

Abstract
We report a dramatically improved total synthesis of two highly selective V600EBRAF inhibitors, PLX4720 and PLX4032, that leverages microwave-assisted organic synthesis (MAOS). Compared with previously reported approaches, our novel MAOS method significantly reduces overall reaction time without compromising yield. In addition to providing a gram-scale route to these compounds for preclinical oncology research, we anticipate this approach could accelerate the synthesis of azaindoles in high-throughput, library-based formats.
Keywords: PLX4720, PLX4032, BRAF, MAOS, Melanoma
As mutations in vital genes accrue, normal programs of cell proliferation, differentiation, and death are recast, forming the basis of cancer. Of the known protein kinases, the BRAF paralog of the rapidly growing fibrosarcoma (RAF) family of proteins is the most frequently mutated in human cancer1. Activating somatic mutations in BRAF occur in malignant melanomas (50%), ovarian cancer (30%), thyroid cancer (30%), colorectal cancer (CRC) (15%), and less frequently in other cancer types2,3. While several mutations in BRAF have been reported, the most common mutation substitutes valine for glutamic acid at codon 600 (V600EBRAF) in the activation segment of the kinase. This particular mutation accounts for greater than 90% of BRAF mutations in cancer 2. V600EBRAF is constitutively activated, as are its associated downstream effectors within the mitogen-activated protein kinase (MAPK) pathway4. Clinically, tumour expression of V600EBRAF correlates with elevated proliferation, aggressiveness, and poor prognosis5. Since growth and proliferation of tumours expressing V600EBRAF tend to rely upon MAPK pathway activity, pharmacological inhibition of V600EBRAF represents an attractive therapeutic approach in oncology6.
Small-molecule inhibition of BRAF in oncology has been historically approached using pan-kinase inhibitors7,8, such as sorafenib (Nexavar) (Figure 1). However, for a variety of postulated reasons9, this approach has led to disappointing outcomes in V600EBRAF-dependent tumours such as melanoma10,11.
Figure 1.
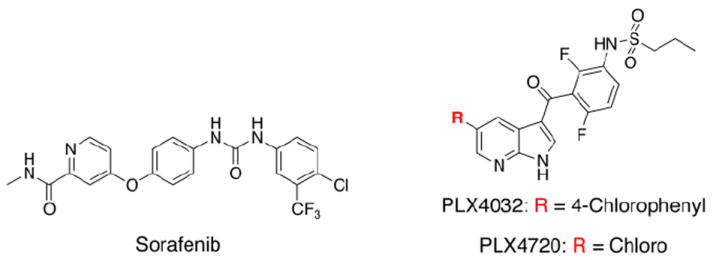
Chemical structures of select BRAF inhibitors.
An encouraging alternative approach to pan-kinase inhibitors that has shown recent success has been the clinical development of V600EBRAF-selective small-molecule inhibitors. Currently, two of the most promising selective inhibitors include PLX472012 and its clinically used analogue, PLX4032 (vemurafinib)13. Uniquely, these drugs selectively inhibit V600EBRAF kinase at low nanomolar concentrations and, accordingly, attenuate associated MAPK pathway activity in V600EBRAF tumours12-17. Recently, PLX4032 has been approved by the FDA for treatment of late-stage V600EBRAF-positive melanoma18, and continues to be evaluated in other single-agent and combination settings19-21.
Our laboratory’s interest in developing and validating predictive imaging biomarkers to reflect tumour response to V600EBRAF inhibition required milligram- to gram-scale quantities of PLX4032 and PLX4720 suitable for preclinical in vivo studies. We found published production-scale syntheses of these compounds, yet these methods were inappropriate for typical academic research laboratories, as well as being time and labour intensive12,13. These compounds could also be purchased commercially, but only at great expense given the scale required for our research activities. To circumvent these limitations, we developed a rapid, in-house approach to synthesize PLX4720 and PLX4032 that capitalized upon the advantages of microwave-assisted organic synthesis (MAOS). MAOS employs non-classical heating via microwaves in lieu of traditional thermal convection or conduction. Commonly, MAOS reaction times are dramatically reduced, reaction efficiencies are increased, and material and labour costs are reduced22,23.
In this study, MAOS was successfully adapted to each of the traditional syntheses reported by Tsai et al.12 and Bollag et al.13 (Table 1), with MAOS offering significant advantages in all steps required to synthesize PLX4720 and four of the six steps required to synthesize PLX4032. We herein report dramatically reduced reaction times required for synthesis of the drugs while achieving comparable, or in most cases, improved yields. The divergent synthesis developed within for this study is shown in Scheme 1. In Table 1, we report conditions, reaction times, and yields described in the original literature (refs. a,b) and MAOS application. Assuming overnight to be 16 hours, for PLX4720, MAOS resulted in a 91% reduction in overall reaction time (87 hours to 6 hours). For PLX4032, four of the six steps were amenable to MAOS and resulted in a 33% reduction in reaction time (141 hours to 94 hours). Successful gram-scale MAOS was carried out for select intermediates (3, 6b) to ensure scalability of the developed method. Full synthetic methodology and characterization data can be found in Supplementary Data.
Table 1.
| MAOS of N-(3,5-difluorophenyl)propane-1-sulfonamide (3).
| |||||
| Entry | MAOS | Reaction Conditions | Temperature | Time | Yield |
|
| |||||
| 1 | No | Pyridine, DMAP, CH2Cl2 | Reflux | 16 h | Quant.a |
| 2 | Yes | Pyridine, DMAP, CH2Cl2 | 100 °C | 30 min | 89% |
|
| |||||
| MAOS of N-(3-5-difluoro-4-formylphenyl)propane-1-sulfonamide (5).
| |||||
| Entry | MAOS | Reaction Conditions | Temperature | Time | Yield |
|
| |||||
| 3 | No | LDA, THF | -78 °C/RT | 5/16 h | 51%a |
| 4 | Yes | LHMDS, THF | 0/110 °C | 30 min/1 h | 56% |
|
| |||||
| MAOS of 5-(4-chlorophenyl)-1H-pyrrolo[2,3-b]pyridine (6b).
| |||||
| Entry | MAOS | Reaction Conditions | Temperature | Time | Yield |
|
| |||||
| 5 | No | K2CO3, Pd(PPh3)2Cl2, DME | Reflux | 16 h | 81%b |
| 6 | Yes | K2CO3, Pd(PPh3)2Cl2, DME | 130 °C | 30 min | 76% |
|
| |||||
| MAOS of N-(3-((5-chloro-1H-pyrrolo[2,3-b]pyridine-3-yl)(hydroxyl)methyl)2,4-difluorophenyl)propane-1-sulfonamide (7a).
| |||||
| Entry | MAOS | Reaction Conditions | Temperature | Time | Yield |
|
| |||||
| 7 | No | K2CO3, MeOH:Water | RT | 48 h | 88%a |
| 8 | Yes | K2CO3, MeOH:Water | 130 °C | 30 min | 88% |
|
| |||||
| Synthesis of N-(3-((5-(4-chlorophenyl)-1H-pyrrolo[2,3-b]pyridin-3-yl)(hydroxy)methyl)-2,4-difluorophenyl)propane-1-sulfonamide (7b).
| |||||
| Entry | MAOS | Reaction Conditions | Temperature | Time | Yield |
|
| |||||
| 9 | No | KOH, MeOH | RT | 72 h | NAc |
| 10 | No | HBr (aq), AcOH | RT | 16 h | NAc |
| 11 | No | KOH, MeOH | RT | 72 h | NAd |
| 12 | No | HBr (aq), AcOH | RT | 16 h | 44%e |
|
| |||||
| MAOS of N-(5-chloro-1H-pyrrolo[2,3-b]pyridine-3-carbonyl)-2,4-difluorophenyl)propane-1-sulfonoamide (8a, PLX4720).
| |||||
| Entry | MAOS | Reaction Conditions | Temperature | Time | Yield |
|
| |||||
| 13 | No | DDQ, H2O:1,4-Dioxane | RT | 2 h | 90%a |
| 14 | Yes | DDQ, H2O:1,4-Dioxane | 100 °C | 10 min | 87% |
|
| |||||
| MAOS of N-(3-(5-(4-chlorophenyl)-1H-pyrrolo[2,3-b]pyridine-3-carbonyl)-2,4-difluorophenyl)propane-1-sulfonamide (8b, PLX4032).
| |||||
| Entry | MAOS | Reaction Conditions | Temperature | Time | Yield |
|
| |||||
| 15 | No | DDQ, H2O:1,4-Dioxane | RT | 2 h | 45%f |
| 16 | Yes | DDQ, H2O:1,4-Dioxane | 100 °C | 10 min | 92% |
See Tsai et al. 2008.
See Bollag et al. 2010.
See Bollag et al. 2010, combined yield with entry 15.
Combined yield with entry 12.
Combined yield with entry 11.
See Bollag et al. 2010, combined yield with entries 9 & 10.
Scheme 1.
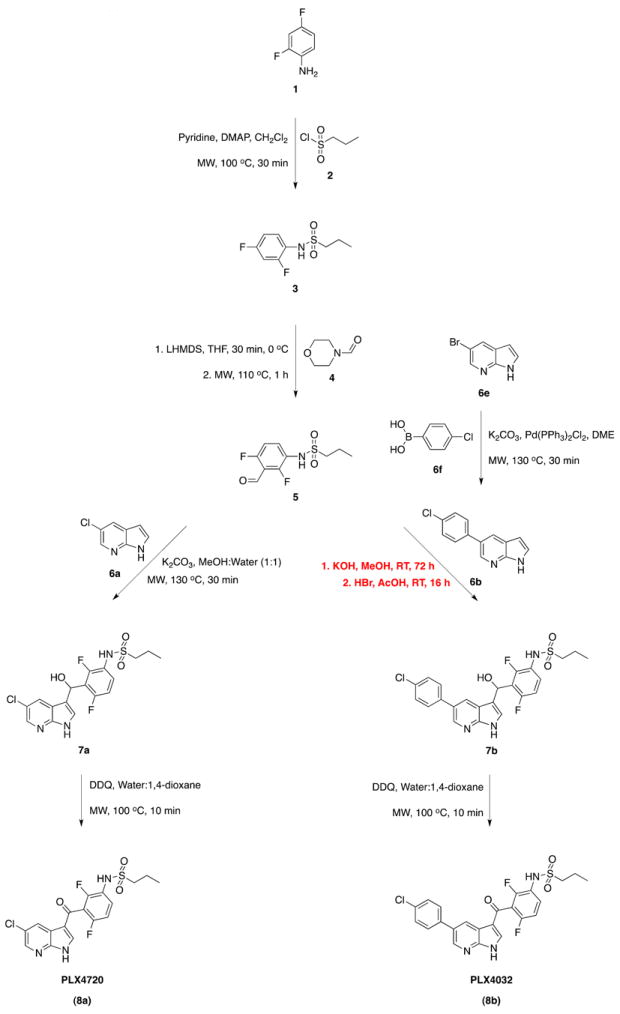
MAOS of PLX4720 and PLX4032 (MW = microwaves).
1. Synthesis of PLX4720 (8a)
1.1 MAOS of N-(3,5-difluorophenyl)propane-1-sulfonamide (3)
The divergent synthesis begins with formation of 3 by reaction of 2,4-difluoroaniline (1) and propane-1-sulfonyl chloride (2) in anhydrous methylene chloride, dimethyaminopyridine (DMAP), and pyridine. Previous studies carried out this reaction at room temperature overnight with a quantitative yield (entry 1, Table 1)12. Microwave irradiation at 100 °C reduced the reaction time to 30 minutes. Subsequent flash chromatography on silica gel gave 3 in a comparable yield of 89% (entry 2, Table 1).
1.2 MAOS of N-(3-5-difluoro-4-formylphenyl)propane-1-sulfonamide (5)
Synthesis of 5 features a two-step formylation of 3 with morpholine-4-carbaldehyde (4). Previously12, in situ generation of lithium diisopropylamide (LDA) using n-butyllithium in THF and diisoproplyamine was carried out at 0 °C for one hour. This was followed by deprotonation of 3 with this LDA solution for four hours. Subsequent formylation with morpholine-4-carbaldehyde (4) at -78 °C for four hours, and a further 16 hours at room temperature gave 5 in 51% yield (entry 1, Table 2). In this study, given the temperature requirements of the deprotonation, we chose not to pursue MAOS. Moreover, we attained comparable yields using commercially available lithium bis(trimethylsilyl)amide (LHMDS) in place of LDA. The subsequent formylation, however, was adaptable to MAOS via microwave irradiation at 100 °C for one hour, with a comparable yield of 56% (entry 4, Table 1).
1.3 MAOS of N-(3-((5-chloro-1H-pyrrolo[2,3-b]pyridine-3-yl)(hydroxyl)methyl)2,4-difluorophenyl)propane-1-sulfonamide (7a)
The PLX4720 intermediate 7a was originally synthesized in 88% from the reaction of 5 with the azaindole core 5-chloro-1H-pyrrolo[2,3-b]pyridine (6a) in methanol:water (1:1) for 48 hours at room temperature (entry 7, Table 1)12. Optimization with microwave irradiation resulted in final reaction conditions of 130 °C in the same solvent system for only a fraction of the original time (30 minutes). Purification by flash chromatography on silica gel gave 7a in a matching yield of 88% (entry 8, Table 1).
1.4 MAOS of N-(5-chloro-1H-pyrrolo[2,3-b]pyridine-3-carbonyl)-2,4-difluorophenyl)propane-1-sulfonoamide (8a, PLX4720)
From compound 7a, reported oxidation to PLX4720 (8a) utilized 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) in a mixture of water and 1,4-dioxane at room temperature for two hours, with a yield of 90% (entry 13, Table 1)12. Under MAOS, an optimized reaction temperature of 100 °C was achieved with a reduction in reaction time to 10 minutes, a factor of 12, while still achieving a comparable yield of 87% (entry 14, Table 1).
2 . Synthesis of PLX4320 (8b)
2.1 MAOS of 5-(4-chlorophenyl)-1H-pyrrolo[2,3-b]pyridine (6b)
Suzuki coupling of 5-bromo-1H-pyrrolo[2,3-b]pyridine (6e) with 4-(chlorophenyl)boronic acid (6f) in the presence of K2CO3 and Pd(PPh3)2Cl2 in 1,2-dimethoxyethane (DME) for 30 minutes at 130 °C secured the necessary PLX4032 azaindole core (6b) (entry 6, Table 1). Previous synthesis of this intermediate required overnight reflux, with a yield of 81% (entry 5, Table 1)13, thus underscoring the effectiveness of MAOS in reducing overall reaction time.
2.2 Synthesis of N-(3-((5-(4-chlorophenyl)-1H-pyrrolo[2,3-b]pyridin-3-yl)(hydroxy)methyl)-2,4-difluorophenyl)propane-1-sulfonamide (7b)
Coupling of the respective azaindole cores of PLX4032 (6b) and PLX4720 (6a) with intermediate (5) is where the synthesis diverges. For PLX4032, unlike PLX4720, application of MAOS of 6b with 5 in the presence of KOH and methanol proved less advantageous (entry 11, Table 1), frequently yielding mixed by-products. Moreover, deprotection of the methyl ether intermediate (structure not shown) with aqueous hydrogen bromide and acetic acid to the final product (7b) (entry 12, Table 1) was also problematic under microwave irradiation, primarily yielding by-products. Modification of both reaction conditions marginally affected applicability of microwave irradiation. Accordingly, MAOS was not pursued further, in favour of the published methodology of Bollag et al.13. Nevertheless, the combined yield of these two steps in our hands was 44%, compared to Bollag’s 45%, which also included the final synthetic step (entry 15, Table 1).
2.3 MAOS of N-(3-(5-(4-chlorophenyl)-1H-pyrrolo[2,3-b]pyridine-3-carbonyl)-2,4-difluorophenyl)propane-1-sulfonamide (8b, PLX4032)
Bollag et al.13 reported a final oxidation of precursor 7b to PLX4032 (8b) analogous to Tsai et al.12. The exact yield of this step is unknown as it was reported in combination with the two preceding reactions (entry 15, Table 1). Nonetheless, the MAOS conditions developed for the final-step oxidation of PLX4720 (8a) were also employed towards PLX4032 (8b). Irradiation of 7b at 100 °C for 10 minutes gave a 92% yield of the final PLX4032 (8b) (entry 16, Table 1).
In summary, we report optimized, gram-scale syntheses of PLX4720 and PLX4032 that leverage MAOS. Where applicable, the MAOS protocol reported herein significantly improves overall reaction times while maintaining or even improving synthetic yields. We envision this methodology could potentially be extended not only to the synthesis of PLX4032, PLX4720, but to other novel azaindoles as well.
Supplementary Material
Acknowledgments
The authors wish to gratefully acknowledge funding from the National Cancer Institute (NCI): 1R01 CA140628, 1RC1 CA145138-01, K25 CA127349, 1P50 CA128323 (Vanderbilt ICMIC Program), 2P50 CA095103 (Vanderbilt SPORE in GI Cancer), 5P30 DK058404, and the Kleberg Foundation. The authors wish to thank Dr. Yiu-Yin Cheung for helpful discussions.
Footnotes
Supplementary Material
Supplementary data associated with this article can be found in the online version at
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Greenman C, Stephens P, Smith R, Dalgliesh GL, Hunter C, Bignell G, Davies H, Teague J, Butler A, Stevens C, Edkins S, O’Meara S, Vastrik I, Schmidt EE, Avis T, Barthorpe S, Bhamra G, Buck G, Choudhury B, Clements J, Cole J, Dicks E, Forbes S, Gray K, Halliday K, Harrison R, Hills K, Hinton J, Jenkinson A, Jones D, Menzies A, Mironenko T, Perry J, Raine K, Richardson D, Shepherd R, Small A, Tofts C, Varian J, Webb T, West S, Widaa S, Yates A, Cahill DP, Louis DN, Goldstraw P, Nicholson AG, Brasseur F, Looijenga L, Weber BL, Chiew YE, DeFazio A, Greaves MF, Green AR, Campbell P, Birney E, Easton DF, Chenevix-Trench G, Tan MH, Khoo SK, Teh BT, Yuen ST, Leung SY, Wooster R, Futreal PA, Stratton MR. Nature. 2007;446:153. doi: 10.1038/nature05610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, Davis N, Dicks E, Ewing R, Floyd Y, Gray K, Hall S, Hawes R, Hughes J, Kosmidou V, Menzies A, Mould C, Parker A, Stevens C, Watt S, Hooper S, Wilson R, Jayatilake H, Gusterson BA, Cooper C, Shipley J, Hargrave D, Pritchard-Jones K, Maitland N, Chenevix-Trench G, Riggins GJ, Bigner DD, Palmieri G, Cossu A, Flanagan A, Nicholson A, Ho JW, Leung SY, Yuen ST, Weber BL, Seigler HF, Darrow TL, Paterson H, Marais R, Marshall CJ, Wooster R, Stratton MR, Futreal PA. Nature. 2002;417:949. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 3.Garnett MJ, Marais R. Cancer Cell. 2004;6:313. doi: 10.1016/j.ccr.2004.09.022. [DOI] [PubMed] [Google Scholar]
- 4.Satyamoorthy K, Li G, Gerrero MR, Brose MS, Volpe P, Weber BL, Van Belle P, Elder DE, Herlyn M. Cancer Res. 2003;63:756. [PubMed] [Google Scholar]
- 5.Yokota T, Ura T, Shibata N, Takahari D, Shitara K, Nomura M, Kondo C, Mizota A, Utsunomiya S, Muro K, Yatabe Y. Br J Cancer. 2011;104:856. doi: 10.1038/bjc.2011.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sharma A, Trivedi NR, Zimmerman MA, Tuveson DA, Smith CD, Robertson GP. Cancer Res. 2005;65:2412. doi: 10.1158/0008-5472.CAN-04-2423. [DOI] [PubMed] [Google Scholar]
- 7.Flaherty KT. Annu Rev Med. 2011 doi: 10.1146/annurev-med-050410-105655. [DOI] [PubMed] [Google Scholar]
- 8.Perez-Lorenzo R, Zheng B. Biosci Rep. 2012;32:25. doi: 10.1042/BSR20110068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fedorenko IV, Paraiso KH, Smalley KS. Biochem Pharmacol. 2011;82:201. doi: 10.1016/j.bcp.2011.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eisen T, Ahmad T, Flaherty KT, Gore M, Kaye S, Marais R, Gibbens I, Hackett S, James M, Schuchter LM, Nathanson KL, Xia C, Simantov R, Schwartz B, Poulin-Costello M, O’Dwyer PJ, Ratain MJ. Br J Cancer. 2006;95:581. doi: 10.1038/sj.bjc.6603291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hauschild A, Agarwala SS, Trefzer U, Hogg D, Robert C, Hersey P, Eggermont A, Grabbe S, Gonzalez R, Gille J, Peschel C, Schadendorf D, Garbe C, O’Day S, Daud A, White JM, Xia C, Patel K, Kirkwood JM, Keilholz U. J Clin Oncol. 2009;27:2823. doi: 10.1200/JCO.2007.15.7636. [DOI] [PubMed] [Google Scholar]
- 12.Tsai J, Lee JT, Wang W, Zhang J, Cho H, Mamo S, Bremer R, Gillette S, Kong J, Haass NK, Sproesser K, Li L, Smalley KS, Fong D, Zhu YL, Marimuthu A, Nguyen H, Lam B, Liu J, Cheung I, Rice J, Suzuki Y, Luu C, Settachatgul C, Shellooe R, Cantwell J, Kim SH, Schlessinger J, Zhang KY, West BL, Powell B, Habets G, Zhang C, Ibrahim PN, Hirth P, Artis DR, Herlyn M, Bollag G. Proc Natl Acad Sci U S A. 2008;105:3041. doi: 10.1073/pnas.0711741105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bollag G, Hirth P, Tsai J, Zhang J, Ibrahim PN, Cho H, Spevak W, Zhang C, Zhang Y, Habets G, Burton EA, Wong B, Tsang G, West BL, Powell B, Shellooe R, Marimuthu A, Nguyen H, Zhang KY, Artis DR, Schlessinger J, Su F, Higgins B, Iyer R, D’Andrea K, Koehler A, Stumm M, Lin PS, Lee RJ, Grippo J, Puzanov I, Kim KB, Ribas A, McArthur GA, Sosman JA, Chapman PB, Flaherty KT, Xu X, Nathanson KL, Nolop K. Nature. 2010;467:596. doi: 10.1038/nature09454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Joseph EW, Pratilas CA, Poulikakos PI, Tadi M, Wang W, Taylor BS, Halilovic E, Persaud Y, Xing F, Viale A, Tsai J, Chapman PB, Bollag G, Solit DB, Rosen N. Proc Natl Acad Sci U S A. 2010;107:14903. doi: 10.1073/pnas.1008990107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sondergaard JN, Nazarian R, Wang Q, Guo D, Hsueh T, Mok S, Sazegar H, MacConaill LE, Barretina JG, Kehoe SM, Attar N, von Euw E, Zuckerman JE, Chmielowski B, Comin-Anduix B, Koya RC, Mischel PS, Lo RS, Ribas A. Journal of translational medicine. 2010;8:39. doi: 10.1186/1479-5876-8-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Whittaker S, Kirk R, Hayward R, Zambon A, Viros A, Cantarino N, Affolter A, Nourry A, Niculescu-Duvaz D, Springer C, Marais R. Science translational medicine. 2010;2:35ra41. doi: 10.1126/scitranslmed.3000758. [DOI] [PubMed] [Google Scholar]
- 17.Yang H, Higgins B, Kolinsky K, Packman K, Go Z, Iyer R, Kolis S, Zhao S, Lee R, Grippo JF, Schostack K, Simcox ME, Heimbrook D, Bollag G, Su F. Cancer Res. 2010;70:5518. doi: 10.1158/0008-5472.CAN-10-0646. [DOI] [PubMed] [Google Scholar]
- 18.Luke JJ, Hodi FS. Clin Cancer Res. 2011 doi: 10.1158/1078-0432.CCR-11-2197. [DOI] [PubMed] [Google Scholar]
- 19.NIH. 2012 http://clinicaltrials.gov/
- 20.Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, Dummer R, Garbe C, Testori A, Maio M, Hogg D, Lorigan P, Lebbe C, Jouary T, Schadendorf D, Ribas A, O’Day SJ, Sosman JA, Kirkwood JM, Eggermont AM, Dreno B, Nolop K, Li J, Nelson B, Hou J, Lee RJ, Flaherty KT, McArthur GA. N Engl J Med. 2011;364:2507. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Flaherty KT, Puzanov I, Kim KB, Ribas A, McArthur GA, Sosman JA, O’Dwyer PJ, Lee RJ, Grippo JF, Nolop K, Chapman PB. N Engl J Med. 2010;363:809. doi: 10.1056/NEJMoa1002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dallinger D, Kappe CO. Chemical Reviews. 2007;107:2563. doi: 10.1021/cr0509410. [DOI] [PubMed] [Google Scholar]
- 23.Tang D, Buck JR, Hight MR, Manning HC. Tetrahedron Lett. 2010;51:4595. doi: 10.1016/j.tetlet.2010.06.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.