

Abstract
The level and integrity of glutamate transmission during critical periods of postnatal development plays an important role in the refinement of pyramidal neuron dendritic arbor, synaptic plasticity, and cognition. Presently, it is not clear how excitatory transmission via the two predominant isoforms of the vesicular glutamate transporter (VGLUT1 and VGLUT2) participate in this process. To assess a neurodevelopmental role for VGLUT2 in pyramidal neuron maturation, we generated recombinant VGLUT2 knock-out mice and inactivated VGLUT2 throughout development using Emx1-Cre+/+ knock-in mice. We show that VGLUT2 deficiency in corticolimbic circuits results in reduced evoked glutamate transmission, release probability, and LTD at hippocampal CA3-CA1 synapses during a formative developmental period (postnatal days 11–14). In adults, we find a marked reduction in the amount of dendritic arbor across the span of the dendritic tree of CA1 pyramidal neurons and reduced long-term potentiation and levels of synaptic markers spinophilin and VGLUT1. Loss of dendritic arbor is accompanied by corresponding reductions in the number of dendritic spines, suggesting widespread alterations in synaptic connectivity. Conditional VGLUT2 knock-out mice exhibit increased open-field exploratory activity yet impaired spatial learning and memory, endophenotypes similar to those of NMDA receptor knock-down mice. Remarkably, the impairment in learning can be partially restored by selectively increasing NMDA receptor-mediated glutamate transmission in adult mice by prolonged treatment with d-serine and a d-amino acid oxidase inhibitor. Our data indicate that VGLUT2 expression is pivotal to the proper development of mature pyramidal neuronal architecture and plasticity, and that such glutamatergic deficiency leads to cognitive malfunction as observed in several neurodevelopmental psychiatric disorders.
Introduction
A large body of work has revealed a close association between activity-dependent plasticity, such as long-term depression (LTD) and long-term potentiation (LTP), dendrite refinement, and cognition (Nägerl et al., 2004; Cline and Haas, 2008). Alterations in dendritic structure and plasticity lead to cognitive impairment in some models of neurodevelopmental psychiatric disorders (Broadbelt et al., 2002; Zoghbi, 2003; Asaka et al., 2006; Ramsey, 2009; Penzes et al., 2011; Na et al., 2012). Recent work suggests that deficits in glutamate release might also contribute to impaired dendritic structure and cognition (Sorensen and Rubel, 2006; Chao et al., 2007; Oni-Orisan et al., 2008; Jentsch et al., 2009; Wallén-Mackenzie et al., 2009; Balschun et al., 2010; Sorensen and Rubel, 2011). Glutamate release from most excitatory synapses in mammalian brain relies on the vesicular transporter isoforms VGLUT1 and VGLUT2 (Fremeau et al., 2004a). Evidence for specialized physiological roles for VGLUT1 and VGLUT2 include the following: (1) presence of only a single VGLUT isoform in lower organisms (Lee et al., 1999; Daniels et al., 2004); (2) differential distribution in neurons where LTP and LTD are generally studied (Fremeau et al., 2001; Herzog et al., 2001; Varoqui et al., 2002); (3) differential release probability when expressed in hippocampal pyramidal neurons in vitro (Weston et al., 2011); (4) differential activity-dependent regulation in neocortical neuronal networks in vitro (De Gois et al., 2005; Erickson et al., 2006; Doyle et al., 2010); (5) differential transcriptional response to psychiatric drugs in vivo (Moutsimilli et al., 2005; Tordera et al., 2005; Moutsimilli et al., 2008); and (6) differential behavioral effects of gene deletion relevant to psychiatric/cognitive illness (Moechars et al., 2006; Tordera et al., 2007; Wallén-Mackenzie et al., 2009; Balschun et al., 2010).
VGLUT1 is the predominant transporter in adult pyramidal neurons (Kaneko and Fujiyama, 2002), while VGLUT2 is the major isoform in thalamocortical sensory projections (Fujiyama et al., 2001). An important window for the maturation of VGLUT1-encoded transmission in pyramidal neurons occurs during the second and third postnatal weeks in rodents (Bolshakov and Siegelbaum, 1995; Wasling et al., 2004; Wilson et al., 2005). We hypothesize that VGLUT2 plays an important role in the maturation of cortical VGLUT1 transmission as it is expressed in a considerable number of VGLUT1-containing pyramidal neurons and synapses during critical periods of developmental plasticity (Boulland et al., 2004; Fremeau et al., 2004b; De Gois et al., 2005; Herzog et al., 2006; Nakamura et al., 2007). To assess a role for VGLUT2-encoded transmission in pyramidal neuron maturation and plasticity, we generated recombinant VGLUT2 knock-out mice and inactivated VGLUT2 using Emx1-Cre+/+ knock-in mice. Emx1-Cre is expressed in multiple areas during development that have roles in processing limbic information in adults, including the cingulate, prefrontal, temporal, and sensory association cortices, and in limbic structures involved in learning and memory, including the hippocampal formation (Gorski et al., 2002). Here, we examined the electrophysiological properties of young and adult hippocampal CA3-CA1 connections, dendritic structure of adult CA1 pyramidal neurons, and spatial learning and memory in control and VGLUT2-deficient mice. Our results support a critical neurodevelopmental role for VGLUT2 in the postnatal maturation of pyramidal neuron plasticity, dendritic refinement, and cognition.
Materials and Methods
Generation of VGLUT2 recombinant mice.
A 4.5 kb fragment upstream and a 3.9 kb fragment downstream of exon 2 of the murine (C57BL/6J) Slc17a6 gene that encodes VGLUT2 were amplified by high-fidelity Platinum Pfx Polymerase DNA (Epicenter Technologies) PCR from a bacterial artificial chromosome (BAC) mouse genomic clone (Invitrogen) and correspond to homologous 5′ and 3′ VGLUT2 arms, respectively. A 782 bp floxed region containing exon 2 was also obtained by PCR. These fragments were inserted into the conditional targeting vector pVBFRTCKR (Vega Biolabs) such that exon 2 is floxed by LoxP sites and the neomycin resistance gene is floxed by Frt sites. Primary sequences of exons 1–3 were verified in the targeting construct by direct sequencing before electroporation into mouse embryonic stem (ES) cells that were an F1 hybrid of C57BL/6J and 129 Sv mice.
We analyzed the DNA from initial ES cell transformants by Southern blotting to choose a sample for blastocyst injection by standard techniques. We distinguished the pattern of labeling in the normal genome from the homologous recombination using two probes (32P-dCTP labeled by random priming) and three different restriction digestions (BstEII, NsiI, and NdeI). Thirteen positive ES cell recombinants were identified by Southern analysis. The two cDNA probes were made against intronic sequences and amplified by PCR. For probe1 the outside primers were 5′-ctaatggtggaagagaggtgatgg-3′ (forward) and 5′-tctgagtctgagatgatgatgatgc-3′ (reverse); inside primers were 5′-ccgaattcaaggcttttacaactatggctaag-3′ (forward) and 5′-gcggatccagatggaactagacacgataggaag-3′ (reverse). For probe2 the outside primers were 5′-cgagaatgcttcttatccctcttc-3′ (forward) and 5′-atatgcagcctctgtgtagctatc-3′ (reverse); inside primers were 5′-ccgaattcaggaagtaagatgggatttcagagg-3′ (forward) and 5′-gcggatcctgatattcaagatggtagcaatgcc-3′ (reverse). BamHI and EcoRI restriction sites (in italics) were added to the 5′ ends of the inside set of primers to facilitate subcloning into a riboprobe vector (pBluescript). Sizes of wild-type genomic fragments cut by BstEII, NsiI, and NdeI are predicted to be 18.2, 6, and 5.9 kb, respectively. As new restriction sites are engineered into the vector targeting sequences (i.e., NsiI, NdeI, and BstEII at the 3′ LoxP site), new recombinant fragments can be predicted in the transformants. Following karyotyping and verification of normal chromosomal number in four of these samples, one clone was chosen and used for mouse blastocyst injection. Recombinant ES cells were injected into blastocysts at the Human Embryonic Stem Cell Core Facility at Yale University, New Haven, CT (courtesy of Dr. Tim Nolti) that were implanted into pseudo-pregnant females and resulted in four chimeric mice. Tail DNA from chimeric heterozygotic offspring was isolated using the DirectPCR Lysis Reagent (Viagen Biotec), digested with NdeI, and analyzed by Southern blotting using probe2 as described above in this paragraph.
The positive selectable marker at flanking Frt sites was excised by crossing chimeric mice to mice that express germ line flp recombinase (Farley et al., 2000). To show that the placement of the LoxP sites keeps the conditional allele fully functional for recombination by Cre recombinase and that they are in a position suitable for the deletion of exon 2, we have mated F1 offspring from the chimera/flp mice (VGLUT2+/fx) to a Cre-deleter strain (Schwenk et al., 1995) to obtain VGLUT2Δ/+ offspring with a single functional VGLUT2 allele. Then, we bred VGLUT2Δ/+ heterozygotes to each other and harvested embryonic day (E)16 embryos. Whole brains from offspring of VGLUT2Δ/+ heterozygotes were homogenized in cold PBS containing 5 μg/ml pepstatin, 5 μg/ml aprotinin, 5 μg/ml leupeptin, and 1 mm phenylmethylsulfonyl fluoride, subjected to brief sonication, and then centrifuged at 10,000 × g for 20 min. The supernatants were subsequently centrifuged at 100,000 × g for 30 min, the pellets were dissolved in SDS-containing sample buffer, and the membrane protein content was quantified. Vesicle-enriched membrane samples from VGLUT2+/+, VGLUT2Δ/+, and VGLUT2Δ/Δ mice (10 μg protein) were separated by SDS-PAGE for Western blotting and probed with anti-VGLUT2 antibodies as described (Varoqui et al., 2002).
To selectively silence VGLUT2 in corticolimbic circuits, VGLUT2fx/fx mice were bred with Emx1-Cre+/+ knock-in mice (Gorski et al., 2002) that are available from The Jackson Laboratory (stock no. 005628). Genotyping for the Emx1 or by default the Emx1-Cre knock-in, allele was determined by PCR using tail DNA and two primers sets: wild type (wt), 5′-aaggtgtggttccagaatcg-3′ (forward) and 5′-ctctccaccagaaggctgag-3′ (reverse) (378 bp); Cre, 5′-gcggtctggcagtaaaaactatc-3′ (forward) and 5′-gtgaaacagcattgctgtcactt-3′ (reverse) (100 bp). The 378 bp band is found only in the wild-type Emx1 locus and is lost in the Emx1-Cre genome because the 3′ downstream primer is in noncoding sequences of Emx1 that have been replaced by internal ribosome entry site (IRES) sequences followed by the coding sequences of Cre recombinase. Thus, Emx1 coding sequences in the Emx1-Cre gene locus do not differ between Emx-Cre+/+/VGLUT2fx/fx and Emx1-Cre+/−/VGLUT2fx/fx and are identical in VGLUT2fx/fx mice (Gorski et al., 2002). In addition, no Emx1 gene dosage effects occur between Emx-Cre+/+/VGLUT2fx/fx, Emx1-Cre+/−/VGLUT2fx/fx, and VGLUT2fx/fx mice (Gorski et al., 2002). Unexpectedly, some offspring of VGLUT2fx/fx breeding with the Emx1-Cre+/+ strain gave rise to null alleles (VGLUT2Δ allele), indicating occasional germline expression of Cre, although VGLUT2Δ/+ mice do not display any discernible electrophysiological phenotype or any change in hippocampus-dependent types of learning (Moechars et al., 2006; Balschun et al., 2010), and VGLUT2Δ/Δ mice die at birth.
To assess the efficiency of Cre-mediated recombination in vitro, primary cortical/hippocampal cultures were prepared from Emx1-Cre+/+/VGLUT2fx/fx and VGLUT2fx/fx embryos (E18) and treated with gabazine as described (Doyle et al., 2010). Fully differentiated neurons were harvested and disrupted using brief pulses from a probe sonicator, and total RNA was isolated using the Qiagen RNeasy MINI kit according to the manufacturer's instructions. RNA (1 μg) was reversed-transcribed using Bio-Rad iScript cDNA synthesis kit (oligo(dT) plus random hexamers) (Bio-Rad). The mRNA region of VGLUT2 between exons 1 and 3 was amplified using the following primer set 5′-ggaattccagtctttacaagaatggagtcg-3′ (forward) and 5′-ctggatccagaagaacgatccgtggatcat-3′ (reverse).
To assess the specificity of Cre-mediated recombination, in vivo Emx1-Cre+/+ mice were bred with Z/EG mice (Novak et al., 2000), which are available from The Jackson Laboratory (stock no. 004178). Genotyping for one or two copies of Emx1-Cre was determined as above. The primer sets, 5′-aagttcatctgcaccaccg-3′ (forward) and 5′tccttgaagaagatggtgcg-3′ (reverse), were used to detect the presence of enhanced green fluorescent protein (EGFP).
Mouse care.
Animals were maintained in a specific pathogen-free facility. Mice were group housed in a climate-controlled animal colony with a 12 h light/dark cycle with access to food and water ad libitum. Adequate measures were taken to minimize pain or discomfort. All procedures using animals were approved by Louisiana State University Health Sciences Center Institutional Animal Care Committee (New Orleans, LA) and are consistent with recommendations by the Panel of Euthanasia of the American Veterinary Medical Association.
Behavioral analysis.
The Morris water maze test (Morris, 1984) was used to evaluate spatial learning and memory as described previously (Tang et al., 1999). A circular water tank (diameter 120 cm and 75 cm in height) was filled with water that was made opaque with nontoxic white paint. A black curtain surrounded the pool and four visual cues were present (four standing lamps). The investigator was behind the curtain. A round platform (diameter 15 cm) was placed in the center of a given quadrant of the pool and was hidden 1 cm beneath the surface of the water. Training was conducted each day for 6–7 days (6–7 sessions), and each session consisted of four trials. In every trial the mouse was placed into the water facing the wall of the pool and allowed to search for the platform. The trial ended either when an animal climbed onto the platform or when a maximum of 60 s elapsed. At the end of each trial the mouse was allowed to rest on the platform for 15 s. Mice that failed to find the platform were placed on the platform and allowed to sit there for 15 s. In a block of four trials the starting position was varied pseudo-randomly among four positions. An interval of 2 h was set between two trials. Navigation was recorded by a video camera and the task performances, which included swimming paths, swimming speed, and time spent in each quadrant, were recorded and analyzed by a PolyTrack video tracking system (San Diego Instruments). A probe test was conducted 24 h after the completion of the training. In this test, the platform was removed from the pool and the task performances were recorded for 60 s. The time spent in each quadrant and the number of times the animal crossed the location where the platform was previously located were considered indices for their memory retention.
Novelty-induced exploratory activity was measured in an open-field Plexiglas chamber (41 × 41 cm). Mice were removed from their home cage and allowed to explore the open field chamber once. Forward movements (locomotion) were measured by the number of interruptions of infrared beams in the chamber, which were counted and transferred to a microcomputer via an interface. For open-field activity, forward locomotion scores were summed every 5 min for a duration of 1 h, or summed for 20 min.
Dendritic analysis.
Adult animals were killed by CO2 inhalation. Directly after sacrifice, the brains were excised, split into two hemispheres, and processed using a modified Golgi–Cox stain (Rapid Golgi; FD NeuroTechnologies). After impregnation, coronal sections (100 μm) were obtained using a cryostat, mounted on gelatin-coated glass slides, and then developed as per the manufacturer's instructions. Sections were counterstained with cresyl violet (Sigma-Aldrich), dehydrated, and cleared with xylenes. Slides were coverslipped using Permount (Fisher Scientific) and allowed to dry. For quantitative analyses, neurons from the CA1 region were imaged through the z-plane (Zeiss Axiovert 200M, Oberkochen, Germany) to allow for accurate reconstruction of the whole neuron in three dimensions using camera lucida software (Neurolucida; MBF Biosciences). Digital images were collected at 20× for quantification of dendritic parameters and at 100.8× for visualization of dendritic spines. For analyses of dendrite morphology, the following parameters were measured: number of primary dendrites; number of branch points and branch tips; total arbor per cell; average segment length; total number of dendritic segments. We have used these parameters previously to provide a reliable estimate of the length and complexity of dendritic arbor (Prithviraj and Inglis, 2008). Dendrites were also analyzed according to branch order (where a primary dendrite emanates directly from the cell body) and by Sholl analyses to provide estimates of intrinsic geometry and spatial distribution of dendritic trees. In a subset of neurons, the number of dendritic spines, spines per segment, and spine density per 10 μm dendritic length were compared between the two groups of mice.
Electrophysiological analysis.
Transverse acute hippocampal slices (350–400 μm) were prepared from young postnatal (P11–14) and adult (P45–60) mice. After decapitation, the brains were rapidly removed and cut in artificial CSF (ACSF) composed of (in mm) 125 NaCl, 2.5 KCl, 1 MgCl2, 25 NaHCO3, 1.25 NaH2PO4, 2 CaCl2, 25 glucose, 3 pyruvic acid, and 1 ascorbic acid. Slices were transferred to a holding chamber in an incubator containing oxygenated (95% O2, 5% CO2) ACSF at 34°C for 0.5–1 h and then maintained in an incubator containing oxygenated ACSF at room temperature for at least 1 h before recordings. Slices were then transferred to a nylon mesh interface recording chamber maintained at 30°C and constantly perfused with oxygenated ACSF (∼1 ml/min) containing (in mm) 120 NaCl, 3 KCl, 2 MgSO4, 2 CaCl2, 1.2 NaH2P04, 23 NaHCO3, and 11 glucose.
Hippocampal CA1 field potentials were evoked by constant current stimuli (200 μs) applied via bipolar stimulus electrodes to CA3 Schaffer collateral commissural projections. Field potentials were recorded via an extracellular glass microelectrode filled with ACSF placed in the stratum radiatum of CA1. LTP, paired-pulse facilitation (PPF), and input–output (I/O) function were measured as described previously (Fan et al., 2010). Evoked fEPSPs were amplified, digitized (Axoclamp-2B, DigiData 1320), and analyzed using Axon Clampfit 8.2. Hippocampal Schaffer collateral LTP was induced by a theta-burst stimulation (TBS) consisting of a series of 10 bursts of 5 stimuli at 100 Hz (200 ms interburst interval, which was repeated three times). The baseline stimulation strength was set to provide fEPSP with an amplitude of ∼30% from the subthreshold maximum derived from the input–output function. Paired-pulse stimulation was induced by delivering two pulses with an interpulse interval of 30–330 ms. The paired-pulse ratio (PPR) was calculated as fEPSP2/fEPSP1. LTD was induced by low-frequency stimulation (LFS) consisting of 900 shocks at 1 Hz for 15 min.
For whole-cell voltage-clamp analysis, sagittal hippocampal slices were prepared from young (P12-P16) mice. Slices were perfused and recorded at room temperature in a bicarbonate-buffered ACSF containing (in mm) 124 NaCl, 5 KCl, 26 NaHCO3, 1.23 NaH2PO4, 3 MgCl2, 2 CaCl2, and 10 glucose and bubbled constantly with 95% O2 and 5% CO2. Recordings were made using glass microelectrodes (Warner Instruments) filled with a cesium-based intracellular solution containing (in mm): 10 CsCl, 105 CsMeSO3, 8 NaCl, 0.5 ATP, 10 HEPES, 5 glucose, 2 MgCl2, and 3 EGTA (pH 7.3). Pipette resistances ranged from 3–13 MΩ. Series access resistances ranged from ∼7 to 35 MΩ and were monitored for consistency during recordings. Spontaneous AMPAR-mediated mEPSCs were isolated by voltage-clamping neurons at −65 mV in the presence of TTX (0.5 μm) and picrotoxin (50 μm). mEPSCs were acquired and analyzed using Igor Pro software (WaveMetrics) as described (Hall et al., 2007; Wang et al., 2011).
Biochemical analysis.
The expression of various synaptic markers in adult hippocampus was examined by Western blotting. Mouse hippocampi were dissected and homogenized in CelLytic MT Mammalian Tissue Lysis/Extraction Reagent (Sigma-Aldrich), briefly sonicated, and centrifuged at 10,000 × g for 10 min. Supernatants were collected and proteins were quantitated using the Bradford assay and then solubilized in Laemmli SDS sample buffer, size-fractionated on 10% precise Tris-HCl polyacrylamide gels, and electrophoretically transferred to nitrocellulose membrane using standard protocols. Primary antibodies used were rabbit anti-VGLUT1 (1:1500, (Varoqui et al., 2002), guinea pig anti-VGLUT2 (1:3000, (Schäfer et al., 2002), rabbit anti-VIAAT (vesicular inhibitory amino acid transporter, also known as vesicular GABA transporter VGAT) (1:3000, a kind gift of Dr. B. Gasnier, Paris, France), mouse PSD95 (1:1000, Sigma-Aldrich), rabbit anti-spinophilin (1:1000, Millipore), and mouse anti-β-actin (1:1000, Abcam). Secondary antibodies used were horseradish peroxidase-conjugated anti-rabbit, anti-guinea pig, or anti-mouse (1:8000; Sigma-Aldrich), followed by enhanced chemiluminescence (West Pico, Pierce) and exposure to Hyperfilm ECL (GE Healthcare). Individual bands were visualized by the enhanced chemiluminescence method (West Pico, Pierce), quantitated by densitometry MultiGauge software analysis, and normalized to a loading control (β-actin) or to PSD95, which did not differ between groups.
Immunohistochemistry.
Adult and 18-day-old Emx1-Cre+/+/VGLUT2fx/fx, Emx1-Cre+/−/VGLUT2fx/fx, and VGLUT2fx/fx mice were anesthetized with a mixture of ketamine and xylazine (100 mg/kg; 10 mg/kg), cardiac perfused with cold PBS followed by 4% paraformaldehyde (PFA) in PBS, and the brains were removed and divided into two hemispheres, postfixed by immersion in 4% PFA/PBS overnight, equilibrated with 20% sucrose/PBS, cut obliquely at 30° angle for parasagittal sectioning, and frozen in OCT. Alternatively, E16-E19 mice from VGLUT2Δ/+ × VGLUT2Δ/+ embryos were killed by decapitation and the brains removed, immersion fixed in 4% PAF/PBS overnight, equilibrated with 20% sucrose/PBS, and frozen in OTC for coronal sectioning. Emx1-Cre+/−/Z-EG × Emx1-Cre+/−/Z-EG offspring (P0 mice) were processed the same way except that immersion fixation in 4% PAF/PBS was for only 6 h to preserve the autofluorescence of EGFP. Section (35 μm) were cut and then floated onto PBS, postfixed in methanol at −20°C (5 min), washed in PBS, and then transferred to a 24-well chamber plate containing 400 μl of 10% (v/v) normal goat serum (NGS) and 3% bovine serum albumin (BSA) in 0.1 M Tris base/0.3 m NaCl (TBS) and 0.25% (w/v) Triton X-100, pH 7.4 (blocking buffer). Sections were incubated in blocking buffer overnight (4°C) and then incubated again overnight (4°C) in blocking buffer (200 μl) containing the primary antibodies guinea pig anti-VGLUT2 (1:5000), rabbit anti-VGLUT1 (1:5000), and/or rabbit anti-VIAAT (1:4000). Sections were then diluted tenfold with TBS (1 h, room temperature) and washed once (15 min) with TBS containing 0.2% BSA/0.1%Tween-20 followed by three rinses in TBS. Sections were then incubated in buffer containing 3% NGS/1% BSA/TBS for 1 h and then incubated in same buffer with species-specific and highly cross-adsorbed secondary antibodies coupled to Alexa Fluor 488 or 594 (Invitrogen) diluted 1:300 and 4′,6-diamidino-2-phenylindole (DAPI, Invitrogen) diluted to 0.5 μg/ml for 30 min at room temperature. Sections were then diluted tenfold with TBS, washed (15 min) two times with TBS containing 0.2% BSA/0.1% Tween 20 followed by three rinses in TBS. Sections were transferred to glass slides, mounted with Prolong Gold antifade reagent (Invitrogen), and viewed. For EGFP sections from Emx1-Cre+/−/Z/EG offspring, the methanol postfixation step and antibody reactions were omitted and they were cut coronally, floated onto PBS, mounted, and viewed directly. All images were obtained from a Leica DMRXA automated upright epifluorescent microscope, a Sensicam QE charge-coupled devise digital camera (PCO-TECH), and suitable filter sets (Chroma sets 4001 and 31004; Chroma Technology). Images were deconvolved using SlideBook 5 software (Intelligent Imaging Innovations).
Statistical Analysis.
Statistical comparisons were made by one-way, two-way, or repeated measures ANOVA with appropriate post hoc analyses or by Student's t test as indicated. I/O function and paired-pulse ratios (PPRs) for the two genotypes were analyzed in nested ANOVA. Interaction means (genotype × stimulus) were separated after the finding of a significant overall F test in the ANOVA as well as a significant effect of genotype in the nested term tests of the ANOVA model. Separation of genotype by stimulus interaction means was conducted by post hoc protected t tests on least square means (Milliken and Johnson, 1984) using α level adjustment for multiple comparisons by a method of simulation (Edwards and Berry, 1987). Statistics reported included mean ± SEM Statistical significance was set at p < 0.05.
Results
Emx1-Cre+/+/VGLUT2fx/fx mice are viable but developmentally delayed
We have used the Cre/LoxP and flp recombinase systems combined with VGLUT2 gene targeting to generate conditional VGLUT2 knock-out mice (VGLUT2fx/fx). We used homologous recombination in embryonic stem cells to produce a conditional allele of the mouse Slc17a6 gene encoding VGLUT2 with exon 2 surrounded by LoxP sites (Fig. 1A). Exon 2 contains part of the N terminus, the entire first transmembrane, and a portion of the first vesicle luminal domain including the predicted N-glycosylation sites. We analyzed the DNA from initial ES cell transformants by Southern blotting, which gave the expected reconstruction patterns (Fig. 1B). Chimeric mice were bred with flp mice (Farley et al., 2000) that express flp recombinase to excise the neomycin gene that was floxed by Frt sites (Fig. 1A). To show that the placement of the LoxP sites keeps the conditional allele fully functional for recombination by Cre recombinase, and that they are in a position suitable for the deletion of exon 2, we have mated F1 offspring from the chimera/flp mice (VGLUT2+/fx) to a Cre-deleter strain (Schwenk et al., 1995) to obtain VGLUT2Δ/+ offspring with a single functional VGLUT2 allele. Previous studies have shown that global VGLUT2Δ/Δ knock-out mice die at birth due to respiratory failure (Moechars et al., 2006; Wallén-Mackenzie et al., 2006). So, to assess the loss of VGLUT2 protein we crossed VGLUT2Δ/+ heterozygote mice and processed the brains from these E16-E19 embryos for Western blot and immunohistochemical analysis. Vesicle-enriched membrane fractions from harvested brains revealed that a VGLUT2 immunoreactive band (∼60 kDa) in wild-type VGLUT2+/+ mice is less abundant in VGLUT2Δ/+ mice and is not present in VGLUT2Δ/Δ mice (Fig. 1C). In addition, Cre-mediated recombination eliminated VGLUT2 expression in the brains from VGLUT2Δ/Δ mice by immunohistochemistry (IHC) (Fig. 1D).
Figure 1.
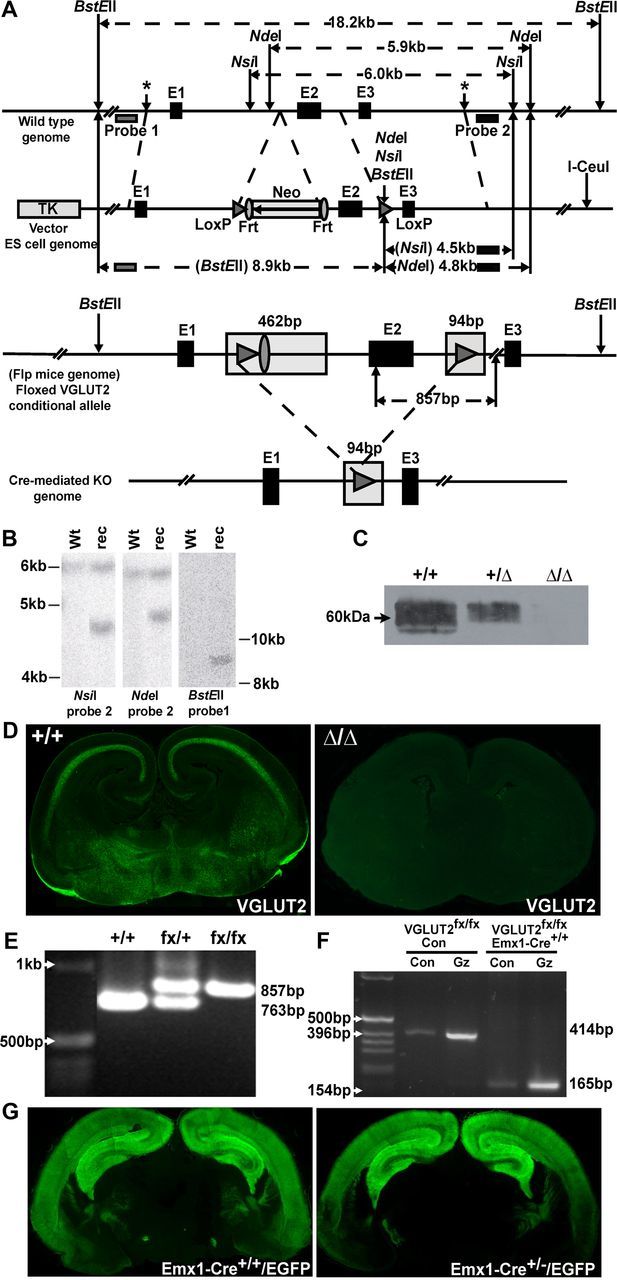
VGLUT2 gene targeting and inactivation of VGLUT2 in Emx1-Cre+/+/VGLUT2fx/fx mice. A, Top, A targeting construct engineered with LoxP sites flanking exon 2 of the Slc17a6 gene encoding VGLUT2 was inserted into the endogenous VGLUT2 locus by homologous recombination. Wild-type genome DNA schematic surrounding exons (E)1–3 is shown. The locations of Probe 1 and Probe 2 that recognize wild-type sequence are indicated. Approximate sites of insertion of vector targeting sequences (*, LoxP-Frt, and LoxP) in wild-type genome are shown. The vector cassette (pVBFRTCKR01) with the thymidine kinase promoter (TK) and I-CeuI vector linearization site are shown. The selectable marker (Neo, neomycin gene) is flanked by Frt sites. Exon 2 (E2), including this Frt region, is flanked by LoxP sites. Relative distances between sites in relation to the wild-type genome are marked and indicated in kilobase pairs (kb). New restriction sites (NsiI, NdeI, BstEII) introduced in the downstream LoxP site of the conditional allele are indicated (arrow), and the predicted sizes of fragments following restriction in the wild-type genome and conditional allele are shown. A, Bottom, Expanded view of floxed conditional VGLUT2 allele in flp+ mice whereby excision of the neomycin gene (Neo) has taken place, leaving one Frt site remaining in genome. Note: additional DNA sequences are added to intronic regions flanking exon 2 of VGLUT2 (shaded). B, Southern blot screening of ES cell genomic DNA from wild-type (Wt) and recombinant (rec) sample using NdeI and NsiI with probe 2 and BstEII with probe 1. In the normal genome, NsiI digestion gives rise to a 6.0 kb fragment and NdeI digestion gives rise to a 5.9 kb fragment hybridizing to probe 2. Instead, homologous recombination reveals bands of 4.5 kb and 4.8 kb following NsiI and NdeI digestion, respectively. BstEII digestion gives an 8.9 kb band in the recombinant (rec). C, Loss of VGLUT2 protein expression in VGLUT2Δ/Δ mice. Western blot of enriched whole brain vesicles prepared from E16-E19 wild-type (+/+), heterozygote (Δ/+), and unconditional VGLUT2 knock-out (Δ/Δ) mice. D, Immunostaining of wild-type mouse brain (+/+, left) shows VGLUT2 immunoreactivity absent from the unconditional VGLUT2 knock-out (Δ/Δ, right). E, Genotypes of wild-type (+/+), heterozygotes (fx/+), and homozygous (fx/fx) mice were verified by PCR using DNA extracted from tail samples and PCR amplification, revealing an 857 bp fragment in the floxed VGLUT2 conditional allele and a 763 bp fragment in the wild-type allele. F, VGLUT2 mRNA is inactivated by excision of exon 2 in cortical/hippocampal neurons in vitro from Emx1-Cre+/+/VGLUT2fx/fx mice. In VGLUT2fx/fx control cultures, a 414 bp fragment can be amplified using cRNA primers in exon 1 and exon 3, whereas only a 165 bp fragment was amplified from Emx1-Cre+/+/VGLUT2fx/fx cultures. Con, Control; GZ, gabazine. G, EGFP autofluorescence in coronal brain sections from Emx1-Cre+/+/Z-EG and Emx-Cre+/−/Z-EG mice shows the vast majority of EGFP+ neurons are found in cortical/hippocampal regions, demonstrating the specificity of recombination in vivo.
VGLUT2fx/+ mice were backcrossed with C57BL/6J mice for five generations and then bred with each other to produce homozygous VGLUT2fx/fx mice. Genotyping of offspring from VGLUT2fx/+ crosses reveals DNA fragments of defined size (Fig. 1E) by PCR corresponding to wild-type (+/+, 763 bp), heterozygote (fx/+, 763 bp and 857 bp), and homozygote (fx/fx, 857 bp) mice. VGLUT2fx/fx mice were subsequently bred with Emx1-Cre+/+ knock-in mice to inactivate VGLUT2 mRNA before birth (E9.5-E11.5) in corticolimbic areas (Gorski et al., 2002). We used Emx1-Cre+/−/VGLUT2fx/fx mice as breeding pairs to produce Emx1-Cre+/+/VGLUT2fx/fx, Emx1-Cre+/−/VGLUT2fx/fx, and VGLUT2fx/fx control mice that were generated at the expected Mendelian ratios. Emx1-Cre+/+/VGLUT2fx/fx mice are indistinguishable from Emx1-Cre+/−/VGLUT2fx/fx and VGLUT2fx/fx mice with regard to survival. However, Emx1-Cre+/+/VGLUT2fx/fx mice are smaller in size at P14 (Emx1-Cre+/+/VGLUT2fx/fx mice, 5.95 ± 0.5 g; VGLUT2fx/fx mice, 7.55 ± 0.6 g). Body weights of adult (P45–60) Emx1-Cre+/+/VGLUT2fx/fx mice, however, were not different from VGLUT2fx/fx mice (Emx1-Cre+/+/VGLUT2fx/fx mice, 20.6 ± 2.1 g; VGLUT2fx/fx mice, 23.0 ± 1.9 g).
To assess the efficiency of recombination (Fig. 1F) we prepared dissociated neuronal cultures from cortex and hippocampus dissections of brains from Emx1-Cre+/+/VGLUT2fx/fx and VGLUT2fx/fx embryos (E18) and pharmacologically treated mature neurons (day in vitro 18) with the GABAA-receptor antagonist gabazine (20 μm; 24 h) to increase neural network activity and trigger the induction of VGLUT2 biosynthesis (De Gois et al., 2005; Doyle et al., 2010). VGLUT2 mRNA expression was assessed by RT-PCR using primers that amplify the region between exon 1 and exon 3 of the VGLUT2 open-reading frame. A 414 bp PCR fragment was obtained from VGLUT2fx/fx control mice, which corresponds to wild-type VGLUT2 and contains exon 2. Only a shorter 165 bp fragment was induced by prolonged activity in cultures from Emx1-Cre+/+/VGLUT2fx/fx mice (Fig. 1F). Subcloning and direct sequencing of the shorter VGLUT2 amplicon indicated that it contained a stop codon in the open reading frame; the predicted amino acid sequence thus encodes a frame-shifted polypeptide (41 amino acids), thereby inactivating the VGLUT2 mRNA. Hence, while VGLUT2 mRNA is still expressed in cortical/hippocampal excitatory neurons from Emx1-Cre+/+/VGLUT2fx/fx mice, it has been inactivated and does not produce VGLUT2 protein.
Previous studies characterizing the specificity of recombination using Emx1-Cre mice have employed the R26R strain (Soriano, 1999) in which Cre-mediated recombination enables expression of lacZ-encoded β-galactosidase in corticolimbic areas (Gorski et al., 2002). Since Rosa26 and Emx1 are on the same chromosome and too near one another (13 centimorgan separation) for rare recombination events, we have used Z-EG, a double reporter mouse line that expresses EGFP upon Cre-mediated excision (Novak et al., 2000) to examine the specificity of recombination in Emx1-Cre+/+ mice. We find that high levels of EGFP autofluorescence in coronal sections from newborn Emx1-Cre+/+/Z-EG and Emx1-Cre+/−/Z-EG mice appeared to be similarly confined to cortical and hippocampal regions (Fig. 1G).
Impairment in spatial learning, yet increased novelty-exploratory activity, in Emx1-Cre+/+/VGLUT2fx/fx mice
To assess a physiological role for VGLUT2 in cognition, we have used the Morris water maze test to evaluate spatial learning and reference memory in adult Emx1-Cre+/+/VGLUT2fx/fx and Emx1-Cre+/−/VGLUT2fx/fx mice. Performance in this test relies on several cognitive functions, including learning, working and long-term memory, memory retention, and attention (Morris, 1984). Previous work has shown that spatial learning curves are not impaired in global VGLUT2+/− mice (Moechars et al., 2006). In addition, spatial learning is normal in CaMKIIαCre+/VGLUT2fx/fx mice where an escalating deletion of VGLUT2 occurs in cortical/hippocampal neurons from P15–30 (Minichiello et al., 1999; Wallén-Mackenzie et al., 2009), although spatial memory is impaired in adults (Wallén-Mackenzie et al., 2009). Our controls included VGLUT2fx/fx mice and Emx1-Cre+/+ mice, which did not differ. We trained Emx-Cre+/+/VGLUT2fx/fx mice, Emx1-Cre+/−/VGLUT2fx/fx mice, and control mice with a 6 day training protocol to find a hidden platform submerged under water. Interestingly, we found that Emx1-Cre+/+/VGLUT2fx/fx mice could not learn to navigate the maze and exhibited a significant impairment in spatial learning throughout the training period (Fig. 2A; F(2,305) = 12.418, p = 0.001). Since Emx1-Cre+/+/VGLUT2fx/fx mice failed the spatial navigation learning test, they similarly failed the test for spatial reference memory (Fig. 2C–E). Spatial memory was tested 24 h after the last training period by allowing the mice to search the pool in which the platform was removed. Emx1-Cre+/+/VGLUT2fx/fx mice did not spend more time in the quadrant where the platform was previously located (Fig. 2C; p = 0.015), and they crossed the area where the platform was previously located only 0.8 ± 0.3 times compared to control mice that crossed the area 3.7 ± 0.4 times (Fig. 2D; p = 0.001). In contrast, acquisition training showed comparable learning performance in Emx1-Cre+/−/VGLUT2fx/fx mice and in control mice (Fig. 2A). A trend for a decrease in the amount of time spent in the target quadrant (Fig. 2C; p = 0.071) was observed for Emx1-Cre+/−/VGLUT2fx/fx mice, and a significant decrease in the number of platform crossings (Fig. 2D; p = 0.037) during the memory probe test was observed for Emx1-Cre+/−/VGLUT2fx/fx mice (2.4 ± 0.3) compared to control mice (3.7 ± 0.4). These results indicate that Emx1-Cre+/−/VGLUT2fx/fx mice are similar to the CaMKIIα-Cre+/−/VGLUT2fx/fx mice described previously (Wallén-Mackenzie et al., 2009), as they exhibit a minor cognitive impairment in spatial reference memory. Thigmotaxis (i.e., wall swimming) was observed in many mice during the first couple days of training, but most Emx1-Cre+/+/VGLUT2fx/fx mice used a search strategy involving repetitive looping paths with persistent swimming around the outer 15 cm of the pool or a random search strategy during the probe test (Fig. 2E). Emx1-Cre+/+/VGLUT2fx/fx mice did not exhibit motor disturbances, as swimming speed and swimming distance for Emx1-Cre+/+/VGLUT2fx/fx mice was not different from those of Emx1-Cre+/−/VGLUT2fx/fx and control mice (Fig. 2B,E). Furthermore, we measured novelty-induced exploratory activity in open-field chambers and found that Emx1-Cre+/+/VGLUT2fx/fx mice displayed higher forward locomotion levels throughout a 1 h period, compared to control mice (Fig. 2F). This novelty-induced locomotion was reduced by 2 h in both groups of mice.
Figure 2.

Impairment of spatial learning yet increased novelty–exploratory activity in Emx1-Cre+/+/VGLUT2fx/fx mice. A, We used the Morris water maze to assess spatial learning. Comparison of the latency among Emx1-Cre+/+/VGLUT2fx/fx, Emx1-Cre+/−/VGLUT2fx/fx, and control VGLUT2fx/fx (Wt) mice shows impairment of spatial learning in Emx1-Cre+/+/VGLUT2fx/fx mice. A similar decrease in escape latency occurred for Emx1-Cre+/−/VGLUT2fx/fx mice and Wt mice during training. Data are presented as the mean ± SEM (n = 14–21 in each group; males and females); **p < 0.001; repeated-measures ANOVA with a LSD post hoc test. B, Comparison of swimming speed during training among Emx1-Cre+/+/VGLUT2fx/fx, Emx1-Cre+/−/VGLUT2fx/fx, and control mice showed no differences between groups. C, Control mice spent significantly more time in the quadrant where the platform was previously located in a probe test for spatial reference memory. *p < 0.05; ANOVA. In contrast, Emx1-Cre+/+/VGLUT2fx/fx and Emx1-Cre+/−/VGLUT2fx/fx mice showed no preference for any quadrant. D, During the probe test, control mice crossed the platform more often than Emx1-Cre+/−/VGLUT2fx/fx mice and Emx1-Cre+/+/VGLUT2fx/fx mice; *p < 0.05, **p < 0.01, significant difference from control mice; ANOVA. E, Swimming traces during the probe test. F, Emx1-Cre+/+/VGLUT2fx/fx mice retain higher forward locomotion level throughout a 1 h period compared to control mice using a novelty exploration test in an open-field chamber. Error bars represent SEM; *p < 0.05; Student's t test.
VGLUT2-deficiency in Emx1-Cre+/+/VGLUT2fx/fx mice results in reduced dendritic arbor and reduced number of spines in adult CA1 hippocampal pyramidal neurons
Aberrant dendritic structure of pyramidal neurons is associated with deficits in learning (Penzes et al., 2011). To determine whether the deficit in spatial learning in Emx1-Cre+/+/VGLUT2fx/fx mice was associated with aberrant dendritic structure, we used the Golgi–Cox stain to visualize neuronal architecture and spine distribution. We show that adult Emx1-Cre+/+/VGLUT2fx/fx mice exhibit substantial reductions in the total amount of dendritic arbor in CA1 pyramidal neurons in vivo. Reduction of dendritic length was found in both basal and apical dendrites (Fig. 3A–C; Table 1). Notably, there was no reduction in the number of dendritic segments; rather, loss of arbor was due to a reduction in the length of segments across the span of the dendritic tree. However, basal dendrites of Emx1-Cre+/+/VGLUT2fx/fx mice show a greater reduction in arbor over most of the dendritic tree (F(18,1044) = 4.345, p = 0.001). We also observed a trend toward a decrease in dendrite complexity in basal dendrites (Table 1), as indicated by the total number of interbranch dendritic segments (p = 0.054). These changes were accompanied by a decrease in the average segment length (Fig. 3C; Table 1).
Figure 3.

VGLUT2-deficiency results in reduced dendritic arbor and reduced number of spines in adult CA1 hippocampal pyramidal neurons. A, Golgi–Cox impregnated neurons and their corresponding digital camera lucida reconstructions. Scale bar represents 50 μm for whole neurons and 10 μm for spine images. Wt, Wild type. B, Basal dendrites of Emx1-Cre+/+/VGLUT2fx/fx mice show a significant reduction in arbor over most of the dendritic tree. Apical dendrites show incremental decreases in arbor over the tree. C, Basal and apical dendrites have significantly reduced total arbor and decreased average segment length. D, Both the basal and apical dendrites show reductions in the number of spines. E, Spine density remains unchanged across groups. Results for each group are obtained from 30 neurons among 6 animals. Error bars represent SEM; *p < 0.05, **p < 0.01; two-way repeated-measures ANOVA with a Scheffé post hoc or a Student's t test.
Table 1.
Effects of VGLUT2 deficiency on the dendrite arbor of adult CA1 pyramidal neurons
| Neuron parameter | VGLUT2fx/fx | Emx1-Cre+/+/VGLUT2fx/fx | t; p |
|---|---|---|---|
| Basal dendrites | |||
| Total dendrite arbor per cell (μm) | 1095 ± 73 | 810.7 ± 65.5** | t = −2.889; p = 0.005 |
| Total number of dendritic segments | 39.3 ± 2.3 | 33.7 ± 1095.3† | t = −1.965; p = 0.054 |
| Average length of segment (μm) | 26.8 ± 1.4 | 22.8 ± 1.6 | t = −1.921; p = 0.060 |
| Branch points | 17.9 ± 1.1 | 15.3 ± 0.9 | t = −1.837; p = 0.071 |
| Branch tips | 21.4 ± 1.1 | 18.4 ± 0.9* | t = −2.077; p = 0.042 |
| Apical dendrites | |||
| Total dendrite arbor per cell (μm) | 1270 ± 127 | 996.0 ± 72.5* | t = −2.168; p = 0.034 |
| Total number of dendritic segments | 48.7 ± 3.4 | 42.7 ± 2.7 | t = −1.374; p = 0.175 |
| Average length of segment (μm) | 33.0 ± 3.8 | 23.9 ± 1.4* | t = −2.239; p = 0.029 |
| Branch points | 23.8 ± 1.7 | 20.8 ± 1.3 | t = −1.397; p = 0.168 |
| Branch tips | 25 ± 1.7 | 21.9 ± 1.3 | t = −1.418; p = 0.162 |
Numbers represent mean ± SEM of parameters from VGLUT2fx/fx (n = 30, 6 animals) and Emx1-Cre+/+/ VGLUT2fx/fx (n = 30, 6 animals) mice. Significance was determined at p < 0.05 using the Student's t test.
*Significant difference from wild-type with p < 0.05;
**Significant difference from wild-type with p < 0.01;
†Difference trending to significance (p < 0.06).
Loss of dendritic arbor was accompanied by corresponding reductions in the number of dendritic spines, suggesting widespread alterations in the connectivity of hippocampal pyramidal neurons. Both the basal and apical dendrites show reductions in the number of spines (basal, F(14,714) = 2.055, p = 0.012; apical, F(16,912) = 2.155, p = 0.005). The Emx1-Cre+/+/VGLUT2fx/fx mice maintained a spine density equivalent to that of the VGLUT2fx/fx (wild type) controls (Table 2). Sholl analyses revealed that the reduction in dendritic arbor contributed to a decrease in the number of spines, especially in the basal dendrites (Fig. 3D,E), due to the overall loss of dendritic length. Previous work has revealed that deficient glutamatergic input early in postnatal development results in CA1 pyramidal neurons with reduced total length of the dendritic tree (Groc et al., 2002), while enhanced synaptic glutamate activity promotes dendritic arbor elaboration (Cline and Haas, 2008). Our observations suggest that a deficiency of VGLUT2-encoded glutamate transmission in Emx1-Cre+/+/VGLUT2fx/fx mice during postnatal development has long-term consequences on the maturation of pyramidal neuron dendritic arbor. These changes in dendrite architecture imply a decreased synaptic connectivity in adult mice that likely contributes to the deficit in spatial learning observed.
Table 2.
Effects of VGLUT2 deficiency on the number and distribution of spines on CA1 pyramidal neurons
| Spine parameter | VGLUT2fx/fx | Emx1-Cre+/+/VGLUT2fx/fx | t; p |
|---|---|---|---|
| Basal dendrites | |||
| Number of spines per segment | 55.9 ± 3.4 | 51.9 ± 5.4 | t = −0.620; p = 0.538 |
| Spine density per 10 μm dendrite length | 18.1 ± 0.7 | 17.7 ± 0.9 | t = −0.383; p = 0.703 |
| Apical dendrites | |||
| Number of spines per segment | 47.2 ± 3.2 | 41.4 ± 4.0 | t = −1.139; p = 0.259 |
| Spine density per 10 μm dendrite length | 19.2 ± 0.8 | 17.4 ± 0.9 | t = −1.513; p = 0.136 |
Numbers represent mean ± SEM of parameters from VGLUT2fx/fx (basal n = 26, apical n = 30; 6 animals) and Emx1-Cre+/+/VGLUT2fx/fx (basal n = 27, apical n = 30; 6 animals) mice. Significance was determined at p < 0.05 using the Student's t test.
VGLUT2-deficiency in Emx1-Cre+/+/VGLUT2fx/fx mice results in reduced evoked basal transmission, increased paired-pulse ratio, and reduced LTD in young CA3-CA1 connections
VGLUT2 loads synaptic vesicles with glutamate to be made available for release from synapses (Takamori et al., 2001). To test whether VGLUT2 deficiency results in altered basal synaptic glutamatergic transmission in young CA3–CA1 connections, we measured the input–output function in acute hippocampal slices (P11–14) from Emx1-Cre+/+/VGLUT2fx/fx mice and control VGLUT2fx/fx mice (wild type) by field potential recordings. We found that the I/O function (Fig. 4A) was lower in Emx1-Cre+/+/VGLUT2fx/fx mice compared to that of control mice (F(1,45) = 13.26; p = 0.001). To determine whether the reduction in basal excitatory transmission observed was associated with a presynaptic change in glutamate release probability at pyramidal neuron synapses, we assessed glutamate release probability by PPF, whereby the synaptic response following the second stimulus in a stimulus pair is larger than following the first stimulus. A change in the ratio of the second to the first response (PPR) is indicative of a change in release probability. We find that PPR is increased (Fig. 4B) at P11–14 CA3-CA1 synapses in Emx1-Cre+/+/VGLUT2fx/fx mice compared to VGLUT2fx/fx control mice (F(1,21) = 8.2; p = 0.009). An increased PPR is indicative of a decreased evoked glutamate release probability in young VGLUT2-deficient Emx1-Cre+/+/VGLUT2fx/fx CA3-CA1 hippocampal synapses.
Figure 4.

VGLUT2-deficiency results in reduced basal transmission, increased paired-pulse ratio, and reduced LTD in young CA3-CA1 connections. A, Representative fEPSP waveforms recorded at in the stratum radiatum of the CA1 region from hippocampal CA3–CA1 connections in young (P11–14) control VGLUT2fx/fx (Wt, wild type) and Emx1-Cre+/+/VGLUT2fx/fx mice. Input–output function (Wt, 29 recordings/6 animals; Emx1-Cre+/+/VGLUT2fx/fx, 18 recordings/5 animals). Stimulus intensity was normalized to the maximum intensity. B, Representative fEPSP traces. fEPSPs were induced by a paired-pulse protocol with varying interpulse intervals. Paired-pulse ratio (fEPSP2/fEPSP1) in control and Emx1-Cre+/+/VGLUT2fx/fx mice (Wt, 19 recordings/4 animals; Emx1-Cre+/+/VGLUT2fx/fx, 25 recordings/5 animals). C, Representative traces of fEPSPs recorded before (1) and after (2) LFS by stimulating the Schaffer collateral pathway in acute hippocampal slices from young (P11–14) animals. Time courses of fEPSP slopes are also shown. D, Representative traces of fEPSPs recorded before and after LFS by stimulating the Schaffer collateral pathway in acute hippocampal slices from adult (P45–60) animals. Time courses of fEPSP slopes are also shown. E, Mean values of the potentiation of fEPSPs averaged from 55 to 60 min following LFS (Wt, 11 recordings/5 animals; Emx1-Cre+/+/VGLUT2fx/fx, 10 recordings/5 animals). F, Mean values of the potentiation of fEPSPs averaged from 55 to 60 min following TBS (Wt, 7 recordings/4 animals; Emx1-Cre+/+/VGLUT2fx/fx, 10 recordings/4 animals). Error bars represent SEM; **p < 0.01; ANOVA.
In adults, synapses with high evoked release probability (i.e., VGLUT2-encoded neurons) more easily express some forms of synaptic plasticity, such as LTD, while those synapses with inherent low release probability (i.e., VGLUT1-encoded neurons) more easily express LTP (Varoqui et al., 2002). In contrast to the adult, LTD can be induced in CA3–CA1 connections by low-frequency stimulation (LFS) only during the second and third postnatal weeks of development (Dudek and Bear, 1992, 1993; Milner et al., 2004; Le Roux et al., 2007; Gonzalez-Burgos et al., 2008). To determine whether VGLUT2 transmission might contribute to age-dependent LTD, we measured LTD by field stimulation using a LFS protocol (1 Hz; 900 pulses). We verified that LTD is observed in young control hippocampal CA3–CA1 synapses (Fig. 4C), but not in the adult synapses (Fig. 4D). Importantly, we show that LTD was reduced in young Emx1-Cre+/+/VGLUT2fx/fx mice (Fig. 4C) compared to control mice (F(1,19) = 20.9; p = 0.001). Taken together, our electrophysiology data indicate that VGLUT2 deficiency in Emx1-Cre+/+/VGLUT2fx/fx mice results in persistent glutamatergic hypofunction during postnatal development that likely contributes to the deficit in the dendritic refinement of CA1 pyramidal neurons.
To assess the strength and frequency of spontaneous quantal synaptic transmission onto CA1 cells, AMPAR-mediated miniature excitatory postsynaptic currents (AMPA-mEPSCs) were recorded from CA1 pyramidal neurons in young slices. We found no significant difference in the mean amplitude of events in response to conditional knock-out of VGLUT2. We did observe a trend toward decreased interevent intervals in Emx1-Cre+/+/VGLUT2fx/fx neurons compared to control VGLUT2fx/fx neurons (8.7 ± 0.68 s, n = 41 vs 10.2 ± 0.88 s, n = 24; p = 0.16; t = 1.42; df = 63). This translated into a minor but significant increase in the frequency of AMPAR-mEPSCs in Emx1-Cre+/+/VGLUT2fx/fx neurons compared to controls (11.8 ± 0.75 events per 100 s vs 9.4 ± 0.83 events per 100 s; p = 0.04; t = −2.1). While the spontaneous release events are low, these data are consistent with a minor increase in the probability of spontaneous synaptic release from, or an increased number of, presynaptic inputs onto these neurons in Emx1-Cre+/+/VGLUT2fx/fx mice compared to VGLUT2fx/fx mice. Together with results from the PPR experiment that show a decrease in release probability by evoked release in Emx1-Cre+/+/VGLUT2fx/fx mice, these data may indicate that there could be distinct functional consequences of conditional VGLUT2 deficiency in relation to the probability of spontaneous versus synchronized synaptic glutamate release onto CA1 pyramidal neurons (Ramirez and Kavalali, 2011).
Evoked basal synaptic transmission and LTP are reduced in CA3-CA1 synapses in adult Emx1-Cre+/+/VGLUT2fx/fx mice
Persistent hypoglutamatergic function during postnatal development and diminished dendritic arbor may lead to altered synaptic plasticity in adults. Here, we measured basal excitatory transmission in adult Emx1-Cre+/+/VGLUT2fx/fx mice and control VGLUT2fx/fx mice by field potential recordings. We found that the evoked basal excitatory transmission, assessed by I/O function, was lower in adult Emx1-Cre+/+/VGLUT2fx/fx mice than in control mice (Fig. 5A; F(1,17) = 4.48; p = 0.04), similar to what we saw in young Emx1-Cre+/+/VGLUT2fx/fx mice (Fig. 4A). In contrast to the reduction in PPR observed in young Emx1-Cre+/+/VGLUT2fx/fx mice (Fig. 4B), no differences in PPR were observed between adult Emx1-Cre+/+/VGLUT2fx/fx mice and control mice (Fig. 5B). We also examined LTP in both young and adult Emx1-Cre+/+/VGLUT2fx/fx mice and control mice. Previous work has shown that LTP is neither impaired in global VGLUT2+/− mice nor in CaMKIIαCre+/VGLUT2fx/fx mice (Moechars et al., 2006; Wallen-Mackenzie et al., 2009). In contrast to the difference in LTD observed in young Emx1-Cre+/+/VGLUT2fx/fx and VGLUT2fx/fx mice (Fig. 4C), there were no differences in LTP between young Emx1-Cre+/+/VGLUT2fx/fx and VGLUT2fx/fx mice (Fig. 5C). However, in adult mice, LTP was reduced in Emx1-Cre+/+/VGLUT2fx/fx mice compared to controls (Fig. 5D; F(1,18) = 6.55; p = 0.02). Together, our results suggest that the reduction in evoked basal excitatory transmission and plasticity (LTP) in adult CA3-CA1 connections reflects a reduction in the degree of synaptic connectivity of VGLUT1-encoded axon terminals and CA1 dendritic spines.
Figure 5.

Basal synaptic transmission and LTP are reduced in CA3–CA1 synapses in adult Emx1-Cre+/+/VGLUT2fx/fx mice. A, Representative fEPSP waveforms recorded at in the stratum radiatum of the CA1 region following Schaffer collateral stimulation in adult (P45–60) control VGLUT2fx/fx (Wt, Wild type) and Emx1-Cre+/+/VGLUT2fx/fx mice. Input–output function is shown for Wt (15 recordings/4 animals) and Emx1-Cre+/+/VGLUT2fx/fx (18 recordings/4 animals) mice. B, Representative fEPSP waveforms. Paired-pulse ratio (fEPSP2/fEPSP1) is shown for adult Wt (13 recordings/4 animals) and Emx1-Cre+/+/VGLUT2fx/fx (15 recordings/4 animals) mice. C, Representative traces of fEPSPs recorded before (1) and after (2) TBS by stimulating CA3-CA1 connections in acute hippocampal slices from young (P11–14) animals. Time course of fEPSP slopes is also shown. D, Representative traces of fEPSPs recorded before and after TBS by stimulating Schaffer collateral pathway in acute hippocampal slices from adult (P45–60) animals. Time course of fEPSP slopes are also shown. E, Mean values of the potentiation of fEPSPs averaged from 55 to 60 min following TBS (Wt, 8 recordings/4 animals; Emx1-Cre+/+/VGLUT2fx/fx, 12 recordings/4 animals). F, Mean values of the potentiation of fEPSPs averaged from 55 to 60 min following TBS (Wt, 16 recordings/5 animals; Emx1-Cre+/+/VGLUT2fx/fx, 16 recordings/5 animals). Error bars represent SEM; *p < 0.05; ANOVA.
Reduced hippocampal expression of VGLUT1 and spinophilin in adult Emx1-Cre+/+/VGLUT2fx/fx mice
Recent work indicates that VGLUT1+/− mice display impaired LTP in the CA1 region (Balschun et al., 2010). We evaluated whether the expression levels of various synaptic markers were altered in Emx1-Cre+/+/VGLUT2fx/fx mice compared to control VGLUT2fx/fx mice. By Western analysis, VGLUT1 and spinophilin (a marker for dendritic spines) levels were reduced in the hippocampus of adult Emx1-Cre+/+/VGLUT2fx/fx mice compared to control mice (Fig. 6). Levels of VIAAT, GAD67, PSD95, and β-actin were not different between adult Emx1-Cre+/+/VGLUT2fx/fx and control mice (Fig. 6). We also assessed the regional distribution of VGLUT1 in the hippocampus of adult Emx1-Cre/VGLUT2fx/fx haplotypes and control mice by IHC (Fig. 7A–C). As expected, all staining of VGLUT1 was observed as discrete puncta, and no cell body labeling occurred. Previously, we were able to quantitate the number and the level of fluorescence in individual VGLUT1-containing synaptic puncta in primary hippocampal and cortical neuronal cultures (De Gois et al., 2005; Wilson et al., 2005). However, because of their small size and incredible packing density, we were not able to resolve individual VGLUT1 synaptic puncta in thin sections in situ by fluorescence microscopy. An intact basic structure of the hippocampus including the trisynaptic circuit—the dentate gyrus (DG) and pyramidal neuron subfields (CA1–4)—was clearly present in all mice.
Figure 6.

Reduced hippocampal expression of VGLUT1 and spinophilin in adult Emx1-Cre+/+/VGLUT2fx/fx mice. A, Western blot of vesicle-enriched membranes (milligram membrane protein) from the following: (1) VGLUT1 (2 μg); (2) VIAAT (4 μg); (3) spinophilin (50 μg); (4) GAD67 (50 μg); (5) PSD95 (50 μg); and (6) β-actin (4 μg). Total hippocampal protein extracts were prepared from three mice in each group. B, Quantitation of bands by densitometry. Results are expressed as a ratio of VGLUT1, VIAAT, spinophilin, and GAD67 against β-actin or against PSD95, which did not differ between adult Emx1-Cre+/+/VGLUT2fx/fx and control VGLUT2fx/fx mice. Error bars represent SEM; *p < 0.05; Student's t test.
Figure 7.
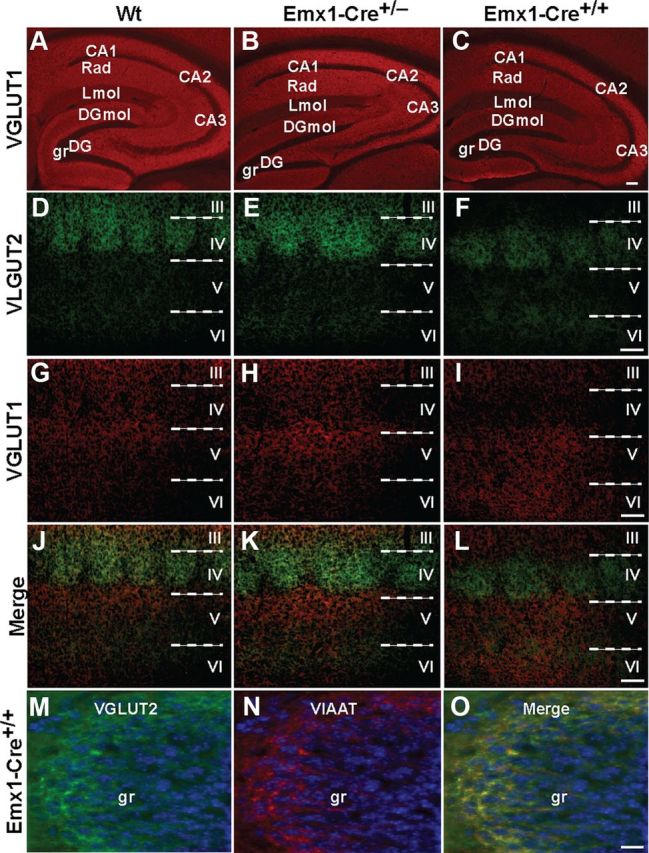
Representative examples of VGLUT1, VGLUT2, and VIAAT expression in the hippocampus and cerebral cortex of adult Emx1-Cre+/+/VGLUT2fx/fx, Emx1-Cre+/−/VGLUT2fx/fx, and control VGLUT2fx/fx mice. A–C, These micrographs show robust immunofluorescence signals for VGLUT1 in the hippocampus. Note: although cell nuclei are unlabeled, an intact structure of the basic trisynaptic circuit is evident in conditional VGLUT2 knock-out mice. Rad, Striatum radiatum; grDG, supragranular region of dentate gyrus; Lmol, stratum lacunosum-molecular; DGmol, molecular layer of the dentate gyrus. Scale bar, 100 μm. D–L, Distributions of VGLUT1 and VGLUT2 immunoreactivities in area S1 of the neocortex. As expected, the highest levels of VGLUT2 expression are seen in layer IV of area S1 barrel subfield, followed by layers II/III and the border between layers V and VI in control VGLUT2fx/fx (Wt, Wild type) mice. Similar immunostaining for VGLUT2 is observed in control and Emx1-Cre+/−/VGLUT2fx/fx mice. However, VGLUT2 immunofluorescence is noticeably reduced in layer IV and absent in layer III in Emx1-Cre+/+/VGLUT2fx/fx mice. Scale bar, 100 μm. M–O, VGLUT2 expression is retained in the supragranular region (gr) of the dentate gyrus in Emx1-Cre+/+/VGLUT2fx/fx mice, where it colocalizes with VIAAT. Scale bar, 20 μm.
Regional differences in VGLUT2 depletion in Emx1-Cre+/+/VGLUT2fx/fx and Emx1-Cre+/−/VGLUT2fx/fx mice
We hypothesized that the cognitive differences observed between Emx1-Cre+/+/VGLUT2fx/fx and Emx1-Cre+/−/VGLUT2fx/fx mice (Fig. 2) may be due to the differential and incomplete loss of intrinsic cortical/hippocampal VGLUT2 synapses or to a loss of VGLUT2 synapses originating in other regions of the brain (e.g., limbic/sensory projections). We therefore examined the expression of VGLUT2 in Emx1-Cre+/+/VGLUT2fx/fx, Emx1-Cre+/−/VGLUT2fx/fx, and control VGLUT2fx/fx mice by IHC. First, we examined the integrity of VGLUT2-encoded thalamocortical projections to the primary somatosensory cortex in adult (90 d) mice. VGLUT2-encoded thalamic axons provide a sensorimotor subsystem important for the selection of behavioral search strategies required to solve the Morris water maze task (Cain, 1998). The strongest VGLUT2 fluorescence was observed in the area S1 barrel subfield (layer IV) in all mice (Fig. 7D–F), as reported previously (Nakamura et al., 2007). While VGLUT2 levels appeared to be reduced in layer IV of the somatosensory cortex in Emx1-Cre+/+/VGLUT2fx/fx mice, the barrelettes can be visualized in all mice. The reduction of VGLUT2 in layer IV may be due the loss of intrinsic cortical expression of VGLUT2 in nonpyramidal excitatory neurons in layer IV (De Gois et al., 2005), which integrate incoming sensory input to cortical association (layer II/III) and output (layer V) pyramidal neurons. Indeed, layer IV excitatory neurons project to other layer IV neurons and to pyramidal and inhibitory neurons in layer II/III (Feldmeyer et al., 1999, 2002). Indeed, VGLUT2 immunolabeling in layer II/III is conspicuously absent in Emx1-Cre+/+/VGLUT2fx/fx mice, compared to Emx1-Cre+/−/VGLUT2fx/fx and control mice (Fig. 7D–L). The boundary between layers V and VI also showed VGLUT2 immunoreactive puncta in all mice.
Previous studies have shown that hippocampal theta activity is important for spatial learning and that extrinsic input into the hippocampus from the medial supramammillary nucleus is involved in the control of the frequency of theta (Pan and McNaughton, 2004). The medial supramammillary nucleus supplies a glutamatergic/GABAergic projection dually encoded by VGLUT2 and VIAAT into the outer part of the granule cell layer (g) and the immediately adjacent part of the inner zone of the molecular layer of the dentate gyrus (DGmol) (Fremeau et al., 2001; Atkinson et al., 2004; Halasy et al., 2004; Boulland et al., 2009). Here, we find a VGLUT2/VIAAT coexpressing phenotype existing in large terminals in the supragranular region of the dentate gyrus in control mice that is preserved in Emx-Cre+/+/VGLUT2fx/fx mice (Fig. 7M, 8A–L). While the highest density of VGLUT2-positive puncta were observed in the supragranular layer of the dentate gyrus in adult mice, an intermediate level was observed in the CA2 pyramidal and oriens layers, and lower levels were found in the stratum lacunosum-moleculare (Lmol) of the CA1 field of all mice, as described previously (Fremeau et al., 2001; Atkinson et al., 2004; Halasy et al., 2004; Herzog et al., 2006). The most likely source of VGLUT2-positive boutons in the stratum lacunosum-moleculare of the CA1 field in adult mice is the nucleus reunions thalami (Herkenham, 1978). These results suggest that the effects on synaptic transmission in adults are not due to an acute lack of VGLUT2 but instead are the result of the early postnatal developmental VGLUT2 deficiency.
Figure 8.
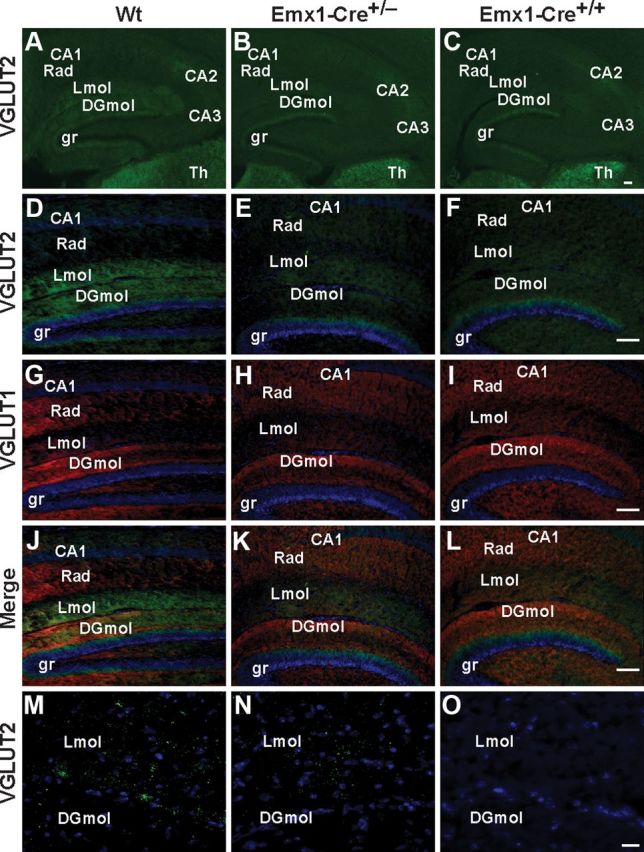
Representative examples of VGLUT1 and VGLUT2 expression in the hippocampus of Emx1-Cre+/+/VGLUT2fx/fx, Emx1-Cre+/−/VGLUT2fx/fx, and VGLUT2fx/fx mice at P18. A–C, Selective reduction of VGLUT2 expression in hippocampal subfields in conditional VGLUT2 knock-out mice. VGLUT2 immunofluorescence is strong in the supragranular region (gr) of the dentate gyrus, the stratum lacunosum-moleculare (Lmol), and the molecular layer of the dentate gyrus (DGmol) in VGLUT2fx/fx (Wt, Wild type) mice. Note: loss of VGLUT2 expression in DGmol in both Emx1-Cre lines. Note: Emx1-Cre+/− mice retain more expression of VGLUT2 in Lmol than Emx1-Cre+/+ mice. Note: high VGLUT2 expression in the supragranular region of the dentate gyrus and in the thalamus (Th) across all genotypes. Scale bar, 100 μm. D–L, Higher power micrographs reveal VGLUT2 (green), VGLUT1 (red), and merged images, together with the DAPI nuclear labeling (blue) Scale bar, 100 μm. M–O, Deconvolution microscopy and projection images show a clear reduction in the number of VGLUT2 puncta in the Lmol fields from Emx1-Cre+/−VGLUT2fx/fx mice and their absence in Emx1-Cre+/+/VGLUT2fx/fx mice. Scale bar, 20 μm.
A comparison of VGLUT2 expression in the hippocampal subfields from young mice revealed important differences between Emx1-Cre+/+/VGLUT2fx/fx, Emx1-Cre+/−/VGLUT2fx/fx, and control mice. We examined mice at P18 because, at this age, the pups have left the nest and have begun to actively explore their environment. In control VGLUT2fx/fx mice, VGLUT2 antibodies strongly stained small varicosities in the DGmol and in the Lmol (Fig. 8A,D,G,J,M). At high magnification we also observe thin and small VGLUT2-positive varicosities in the stratum radiatum and stratum oriens layers of the CA1 region of the hippocampus (data not shown) as described previously (Herzog et al., 2006). VGLUT2 expression in these CA3 projections to basal and proximal apical dendrites of CA1 neurons are not present in Emx1-Cre+/+/VGLUT2fx/fx mice. The most obvious differences in VGLUT2 expression between Emx1-Cre+/+/VGLUT2fx/fx and Emx1-Cre+/−/VGLUT2fx/fx are seen in DGmol and Lmol, which predominantly contain the terminal fields of layer II and layer III entorhinal cortex inputs, respectfully. In both Emx1-Cre+/−/VGLUT2fx/fx and Emx1-Cre+/+/VGLUT2fx/fx mice, VGLUT2-expressing puncta are lost in the DGmol (Fig. 8). However, in the Lmol, an intermediate level of VGLUT2-expressing puncta, is present in Emx1-Cre+/−/VGLUT2fx/fx mice compared to control mice. VGLUT2-expressing puncta are greatly reduced in young Lmol in Emx1-Cre+/+/VGLUT2fx/fx mice (Fig. 8M–O). Together, these results suggest that a differential loss of VGLUT2 synaptic puncta in the dendritic fields of CA1 pyramidal neurons during development between Emx1-Cre+/−/VGLUT2fx/fx, Emx1-Cre+/+/VGLUT2fx/fx, and VGLUT2fx/fx mice may contribute to the differences in dendritic refinement, electrophysiological properties, and cognitive impairment observed.
d-Serine administration, together with d-amino acid oxidase inhibition, enhances learning in adult Emx1-Cre+/+/VGLUT2fx/fx mice
A current approach to improve cognitive deficits seen in neurodevelopmental disorders is to tap into mechanisms of glutamatergic synaptic plasticity in the adult (Ehninger et al., 2008). In this regard, allosteric modulation at the NMDA receptor is considered to be an attractive target (Moskal et al., 2011; Moghaddam and Javitt, 2012). To assess the reparability of the spatial learning deficit, adult Emx1-Cre+/+/VGLUT2fx/fx mice were injected each day for 2 weeks (i.p.) with saline, the atypical antipsychotic drug clozapine (1 mg/kg), or d-serine (600 mg/kg) plus 4H–thieno-[3,2-b]pyrrole-5-carboxylic acid (compound 8; 50 mg/kg) before behavioral analysis and 1 h before behavioral testing each day (Fig. 9). d-Serine increases NMDA-mediated glutamate receptor function (Coyle and Tsai, 2004; Hashimoto et al., 2009). Compound 8 is a novel d-amino acid oxidase (DAAO) inhibitor that prevents peripheral metabolism and degradation of d-serine (Smith et al., 2009). One day before training in the Morris water maze, open-field activity was measured in a novel chamber (Fig. 9A). Forward locomotion scores (20 min) showed a trend for decreased activity in both clozapine- and d-serine/compound 8-treated Emx1-Cre+/+/VGLUT2fx/fx mice in this test of exploratory activity (1 h). The mice were retested in the same chamber 6 h later and displayed no significant differences, indicating that the doses of drugs used did not interfere with motor behavior. In the Morris water maze, increased learning performance was observed in Emx1-Cre+/+/VGLUT2fx/fx mice that received d-serine/compound 8 (F(2,168) = 6.082; p = 0.027), as they displayed shorter escape latency during a 7 day training period (Fig. 9B). Spatial learning was not improved in clozapine-treated compared to saline-treated Emx1-Cre+/+/VGLUT2fx/fx mice (Fig. 9B), which was not different from uninjected Emx1-Cre+/+/VGLUT2fx/fx mice (Fig. 2A). Interestingly, the probe test revealed that d-serine/compound 8-treated mice did not spend more time searching the quadrant where the platform was previously located (Fig. 9C), similarly as Emx1-Cre+/+/VGLUT2fx/fx mice (Fig. 2C), indicating that spatial reference memory was still impaired. However, a significant increase in the number of crossings of the platform area was observed in Emx1-Cre+/+/VGLUT2fx/fx mice treated with d-serine/compound 8 (p = 0.03) compared to saline-treated or clozapine-treated mice (Fig. 9D), indicating that nonspatial, systematic strategies may have been used. Collectively, our results suggest that augmentation of NMDA receptor function via d-serine/compound 8 may increase learning performance in Emx1-Cre+/+/VGLUT2fx/fx mice by increasing behavioral plasticity and altering search strategy, although longer training or stronger visual cues may also facilitate spatial learning and memory performance.
Figure 9.

d-Serine administration, together with d-amino acid oxidase inhibition, enhances learning in adult Emx1-Cre+/+/VGLUT2fx/fx mice. Adult Emx1-Cre+/+/VGLUT2fx/fx mice (P60–80, males/females) were treated with daily injections of saline, clozapine (1 mg/kg), or d-serine (600 mg/kg) and the DAAO inhibitor (compound 8; 50 mg/kg) for 2 weeks in the home cage and then each day 1 h before training or testing. A, The doses of these compounds used did not adversely affect locomotor activity at 1 or 6 h postinjection in an open-field chamber. B, In the Morris water maze test there was no significant improvement in the escape latency between the saline-injected and clozapine-injected groups during 7 d of training. However, the animals in the d-serine/compound 8-injected group had significantly lower latency and time to reach the hidden platform; *p < 0.05; repeated-measures ANOVA with a LSD post hoc test. C, No significant preference for the quadrant where the platform was previously located in the probe test was observed for all groups. D, A comparison of the number of times the mice crossed the location where the platform was previously hidden revealed that d-serine/compound 8-treated mice showed significant improvement when compared to saline and clozapine-treated animals. Error bars represent SEM; *p < 0.05; ANOVA.
Discussion
The present data indicate that VGLUT2 plays an important role in the maturation of CA1 pyramidal neurons—a major output of the hippocampus and critically involved in spatial learning. The persistent reduction in synchronized glutamate synaptic transmission, high probability evoked glutamate release, and LTD in developing CA3–CA1 connections of VGLUT2-deficient Emx1-Cre+/+/VGLUT2fx/fx mice likely contribute to the curtailment in dendritic refinement of CA1 pyramidal neurons. The loss of VGLUT2 synaptic input during development is not confined to the CA3-CA1 projections onto basal and proximal apical dendrites of CA1 pyramidal neurons however, and is also evident in stratum lacunosum-moleculare, where the distal apical dendrites are located. Thus, a reduction in dendritic branch length and spine number exists throughout the length of the dendritic tree. The changes in dendrite architecture, together with the reduction in hippocampal expression of VGLUT1 and spinophilin, imply a decreased synaptic connectivity and impoverished neuropil that likely contributes to the impairment in LTP, spatial learning, and memory observed in adult mice. Despite these structural and functional changes, the learning deficit in Emx1-Cre+/+/VGLUT2fx/fx mice is partially reparable in adult mice by augmenting NMDA receptor function with a prolonged administration of d-serine together with DAAO inhibition. Our data are the first to demonstrate a neurodevelopmental role for VGLUT2 in the maturation of pyramidal neuron dendritic arbor and synaptic plasticity, and that such VGLUT2 deficiency in Emx1-Cre+/+/VGLUT2fx/fx mice leads to cognitive malfunction.
A role for VGLUT2 in the development of VGLUT1-encoded transmission and plasticity and in pyramidal neuron dendritic refinement is intriguing, because hypoglutamatergic function and excitatory/inhibitory imbalance during early periods of neuronal maturation have relevance to several neurodevelopmental psychiatric/cognitive illnesses, including autism spectrum and schizophrenia-related disorders (Javitt and Zukin, 1991; Carlsson, 1998; Dani et al., 2005; Smith et al., 2011; Choudhury et al., 2012; Moghaddam and Javitt, 2012; Na et al., 2012). In Emx1-Cre+/+/VGLUT2fx/fx mice, we find a decrease in evoked basal excitatory transmission (input/output function) and a decrease in release probability (increased PPR) in young (P11–14) CA3-CA1 connections. High glutamate release probability is characteristic of developing pyramidal neurons in vivo (Bolshakov and Siegelbaum, 1995; Dobrunz and Stevens, 1997; Hsia et al., 1998; Wasling et al., 2004). Recent work has established that VGLUT2-expressing vesicles have higher release probability in hippocampal pyramidal neurons than vesicles containing only VGLUT1 in vitro (Weston et al., 2011). VGLUT2-encoded sensory neurons also display high release probability and LTD induction by LFS protocols in adults (Thomson, 2000; Amitai, 2001). Importantly, LTD induced by LFS can only be recorded in young CA3–CA1 synapses from mice aged <3 weeks (Milner et al., 2004) when VGLUT2 is transiently expressed. Our results support the notion that VGLUT2 contributes to high release probability in young CA3-CA1 connections and that LTD induced by LFS may involve plasticity of VGLUT2-encoded vesicle release in vivo.
Glutamatergic signaling plays a large role in the refinement of pyramidal neuron dendritic arbor (Cline, 2001; Wong and Ghosh, 2002). In vivo studies indicate that synaptic glutamate activity promotes dendritic arbor elaboration at early stages of brain development (Cline and Haas, 2008; Sorensen and Rubel, 2011). On the other hand, synaptic inactivity (or repetitive LTD) early in postnatal development results in CA1 pyramidal neurons with reduced total length of the basal dendritic tree in vivo (Groc et al., 2002; Monfils and Teskey, 2004) and synaptic pruning in vitro (Shinoda et al., 2010). Importantly, abnormal pruning of synapses is thought to result in neurodevelopmental psychiatric/cognitive illnesses (Keshavan et al., 1994; Johnston et al., 2003; Penzes et al., 2011). Emx1-Cre+/+/VGLUT2fx/fx mice displayed a persistent depression of evoked glutamate transmission and LTD at CA3–CA1 connections throughout development and a reduction in basal and apical dendritic branch length and number of spines on CA1 dendrites in adults. This indicates a neurodevelopmental role for VGLUT2 in the structural maturation and refinement of CA1 pyramidal neuron dendritic arbor.
While LTD is thought to play a critical role in dendritic pruning during development, LTP is regarded as a cellular model for learning and memory (Lynch, 2004; Kemp and Manahan-Vaughan, 2007). Interestingly, recent work shows that decreased VGLUT1 in global VGLUT1+/− knockdown mice results in decreased LTP and spatial reversal learning in adults (Balschun et al., 2010). Similarly, reduced expression of LTP in adult CA3–CA1 connections and reduced levels of VGLUT1 and spinophilin were observed in the hippocampus of adult Emx1-Cre+/+/VGLUT2fx/fx mice. While the learning deficit in Emx1-Cre+/+/VGLUT2fx/fx mice is more severe than that in global VGLUT1(+/−) mice, reduced glutamate signaling throughout development from either VGLUT1 or VGLUT2 results in impaired learning in the adult. Importantly, our results support a neurodevelopmental role for VGLUT2 in the maturation of VGLUT1-encoded synaptic connectivity and long-term synaptic plasticity.
The cognitive deficit in Emx-Cre+/+/VGLUT2fx/fx mice is greater than that in Emx1-Cre+/−/VGLUT2fx/fx mice, which may reflect a greater degree of VGLUT2-encoded glutamatergic hypofunction in specific corticolimbic circuits during development. Since Cre recombinase is in the second position of the bicistronic Emx1-Cre mRNA, it likely exhibits reduced expression (Mizuguchi et al., 2000) compared to Emx1 (which is expressed at wild-type levels) that could be critical for extinguishing gene expression in certain neurons (Tsien et al., 1996). Indeed, lower levels of Cre-mediated recombination in Emx1-Cre+/− mice have been detected in layer IV of the neocortex and in various subcortical limbic structures, including the anterior thalamic nuclei, the lateral septum, and in various subdivisions of the amygdala (Gorski et al., 2002). Thalamocortical projections terminate in layer IV (Fujiyama et al., 2001). Layer IV nonpyramidal glutamatergic neurons send their axon terminals to other layer IV neurons and also to pyramidal neurons and inhibitory interneurons that are predominantly in layer III (Feldmeyer et al., 1999, 2002). The selective loss of VGLUT2 in the VGLUT1/VGLUT2-coexpressing nonpyramidal excitatory neurons of layer IV (De Gois et al., 2005) in Emx1-Cre+/+/VGLUT2fx/fx mice could disrupt the plastic interface that exists between subcortical sensory input (VGLUT2-encoded) and cortical association and output (VGLUT1-encoded) neurons, which would then impair cortical integration of sensory information—and should impair learning. Additionally, adult animals with extensive CA3 lesions that impair Schaffer collateral transmission to CA1 pyramidal neurons still perform well on spatial recognition tasks (Brun et al., 2002). This might suggest that loss of additional excitatory input to CA1 neurons in Emx1-Cre+/+/VGLUT2fx/fx mice, such as via the stratum lacunosum-moleculare—the target of the temporammonic pathway (Skelton and McNamara, 1992; Sybirska et al., 2000; Otmakhova et al., 2002; Speed and Dobrunz, 2009)—may reduce signal convergence of sensory input onto CA1 dendrites and contribute to perturbed learning. However, in Emx1-Cre+/+/VGLUT2fx/fx mice the reduction in glutamatergic transmission occurs throughout development. Nonetheless, the contribution of both CA3–CA1 and TA–CA1 inputs during development may play a role in CA1 neuron functional maturation and structural refinement (Speed and Dobrunz, 2009).
Pharmacological blockade of NMDA receptors during the second week of development, a prolonged reduction of NMDA-dependent glutamate neurotransmission in adults, and genetic NMDA knock-down mice represent animal models of glutamatergic hypofunction that increase exploratory activity yet induce cognitive deficits (Javitt and Zukin, 1991; Sakimura et al., 1995; Carlsson, 1998; Jentsch and Roth, 1999; Mohn et al., 1999; Stefani and Moghaddam, 2005; Nakatani-Pawlak et al., 2009; Ramsey, 2009). Likewise, our study shows that Emx1-Cre+/+/VGLUT2fx/fx mice displayed increased novelty-induced, open-field activity and yet a profound deficit in spatial learning and memory. Since Emx1-Cre+/+/VGLUT2fx/fx mice have hypoglutamatergic transmission in pyramidal neurons as a core feature, both during development (via VGLUT2) and in the adult (via VGLUT1), it is reasonable to presume that drugs that selectively increase synaptic glutamatergic function may increase learning performance. Indeed, tapping into adult mechanisms of cellular, structural, and behavioral plasticity may result in substantial improvements in function in neurodevelopmental cognitive disorders (Silva and Ehninger, 2009). For example, compounds that potentiate NMDA receptor-mediated neurotransmission (Johnson and Ascher, 1987; Bergeron et al., 1998; Martina et al., 2004) through the d-serine/glycine binding site within the NMDA receptor complex improve some aspects of cognitive dysfunction in humans with, and in animal models of, psychiatric/cognitive illness (Goff et al., 1996; Carlsson, 1998; Coyle and Tsai, 2004; Javitt, 2009; Modi and Young, 2011; Moskal et al., 2011) despite the lack of any evidence that NMDA receptors are per se dysfunctional. Coadministration of d-serine with a DAAO inhibitor (e.g., compound 8) potentiates the efficacy of d-serine administration to increase NMDA receptor function (Ferraris et al., 2008; Hashimoto et al., 2009; Smith et al., 2009, 2010). Here, we have shown that learning performance was modestly improved in adult Emx1-Cre+/+/VGLUT2fx/fx mice following a 2-week treatment with d-serine, together with a DAAO inhibitor. In humans and in preclinical models of psychiatric disease, the efficacy of drug therapies on cognitive impairments in adults is also rather limited, perhaps due to the intractability of reversing neurodevelopmental disorders in adults (Ehninger et al., 2008). Future efforts to improve VGLUT2-mediated transmission via NMDA-positive modulation during formative periods of pyramidal neuron development could further improve learning ability in adult Emx1-Cre+/+/VGLUT2fx/fx mice. In conclusion, our strategy to elicit VGLUT2 hypofunction during a time when VGLUT2-mediated glutamate transmission is critical to the structural and functional maturation of pyramidal neurons provides further insights into the mechanisms of learning in general, and to cellular and behavioral manifestations of neurodevelopmental cognitive aberrations in psychiatric disease.
Footnotes
This work was supported by National Institutes of Health Grant R01 NS036936, a NARSAD Independent Investigator Award from the Brain and Behavior Research Foundation, and a Research Enhancement Fund Grant from Louisiana State University Health Science Center to J.D.E. National Institute of Health Grant R01 NS076815 supported C.C. National Institute of Health COBRE Grant P20RR016816 supported F.M.I. National Science Foundation CAREER Award 0952455 supported B.J.H. We thank Dr. Hilary Thompson (Department of Biostatistics, Louisiana State University Health Sciences Center, New Orleans, LA) for extensive consultation and assistance with the statistical analyses.
The authors declare no competing financial interests.
References
- Amitai Y. Thalamocortical synaptic connections: efficacy, modulation, inhibition and plasticity. Rev Neurosci. 2001;12:159–173. doi: 10.1515/revneuro.2001.12.2.159. [DOI] [PubMed] [Google Scholar]
- Asaka Y, Jugloff DG, Zhang L, Eubanks JH, Fitzsimonds RM. Hippocampal synaptic plasticity is impaired in the Mecp2-null mouse model of Rett syndrome. Neurobiol Dis. 2006;21:217–227. doi: 10.1016/j.nbd.2005.07.005. [DOI] [PubMed] [Google Scholar]
- Atkinson L, Batten TF, Moores TS, Varoqui H, Erickson JD, Deuchars J. Differential co-localisation of the P2X7 receptor subunit with vesicular glutamate transporters VGLUT1 and VGLUT2 in rat CNS. Neuroscience. 2004;123:761–768. doi: 10.1016/j.neuroscience.2003.08.065. [DOI] [PubMed] [Google Scholar]
- Balschun D, Moechars D, Callaerts-Vegh Z, Vermaercke B, Van Acker N, Andries L, D'Hooge R. Vesicular glutamate transporter VGLUT1 has a role in hippocampal long-term potentiation and spatial reversal learning. Cereb Cortex. 2010;20:684–693. doi: 10.1093/cercor/bhp133. [DOI] [PubMed] [Google Scholar]
- Bergeron R, Meyer TM, Coyle JT, Greene RW. Modulation of N-methyl-d-aspartate receptor function by glycine transport. Proc Natl Acad Sci U S A. 1998;95:15730–15734. doi: 10.1073/pnas.95.26.15730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolshakov VY, Siegelbaum SA. Regulation of hippocampal transmitter release during development and long-term potentiation. Science. 1995;269:1730–1734. doi: 10.1126/science.7569903. [DOI] [PubMed] [Google Scholar]
- Boulland JL, Qureshi T, Seal RP, Rafiki A, Gundersen V, Bergersen LH, Fremeau RT, Jr, Edwards RH, Storm-Mathisen J, Chaudhry FA. Expression of the vesicular glutamate transporters during development indicates the widespread corelease of multiple neurotransmitters. J Comp Neurol. 2004;480:264–280. doi: 10.1002/cne.20354. [DOI] [PubMed] [Google Scholar]
- Boulland JL, Jenstad M, Boekel AJ, Wouterlood FG, Edwards RH, Storm-Mathisen J, Chaudhry FA. Vesicular glutamate and GABA transporters sort to distinct sets of vesicles in a population of presynaptic terminals. Cereb Cortex. 2009;19:241–248. doi: 10.1093/cercor/bhn077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broadbelt K, Byne W, Jones LB. Evidence for a decrease in basilar dendrites of pyramidal cells in schizophrenic medial prefrontal cortex. Schizophr Res. 2002;58:75–81. doi: 10.1016/s0920-9964(02)00201-3. [DOI] [PubMed] [Google Scholar]
- Brun VH, Otnass MK, Molden S, Steffenach HA, Witter MP, Moser MB, Moser EI. Place cells and place recognition maintained by direct entorhinal-hippocampal circuitry. Science. 2002;296:2243–2246. doi: 10.1126/science.1071089. [DOI] [PubMed] [Google Scholar]
- Cain DP. Testing the NMDA, long-term potentiation, and cholinergic hypothesis of spatial learning. Neurosci Biobehav Rev. 1998;22:181–193. doi: 10.1016/s0149-7634(97)00005-5. [DOI] [PubMed] [Google Scholar]
- Carlsson ML. Hypothesis: is infantile autism a hypoglutamatergic disorder? Relevance of glutamate - serotonin interactions for pharmacotherapy. J Neural Transm. 1998;105:525–535. doi: 10.1007/s007020050076. [DOI] [PubMed] [Google Scholar]
- Chao HT, Zoghbi HY, Rosenmund C. MeCP2 controls excitatory synaptic strength by regulating glutamatergic synapse number. Neuron. 2007;56:58–65. doi: 10.1016/j.neuron.2007.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhury PR, Lahiri S, Rajamma U. Glutamate mediated signaling in the pathophysiology of autism spectrum disorders. Pharmacol Biochem Behav. 2012;100:841–849. doi: 10.1016/j.pbb.2011.06.023. [DOI] [PubMed] [Google Scholar]
- Cline H, Haas K. The regulation of dendritic arbor development and plasticity by glutamatergic synaptic input: a review of the synaptotrophic hypothesis. J Physiol. 2008;586:1509–1517. doi: 10.1113/jphysiol.2007.150029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cline HT. Dendritic arbor development and synaptogenesis. Curr Opin Neurobiol. 2001;11:118–126. doi: 10.1016/s0959-4388(00)00182-3. [DOI] [PubMed] [Google Scholar]
- Coyle JT, Tsai G. The NMDA receptor glycine modulatory site: a therapeutic target for improving cognition and reducing negative symptoms in schizophrenia. Psychopharmacology (Berl) 2004;174:32–38. doi: 10.1007/s00213-003-1709-2. [DOI] [PubMed] [Google Scholar]
- Dani VS, Chang Q, Maffei A, Turrigiano GG, Jaenisch R, Nelson SB. REduced cortical activity due to a shift in the balance between excitation and inhibition in a mouse model of Rett syndrome. Proc Natl Acad Sci U S A. 2005;102:12560–12565. doi: 10.1073/pnas.0506071102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels RW, Collins CA, Gelfand MV, Dant J, Brooks ES, Krantz DE, DiAntonio A. Increased expression of the Drosophila vesicular glutamate transporter leads to excess glutamate release and a compensatory decrease in quantal content. J Neurosci. 2004;24:10466–10474. doi: 10.1523/JNEUROSCI.3001-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Gois S, Schäfer MK, Defamie N, Chen C, Ricci A, Weihe E, Varoqui H, Erickson JD. Homeostatic scaling of vesicular glutamate and GABA transporter expression in rat neocortical circuits. J Neurosci. 2005;25:7121–7133. doi: 10.1523/JNEUROSCI.5221-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobrunz LE, Stevens CF. Heterogeneity of release probability, facilitation, and depletion at central synapses. Neuron. 1997;18:995–1008. doi: 10.1016/s0896-6273(00)80338-4. [DOI] [PubMed] [Google Scholar]
- Doyle S, Pyndiah S, De Gois S, Erickson JD. Excitation-transcription coupling via calcium/calmodulin-dependent protein kinase/ERK1/2 signaling mediates the coordinate induction of VGLUT2 and Narp triggered by a prolonged increase in glutamatergic synaptic activity. J Biol Chem. 2010;285:14366–14376. doi: 10.1074/jbc.M109.080069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudek SM, Bear MF. Homosynaptic long-term depression in area CA1 of hippocampus and effects of N-methyl-d-aspartate receptor blockade. Proc Natl Acad Sci U S A. 1992;89:4363–4367. doi: 10.1073/pnas.89.10.4363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudek SM, Bear MF. Bidirectional long-term modification of synaptic effectiveness in the adult and immature hippocampus. J Neurosci. 1993;13:2910–2918. doi: 10.1523/JNEUROSCI.13-07-02910.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards D, Berry JJ. The efficiency of simulation-based multiple comparisons. Biometrics. 1987;43:913–928. [PubMed] [Google Scholar]
- Ehninger D, Li W, Fox K, Stryker MP, Silva AJ. Reversing neurodevelopmental disorders in adults. Neuron. 2008;60:950–960. doi: 10.1016/j.neuron.2008.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson JD, De Gois S, Varoqui H, Schafer MK, Weihe E. Activity-dependent regulation of vesicular glutamate and GABA transporters: a means to scale quantal size. Neurochem Int. 2006;48:643–649. doi: 10.1016/j.neuint.2005.12.029. [DOI] [PubMed] [Google Scholar]
- Fan N, Yang H, Zhang J, Chen C. Reduced expression of glutamate receptors and phosphorylation of CREB are responsible for in vivo Delta9-THC exposure-impaired hippocampal synaptic plasticity. J Neurochem. 2010;112:691–702. doi: 10.1111/j.1471-4159.2009.06489.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farley FW, Soriano P, Steffen LS, Dymecki SM. Widespread recombinase expression using FLPeR (Flipper) mice. Genesis. 2000;28:106–110. [PubMed] [Google Scholar]
- Feldmeyer D, Egger V, Lubke J, Sakmann B. Reliable synaptic connections between pairs of excitatory layer 4 neurones within a single ‘barrel’ of developing rat somatosensory cortex. J Physiol. 1999;521:169–190. doi: 10.1111/j.1469-7793.1999.00169.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldmeyer D, Lübke J, Silver RA, Sakmann B. Synaptic connections between layer 4 spinay neuron-layer 2/3 pyramidal cell pairs in juvenile rat barrel cortex: physiology and anatomy of interlaminar signalling with a cortical column. J Physiol. 2002;538:803–822. doi: 10.1113/jphysiol.2001.012959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferraris D, Duvall B, Ko YS, Thomas AG, Rojas C, Majer P, Hashimoto K, Tsukamoto T. Synthesis and biological evaluation of d-amino acid oxidase inhibitors. J Med Chem. 2008;51:3357–3359. doi: 10.1021/jm800200u. [DOI] [PubMed] [Google Scholar]
- Fremeau RT, Jr, Troyer MD, Pahner I, Nygaard GO, Tran CH, Reimer RJ, Bellocchio EE, Fortin D, Storm-Mathisen J, Edwards RH. The expression of vesicular glutamate transporters defines two classes of excitatory synapse. Neuron. 2001;31:247–260. doi: 10.1016/s0896-6273(01)00344-0. [DOI] [PubMed] [Google Scholar]
- Fremeau RT, Jr, Voglmaier S, Seal RP, Edwards RH. VGLUTs define subsets of excitatory neurons and suggest novel roles for glutamate. Trends Neurosci. 2004a;27:98–103. doi: 10.1016/j.tins.2003.11.005. [DOI] [PubMed] [Google Scholar]
- Fremeau RT, Jr, Kam K, Qureshi T, Johnson J, Copenhagen DR, Storm-Mathisen J, Chaudhry FA, Nicoll RA, Edwards RH. Vesicular glutamate transporters 1 and 2 target to functionally distinct synaptic release sites. Science. 2004b;304:1815–1819. doi: 10.1126/science.1097468. [DOI] [PubMed] [Google Scholar]
- Fujiyama F, Furuta T, Kaneko T. Immunocytochemical localization of candidates for vesicular glutamate transporters in the rat cerebral cortex. J Comp Neurol. 2001;435:379–387. doi: 10.1002/cne.1037. [DOI] [PubMed] [Google Scholar]
- Goff DC, Tsai G, Manoach DS, Flood J, Darby DG, Coyle JT. d-Cycloserine added to clozapine for patients with schizophrenia. Am J Psychiatry. 1996;153:1628–1630. doi: 10.1176/ajp.153.12.1628. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Burgos G, Kroener S, Zaitsev AV, Povysheva NV, Krimer LS, Barrionuevo G, Lewis DA. Functional maturation of excitatory synapses in layer 3 pyramidal neurons during postnatal development of the primate prefrontal cortex. Cereb Cortex. 2008;18:626–637. doi: 10.1093/cercor/bhm095. [DOI] [PubMed] [Google Scholar]
- Gorski JA, Talley T, Qiu M, Puelles L, Rubenstein JL, Jones KR. Cortical excitatory neurons and glia, but not GABAergic neurons, are produced in the Emx1-expressing lineage. J Neurosci. 2002;22:6309–6314. doi: 10.1523/JNEUROSCI.22-15-06309.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groc L, Petanjek Z, Gustafsson B, Ben-Ari Y, Hanse E, Khazipov R. In vivo blockade of neural activity alters dendritic development of neonatal CA1 pyramidal cells. Eur J Neurosci. 2002;16:1931–1938. doi: 10.1046/j.1460-9568.2002.02264.x. [DOI] [PubMed] [Google Scholar]
- Halasy K, Hajszan T, Kovács EG, Lam TT, Leranth C. Distribution and origin of vesicular glutamate transporter 2-immunoreactive fibers in the rat hippocampus. Hippocampus. 2004;14:908–918. doi: 10.1002/hipo.20006. [DOI] [PubMed] [Google Scholar]
- Hall BJ, Ripley B, Ghosh A. NR2B signaling regulates the development of synaptic AMPA receptor current. J Neurosci. 2007;27:13446–13456. doi: 10.1523/JNEUROSCI.3793-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto K, Fujita Y, Horio M, Kunitachi S, Iyo M, Ferraris D, Tsukamoto T. Co-administration of a d-amino acid oxidase inhibitor potentiates the efficacy of d-serine in attenuating prepulse inhibition deficits after administration of dizocilpine. Biol Psychiatry. 2009;65:1103–1106. doi: 10.1016/j.biopsych.2009.01.002. [DOI] [PubMed] [Google Scholar]
- Herkenham M. The connections of the nucleus reuniens thalami: evidence for a direct thalamo-hippocampal pathway in the rat. J Comp Neurol. 1978;177:589–610. doi: 10.1002/cne.901770405. [DOI] [PubMed] [Google Scholar]
- Herzog E, Bellenchi GC, Gras C, Bernard V, Ravassard P, Bedet C, Gasnier B, Giros B, El Mestikawy S. The existence of a second vesicular glutamate transporter specifies subpopulations of glutamatergic neurons. J Neurosci. 2001;21:RC181. doi: 10.1523/JNEUROSCI.21-22-j0001.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzog E, Takamori S, Jahn R, Brose N, Wojcik SM. Synaptic and vesicular co-localization of the glutamate transporters VGLUT1 and VGLUT2 in the mouse hippocampus. J Neurochem. 2006;99:1011–1018. doi: 10.1111/j.1471-4159.2006.04144.x. [DOI] [PubMed] [Google Scholar]
- Hsia AY, Malenka RC, Nicoll RA. Development of excitatory circuitry in the hippocampus. J Neurophysiol. 1998;79:2013–2024. doi: 10.1152/jn.1998.79.4.2013. [DOI] [PubMed] [Google Scholar]
- Javitt DC. Glycine transport inhibitors for the treatment of schizophrenia: symptom and disease modification. Curr Opin Drug Discov Devel. 2009;12:468–478. [PubMed] [Google Scholar]
- Javitt DC, Zukin SR. Recent advances in the phencyclidine model of schizophrenia. Am J Psychiatry. 1991;148:1301–1308. doi: 10.1176/ajp.148.10.1301. [DOI] [PubMed] [Google Scholar]
- Jentsch JD, Roth RH. The neuropsychopharmacology of phencyclidine: from NMDA receptor hypofunction to the dopamine hypothesis of schizophrenia. Neuropsychopharmacology. 1999;20:201–225. doi: 10.1016/S0893-133X(98)00060-8. [DOI] [PubMed] [Google Scholar]
- Jentsch JD, Trantham-Davidson H, Jairl C, Tinsley M, Cannon TD, Lavin A. Dysbindin modulates prefrontal cortical glutamatergic circuits and working memory function in mice. Neuropsychopharmacology. 2009;34:2601–2608. doi: 10.1038/npp.2009.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson JW, Ascher P. Glycine potentiates the NMDA response in cultured mouse brain neurons. Nature. 1987;325:529–531. doi: 10.1038/325529a0. [DOI] [PubMed] [Google Scholar]
- Johnston MV, Mullaney B, Blue ME. Neurobiology of Rett syndrome. J Child Neurol. 2003;18:688–692. doi: 10.1177/08830738030180100501. [DOI] [PubMed] [Google Scholar]
- Kaneko T, Fujiyama F. Complementary distribution of vesicular glutamate transporters in the central nervous system. Neurosci Res. 2002;42:243–250. doi: 10.1016/s0168-0102(02)00009-3. [DOI] [PubMed] [Google Scholar]
- Kemp A, Manahan-Vaughan D. Hippocampal long-term depression: master or minion in declarative memory processes? Trends Neurosci. 2007;30:111–118. doi: 10.1016/j.tins.2007.01.002. [DOI] [PubMed] [Google Scholar]
- Keshavan MS, Anderson S, Pettegrew JW. Is schizophrenia due to excessive synaptic pruning in the prefrontal cortex? The Feinberg hypothesis revisited. J Psychiatr Res. 1994;28:239–265. doi: 10.1016/0022-3956(94)90009-4. [DOI] [PubMed] [Google Scholar]
- Lee RY, Sawin ER, Chalfie M, Horvitz HR, Avery L. EAT-4, a homolog of a mammalian sodium-dependent inorganic phosphate cotransporter, is necessary for glutamatergic neurotransmission in Caenorhabditis elegans. J Neurosci. 1999;19:159–167. doi: 10.1523/JNEUROSCI.19-01-00159.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Roux N, Amar M, Moreau A, Fossier P. Involvement of NR2A- or NR2B-containing N-methyl-d-aspartate receptors in the potentiation of cortical layer 5 pyramidal neurone inputs depends on the developmental stage. Eur J Neurosci. 2007;26:289–301. doi: 10.1111/j.1460-9568.2007.05671.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch MA. Long-term potentiation and memory. Physiol Rev. 2004;84:87–136. doi: 10.1152/physrev.00014.2003. [DOI] [PubMed] [Google Scholar]
- Martina M, Gorfinkel Y, Halman S, Lowe JA, Periyalwar P, Schmidt CJ, Bergeron R. Glycine transporter type 1 blockade changes NMDA receptor-mediated responses and LTP in hippocampal CA1 pyramidal cells by altering extracellular glycine levels. J Physiol. 2004;557:489–500. doi: 10.1113/jphysiol.2004.063321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milliken G, Johnson D. Analysis of messy data, volume I: designed experiments. New York: Van Nostrand Reinhold; 1984. p. 463. [Google Scholar]
- Milner AJ, Cummings DM, Spencer JP, Murphy KP. Bi-directional plasticity and age-dependent long-term depression at mouse CA3-CA1 hippocampal synapses. Neurosci Lett. 2004;367:1–5. doi: 10.1016/j.neulet.2004.04.056. [DOI] [PubMed] [Google Scholar]
- Minichiello L, Korte M, Wolfer D, Kühn R, Unsicker K, Cestari V, Rossi-Arnaud C, Lipp HP, Bonhoeffer T, Klein R. Essential role for TrkB receptors in hippocampus-mediated learning. Neuron. 1999;24:401–414. doi: 10.1016/s0896-6273(00)80853-3. [DOI] [PubMed] [Google Scholar]
- Mizuguchi H, Xu Z, Ishii-Watabe A, Uchida E, Hayakawa T. IRES-dependent second gene expression is significantly lower than cap-dependent first gene expression in a bicistronic vector. Mol Ther. 2000;1:376–382. doi: 10.1006/mthe.2000.0050. [DOI] [PubMed] [Google Scholar]
- Modi ME, Young LJ. d-Cycloserine facilitates socially reinforced learning in an animal model relevant to autism spectrum disorders. Biol Psychiatry. 2011;70:298–304. doi: 10.1016/j.biopsych.2011.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moechars D, Weston MC, Leo S, Callaerts-Vegh Z, Goris I, Daneels G, Buist A, Cik M, van der Spek P, Kass S, Meert T, D'Hooge R, Rosenmund C, Hampson RM. Vesicular glutamate transporter VGLUT2 expression levels control quantal size and neuropathic pain. J Neurosci. 2006;26:12055–12066. doi: 10.1523/JNEUROSCI.2556-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moghaddam B, Javitt D. From revolution to evolution: The glutamate hypothesis of schizophrenia and its implication for treatment. Neuropsychopharmacology. 2012;Rev37:4–15. doi: 10.1038/npp.2011.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohn AR, Gainetdinov RR, Caron MG, Koller BH. Mice with reduced NMDA receptor expression display behaviors related to schizophrenia. Cell. 1999;98:427–436. doi: 10.1016/s0092-8674(00)81972-8. [DOI] [PubMed] [Google Scholar]
- Monfils MH, Teskey GC. Induction of long-term depression is associated with decreased dendritic length and spine density in layers III and V of sensorimotor neocortex. Synapse. 2004;53:114–121. doi: 10.1002/syn.20039. [DOI] [PubMed] [Google Scholar]
- Morris R. Developments of a water-maze procedure for studying spatial learning in the rat. J Neurosci Methods. 1984;11:47–60. doi: 10.1016/0165-0270(84)90007-4. [DOI] [PubMed] [Google Scholar]
- Moskal JR, Burgdorf J, Kroes RA, Brudzynski SM, Panksepp J. A novel NMDA receptor glycine-site partial agonist, GLYX-13, has therapeutic potential for the treatment of autism. Neurosci Biobehav Rev. 2011;35:1982–1988. doi: 10.1016/j.neubiorev.2011.06.006. [DOI] [PubMed] [Google Scholar]
- Moutsimilli L, Farley S, Dumas S, El Mestikawy S, Giros B, Tzavara ET. Selective cortical VGLUT1 increase as a marker for antidepressant activity. Neuropharmacology. 2005;49:890–900. doi: 10.1016/j.neuropharm.2005.06.017. [DOI] [PubMed] [Google Scholar]
- Moutsimilli L, Farley S, El Khoury MA, Chamot C, Sibarita JB, Racine V, El Mestikawy S, Mathieu F, Dumas S, Giros B, Tzavara ET. Antipsychotics increase vesicular glutamate transporter 2 (VGLUT2) expression in thalamolimbic pathways. Neuropharmacology. 2008;54:497–508. doi: 10.1016/j.neuropharm.2007.10.022. [DOI] [PubMed] [Google Scholar]
- Na ES, Nelson ED, Kavalali ET, Monteggia LM. The impact of MeCP2 loss- or gain-of-function on synaptic plasticity. Neuropsychopharmacology. 2012 doi: 10.1038/npp.2012.116. Advance online publication. Retrieved July 11, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nägerl UV, Eberhorn N, Cambridge SB, Bonhoeffer T. Bidirectional activity-dependent morphological plasticity in hippocampal neurons. Neuron. 2004;44:759–767. doi: 10.1016/j.neuron.2004.11.016. [DOI] [PubMed] [Google Scholar]
- Nakamura K, Watakabe A, Hioki H, Fujiyama F, Tanaka Y, Yamamori T, Kaneko T. Transiently increased colocalization of vesicular glutamate transporters 1 and 2 at single axon terminals during postnatal development of mouse neocortex: a quantitative analysis with correlation coefficient. Eur J Neurosci. 2007;26:3054–3067. doi: 10.1111/j.1460-9568.2007.05868.x. [DOI] [PubMed] [Google Scholar]
- Nakatani-Pawlak A, Yamaguchi K, Tatsumi Y, Mizoguchi H, Yoneda Y. Neonatal phencyclidine treatment in mice induces behavioral, histological and neurochemical abnormalities in adulthood. Biol Pharm Bull. 2009;32:1576–1583. doi: 10.1248/bpb.32.1576. [DOI] [PubMed] [Google Scholar]
- Novak A, Guo C, Yang W, Nagy A, Lobe CG. Z/EG, a double reporter mouse line that expresses enhanced green fluorescent protein upon Cre-mediated excision. Genesis. 2000;28:147–155. [PubMed] [Google Scholar]
- Oni-Orisan A, Kristiansen LV, Haroutunian V, Meador-Woodruff JH, McCullumsmith RE. Altered vesicular glutamate transporter expression in the anterior cingulate cortex in schizophrenia. Biol Psychiatry. 2008;63:766–775. doi: 10.1016/j.biopsych.2007.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otmakhova NA, Otmakhov N, Lisman JE. Pathway-specific properties of AMPA and NMDA-mediated transmission in CA1 hippocampal pyramidal cells. J Neurosci. 2002;22:1199–1207. doi: 10.1523/JNEUROSCI.22-04-01199.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan WX, McNaughton N. The supramammillary area: its organization, functions and relationship to the hippocampus. Prog Neurobiol. 2004;74:127–166. doi: 10.1016/j.pneurobio.2004.09.003. [DOI] [PubMed] [Google Scholar]
- Penzes P, Cahill ME, Jones KA, VanLeeuwen JE, Woolfrey KM. Dendritic spine pathology in neuropsychiatric disorders. Nat Neurosci. 2011;14:285–293. doi: 10.1038/nn.2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prithviraj R, Inglis FM. Expression of the N-methyl-d-aspartate receptor subunit NR3B regulates dendrite morphogenesis in spinal motor neurons. Neuroscience. 2008;155:145–153. doi: 10.1016/j.neuroscience.2008.03.089. (PMID:18541382) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez DM, Kavalali ET. Differential regulation of spontaneous and evoked neurotransmitter release at central synapses. Curr Opin Neurobiol. 2011;21:275–282. doi: 10.1016/j.conb.2011.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsey AJ. NR1 knockdown mice as a representative model of the glutamate hypothesis of schizophrenia. Prog Brain Res. 2009;179:51–58. doi: 10.1016/S0079-6123(09)17906-2. [DOI] [PubMed] [Google Scholar]
- Sakimura K, Kutsuwada T, Ito I, Manabe T, Takayama C, Kushiya E, Yagi T, Aizawa S, Inoue Y, Sugiyama H. Reduced hippocampal LTP and spatial learning in mice lacking NMDA receptor epsilon 1 subunit. Nature. 1995;373:151–155. doi: 10.1038/373151a0. [DOI] [PubMed] [Google Scholar]
- Schäfer MK, Varoqui H, Defamie N, Weihe E, Erickson JD. Molecular cloning and functional identification of mouse vesicular glutamate transporter 3 and its expression in subsets of novel excitatory neurons. J Biol Chem. 2002;277:50734–50748. doi: 10.1074/jbc.M206738200. [DOI] [PubMed] [Google Scholar]
- Schwenk F, Baron U, Rajewsky K. A cre-transgenic mouse strain for the ubiquitous deletion of LoxP-flanked gene segments including deletion in germ cells. Nucleic Acids Res. 1995;23:5080–5081. doi: 10.1093/nar/23.24.5080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinoda Y, Tanaka T, Tominaga-Yoshino K, Ogura A. Persistent synapse loss induced by repetitive LTD in developing rat hippocampal neurons. PLoS One. 2010;5:e10390. doi: 10.1371/journal.pone.0010390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva AJ, Ehninger D. Adult reversal of cognitive phenotypes in neurodevelopmental disorders. J Neurodev Disord. 2009;1:150–157. doi: 10.1007/s11689-009-9018-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skelton RW, McNamara RK. Bilateral knife cuts to the perforant path disrupt spatial learning in the Morris water maze. Hippocampus. 1992;2:73–80. doi: 10.1002/hipo.450020110. [DOI] [PubMed] [Google Scholar]
- Smith SE, Zhou YD, Zhang G, Jin Z, Stoppel DC, Anderson MP. Increased gene dosage of Ube3a results in autism traits and decreased glutamate synaptic transmission in mice. Sci Transl Med. 2011;3:103ra197. doi: 10.1126/scitranslmed.3002627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SM, Uslaner JM, Yao L, Mullins CM, Surles NO, Huszar SL, McNaughton CH, Pascarella DM, Kandebo M, Hinchliffe RM, Sparey T, Brandon NJ, Jones B, Venkatraman S, Young MB, Sachs N, Jacobson MA, Hutson PH. The behavioral and neurochemical effects of a novel d-amino acid oxidase inhibitor compound 8 [4H-thieno [3,2-b]pyrrole-5-carboxylic acid] and d-serine. J Pharmacol Exp Ther. 2009;328:921–930. doi: 10.1124/jpet.108.147884. [DOI] [PubMed] [Google Scholar]
- Smith SM, Uslaner JM, Hutson PH. The therapeutic potential of d-amino acid oxidase (DAAO) inhibitors. Open Med Chem J. 2010;4:3–9. doi: 10.2174/1874104501004020003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorensen SA, Rubel EW. The level and integrity of synaptic input regulates dendrite structure. J Neurosci. 2006;26:1539–1550. doi: 10.1523/JNEUROSCI.3807-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorensen SA, Rubel EW. Relative input strength rapidly regulates dendritic stucture of chick auditory brainstem neurons. J Comp Neurol. 2011;519:2838–2851. doi: 10.1002/cne.22656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 1999;21:70–71. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- Speed HE, Dobrunz LE. Developmental changes in short-term facilitation are opposite at temporammonic synapses compared to Schaffer collateral synapses onto CA1 pyramidal cells. Hippocampus. 2009;19:187–204. doi: 10.1002/hipo.20496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefani MR, Moghaddam B. Transient N-methyl-d-aspartate receptor blockade in early development causes lasting cognitive deficits relevant to schizophrenia. Biol Psychiatry. 2005;57:433–436. doi: 10.1016/j.biopsych.2004.11.031. [DOI] [PubMed] [Google Scholar]
- Sybirska E, Davachi L, Goldman-Rakic PS. Prominence of direct entorhinal-CA1 pathway activation in sensorimotor and cognitive tasks revealed by 2-DG functional mapping in nonhuman primate. J Neurosci. 2000;20:5827–5834. doi: 10.1523/JNEUROSCI.20-15-05827.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takamori S, Rhee JS, Rosenmund C, Jahn R. Identification of differentiation-associated brain-specific phosphate transporter as a second vesicular glutamate transporter (VGLUT2) J Neurosci. 2001;21:RC182. doi: 10.1523/JNEUROSCI.21-22-j0002.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang YP, Shimizu E, Dube GR, Rampon C, Kerchner GA, Zhuo M, Liu G, Tsien JZ. Genetic enhancement of learning and memory in mice. Nature. 1999;401:63–69. doi: 10.1038/43432. [DOI] [PubMed] [Google Scholar]
- Thomson AM. Facilitation, augmentation and potentiation at central synapses. Trends Neurosci. 2000;23:305–312. doi: 10.1016/s0166-2236(00)01580-0. [DOI] [PubMed] [Google Scholar]
- Tordera RM, Pei Q, Sharp T. Evidence for increased expression of the vesicular glutamate transporter, VGLUT1, by a course of antidepressant treatment. J Neurochem. 2005;94:875–883. doi: 10.1111/j.1471-4159.2005.03192.x. [DOI] [PubMed] [Google Scholar]
- Tordera RM, Totterdell S, Wojcik SM, Brose N, Elizalde N, Lasheras B, Del Rio J. Enhanced anxiety, depressive-like behaviour and impaired recognition memory in mice with reduced expression of the vesicular glutamate transporter 1 (VGLUT1) Eur J Neurosci. 2007;25:281–290. doi: 10.1111/j.1460-9568.2006.05259.x. [DOI] [PubMed] [Google Scholar]
- Tsien JZ, Chen DF, Gerber D, Tom C, Mercer EH, Anderson DJ, Mayford M, Kandel ER, Tonegawa S. Subregion- and cell type-restricted gene knockout in mouse brain. Cell. 1996;87:1317–1326. doi: 10.1016/s0092-8674(00)81826-7. [DOI] [PubMed] [Google Scholar]
- Varoqui H, Schäfer MK, Zhu H, Weihe E, Erickson JD. Identification of the differentiation-associated Na+/PI transporter as a novel vesicular glutamate transporter expressed in a distinct set of glutamatergic synapses. J Neurosci. 2002;22:142–155. doi: 10.1523/JNEUROSCI.22-01-00142.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallén-Mackenzie A, Gezelius H, Thoby-Brisson M, Nygård A, Enjin A, Fujiyama F, Fortin G, Kullander K. Vesicular glutamate transporter 2 is required for central respiratory rhythm generation but not for locomotor central pattern generation. J Neurosci. 2006;26:12294–12307. doi: 10.1523/JNEUROSCI.3855-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallén-Mackenzie A, Nordenankar K, Fejgin K, Lagerström MC, Emilsson L, Fredriksson R, Wass C, Andersson D, Egecioglu E, Andersson M, Strandberg J, Lindhe O, Schiöth HB, Chergui K, Hanse E, Långström B, Fredriksson A, Svensson L, Roman E, Kullander K. Restricted cortical and amygdaloid removal of vesicular glutamate transporter 2 in preadolescent mice impacts dopaminergic activity and neuronal circuitry of higher brain function. J Neurosci. 2009;29:2238–2251. doi: 10.1523/JNEUROSCI.5851-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang CC, Held RG, Chang SC, Delpire E, Ghosh A, Hall BJ. A critical role for NMDA receptors containing the GluN2B subunit in cortical development and function. Neuron. 2011;72:789–805. doi: 10.1016/j.neuron.2011.09.023. [DOI] [PubMed] [Google Scholar]
- Wasling P, Hanse E, Gustafsson B. Developmental changes in release properties of the CA3-CA1 glutamate synapse in rat hippocampus. J Neurophysiol. 2004;92:2714–2724. doi: 10.1152/jn.00464.2004. [DOI] [PubMed] [Google Scholar]
- Weston MC, Nehring RB, Wojcik SM, Rosenmund C. Interplay between VGLUT isoforms and endophilin A1 regulates neurotransmitter release and short-term plasticity. Neuron. 2011;69:1147–1159. doi: 10.1016/j.neuron.2011.02.002. [DOI] [PubMed] [Google Scholar]
- Wilson NR, Kang J, Hueske EV, Leung T, Varoqui H, Murnick JG, Erickson JD, Liu G. Presynaptic regulation of quantal size by the vesicular glutamate transporter VGLUT1. J Neurosci. 2005;25:6221–6234. doi: 10.1523/JNEUROSCI.3003-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong RO, Ghosh A. Activity-dependent regulation of dendritic growth and patterning. Nat Rev Neurosci. 2002;3:803–812. doi: 10.1038/nrn941. [DOI] [PubMed] [Google Scholar]
- Zoghbi HY. Postnatal neurodevelopmental disorders: meeting at the synapse? Science. 2003;302:826–830. doi: 10.1126/science.1089071. [DOI] [PubMed] [Google Scholar]