B, Yan X, et al. Anti-beta2-glycoprotein I antibodies in complex with beta2-glycoprotein I can activate platelets in a dysregulated manner via glycoprotein Ib-IX-V. Arthritis Rheum. 2006;54(8):2558–2567. doi: 10.1002/art.21968. [DOI] [PubMed] [Google Scholar]
- 40.Schwarzenbacher R, Zeth K, Diederichs K, et al. Crystal structure of human beta2-glycoprotein I: implications for phospholipid binding and the antiphospholipid syndrome. EMBO J. 1999;18(22):6228–6239. doi: 10.1093/emboj/18.22.6228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bouma B, de Groot PG, van den Elsen JM, et al. Adhesion mechanism of human beta(2)-glycoprotein I to phospholipids based on its crystal structure. EMBO J. 1999;18(19):5166–5174. doi: 10.1093/emboj/18.19.5166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reddel SW, Wang YX, Sheng YH, Krilis SA. Epitope studies with anti-beta 2-glycoprotein I antibodies from autoantibody and immunized sources. J Autoimmun. 2000;15(2):91–96. doi: 10.1006/jaut.2000.0427. [DOI] [PubMed] [Google Scholar]
- 43.Iverson GM, Reddel S, Victoria EJ, et al. Use of single point mutations in domain I of beta 2-glycoprotein I to determine fine antigenic specificity of antiphospholipid autoantibodies. J Immunol. 2002;169(12):7097–7103. doi: 10.4049/jimmunol.169.12.7097. [DOI] [PubMed] [Google Scholar]
- 44.Ioannou Y, Pericleous C, Giles I, Latchman DS, Isenberg DA, Rahman A. Binding of antiphospholipid antibodies to discontinuous epitopes on domain I of human beta(2)-glycoprotein I: mutation studies including residues R39 to R43. Arthritis Rheum. 2007;56(1):280–290. doi: 10.1002/art.22306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ioannou Y, Romay-Penabad Z, Pericleous C, et al. In vivo inhibition of antiphospholipid antibody-induced pathogenicity utilizing the antigenic target peptide domain I of beta2-glycoprotein I: proof of concept. J Thromb Haemost. 2009;7(5):833–842. doi: 10.1111/j.1538-7836.2009.03316.x. [DOI] [PubMed] [Google Scholar]
- 46.Giles IP, Isenberg DA, Latchman DS, Rahman A. How do antiphospholipid antibodies bind beta2-glycoprotein I? Arthritis Rheum. 2003;48(8):2111–2121. doi: 10.1002/art.11101. [DOI] [PubMed] [Google Scholar]
- 47.Roubey RA, Eisenberg RA, Harper MF, Winfield JB. “Anticardiolipin” autoantibodies recognize beta 2-glycoprotein I in the absence of phospholipid: importance of Ag density and bivalent binding. J Immunol. 1995;154(2):954–960. [PubMed] [Google Scholar]
- 48.Agar C, van Os GM, Morgelin M, et al. Beta2-glycoprotein I can exist in 2 conformations: implications for our understanding of the antiphospholipid syndrome. Blood. 2010;116(8):1336–1343. doi: 10.1182/blood-2009-12-260976. [DOI] [PubMed] [Google Scholar]
- 49.van Os GM, Meijers JC, Agar C, et al. Induction of anti-beta2 -glycoprotein I autoantibodies in mice by protein H of Streptococcus pyogenes. J Thromb Haemost. 2011;9(12):2447–2456. doi: 10.1111/j.1538-7836.2011.04532.x. [DOI] [PubMed] [Google Scholar]
- 50.Chamorro AJ, Marcos M, Miron-Canelo JA, Cervera R, Espinosa G. Val247Leu beta2-glycoprotein-I allelic variant is associated with antiphospholipid syndrome: systematic review and meta-analysis. Autoimmun Rev. 2012;11(10):705–712. doi: 10.1016/j.autrev.2011.12.006. [DOI] [PubMed] [Google Scholar]
- 51.de Laat B, Derksen RH, van Lummel M, Pennings MT, de Groot PG. Pathogenic anti-β2-glycoprotein I antibodies recognize domain I of beta2-glycoprotein I only after a conformational change. Blood. 2006;107(5):1916–1924. doi: 10.1182/blood-2005-05-1943. [DOI] [PubMed] [Google Scholar]
- 52.Horkko S, Miller E, Branch DW, Palinski W, Witztum JL. The epitopes for some antiphospholipid antibodies are adducts of oxidized phospholipid and beta2 glycoprotein 1 (and other proteins). Proc Natl Acad Sci U S A. 1997;94(19):10356–10361. doi: 10.1073/pnas.94.19.10356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Passam FH, Giannakopoulos B, Mirarabshahi P, Krilis SA. Molecular pathophysiology of the antiphospholipid syndrome: the role of oxidative post-translational modification of beta 2 glycoprotein I. J Thromb Haemost. 2011;9(Suppl 1):275–282. doi: 10.1111/j.1538-7836.2011.04301.x. [DOI] [PubMed] [Google Scholar]
- 54.Ioannou Y, Zhang JY, Qi M, et al. Novel assays of thrombogenic pathogenicity in the antiphospholipid syndrome based on the detection of molecular oxidative modification of the major autoantigen beta2-glycoprotein I. Arthritis Rheum. 2011;63(9):2774–2782. doi: 10.1002/art.30383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Passam FH, Rahgozar S, Qi M, et al. Beta2 glycoprotein I is a substrate of thiol oxidoreductases. Blood. 2010;116(11):1995–1997. doi: 10.1182/blood-2010-02-271494. [DOI] [PubMed] [Google Scholar]
- 56.Ioannou Y, Zhang JY, Passam FH, et al. Naturally occurring free thiols within beta 2-glycoprotein I in vivo: nitrosylation, redox modification by endothelial cells, and regulation of oxidative stress-induced cell injury. Blood. 2010;116(11):1961–1970. doi: 10.1182/blood-2009-04-215335. [DOI] [PubMed] [Google Scholar]
- 57.Iuliano L, Pratico D, Ferro D, et al. Enhanced lipid peroxidation in patients positive for antiphospholipid antibodies. Blood. 1997;90(10):3931–3935. [PubMed] [Google Scholar]
- 58.Perez-Sanchez C, Ruiz-Limon P, Aguirre MA, et al. Mitochondrial dysfunction in antiphospholipid syndrome: implications in the pathogenesis of the disease and effects of coenzyme Q10 treatment. Blood. 2012;119(24):5859–5870. doi: 10.1182/blood-2011-12-400986. [DOI] [PubMed] [Google Scholar]
- 59.Ferrara DE, Swerlick R, Casper K, et al. Fluvastatin inhibits up-regulation of tissue factor expression by antiphospholipid antibodies on endothelial cells. J Thromb Haemost. 2004;2(9):1558–1563. doi: 10.1111/j.1538-7836.2004.00896.x. [DOI] [PubMed] [Google Scholar]
- 60.Fleming SD, Pope MR, Hoffman SM, et al. Domain V peptides inhibit beta2-glycoprotein I-mediated mesenteric ischemia/reperfusion-induced tissue damage and inflammation. J Immunol. 2010;185(10):6168–6178. doi: 10.4049/jimmunol.1002520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rand JH, Wu XX, Quinn AS, et al. Hydroxychloroquine protects the annexin A5 anticoagulant shield from disruption by antiphospholipid antibodies: evidence for a novel effect for an old antimalarial drug. Blood. 2010;115(11):2292–2299. doi: 10.1182/blood-2009-04-213520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lutters BC, Derksen RH, Tekelenburg WL, Lenting PJ, Arnout J, de Groot PG. Dimers of beta 2-glycoprotein I increase platelet deposition to collagen via interaction with phospholipids and the apolipoprotein E receptor 2′. J Biol Chem. 2003;278(36):33831–33838. doi: 10.1074/jbc.M212655200. [DOI] [PubMed] [Google Scholar]
- 63.Sikara MP, Routsias JG, Samiotaki M, Panayotou G, Moutsopoulos HM, Vlachoyiannopoulos PG. Beta2 glycoprotein I (beta2GPI) binds platelet factor 4 (PF4): implications for the pathogenesis of antiphospholipid syndrome. Blood. 2010;115(3):713–723. doi: 10.1182/blood-2009-03-206367. [DOI] [PubMed] [Google Scholar]
- 64.Devreese KM, Hoylaerts MF. Is there an association between complement activation and antiphospholipid antibody-related thrombosis? Thromb Haemost. 2010;104(6):1279–1281. doi: 10.1160/TH10-06-0410. [DOI] [PubMed] [Google Scholar]
- 65.Shapira I, Andrade D, Allen SL, Salmon JE. Induction of durable remission in recurrent catastrophic antiphospholipid syndrome via inhibition of terminal complement with eculizumab. Arthritis Rheum. 2012;64(8):2719–2723. doi: 10.1002/art.34440. [DOI] [PubMed] [Google Scholar]
- 66.Bell WR, Braine HG, Ness PM, Kickler TS. Improved survival in thrombotic thrombocytopenic purpura-hemolytic uremic syndrome: clinical experience in 108 patients. N Engl J Med. 1991;325(6):398–403. doi: 10.1056/NEJM199108083250605. [DOI] [PubMed] [Google Scholar]
- 67.Levy GG, Nichols WC, Lian EC, et al. Mutations in a member of the ADAMTS gene family cause thrombotic thrombocytopenic purpura. Nature. 2001;413(6855):488–494. doi: 10.1038/35097008. [DOI] [PubMed] [Google Scholar]
- 68.Tsai HM, Lian EC. Antibodies to von Willebrand factor-cleaving protease in acute thrombotic thrombocytopenic purpura. N Engl J Med. 1998;339(22):1585–1594. doi: 10.1056/NEJM199811263392203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zheng XL, Wu HM, Shang D, et al. Multiple domains of ADAMTS13 are targeted by autoantibodies against ADAMTS13 in patients with acquired idiopathic thrombotic thrombocytopenic purpura. Haematologica. 2010;95(9):1555–1562. doi: 10.3324/haematol.2009.019299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bernardo A, Ball C, Nolasco L, Moake JF, Dong JF. Effects of inflammatory cytokines on the release and cleavage of the endothelial cell-derived ultralarge von Willebrand factor multimers under flow. Blood. 2004;104(1):100–106. doi: 10.1182/blood-2004-01-0107. [DOI] [PubMed] [Google Scholar]
- 71.Dong JF, Moake JL, Nolasco L, et al. ADAMTS-13 rapidly cleaves newly secreted ultralarge von Willebrand factor multimers on the endothelial surface under flowing conditions. Blood. 2002;100(12):4033–4039. doi: 10.1182/blood-2002-05-1401. [DOI] [PubMed] [Google Scholar]
- 72.Dong JF, Moake JL, Bernardo A, et al. ADAMTS-13 metalloprotease interacts with the endothelial cell-derived ultra-large von Willebrand factor. J Biol Chem. 2003;278(32):29633–29639. doi: 10.1074/jbc.M301385200. [DOI] [PubMed] [Google Scholar]
- 73.Jin SY, Skipwith CG, Shang D, Zheng XL. von Willebrand factor cleaved from endothelial cells by ADAMTS13 remains ultralarge in size. J Thromb Haemost. 2009;7(10):1749–1752. doi: 10.1111/j.1538-7836.2009.03570.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tsai HM. Shear stress and von Willebrand factor in health and disease. Semin Thromb Hemost. 2003;29(5):479–488. doi: 10.1055/s-2003-44556. [DOI] [PubMed] [Google Scholar]
- 75.Cao W, Krishnaswamy S, Camire RM, Lenting PJ, Zheng XL. Factor VIII accelerates proteolytic cleavage of von Willebrand factor by ADAMTS13. Proc Natl Acad Sci U S A. 2008;105(21):7416–7421. doi: 10.1073/pnas.0801735105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Skipwith CG, Cao W, Zheng XL. Factor VIII and platelets synergistically accelerate cleavage of von Willebrand factor by ADAMTS13 under fluid shear stress. J Biol Chem. 2010;285(37):28596–28603. doi: 10.1074/jbc.M110.131227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chen J, Fu X, Wang Y, et al. Oxidative modification of von Willebrand factor by neutrophil oxidants inhibits its cleavage by ADAMTS13. Blood. 2010;115(3):706–712. doi: 10.1182/blood-2009-03-213967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cao WJ, Niiya M, Zheng XW, Shang DZ, Zheng XL. Inflammatory cytokines inhibit ADAMTS13 synthesis in hepatic stellate cells and endothelial cells. J Thromb Haemost. 2008;6(7):1233–1235. doi: 10.1111/j.1538-7836.2008.02989.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chen J, Reheman A, Gushiken FC, et al. N-acetylcysteine reduces the size and activity of von Willebrand factor in human plasma and mice. J Clin Invest. 2011;121(2):593–603. doi: 10.1172/JCI41062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Spiel AO, Mayr FB, Ladani N, et al. The aptamer ARC1779 is a potent and specific inhibitor of von Willebrand factor mediated ex vivo platelet function in acute myocardial infarction. Platelets. 2009;20(5):334–340. doi: 10.1080/09537100903085927. [DOI] [PubMed] [Google Scholar]
- 81.Mayr FB, Knobl P, Jilma B, et al. The aptamer ARC1779 blocks von Willebrand factor-dependent platelet function in patients with thrombotic thrombocytopenic purpura ex vivo. Transfusion. 2010;50(5):1079–1087. doi: 10.1111/j.1537-2995.2009.02554.x. [DOI] [PubMed] [Google Scholar]
- 82.Ulrichts H, Silence K, Schoolmeester A, et al. Antithrombotic drug candidate ALX-0081 shows superior preclinical efficacy and safety compared with currently marketed antiplatelet drugs. Blood. 2011;118(3):757–765. doi: 10.1182/blood-2010-11-317859. [DOI] [PubMed] [Google Scholar]
- 83.van Loon JE, de Jaegere PP, Ulrichts H, et al. The in vitro effect of the new antithrombotic drug candidate ALX-0081 on blood samples of patients undergoing percutaneous coronary intervention. Thromb Haemost. 2011;106(1):165–171. doi: 10.1160/TH10-12-0804. [DOI] [PubMed] [Google Scholar]
- 84.Holz JB. The TITAN trial: assessing the efficacy and safety of an anti-von Willebrand factor Nanobody in patients with acquired thrombotic thrombocytopenic purpura. Transfus Apher Sci. 2012;46(3):343–346. doi: 10.1016/j.transci.2012.03.027. [DOI] [PubMed] [Google Scholar]
- 85.Jilma-Stohlawetz P, Gilbert JC, Gorczyca ME, Knobl P, Jilma B. A dose ranging phase I/II trial of the von Willebrand factor inhibiting aptamer ARC1779 in patients with congenital thrombotic thrombocytopenic purpura. Thromb Haemost. 2011;106(3):539–547. doi: 10.1160/TH11-02-0069. [DOI] [PubMed] [Google Scholar]
- 86.Jian C, Xiao J, Gong L, et al. Gain-of-function ADAMTS13 variants that are resistant to autoantibodies against ADAMTS13 in patients with acquired thrombotic thrombocytopenic purpura. Blood. 2012;119(16):3836–3843. doi: 10.1182/blood-2011-12-399501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Pos W, Sorvillo N, Fijnheer R, et al. Residues Arg568 and Phe592 contribute to an antigenic surface for anti-ADAMTS13 antibodies in the spacer domain. Haematologica. 2011;96(11):1670–1677. doi: 10.3324/haematol.2010.036327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Jin SY, Skipwith CG, Zheng XL. Amino acid residues Arg(659), Arg(660), and Tyr(661) in the spacer domain of ADAMTS13 are critical for cleavage of von Willebrand factor. Blood. 2010;115(11):2300–2310. doi: 10.1182/blood-2009-07-235101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Pos W, Crawley JT, Fijnheer R, Voorberg J, Lane DA, Luken BM. An autoantibody epitope comprising residues R660, Y661, and Y665 in the ADAMTS13 spacer domain identifies a binding site for the A2 domain of VWF. Blood. 2010;115(8):1640–1649. doi: 10.1182/blood-2009-06-229203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Xiao J, Jin SY, Xue J, Sorvillo N, Voorberg J, Zheng XL. Essential domains of a disintegrin and metalloprotease with thrombospondin type 1 repeats-13 metalloprotease required for modulation of arterial thrombosis. Arterioscler Thromb Vasc Biol. 2011;31(10):2261–2269. doi: 10.1161/ATVBAHA.111.229609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Peipp M, Lammerts van Bueren JJ, Schneider-Merck T, et al. Antibody fucosylation differentially impacts cytotoxicity mediated by NK and PMN effector cells. Blood. 2008;112(6):2390–2399. doi: 10.1182/blood-2008-03-144600. [DOI] [PubMed] [Google Scholar]
- 92.Avouac J, Gossec L, Dougados M. Diagnostic and predictive value of anti-cyclic citrullinated protein antibodies in rheumatoid arthritis: a systematic literature review. Ann Rheum Dis. 2006;65(7):845–851. doi: 10.1136/ard.2006.051391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Li P, Li M, Lindberg MR, Kennett MJ, Xiong N, Wang Y. PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J Exp Med. 2010;207(9):1853–1862. doi: 10.1084/jem.20100239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Dwivedi N, Upadhyay J, Neeli I, et al. Felty's syndrome autoantibodies bind to deiminated histones and neutrophil extracellular chromatin traps. Arthritis Rheum. 2012;64(4):982–992. doi: 10.1002/art.33432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Coudevylle N, Rokas D, Sakarellos-Daitsiotis M, et al. Phosphorylated and nonphosphorylated epitopes of the La/SSB autoantigen: comparison of their antigenic and conformational characteristics. Biopolymers. 2006;84(4):368–382. doi: 10.1002/bip.20458. [DOI] [PubMed] [Google Scholar]
- 96.Pisetsky DS. Antibody responses to DNA in normal immunity and aberrant immunity. Clin Diagn Lab Immunol. 1998;5(1):1–6. doi: 10.1128/cdli.5.1.1-6.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Casciola-Rosen LA, Anhalt G, Rosen A. Autoantigens targeted in systemic lupus erythematosus are clustered in two populations of surface structures on apoptotic keratinocytes. J Exp Med. 1994;179(4):1317–1330. doi: 10.1084/jem.179.4.1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Pisetsky DS, Lipsky PE. Microparticles as autoadjuvants in the pathogenesis of SLE. Nat Rev Rheumatol. 2010;6(6):368–372. doi: 10.1038/nrrheum.2010.66. [DOI] [PubMed] [Google Scholar]
- 99.Turner N, Nolasco L, Moake J. Generation and breakdown of soluble ultralarge von Willebrand factor multimers. Semin Thromb Hemost. 2012;38(1):38–46. doi: 10.1055/s-0031-1300950. [DOI] [PubMed] [Google Scholar]
- 100.Yeh HC, Zhou Z, Choi H, et al. Disulfide bond reduction of von Willebrand factor by ADAMTS-13. J Thromb Haemost. 2010;8(12):2778–2788. doi: 10.1111/j.1538-7836.2010.04094.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Navarrete A-M, Casari C, Legendre P, et al. A murine model to characterize the antithrombotic effect of molecules targeting human von Willebrand factor. Blood. 2012;120(13):2723–2732. doi: 10.1182/blood-2012-03-420042. [DOI] [PubMed] [Google Scholar]