

Abstract
The trimethyl lock is an o-hydroxydihydrocinnamic acid derivative in which unfavorable steric interactions between three pendant methyl groups encourage lactonization to form a hydrocoumarin. This reaction is extremely rapid, even when the electrophile is an amide and the leaving group is an amino group of a small-molecule drug, fluorophore, peptide, or nucleic acid. O-Acylation of the phenolic hydroxyl group prevents reaction, providing a trigger for the reaction. Thus, the release of an amino group from an amide can be coupled to the hydrolysis of a designated ester (or to another chemical reaction that regenerates the hydroxyl group). Trimethyl lock conjugates are easy to synthesize, making the trimethyl lock a highly versatile module for chemical biology and related fields.
1. Introduction
Chemists and biologists often need to control the reactivity of their molecules in a temporal and spatial manner. This need can be met with the “trimethyl lock”, an o-hydroxydihydrocinnamic acid derivative (1) that undergoes rapid lactonization under physiological conditions to yield a dihydrocoumarin (2) (Fig. 1). Its name arises from the three interlocking methyl groups that are responsible for the extraordinary reaction rate. Blocking the phenolic oxygen with a labile moiety (as in 3 or 4) prevents lactonization and provides an enzymatic, chemical, or photolytic trigger. Here, we review the development of the trimethyl lock and describe its utility as a means to release small-molecule drugs, fluorophores, peptides, and nucleic acids.
Fig. 1.

Rapid reaction of a trimethyl lock acid (1) to form a δ-lactone (2). The three interlocking methyl groups are indicated by filled circles in 1. The high reactivity can be used to release an amine or alcohol after enzymatic, chemical, or photolytic deprotection of a trimethyl lock amide (3) or ester (4).
1.1 History
The trimethyl lock was developed during an era fixated on the mechanism by which ATP synthase catalyzes the synthesis of adenosine 5′-triphosphate (ATP) via oxidative phosphorylation. In the 1960’s, Louis A. Cohen (1926–1996) and his coworkers at the National Institute of Health (USA) developed a model to test whether ubihydroquinone, which was then suspected (and now known) to be a key cofactor in the electron-transport chain, could be esterified by protein carboxylates to produce a high-energy intermediate upon oxidation (Fig. 2).1 The high-energy intermediate would be able to activate inorganic phosphate to form a phosphoanhydride (Fig. 2D). Phosphoanhydrides are sufficiently activated to transfer a phosphoryl group to adenosine 5′-diphosphate (ADP), generating ATP. Reduction of ubiquinone by NADH would regenerate the starting materials. A requirement of the model was that a phenolic hydroxyl group must attack an unactivated carboxyl group. Model compounds did not undergo lactonization at an appreciable rate under physiological conditions, until the realization of trimethyl lock 1.2 Although Cohen’s putative high-energy intermediate was never found, his astute appreciation of the special reactivity of the trimethyl lock spawned the numerous advances described herein.
Fig. 2.

Cohen’s model for oxidative phosphorylation,1 which inspired the trimethyl lock. Panel A depicts a presumed mitochondrial protein·ubihydroquinone complex.
1.2. Kinetics
The rate constant for the lactonization of trimethyl lock 1 exceeds that for its bimolecular counterpart by a factor of 3 × 1015 M.3 This lactonization proceeds at a rate that is much faster than that of o-hydroxydihydrocinnamic acids 5–7, which lack pendant methyl groups (Fig. 3).2 Cohen believed that enzymatic catalysis relied, in part, on freezing the substrate into a conformer that can convert directly into the transition state. The interlocking of the methyl groups in the trimethyl lock led to rates of lactonization that matched or even exceeded the rates of enzyme-catalyzed reactions. This satisfying parallel enabled Cohen to explain some of the rate enhancement imparted by enzymes, and he termed this device to effect catalysis: “stereopopulation control”.3–5
Fig. 3.

Relative reactivity of trimethyl lock derivatives.2
An alternative “relief-of-strain” hypothesis proposed that steric hindrance of the abutting methyl groups in trimethyl lock 1 was relieved upon cyclization.6 This model was supported by an unusual discovery—a steric isotope effect. Replacing all six hydrogens in the gem-dimethyl group of compound 1 with deuterium decreased the rate of lactonization by 10%, presumably because deuterium–carbon bonds, which are slightly shorter than hydrogen–carbon bonds, imposed less strain.7 Further in support of the relief-of-strain hypothesis, crystal structures of quinones 8–11 and hydroquinone 12 (Fig. 4) showed that the methyl group distances did not change between the open and closed forms; however, the open forms 8–11 had the unmasked oxygen forced out of the plane of the quinone ring system.8 Some of this strain was relieved in the closed form, 12.
Fig. 4.

Quinone trimethyl locks (8–11) that have known crystal structures and the lactone (12) that forms upon their reduction to the hydroquinone.8
Computational methods have been used to identify the contributors to the high lactonization rate of the trimethyl lock.10 The difference in ground-state free energy between trimethyl lock 1 and lactone 2 was calculated to be 7 kcal/mol, accounting for only a 106-fold increase in the rate of lactonization. Thus, the relief of strain could not be the only factor influencing the rate of lactonization. A version of stereopopulation control could also make a substantial contribution, and the rate acceleration is likely to have both enthalpic and entropic components (1, Fig. 1).11
2. Prodrugs
Although many bioreversible options exist for modifying an alcohol—such as condensation to form an ester, phosphate, or sulfate—few are available for an amine.12,13 Amides hydrolyze too slowly to be useful prodrugs,14–16 unless the amide happens to be a substrate for a specific protease.9 The trimethyl lock provides a general method to mask amines, and this attribute was developed by Ronald Borchardt, a former Cohen postdoctorate. The utility is apparent in the acetyl ester of trimethyl lock 13 (Fig. 5), which releases p-methoxyaniline upon ester hydrolysis. The half-life of 13 in phosphate-buffered saline is t1/2 = 4039 min. Addition of porcine liver esterase provides p-methoxyaniline within a few minutes. In human plasma, trimethyl lock 13 is activated with t1/2 = 54 min; addition of the esterase inhibitor diisopropylfluorophosphate increases the half-life.9
Fig. 5.

First enzymatic activation of a trimethyl lock amide.9
2.1. Water solubility
Paclitaxel (Taxol®), a natural product from the western yew tree, is an inhibitor of mitosis that is used often in cancer chemotherapy.22 Unfortunately, paclitaxel is poorly soluble in water23 and its use requires solubilizing agents, such as polyethoxylated castor oil and ethanol, which can lead to adverse effects.24 Attempts had been made to generate more soluble derivatives of paclitaxel, including phosphorylation of the C-2′ or C-7 hydroxyl group (14 and 15, Fig. 6).17 Although such phosphorylation increases water solubility, the phosphoryl groups are not hydrolyzed by alkaline phosphatase or in plasma, and prodrugs 14 and 15 are ineffective.
Fig. 6.

Phosphoryl prodrugs of paclitaxel (14–17)17–19 and a combretastatin A4 derivative (18).20
In contrast to prodrugs 14 and 15, derivatives of paclitaxel that contain a phosphorylated trimethyl lock (16 and 17)19 are highly efficacious.18 Both derivatives are much more soluble in water (>10 mg/mL at 37 °C) than is paclitaxel (~2 μg/mL), and both are substrates for alkaline phosphatase. Compound 17 was as effective as paclitaxel in an M109 mouse xenograft model, and required no solubilizing agents in its formulation, unlike paclitaxel.
The same phosphorylated trimethyl lock has been appended to a derivative of combretastatin A4, a microtubule disrupting agent (18, Fig. 6).20 Water solubility increased, as did efficacy in a BDF1 mouse xenograft model.
2.2. Tissue specificity
Trimethyl lock activation can be achieved not only by the hydrolysis of a phenyl ester, but by the reduction of a quinone.25–27 Many tumors have high levels of reductase enzymes as well as low oxygen tension.28 Accordingly, temporary inactivation of a cytotoxic drug with a labile quinone trimethyl lock could increase the therapeutic index by targeting the drug specifically to tumors.29 Quinone trimethyl lock derivatives of four cytotoxins (19–22, Fig. 7) isolated from fungi were synthesized and tested for cytotoxicity in vitro.21 Cells with a more reducing cytosol were found to be more vulnerable to these prodrugs.
Fig. 7.

Cytotoxins masked with a quinone trimethyl lock.21
2.3. Oral bioavailability
Ganciclovir is a DNA polymerase inhibitor that is used to treat cytomegalovirus (CMV) infections. Interestingly, ganciclovir itself is a prodrug, and must be phosphorylated by viral thymidine kinase for activity. Ganciclovir has poor oral bioavailability, and requires large doses to be efficacious.30
A prodrug of ganciclovir incorporating the trimethyl lock improves its bioavailability (Fig. 8).30 The strategy here was somewhat atypical in that the trimethyl lock masks an alcohol, adding a second ester bond. Two ganciclovir derivatives were synthesized, and oral bioavailability was measured in rats. Free ganciclovir had a bioavailability of 3.6%, whereas acetyl ester 23 had a bioavailability of 15.6%. Benzyl ether 24 was not bioavailable—no free ganciclovir was detected in the plasma. These data suggest that hydrolysis of the acetyl trigger, rather than the ester proximal to the gem-dimethyl group, led to the release of free ganciclovir from 23.
Fig. 8.

Trimethyl lock derivatives of ganciclovir.30 Acetyl ester 23 is orally bioavailable.
2.4. PEGylation
Poly(ethylene glycol) (PEG) is a water-soluble, flexible polymer that has been used to improve the properties of numerous pharmaceuticals.31 Appending a small-molecule drug to a high molecular weight polymer has two main effects: decreased renal clearance and increased tumor retention.32 The latter effect is due to the greater vascular permeability and reduced lymphatic drainage of tumors compared to normal tissues, leading to an accumulation of macromolecules. Thus, PEG conjugates can lower the efficacious dose of a drug. A cleavable PEG conjugate has the potential to evade renal filtration, to accumulate in a tumor, and to increase therapeutic index if the active drug is released only at the desired location.
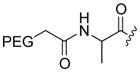
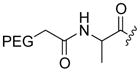
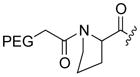
The cytotoxic drug daunorubicin has been linked to a 40-kDa PEG with cleavable trimethyl lock linkers (25–31, Table 1).33 Activation of the trimethyl lock released unmodified daunorubicin. As intended, some of the conjugates showed enhanced stability in plasma. That stability was greater when a methyl group was ortho to the ester linkage (26 and 29), consistent with greater steric occlusion of esterases. A carbonate linkage (30) was cleaved by plasma esterases, but a carbamate linkage (31) was not.
Table 1.
Plasma half-life of PEGylated trimethyl lock daunorubicin.33
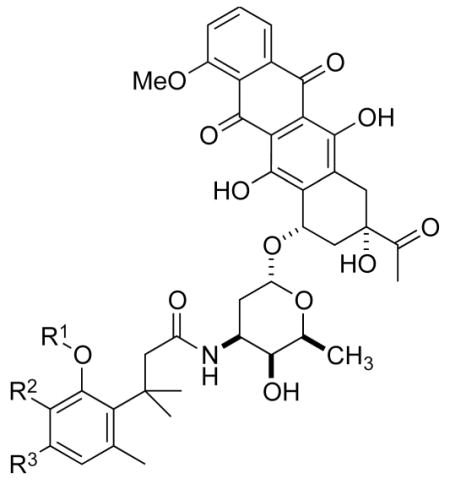
| ||||
|---|---|---|---|---|
| R1 | R2 | R3 | t1/2 (h) rat plasma | |
|
|
||||
| 25 |
|
H | CH3 | 1.9 |
| 26 |
|
CH3 | H | 21 |
| 27 |
|
H | CH3 | 17 |
| 28 |
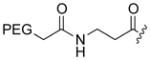
|
H | CH3 | 0.2 |
| 29 |
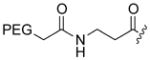
|
CH3 | H | 8 |
| 30 |
|
H | CH3 | 1.1 |
| 31 |
|
H | CH3 | >24 |
2.5. Cyclic peptide
Other benefits of the trimethyl lock are evident in cyclic peptide 32 (Fig. 9).34 The steric bulk of the peptide slowed the rate of ester hydrolysis in human plasma to t1/2 = 508 min.9,34 A significant advantage in using a cyclic prodrug became apparent when comparing the stability of cyclic peptide 32 to its linear counterpart, 33. In plasma, the linear peptide was degraded by proteases with a half-life of only 4 min. Moreover, the linear peptide had poor oral bioavailability. In Caco-2 cells, an in vitro model of intestinal mucosa, the cyclic peptide permeated a monolayer of cells 70-fold more efficiently than did the linear peptide. Whereas some of the poor permeation by the linear peptide was caused by enzymatic degradation, protease inhibitors did not enhance the extent of permeation of 33 to that of 32.34
Fig. 9.

Linearization of a cyclic peptide trimethyl lock.34
3. Resins for solid-phase synthesis
3.1. Peptide-synthesis resins
A carboxyl resin for solid-phase synthesis has been derived from a quinone trimethyl lock. The key step in the synthetic route to the resin was the Claisen rearrangement of allyl hydroquinone 34 (Fig. 10). Cleavage from the resin was initiated with mild reducing agents such as sodium hydrosulfite, which does not affect common protecting groups.35 An additional attractive attribute of resin 35 is that cleavage can be monitored by observing the disappearance of the yellow quinone.
Fig. 10.

Trimethyl lock resin for the synthesis of peptides.35 Starting synthesis at the C terminus required adding an ethanolamine linker, which remained on the product peptide.
A carboxyl resin like 35 is not directly compatible with standard Fmoc-mediated solid-phase peptide synthesis, in which peptides are synthesized starting at their C terminus. Its use required forming an ester with ethanolamine, and the product peptide had an C-terminal ethanolamide. A quinone trimethyl lock overcame this problem (Fig. 11).36 Cleavage of the peptide from the trimethyl lock 36 is done in two high-yielding steps: reduction to the hydroquinone and alkyl transfer catalyzed by tetrabutyl ammonium fluoride (TBAF) to form a cyclic ether rather than a lactone.
Fig. 11.

Soluble model for a trimethyl lock resin for the synthesis of peptides starting at the C terminus without the need for a linker.36
3.2. Oligonucleotide-synthesis resin
A solid-phase resin based on the trimethyl lock has been used for the synthesis of oligonucleotides.37 The nascent oligonucleotide was conjugated through a phosphodiester bond (37, Fig. 12). Upon cleavage of the ester with ammonia, cyclization released an oligonucleotide 3′-phosphate (or 3′-phosphorothioate). This strategy is similar to that in 36,36 though the rate here was enhanced by the better leaving group (i.e., a phosphate monoester), and proceeded spontaneously. Notably, this resin produces oligonucleotides with a 3′-phosphoryl group rather than the 3′-hydroxyl group generated by solid-phase synthesis using standard resins.
Fig. 12.

Trimethyl lock resin for the synthesis of oligonucleotides.37
4. Fluorogenic probes
Phenolic fluorophores such as fluorescein, 7-hydroxycoumarin, and their derivatives, are in widespread use.38 These dyes suffer, however, from two significant limitations. First, their fluorescence relies on the phenolate form and hence decreases at low pH. For example, the pKa of the phenolic hydroxyl group of fluorescein is 6.3.39 Secondly, although their fluorescence can be masked by acylation of the phenolic oxygen, such esters are unstable to spontaneous hydrolysis. The extensive conjugation makes the phenolate an exceptional leaving group. These limitations are necessarily intertwined, as substituents that decrease the pKa of the phenolic hydroxyl group relieve the pH-dependence near physiological pH but enhance the instability of an ensuing phenolic ester.
In contrast to that of phenolic fluorophores, the fluorescence of anilino fluorophores, such as rhodamine and 7-aminocoumarin, is independent of pH. Their fluorescence can likewise be masked by acylation.42 Although the ensuing amides can be substrates for proteases (38, Fig. 13),40 they have limited utility otherwise.
Fig. 13.

Fluorogenic probes that generate rhodamine 110 upon catalysis by a protease (38)40 or esterase (39).41
The trimethyl lock enables chemical biologists to bridge these two types of fluorophores, combining the best attributes of both. In probe 39 (Fig. 13), the ester linkage is insulated from the fluorophore, but cleavage of its otherwise recalcitrant amide bond is coupled to ester hydrolysis.41 This and other rhodamine derivatives can be accessed expeditiously by palladium-catalyzed C–N cross-coupling of fluorescein ditriflates and dihydrocinnamides.43 The initial report of probe 39 has led to a suite of latent fluorophores in which fluorescence generation is triggered by enzymatic and non-enzymatic reactions that include hydrolysis, oxidation, and reduction, as described below.
4.1. Esterase substrates
Desymmetrization of rhodamine 110 by appending only one trimethyl lock led to simplified (i.e., “one-hit”) kinetics, allowing functionalization of the second amino group for additional utility.44 Moreover, converting the second amino group into a urea provides a high quantum yield in the product fluorophore. This urea can be functionalized with maleimide or succinimide linkers (40 and 41, Fig. 14) for conjugation to biomacromolecules.44,45 The ensuing esterase substrates have been used to monitor the cellular internalization of proteins,44,48,49 to label expressed proteins using the HaloTag® system,45 and to image the trafficking of cell-surface receptors using polymers with a multivalent display of ligands.50 The fluorescence of a 7-aminocoumarin and cresyl violet were masked with the same esterase-cleavable trimethyl lock to generate blue and red fluorogenic esterase substrates (42 and 43, Fig. 15),46 as well as a chromogenic esterase substrate 44 with much greater chemical stability than p-nitrophenyl acetate.47
Fig. 14.

Fluorogenic labels based on acetyl trimethyl lock rhodamine.44,45 The image is of three live, unwashed HeLa cells exposed for 1 h to a 40–protein conjugate (10 μM). The nuclei are stained blue with Hoechst 33342.44 The punctate green staining indicates that the conjugate is in endosomes that contain esterase activity, which activates the trimethyl lock. The conjugate is not fluorescent in the extracellular space, which lacks esterase activity. Scale bar: 20 μm.
Fig. 15.

Substrates for esterases based on acetyl trimethyl lock dyes: aminomethyl coumarin (42), cresyl violet (43), and p-nitroaniline (44).46,47
4.2. DT-Diaphorase substrates
Fluorogenic substrates have been developed for a quinone reductase, DT-diaphorase, that might be up-regulated in cancerous cells, making this enzyme a potential drug target.51,52 Di(quinone trimethyl lock) rhodamine 110 derivative 45 and coumarin derivative 46 (Fig. 16) can be used to search for inhibitors. As DT-diaphorase requires NADH as a cofactor. The enzyme and substrates 45 and 46 have been employed in coupled assays for enzymes that generate NADH, including glucose-1-dehydrogenase, glucose-6-phosphate dehydrogenase/phosphoglucomutase, and 3-hydroxybutyrate dehydrogenase. Of note, these assays are insensitive to atmospheric oxygen, distinguishing them from other assays for these enzymes.
Fig. 16.

4.3. Cytochrome P450 substrate
Cytochrome P450 enzymes metabolize many small-molecule chemotherapeutic agents and environmental toxins. Understanding their activity and regulation is important to pharmacology, oncology, and toxicology. Available fluorogenic substrates, 7-ethoxycoumarin and 7-ethoxyresorufin, are dealkylated oxidatively by P450 enzymes to yield a fluorescent molecule. Electrons on the ether oxygens of these substrates delocalize, resulting in high background fluorescence.56 An ethyl ether of the trimethyl lock serves as a superior substrate for the cytochrome P450 isozyme CYP1A1 (47, Fig. 17).53 This substrate revealed the induction of activity in lung cells by a carcinogenic dioxin, as well as its repression by a chemoprotectant, resveratrol.
Fig. 17.

Fluorogenic substrate for a cytochrome P450 enzyme based on O-ethyl trimethyl lock rhodamine.53
4.4. Alkaline phosphatase substrates
Alkaline phosphatase from Escherichia coli is a widely used enzyme for enzyme-linked immunosorbant assays (ELISAs) and as a model for other phosphomonoesterases.57,58 A product of the reaction, inorganic phosphate, is a potent inhibitor of the enzyme (Ki ~1 μM). Accordingly, assays that require a high concentration of substrate are problematic.59 Common substrates for sensitive assays based on luminescence (e.g., luciferin 6′-O-phosphate) and fluorescence (e.g., 3,6-fluorescein diphosphate and 4-methyl umbelliferyl phosphate) are compromised by instability. To overcome this problem, a phosphorylated trimethyl lock has been appended to 6′-aminoluciferin54 and morpholinourea rhodamine55 (48 and 49, Fig. 18) to provide stable substrates for highly sensitive assays.
Fig. 18.

Luminescent and fluorogenic substrates for a phosphatase.54,55
5. Biological switches
5.1. Subcellular localization
The ability to toggle the function of a peptide or protein in cells has the potential to be a powerful tool in chemical biology. Photoswitching can increase the scope further by imposing temporal control over a cellular function. The trimethyl lock has been incorporated into a photoactivated nucleus→cytosol shuttle peptide (50, Fig. 19).60 Exposing cells to ultraviolet light shifts the subcellular localization of a peptide from the nucleus to the cytosol. The key is the bifunctionalization of the trimethyl lock by α-amination of its carboxyl group, essentially transforming the trimethyl lock into an α-amino acid. The hydroxyl group of the trimethyl lock was masked as an o-nitrobenzyl ether, a photocleavable moiety. The N terminus of the phenylalanine derivative was elaborated with a cationic cell-penetrating peptide (R8) sequence and nuclear localization signal (KRKRR). The cleavable C terminus was conjugated to a second peptide containing a nuclear export signal (LARLFSALGV) in two fragments. Fluorescent trimethyl lock 50 was directed to the nucleus of a mammalian cell; but upon irradiation, the nuclear export signal was constituted and fluorescent fragment 51 left the nucleus. Other derivatives of trimethyl lock 50 allowed cleavage with two-photon infrared light.61 A FRET-based assay was developed and used to optimize the amino-acid sequence for the N→O acyl transfer reaction that follows unmasking of the trimethyl lock.62 Finally, the trimethyl lock derivative of phenylalanine was synthesized by an enantioselective route.63
Fig. 19.

Nucleus→cytosol shuttle peptide activated by a photolytic trimethyl lock. The R8 and KRKRR components direct 50 to the nucleus; the constituted LARLFSALGV component directs the nuclear export of 51.60,61
5.2. Nucleic acid unzipping
The α-amino acid trimethyl lock has also been used to synthesize cleavable peptide nucleic acids (PNAs).64 Trimethyl lock 52 (Fig. 20) contains eight thymine nucleobases and hybridizes to (dA)9 to form a duplex with a Tm of 25 °C. Upon cleavage of the p-nitrosulfonyl ester by an exogenous thiol, the PNA splits in half, and the duplex Tm decreases to <10 °C.
Fig. 20.

Sulfonyl trimethyl lock that splits a T8 PNA into two T4 fragments upon activation.64
5.3. Release from liposomes
The trimethyl lock has served as the basis for a liposomal delivery system that releases its contents in a reducing environment (53, Fig. 21).66 A quinone trimethyl lock was used to N-acylate dioleylphosphatidylethanolamine (DOPE), modifying the charge of the lipid. The N-acylated DOPE formed liposomes. Reduction of the quinone trimethyl lock released DOPE, allowing the lipids to transition from a lamellar to an inverted micellar phase.65 The ensuing liposomal instability led to the release of its contents.
Fig. 21.

Quinone trimethyl lock that modifies the charge of a DOPE lipid.65,66
6. Conclusions
The trimethyl lock has been employed in chemical, biological, and pharmacological contexts, providing exceptional stability of conjugates until initiation of a designated reaction triggers rapid scission. The trimethyl lock is a readily accessible and highly adaptable module. Its conjugation to the amino group of a molecule of interest requires only a condensation reaction, and its trigger can be exchanged to alter spatial or temporal aspects of release. The breadth of its demonstrated utility has been remarkable. For example, a trimethyl lock has been used to support the synthesis of a peptide, enhance its bioavailability, image its cellular internalization, and alter its function or localization inside of a cell. No other module provides such versatility, predicating a continuing expansion of its use.
Acknowledgments
The authors thank L. D. Lavis and T. T. Hoang for their critical review of this manuscript. M.N.L was supported by the Medical Scientist Training Program. Work on the trimethyl lock in our laboratory is supported by Grants R01 CA073808 and R01 GM044783 from the NIH.
Notes and references
- 1.Thanassi JW, Cohen LA. Biochim Biophys Acta. 1969;172:389–398. doi: 10.1016/0005-2728(69)90135-2. [DOI] [PubMed] [Google Scholar]
- 2.Milstien S, Cohen LA. Proc Natl Acad Sci USA. 1970;67:1143–1147. doi: 10.1073/pnas.67.3.1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Milstien S, Cohen LA. J Am Chem Soc. 1972;94:9158–9165. doi: 10.1021/ja00781a029. [DOI] [PubMed] [Google Scholar]
- 4.Borchardt RT, Cohen LA. J Am Chem Soc. 1972;94:9166–9174. doi: 10.1021/ja00781a030. [DOI] [PubMed] [Google Scholar]
- 5.Borchardt RT, Cohen LA. J Am Chem Soc. 1972;94:9175–9182. doi: 10.1021/ja00781a031. [DOI] [PubMed] [Google Scholar]
- 6.Winans RE, Wilcox CF. J Am Chem Soc. 1976;98:4281–4285. [Google Scholar]
- 7.Danforth C, Nicholson AW, James JC, Loudon GM. J Am Chem Soc. 1976;98:4275–4281. [Google Scholar]
- 8.Wang B, Nicolaou MG, Liu S, Borchardt RT. Bioorg Chem. 1996;24:39–49. [Google Scholar]
- 9.Amsberry KL, Gerstenberger AE, Borchardt RT. Pharm Res. 1991;8:455–461. doi: 10.1023/a:1015890809507. [DOI] [PubMed] [Google Scholar]
- 10.Karaman R. Res Lett Org Chem. 2009;2009:240253. [Google Scholar]
- 11.Bruice TC, Pandit UK. J Am Chem Soc. 1960;82:5858–5865. [Google Scholar]
- 12.Testa B, Mayer JM. Hydrolysis in Drug and Prodrug Metabolism: Chemistry, Biochemistry and Enzymology. Verlag Helvetica Chimica Acta; Zürich, Switzerland: 2003. [Google Scholar]
- 13.Stella VJ, Borchardt RT, Hageman MJ, Oliyai R, Maag H, Tilley JW, editors. Prodrugs: Challenges and Rewards. Springer; New York, NY: 2007. [Google Scholar]
- 14.Bryant RAR, Hansen DE. J Am Chem Soc. 1996;118:5498–5499. [Google Scholar]
- 15.Radzicka A, Wolfenden R. J Am Chem Soc. 1996;118:6105–6109. [Google Scholar]
- 16.Smith RM, Hansen DE. J Am Chem Soc. 1998;120:8910–8913. [Google Scholar]
- 17.Vyas DM, Wong H, Crosswell AR, Casazza AM, Knipe JO, Mamber SW, Doyle TW. Bioorg Med Chem Lett. 1993;3:1357–1360. [Google Scholar]
- 18.Ueda Y, Mikkilineni AB, Knipe JO, Rose WC, Casazza AM, Vyas DM. Bioorg Med Chem Lett. 1993;3:1761–1766. [Google Scholar]
- 19.Nicolaou MG, Yuan C, Borchardt RT. J Org Chem. 1996;61:8636–8641. doi: 10.1021/jo961069l. [DOI] [PubMed] [Google Scholar]
- 20.Nam NH, Kim Y, You YJ, Hong DH, Kim HM, Ahn BZ. Bioorg Med Chem. 2003;11:1021–1029. doi: 10.1016/s0968-0896(02)00514-x. [DOI] [PubMed] [Google Scholar]
- 21.Weerapreeyakul N, Anorach R, Khuansawad T, Yenjai C, Isaka M. Chem Pharm Bull. 2007;55:930–935. doi: 10.1248/cpb.55.930. [DOI] [PubMed] [Google Scholar]
- 22.Wani MC, Taylor HL, Wall ME, Coggon P, McPhail AT. J Am Chem Soc. 1971;93:2325–2327. doi: 10.1021/ja00738a045. [DOI] [PubMed] [Google Scholar]
- 23.Dordunoo SK, Burt HM. Int J Pharm. 1996;133:191–201. [Google Scholar]
- 24.Rowinsky EK, Cazenave LA, Donehower RC. J Natl Cancer Inst. 1990;82:1247–1259. doi: 10.1093/jnci/82.15.1247. [DOI] [PubMed] [Google Scholar]
- 25.Amsberry KL, Borchardt RT. Pharm Res. 1991;8:323–330. doi: 10.1023/a:1015885213625. [DOI] [PubMed] [Google Scholar]
- 26.Nicolaou MG, Wolfe JL, Schowen RL, Borchardt RT. J Org Chem. 1996;61:6633–6638. doi: 10.1021/jo961069l. [DOI] [PubMed] [Google Scholar]
- 27.Gharat L, Taneja R, Weerapreeyakul N, Rege B, Polli J, Chikhale PJ. Int J Pharm. 2001;219:1–10. doi: 10.1016/s0378-5173(01)00599-3. [DOI] [PubMed] [Google Scholar]
- 28.Lee HH, Palmer BD, Wilson WR, Denny WA. Bioorg Med Chem Lett. 1998;8:1741–1744. doi: 10.1016/s0960-894x(98)00305-9. [DOI] [PubMed] [Google Scholar]
- 29.Naughton DP. Adv Drug Deliver Rev. 2001;53:229–233. doi: 10.1016/s0169-409x(01)00229-0. [DOI] [PubMed] [Google Scholar]
- 30.Dillon MP, Cai H, Maag H. Bioorg Med Chem Lett. 1996;6:1653–1656. [Google Scholar]
- 31.Harris JM, Chess RB. Nat Rev Drug Discov. 2003;2:214–221. doi: 10.1038/nrd1033. [DOI] [PubMed] [Google Scholar]
- 32.Maeda H, Sawa T, Konno T. J Control Release. 2001;74:47–61. doi: 10.1016/s0168-3659(01)00309-1. [DOI] [PubMed] [Google Scholar]
- 33.Greenwald RB, Choe YH, Conover CD, Shum K, Wu D, Royzen M. J Med Chem. 2000;43:475–487. doi: 10.1021/jm990498j. [DOI] [PubMed] [Google Scholar]
- 34.Wang BH, Gangwar S, Pauletti GM, Siahaan TJ, Borchardt RT. J Org Chem. 1997;62:1363–1367. [Google Scholar]
- 35.Zheng A, Shan D, Wang B. J Org Chem. 1999;64:156–161. doi: 10.1021/jo981528d. [DOI] [PubMed] [Google Scholar]
- 36.Shan D, Zheng A, Ballard CE, Wang W, Borchardt RT, Wang B. Chem Pharm Bull. 2000;48:238–244. doi: 10.1248/cpb.48.238. [DOI] [PubMed] [Google Scholar]
- 37.Cheruvallath ZS, Cole DL, Ravikumar VT. Bioorg Med Chem Lett. 2003;13:281–284. doi: 10.1016/s0960-894x(02)00922-8. [DOI] [PubMed] [Google Scholar]
- 38.Lavis LD, Raines RT. ACS Chem Biol. 2008;3:142–155. doi: 10.1021/cb700248m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lavis LD, Rutkoski TJ, Raines RT. Anal Chem. 2007;79:6775–6782. doi: 10.1021/ac070907g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Beija M, Afonso CAM, Martinho JMG. Chem Soc Rev. 2009;38:2410–2433. doi: 10.1039/b901612k. [DOI] [PubMed] [Google Scholar]
- 41.Chandran SS, Dickson KA, Raines RT. J Am Chem Soc. 2005;127:1652–1653. doi: 10.1021/ja043736v. [DOI] [PubMed] [Google Scholar]
- 42.Leytus SP, Melhado LL, Mangel WF. Biochem J. 1983;209:299–307. doi: 10.1042/bj2090299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Grimm JB, Lavis LD. Org Lett. 2011;13:6354–6357. doi: 10.1021/ol202618t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lavis LD, Chao TY, Raines RT. ACS Chem Biol. 2006;1:252–260. doi: 10.1021/cb600132m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Watkins RW, Lavis LD, Kung VM, Los GV, Raines RT. Org Biomol Chem. 2009;7:3969–3975. doi: 10.1039/b907664f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lavis LD, Chao TY, Raines RT. Chem Bio Chem. 2006;7:1151–1154. doi: 10.1002/cbic.200500559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Levine MN, Lavis LD, Raines RT. Molecules. 2008;13:204–211. doi: 10.3390/molecules13020204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Turcotte RF, Lavis LD, Raines RT. FEBS J. 2009;276:4270–4281. doi: 10.1111/j.1742-4658.2009.07098.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chao TY, Lavis LD, Raines RT. Biochemistry. 2010;49:10666–10673. doi: 10.1021/bi1013485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mangold SL, Carpenter RT, Kiessling LL. Org Lett. 2008;10:2997–3000. doi: 10.1021/ol800932w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Huang ST, Lin YL. Org Lett. 2006;8:265–268. doi: 10.1021/ol052635e. [DOI] [PubMed] [Google Scholar]
- 52.Huang ST, Peng YX, Wang KL. Biosens Bioelectron. 2008;23:1793–1798. doi: 10.1016/j.bios.2008.02.015. [DOI] [PubMed] [Google Scholar]
- 53.Yatzeck MM, Lavis LD, Chao TY, Chandran SS, Raines RT. Bioorg Med Chem Lett. 2008;18:5864–5866. doi: 10.1016/j.bmcl.2008.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhou W, Andrews C, Liu J, Shultz JW, Valley MP, Cali JJ, Hawkins EM, Klaubert DH, Bulleit RF, Wood KV. Chem Bio Chem. 2008;9:714–718. doi: 10.1002/cbic.200700644. [DOI] [PubMed] [Google Scholar]
- 55.Levine MN, Raines RT. Anal Bio chem. 2011;418:247–252. doi: 10.1016/j.ab.2011.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Farinas ET, Schwaneberg U, Glieder A, Arnold FH. Adv Synth Catal. 2001;343:601–606. [Google Scholar]
- 57.Porstmann B, Porstmann T, Nugel E, Evers U. J Immunol Methods. 1985;79:27–37. doi: 10.1016/0022-1759(85)90388-6. [DOI] [PubMed] [Google Scholar]
- 58.Cleland WW, Hengge AC. Chem Rev. 2006;106:3252–3278. doi: 10.1021/cr050287o. [DOI] [PubMed] [Google Scholar]
- 59.O’Brien PJ, Herschlag D. Biochemistry. 2002;41:3207–3225. doi: 10.1021/bi012166y. [DOI] [PubMed] [Google Scholar]
- 60.Shigenaga A, Tsuji D, Nishioka N, Tsuda S, Itoh K, Otaka A. Chem Bio Chem. 2007;8:1929–1931. doi: 10.1002/cbic.200700442. [DOI] [PubMed] [Google Scholar]
- 61.Shigenaga A, Yamamoto J, Sumikawa Y, Furuta T, Otaka A. Tetrahedron Lett. 2010;51:2868–2871. [Google Scholar]
- 62.Shigenaga A, Yamamoto J, Hirakawa H, Yamaguchi K, Otaka A. Tetrahedron. 2009;65:2212–2216. [Google Scholar]
- 63.Shigenaga A, Yamamoto J, Nishioka N, Otaka A. Tetrahedron. 2010;66:7367–7372. [Google Scholar]
- 64.Shigenaga A, Yamamoto J, Hirakawa H, Ogura K, Maeda N, Morishita K, Otaka A. Tetrathedron Lett. 2010;51:2525–2528. [Google Scholar]
- 65.Siegel DP, Epand RM. Biophys J. 1997;73:3089–3111. doi: 10.1016/S0006-3495(97)78336-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ong W, Yang Y, Cruciano AC, McCarley RL. J Am Chem Soc. 2008;130:14739–14744. doi: 10.1021/ja8050469. [DOI] [PMC free article] [PubMed] [Google Scholar]