

Epidemic of Cardiometabolic Disease in Developing Nations: A Threat to Global Prosperity
According to the International Diabetes Federation in the year 2011, diabetes mellitus (DM) affects at least 366 million people worldwide, and that number is expected to reach 566 million by the year 2030. Over 99% of all diabetes cases represent type 2 DM with most of these projected to occur in low- to middle-income countries. Technology innovations, globalization with its free movement of food and services, seismic shifts in agrarian practices, and nutritional transition to freely available high-caloric diets have irrevocably altered energy expenditures during work and leisure. These and other factors are helping to foster the continued epidemiological transition occurring across the globe. Scientific effort over the last few decades has focused primarily on components of urbanization such as inactivity and dietary factors. More recent observations have provided additional links between exposure to environmental factors in air/water and propensity to chronic diseases (1). This issue is of importance given the extraordinary confluence of high levels of airborne and water pollutants in urbanized environments. Multiple studies in China, India, and other rapidly urbanizing economies demonstrate a steep gradient in urban–rural prevalence.
This review will summarize recent evidence on how outdoor air pollution may represent an underappreciated yet critical linkage between urbanization and the emergence of cardiometabolic diseases, with a focus on type 2 DM. We define cardiometabolic disease as the confluence of cardiovascular disease and type 2 DM in recognition of the fact that the milieu of diabetes fundamentally alters the pathophysiology of coronary, cerebrovascular, and peripheral arterial disease. Thus, alteration in susceptibility to DM automatically increases the likelihood of cardiovascular disease. Indoor air pollution is not discussed owing to the paucity of data. It should be noted that our current understanding of air pollution–mediated cardiometabolic disease is derived from outdoor air pollution studies, with there being no good reasons to believe that the dose-response relationship to indoor air pollution will be any different. An understanding of potentially reversible environmental factors responsible for this rapid burgeoning of cardiometabolic disorders among developing nations is crucial in order to devise a societal response that is proportionate and adequate (2). In this review, the association between air pollution and type 2 DM is discussed unless this distinction cannot be made in the cited study (typically health registry data sets).
Exposure to Environmental Toxins and Metabolic Disease
Epidemiologic studies that have attempted to investigate environmental factors that accentuate risk for development of cardiometabolic disorders have uncovered a number of factors other than traditional suspects related to diet and exercise. These variables include factors such as stress (mental and emotional), cultural and socioeconomic variables, chronic low-grade infection, and environmental pollutants (Fig. 1). In many instances, these factors are strongly correlated, rendering isolation of cause and effect difficult.
FIG. 1.

A model for development of cardiometabolic disease highlighting importance of gene–environment interactions. (A high-quality digital representation of this figure is available in the online issue.)
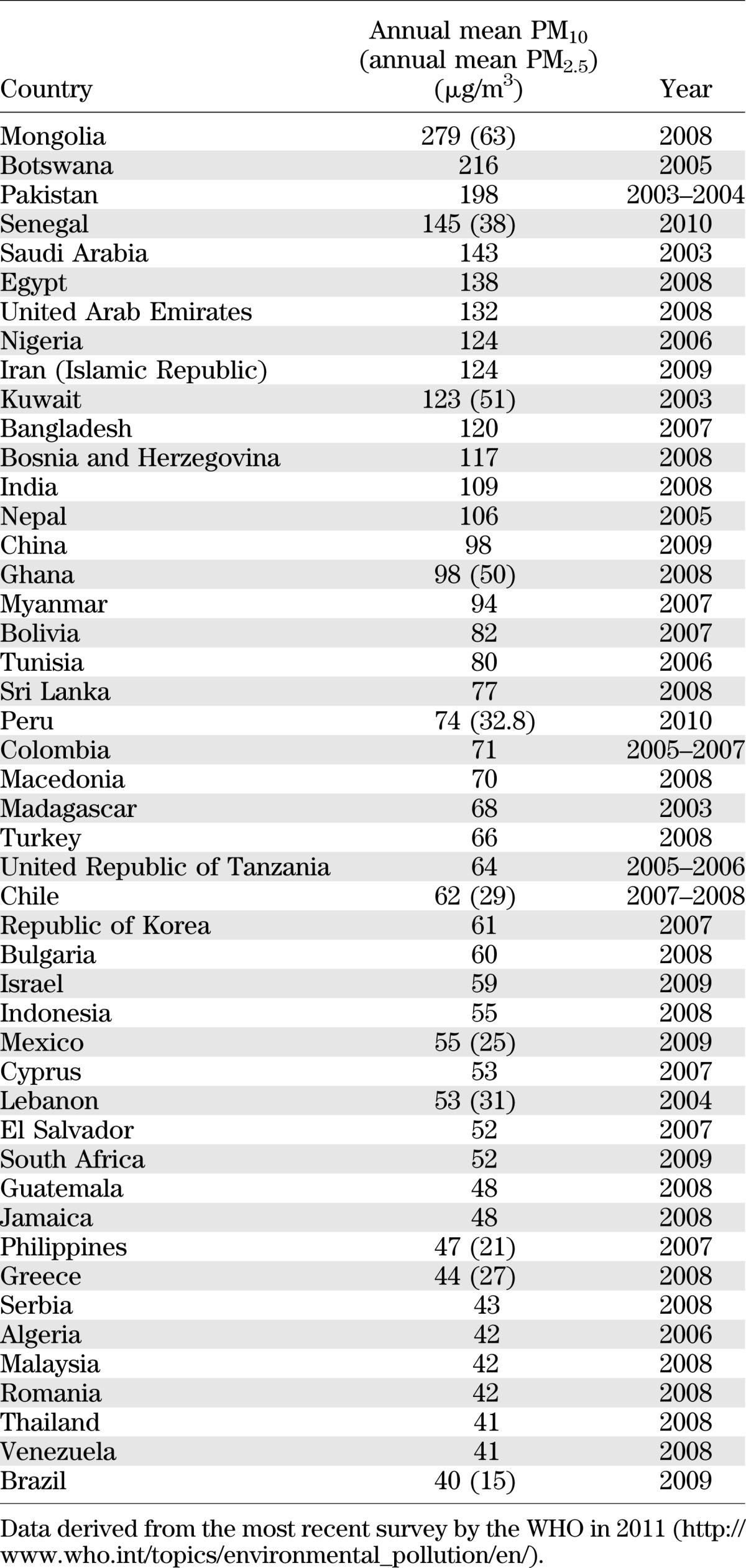
The plausibility that environmental exposures are linked to metabolic disease is exemplified by persistent organic pollutants, toxins that have consistently shown to associate with insulin resistance (IR) and type 2 DM. Prospective cohort studies of subjects exposed to 2,3,7,8-tetrachlorodibenzo-p-dioxin or other persistent organic pollutants in occupational and other settings have reported increased risk of DM and IR (1,3). Air pollution in Asia, Latin America, and Africa is a significant public health burden, especially given the often extraordinarily high concentrations of pollutants (e.g., particulate matter), high population density, and pervasive nature of air pollution. Table 1 lists the top countries for particulate matter (PM) air pollution in the world, all of which have rapidly urbanized populations based on a World Health Organization (WHO) database that reviewed pollution data in >1,100 cities in 91 countries. The mean annual average for the top 10 countries is roughly fivefold higher than the U.S. National Ambient Air Quality Standard of 15 μg/m3 for PM <2.5 μg mass (PM2.5) and the WHO standards of 10 μg/m3. Given the worldwide burden of air-pollution effects and their continuous and omnipresent nature, even small adverse health associations for individuals represent an enormous public health issue that deserves broad changes in public health policy (4).
TABLE 1.
Annual PM data from 47 countries with the highest reported PM10 levels
Epidemiologic Evidence Linking Air Pollution and Type 2 Diabetes
There are now at least six published epidemiologic studies showing some degree of association between PM- or traffic-related air pollutants and DM. At least two other studies have demonstrated a relationship between ambient levels of air pollutants and markers of insulin sensitivity in humans. The main characteristics and principal findings of these studies are summarized in Table 2. In most of these studies, no distinction was made between type 2 DM and type 1, and hence, the overall prevalence of DM is described. Because the vast majority of patients with diabetes are type 2, the associations likely describe an effect of type 2 DM.
TABLE 2.
Epidemiological associations among air pollutants, diabetes, and insulin resistance

Though not all findings from every study were positive, taken together, the majority of observations support an association between air pollution, in particular traffic-related sources, and DM. Nonetheless, not all aspects of this relationship have been consistently reported nor are they fully elucidated at this time. The varying associations noted between studies may relate to numerous differences. These include the population characteristics, risk factors, individual susceptibilities, robustness of the cohort data, prevalence of type 2 DM, technical aspects of the exposure assessment methodologies, pollution types/sources, and the degree and duration of air-pollution exposures. The sex-specific differences seen in some of these studies may relate to true differences in biologic susceptibility, a finding mirrored by observations in the Women’s Health Study that also demonstrated a greater susceptibility of obese women to air pollution–mediated cardiovascular events (5). In contrast, it is also possible that the sex predilection may relate to exposure assessment error, particularly in males, who tend to be more mobile compared with females. Additional considerations include cosegregation of factors such as low socioeconomic status, stress, and poorly characterized pollutants often pervasive in the urban environment.
Type 2 DM and metabolic syndrome as a susceptibility factor for air-pollution effects.
Diabetic patients have previously been shown to be more susceptible to air pollution–induced cardiovascular morbidity and mortality (6,7). A few studies have examined the underlying mechanisms. In a study in Boston-area residents, 6-day moving averages of PM2.5 and black carbon (BC) were associated with decreased vascular reactivity among patients with diabetes (8). Effects were stronger in type 2 than type 1 DM. In another study in Boston involving patients participating in unrelated clinical trials who provided blood samples, BC concentrations from a regional monitoring station were significantly associated with increased levels of inflammatory markers (9). Although many of the estimates were imprecise owing to limited sample size, the overall trend of the point estimates was positive, consistent with epidemiological and experimental data. These data are supported by prospective panel studies in a small population of type 2 DM patients. Flow-mediated dilatation decreased with PM2.5 during the first 24 h. These PM2.5-associated decrements in endothelial function were greater among participants with high hemoglobin A1c, low adiponectin, and elevated myeloperoxidase levels on the examination day (10). Thus, alterations in vascular tone and inflammation may represent potential mechanisms that may explain susceptibility.
Experimental Evidence of Mechanisms of Diabetes Association With Air Pollution
PM2.5 as a mediator of endothelial dysfunction and IR.
Air-pollution exposure alters endothelial function in both animals and humans (11,12). Alterations in endothelial function often precede changes in IR and have been implicated in reduced peripheral glucose uptake (13). In the first experimental investigation directly linking inhalational exposure of PM2.5 with DM, exposure in conjunction with high-fat diet feeding, increased fasting, postprandial glucose, insulin, and Homeostasis Model Assessment-IR (HOMA-IR) measures. The changes in IR measures seen with PM2.5 were incremental to that of high-fat diet alone over a period of 24 weeks (14). The mean concentration of PM2.5 was 60 ± 5 µg/m3 (∼10-fold concentration from ambient levels). Tumor necrosis factor (TNF)-α, interleukin-6 (IL-6), resistin, and leptin levels were elevated following PM2.5 exposure, in keeping with a proinflammatory insulin-resistant state. PM exposure also resulted in elevations in prothrombotic adipokines such as plasminogen activator inhibitor 1 and increased circulating adhesion molecules such as intracellular adhesion molecule-1 and E-selectin. The latter are important in promoting leukocyte adherence in postcapillary venular endothelium (15). PM exposure was associated with impairment in phosphatidylinositol 3-kinase–Akt–endothelial nitric oxide synthase signaling in the aorta and decreased tyrosine phosphorylation of IRS-1 in the liver (15), providing evidence for abnormal insulin signaling in the vasculature.
In subsequent experiments, the effect of PM2.5 exposure early in life with and without concomitant exposure to a high-fat diet was evaluated. C57BL/6 mice fed a normal diet but exposed for 10 weeks exhibited metabolic abnormalities including an increase in HOMA-IR and postprandial glucose that approached those seen with high-fat chow diet–fed mice exposed to filtered air (16). In another study, intratracheal exposure of PM2.5 potentiated IR at the end of 3 weeks in high-fat–fed male Sprague-Dawley rats (17). Taken together, these experiments suggest an important interaction of PM2.5 exposure with high-fat diet, and they raise the possibility that early life may represent a vulnerable period of enhanced susceptibility to PM2.5 exposure effects. There are currently no studies on the effect of air-pollution exposure on β-cell function.
Inflammation including visceral adipose tissue effects of air pollution.
There is evidence that exposure to ambient PM can be associated with elevated systemic proinflammatory biomarkers. Several reports have detailed association between day-to-day variation in acute-phase proteins, such as C-reactive protein (CRP), IL-6, fibrinogen, or white blood cell counts and circulating soluble adhesion molecules as reviewed previously (4). In an analysis of 1,003 myocardial infarction survivors, ambient particle number concentration and PM with diameter <10 μm (PM10) were associated with increased IL-6 and fibrinogen (18). Pollutants associated with primary combustion (e.g., elemental and BC, primary organic carbon) and ultrafine particles rather than PM2.5 appeared to be strongly associated with adverse responses. A positive association between white blood cell count and estimated long-term 1-year exposure to PM10 was reported in the Third National Health and Nutrition Examination Survey (19). Among 4,814 adults in Germany, small increases in annual mean PM2.5 (3.9 μg/m3) were associated with increases in high-sensitivity CRP by 24% and fibrinogen by 4% among men. It is important to note that several studies, including some with improved exposure assessment (20) and large population cohorts (21,22), have not found a relationship between particulate exposure and inflammation. It is thus conceivable that variations in the particulate chemistry and duration/intensity of exposure as well as susceptibility factors may be at play. Subjects with underlying risk factors such as type 2 DM and metabolic syndrome may exhibit stronger associations (19,23,24). As pointed out earlier, type 2 DM may represent a unique susceptibility factor that may potentiate inflammation (8,9).
In experimental animal models, PM2.5 exposure results in an increase in adipose tissue macrophages with a shift to a proinflammatory phenotype characterized by an increase in TNF-α and IL-6 and a decrease in IL-10 gene expression (15). The finding of increased innate immune cells in visceral adipose tissue (VAT) is a pathophysiologic hallmark of type 2 DM. To test the mechanism by which increased proinflammatory monocytes permeate VAT, a transgenic model of yellow fluorescent protein (YFP) expression, driven by a monocyte-lineage promoter (and therefore restricted to monocytes, c-fmsYFP), was employed to follow the migration of cells into the VAT compartment. The animals were initially rendered insulin-resistant with a high-fat diet and then subject to air-pollution exposure. Intravital microscopic studies were conducted to detect leukocyte–endothelial interactions. Pollution exposure resulted in a doubling in the number of endothelial adherent YFP+ cells in mesenteric fat with a sixfold increase in monocytes within adipose (15). Thus, PM mediated adhesion and migration of YFP+ cells into visceral fat depots. These changes in adipose occurred with concomitant low-grade inflammation in the lung (25). In subsequent studies, PM2.5 exposure alone (normal chow diet) resulted in a heightened chemotactic ability of adipose tissue from PM2.5-exposed mice (16). In keeping with the well-known link between air-pollution exposure with oxidant stress, PM2.5 exposure was associated with oxidative stress in VAT and increased phosphorylation of a key cytosolic subunit of NADPH oxidase, and p47. p47phox−/− mice were protected from the effects of PM2.5 exposure and did not exhibit impairment in IR, vascular function, and visceral inflammation in response to PM2.5 (16). Thus, data from experimental animal models suggest that air pollution exposure may direct an innate immune response in VAT.
Hepatic IR and endoplasmic reticulum stress.
Defective insulin signaling in tissues such as the liver are fundamental to the pathogenesis of IR/DM. Increased serine phosphorylation of IRS-1 and decreased tyrosine phosphorylation results in defective phosphatidylinositol 3-kinase–Akt signaling and suppression of insulin-stimulated GLUT4 translocation. PM2.5 exposure decreases phosphorylation of Akt in the liver and skeletal muscle compared with filtered air–exposed control, and these changes were accompanied by hepatic lipid deposition and decreased gluconeogenesis (26,27). Endoplasmic reticulum (ER) stress, also called unfolded protein response (UPR), is an evolutionarily conserved pathway designed to alleviate protein misfolding in response to diverse pathophysiologic stressors (28). In vitro-exposure studies have demonstrated that PM2.5 is capable of inducing ER stress and the UPR in vitro and may be a mechanism by which PM2.5 exerts toxicity. PM exposure results in significant increase in the UPR-associated proteins activating transcription factor (ATF) 4, heat shock proteins 70 and 90, and binding immunoglobulin protein (BiP). In response to inhalational PM2.5, glucose regulatory peptide 94 and BiP increase in lungs and liver compared with minimal induction in aorta and spleen, indicating activation of the ATF6 pathway in these organs (14). ATF6 is one of three key main sensors of ER stress (the others being: inositol requiring 1a and double-stranded RNA-activated protein kinase-like ER kinase [PERK]). Phosphorylated PERK and eukaryotic translation initiation factor 2a were increased in the liver along with induction of C/EBP homologous transcription factor CHOP/GADD153 (14). The latter correlated with apoptosis in the lung and liver. The UPR is known to intersect with a variety of inflammatory and stress-signaling systems including the nuclear factor (NF)-κB and c-Jun N-terminal kinase pathways as well oxidative stress responses, all of which may influence lipid and glucose metabolism. In these studies, a critical role for oxidant stress mediated via NADPH oxidase in activation of the ER stress response was also demonstrated (14). In a subsequent article, Zheng et al. (27) have demonstrated that PM2.5 exposure causes a nonalcoholic steatohepatitis-like phenotype and reduction of hepatic glycogen storage in animals. PM2.5 exposure lead to activation of the inflammatory pathway through c-Jun N-terminal kinase and downregulation of the insulin receptor substrate 1 (IRS1)-mediated signaling and peroxisome proliferator-activated receptor γ2 expression in the liver. These changes were associated with abnormalities in IR and glucose homeostasis (27).
Mitochondrial dysfunction and brown adipose tissue alterations.
Recent data in adult humans suggest an important link between brown adipose tissue (BAT)–mediated thermogenesis and obesity. Defective fatty acid metabolism through β-oxidation in mitochondria lead to accumulation of intracellular metabolites, including fatty-acyl CoA, diacylglycerol, and ceramide in both skeletal muscle and liver contributing to IR/DM (29). Prior studies have shown that both cigarette smoke and hypercholesterolemia greatly increase mitochondrial damage (30). With long-term exposure to PM2.5 (10 months), visible decreases in interscapular BAT and mitochondrial size were noted (25). These changes were accompanied by an increase in excess oxidative and nitrosative stress in BAT, coordinated with phase II antioxidant gene induction, including NF-E2–related factor 2, NAD(P)H quinone oxidoreductase 1, and glutamate-cysteine ligase modifier subunit. BAT expression of Ucp1 and peroxisome proliferator–activated receptor γ coactivator 1α were decreased with PM2.5 exposure, whereas Prdm16, Pgc-1α, and Pparγ2 were significantly decreased in the white adipose tissue, suggestive of downregulation of pathways that modulate insulin sensitivity in adipose (31). Similar results were also seen with a different model of IR (31).
Integrated Model of Mechanisms Involved in Air Pollution–Mediated Diabetes
How may signals perceived in the lung lead to metabolic abnormalities?
The notion that alteration in lung function may lead to metabolic dysfunction is not new (32). Chronic inflammation is a sine qua non for type 2 DM and obesity (metaflammation), is well known to occur with air-pollution exposure, and may represent a potential link between air-pollution exposure and metabolic dysfunction. Type 2 DM in humans and animal models is associated with increased levels of inflammatory cytokines and recruitment and/or activation of innate immune cells in depots such as visceral adipose. Animal models of type 2 DM/obesity have provided strong evidence that diet-induced oxidative stress/inflammation plays a critical role in the pathogenesis of type 2 DM (33). In addition to classical inflammatory pathways, air pollutant–mediated alterations in autonomic balance may further exacerbate systemic insulin resistance via overactivity of the sympathetic nervous system. Numerous pulmonary receptors such as transient receptor potential ankyrin 1 can be stimulated by pollutant inhalation and prompt sympathetic activation through centrally mediated pathways (34). The robust literature linking PM exposure with impaired heart rate variability supports such a mechanism (4). Unpublished observations from one of our studies demonstrate that elevations in ambient levels of fine PM in the Detroit area were associated with worsening of insulin sensitivity among healthy adults over a 5-day period of exposure (S.R., R.D.B.). Reductions in heart rate variability were linked to both exposures and the worsened insulin sensitivity, suggesting that (at least relatively acutely) autonomic pathways might be contributing to PM-induced alterations in metabolic insulin sensitivity. Other potential pathways that have not as of yet been explored may also be involved. Plausible mechanisms that could be impacted by air pollutants include activation of hypothalamic–pituitary–adrenal axis, impaired insulin-sensitive tissue perfusion due to endothelial dysfunction or vasoconstriction, dysfunctional activity of high-density lipoprotein particles, and/or epigenetic alterations of critical modulators of cellular insulin signaling (e.g., PGC-1α).
Toll-like receptors/nucleotide-binding oligomerization domain-like receptors as sensors for environmental signals: links between immunity and type 2 DM.
Toll-like receptors (TLRs) and nucleotide-binding oligomerization domain-like receptors (NLRs) are widely expressed on immune cells, with a substantive body of evidence linking these pathways in experimental and human obesity/DM (35,36). In contrast to dietary signals, the alveolar macrophage and bronchial epithelial cells are principal initial cellular sensors of PM and express TLRs and NLRs. Biologic components intrinsic to PM such as lipopolysaccharide (LPS) and peptides and gaseous copollutants such as ozone can directly activate TLRs (37,38). The NLR Nalp3 has been shown to sense a diversity of particulate components and induce production of IL-1β (39). Although the level of LPS is lower in PM2.5 versus PM10, there are data linking levels of such components with IR. In a recent prospective study by Sun et al. (40) in an urban population in Shanghai, an important predictor for the development of type 2 DM on multivariate analysis after adjustment of most risk factors including CRP was LPS binding protein. LPS binding protein is a better surrogate for LPS in plasma, and emerging studies suggest that this may serve as a surrogate for inflammatory disorders resulting from activation of the innate immune system (38,41). Endogenous danger-associated molecular patterns (DAMPs) that are released in response to PM may represent additional mechanisms for TLR/NLR activation that may potentiate already overactive pathways in obesity/IR. In a recent study, a key role for lipotoxicity-associated ceramide accumulation in the pathogenesis of type 2 DM via activation of Nlp3 was demonstrated (36). A number of DAMPs released in response to PM and/or gaseous components have been demonstrated, including oxidized phospholipid components and hyaluronan fragments (26,37). Oxidation products of palmitoyl-arachidonyl phosphocholine, an abundant phospholipid in lung lavage fluid, has been implicated in a diverse variety of lung-injury signals to activate TLR4 (42). Release of oxidized palmitoyl-arachidonyl phosphocholine may facilitate innate immune activation in the lung and function as a mechanism to release chemokines such as CC chemokine ligand (CCL)-2 that may then secondarily mediate efflux of inflammatory monocytes (43). Ozone exposure in animal models may mediate degradation of hyaluronan, which can then activate TLR4 via myeloid differentiating factor 88 pathways (37). Thus, oxidized phospholipids, hyaluronan fragments, and possibly ceramide as a consequence of diet and/or air-pollution coexposure may represent mechanisms that couple IR/type 2 DM development with air pollution.
How may innate immune signals in the lung secondarily lead to systemic inflammation susceptibility to IR/type 2 DM? In this regard, a number of key pathways have been posited: 1) direct inflammatory/oxidative stress of cells such as alveolar macrophages particularly under conditions of overload typified by continual exposure to PM may release innate immune cytokines such as IL-1, TNF-α, IL-6, and chemokine triggers from the lung, including CCL-2 and CCL-5, that may mediate a cellular response from the bone marrow/spleen (26,44). Oxidant stress may be critical in activation of these pathways (26), with prior studies demonstrating a role for NADPH oxidases in TLR4- and Nalp3-mediated inflammation (39,42,45). 2) Uptake by macrophages of particulates may also lead to presentation via dendritic cells to T cells in secondary lymphoid organs resulting in adaptive immune responses (43). 3) Direct penetration of leechable components such as reactive oxygen species/transition metals and stable organic compounds (e.g., quinines, semiquinones, and aldehydes) into the systemic vasculature has also been hypothesized and may lead to vascular inflammation and potentially IR (46,47). 4) Finally, emerging data support a role for central mechanisms for inflammation via afferent pathways linking the lung with the brain (34). Fig. 2 provides a hypothetical framework for these interactions and illustrates how inhalational stimuli may interact with overnutrition to entrain a state of chronic oxidative stress and inflammation.
FIG. 2.

Hypothesized mechanisms of air pollution–mediated cardiometabolic disease wherein inhalational or nutritional signals either directly or via the generation of signals such as DAMPs may serve to activate innate immune mechanisms such as the TLR and NLR. AP1, activator protein 1; CARD, caspase activation and recruitment domain; IKKb, IκB kinase b; IRAK, interleukin receptor-associated kinase; IRF3, interferon regulatory factor 3; MAPK, mitogen-activated protein kinase; MyD88, myeloid differentiation primary response gene 88; NAFLD, nonalcoholic fatty liver disease; PAMP, pathogen-associated molecular pattern; PAPC, palmitoyl-arachidonyl phosphocholine; RNS, reactive nitrogen species; ROS, reactive oxygen species; TAK, transforming growth factor-β–activated kinase; TBK, TANK-binding kinase 1; TRAF, TNF receptor-associated factor; TRIF, Toll/IL-1 receptor-domain-containing adapter-inducing interferon-β; UCP-1, uncoupling protein-1; WAT, white adipose tissue. (A high-quality digital representation of this figure is available in the online issue.)
Homeostatic mechanisms to prevent exuberant lung inflammation.
Teleologically, it is thought that pattern recognition receptors were meant to represent a crude but critical early-warning system to rapidly sense changes in lung microenvironment but also, equally importantly, to dissipate early to prevent unfettered inflammation. Thus, the notion that continual activation of these receptors may occur in a feed-forward manner and in concert with other stimuli without dissipation may be somewhat simplistic. However, it is also true that as humans, we did not evolve to be continually exposed to dietary and inhalational stimuli over the years, and such chronic exposure in vivo may have very different effects that we insufficiently understand. Alveolar macrophages continually exposed to PM may result in particle overload resulting in a state of perpetual low-grade inflammation. Additional counterregulatory mechanisms that prevent excess TLR activation including proteins and phospholipids in the bronchoalveolar fluid may also be rendered dysfunctional with chronic PM exposure and participate in inflammation. The protein component of bronchoalveolar lavage includes the collectins such as surfactant A and surfactant D, both of which inhibit TLR4 and TLR2 signaling and upregulate anti-inflammatory functions including efferocytosis and dissipation of oxidant stress. Surfactants may play a dual role in the presence of DAMPs and facilitate inflammation (48). Oxidative modification of surfactants and phospholipid components has been reported that may lead to a facilitatory role in air pollution–mediated effects (43,49,50).
Emerging data from prospective human studies suggest a relationship between surfactant proteins (SPs) and propensity to inflammation and atherosclerosis. In one study, serum SP-D concentration was significantly decreased in subjects with obesity and type 2 DM (P = 0.005) and negatively associated with fasting/postload glucose, HbA1C, plasma triglycerides, insulin sensitivity, and inflammation (TNF-R levels) (51). Smoking subjects, in contrast, showed significantly higher serum SP-D concentration than nonsmokers. In the Vancouver Angiography cohort, plasma SP-D levels correlated with cardiovascular mortality, and values >176 ng/mL associated with a fourfold excess risk. Addition to SP-D levels to traditional risk factors improved c-statistic (from 0.76 to 0.78) and net reclassification across all levels of risk (52). In the Dallas Heart Study, increasing levels of SP-B was associated with other traditional cardiac risk factors and higher levels of inflammatory biomarkers. In multivariable analyses after adjusting for risk factors, SP-B remained associated with aortic plaque in smokers (odds ratio 1.87, fourth versus first quartiles; P < 0.0001) (53).
How does one reconcile increases in plasma SP levels in population studies to increase susceptibility? Increased levels of surfactants in plasma seen in smoking and lung inflammation have been hypothesized to indicate translocation from the lung to the circulation with lung damage. Surfactants A and D are assembled as large multimeric units composed of lectin-containing globular domains and a collagenous domain. In the presence of DAMPs, they may exert proinflammatory effects by binding to CD47 (thrombospondin receptor) (48). It is also highly possible that increased levels may indicate oxidatively modified forms of surfactant that are not functional. Surfactants are often assembled as multimers and are well-known to undergo oxidative modification to oligomeric forms. Current assays for SPs do not distinguish between these various forms.
Future Directions
A growing body of evidence has implicated inflammatory responses to diet and environmental factors as a key mechanism that help explain the emerging epidemic in diabetes and cardiovascular disease. Both genetic and environmental factors undoubtedly play a role, although the role of the physical and social environment in determining susceptibility appears to be critical. Nontraditional factors such as air pollution that are pervasive in the urban environment may provide low-level synergism with other dominant factors in accelerating propensity for type 2 DM. Emerging data from both experimental and epidemiologic studies are beginning to provide insights into this association. There are a number of areas that would benefit from further studies and enable additional insights into the mechanisms by which environmental signals modulate susceptibility to metabolic disease. The effects of air-pollution exposure on β-cell function, counterregulatory hormones such as glucagon, and effects on insulinotropic mechanisms deserve further study. The effects of air pollution on hypothalamic mechanisms of appetite and satiety are areas of emerging interest, as it is entirely possible that air pollutants may modulate inflammation in key brain homeostatic centers. In addition, the effects on central autonomic control of peripheral inflammation may represent additional pathways by which environmental triggers may play an important role in determining peripheral inflammation. The societal costs of this link, if indeed true, are staggering given the ubiquitous nature of air pollution and the economic costs of obesity/DM-related complications. Given the already established nature of the links between air pollution and cardiovascular disease and regulations already in place, at least in countries like the U.S. and Europe, these additional links, if they can be established in additional large cohorts, would provide persuasive rationale for limiting exposure to air pollution.
ACKNOWLEDGMENTS
This work was supported by National Institute of Environmental Health Sciences (NIEHS) grants R01-ES-017290, R01-ES-015146, and R01-ES-019616 (to S.R.) and the Environmental Protection Agency–supported Great Lakes Center for Environmental Research (to S.R. and R.D.B.).
No potential conflicts of interest relevant to this article were reported.
S.R. and R.D.B. researched the data and wrote and reviewed the manuscript.
The authors thank Geoffrey Gatts, Wexner Medical Center at The Ohio State University, for excellent editorial assistance and data collection.
REFERENCES
- 1.Carpenter DO. Environmental contaminants as risk factors for developing diabetes. Rev Environ Health 2008;23:59–74 [DOI] [PubMed] [Google Scholar]
- 2.Fuster V, Kelly BB. (Eds.). Institute of Medicine (US) Committee on Preventing the Global Epidemic of Cardiovascular Disease: Meeting the Challenges in Developing Countries. Atlanta, GA, National Academies Press, 2010 [Google Scholar]
- 3.Rignell-Hydbom A, Lidfeldt J, Kiviranta H, et al. Exposure to p,p’-DDE: a risk factor for type 2 diabetes. PLoS ONE 2009;4:e7503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brook RD, Rajagopalan S, Pope CA, 3rd, et al. American Heart Association Council on Epidemiology and Prevention, Council on the Kidney in Cardiovascular Disease, and Council on Nutrition, Physical Activity and Metabolism Particulate matter air pollution and cardiovascular disease: An update to the scientific statement from the American Heart Association. Circulation 2010;121:2331–2378 [DOI] [PubMed] [Google Scholar]
- 5.Miller KA, Siscovick DS, Sheppard L, et al. Long-term exposure to air pollution and incidence of cardiovascular events in women. N Engl J Med 2007;356:447–458 [DOI] [PubMed] [Google Scholar]
- 6.Zanobetti A, Schwartz J. Cardiovascular damage by airborne particles: are diabetics more susceptible? Epidemiology 2002;13:588–592 [DOI] [PubMed] [Google Scholar]
- 7.Goldberg MS, Burnett RT, Yale JF, Valois MF, Brook JR. Associations between ambient air pollution and daily mortality among persons with diabetes and cardiovascular disease. Environ Res 2006;100:255–267 [DOI] [PubMed] [Google Scholar]
- 8.O’Neill MS, Veves A, Zanobetti A, et al. Diabetes enhances vulnerability to particulate air pollution-associated impairment in vascular reactivity and endothelial function. Circulation 2005;111:2913–2920 [DOI] [PubMed] [Google Scholar]
- 9.O’Neill MS, Veves A, Sarnat JA, et al. Air pollution and inflammation in type 2 diabetes: a mechanism for susceptibility. Occup Environ Med 2007;64:373–379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schneider A, Neas L, Herbst MC, et al. Endothelial dysfunction: associations with exposure to ambient fine particles in diabetic individuals. Environ Health Perspect 2008;116:1666–1674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mills NL, Törnqvist H, Robinson SD, et al. Diesel exhaust inhalation causes vascular dysfunction and impaired endogenous fibrinolysis. Circulation 2005;112:3930–3936 [DOI] [PubMed] [Google Scholar]
- 12.Sun Q, Wang A, Jin X, et al. Long-term air pollution exposure and acceleration of atherosclerosis and vascular inflammation in an animal model. JAMA 2005;294:3003–3010 [DOI] [PubMed] [Google Scholar]
- 13.Baron AD, Steinberg HO, Chaker H, Leaming R, Johnson A, Brechtel G. Insulin-mediated skeletal muscle vasodilation contributes to both insulin sensitivity and responsiveness in lean humans. J Clin Invest 1995;96:786–792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Laing S, Wang G, Briazova T, et al. Airborne particulate matter selectively activates endoplasmic reticulum stress response in the lung and liver tissues. Am J Physiol Cell Physiol 2010;299:C736–C749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sun Q, Yue P, Deiuliis JA, et al. Ambient air pollution exaggerates adipose inflammation and insulin resistance in a mouse model of diet-induced obesity. Circulation 2009;119:538–546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xu X, Yavar Z, Verdin M, et al. Effect of early particulate air pollution exposure on obesity in mice: role of p47phox. Arterioscler Thromb Vasc Biol 2010;30:2518–2527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yan YH, Chou CC, Lee CT, Liu JY, Cheng TJ. Enhanced insulin resistance in diet-induced obese rats exposed to fine particles by instillation. Inhal Toxicol 2011;23:507–519 [DOI] [PubMed] [Google Scholar]
- 18.Rückerl R, Phipps RP, Schneider A, et al. Ultrafine particles and platelet activation in patients with coronary heart disease—results from a prospective panel study. Part Fibre Toxicol 2007;4:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen JC, Schwartz J. Metabolic syndrome and inflammatory responses to long-term particulate air pollutants. Environ Health Perspect 2008;116:612–617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sullivan JH, Hubbard R, Liu SL, et al. A community study of the effect of particulate matter on blood measures of inflammation and thrombosis in an elderly population. Environ Health 2007;6:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Steinvil A, Kordova-Biezuner L, Shapira I, Berliner S, Rogowski O. Short-term exposure to air pollution and inflammation-sensitive biomarkers. Environ Res 2008;106:51–61 [DOI] [PubMed] [Google Scholar]
- 22.Diez Roux AV, Auchincloss AH, Astor B, et al. Recent exposure to particulate matter and C-reactive protein concentration in the multi-ethnic study of atherosclerosis. Am J Epidemiol 2006;164:437–448 [DOI] [PubMed] [Google Scholar]
- 23.Dubowsky SD, Suh H, Schwartz J, Coull BA, Gold DR. Diabetes, obesity, and hypertension may enhance associations between air pollution and markers of systemic inflammation. Environ Health Perspect 2006;114:992–998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zeka A, Sullivan JR, Vokonas PS, Sparrow D, Schwartz J. Inflammatory markers and particulate air pollution: characterizing the pathway to disease. Int J Epidemiol 2006;35:1347–1354 [DOI] [PubMed] [Google Scholar]
- 25.Xu X, Liu C, Xu Z, et al. Long-term exposure to ambient fine particulate pollution induces insulin resistance and mitochondrial alteration in adipose tissue. Toxicol Sci 2011;124:88–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kampfrath T, Maiseyeu A, Ying Z, et al. Chronic fine particulate matter exposure induces systemic vascular dysfunction via NADPH oxidase and TLR4 pathways. Circ Res 2011;108:716–726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zheng Z, Xu X, Zhang X, et al. Exposure to ambient particulate matter induces a non-alcoholic steatohepatitis-like phenotype and impairs hepatic glucose metabolism. J Hepatol. 15 August 2012 [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science 2011;334:1081–1086 [DOI] [PubMed] [Google Scholar]
- 29.Lowell BB, Shulman GI. Mitochondrial dysfunction and type 2 diabetes. Science 2005;307:384–387 [DOI] [PubMed] [Google Scholar]
- 30.Knight-Lozano CA, Young CG, Burow DL, et al. Cigarette smoke exposure and hypercholesterolemia increase mitochondrial damage in cardiovascular tissues. Circulation 2002;105:849–854 [DOI] [PubMed] [Google Scholar]
- 31.Xu Z, Xu X, Zhong M, et al. Ambient particulate air pollution induces oxidative stress and alterations of mitochondria and gene expression in brown and white adipose tissues. Part Fibre Toxicol 2011;8:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sandler M, Bunn AE, Stewart RI. Pulmonary function in young insulin-dependent diabetic subjects. Chest 1986;90:670–675 [DOI] [PubMed] [Google Scholar]
- 33.Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. J Clin Invest 2006;116:1793–1801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Simon SA, Liedtke W. How irritating: the role of TRPA1 in sensing cigarette smoke and aerogenic oxidants in the airways. J Clin Invest 2008;118:2383–2386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS. TLR4 links innate immunity and fatty acid-induced insulin resistance. J Clin Invest 2006;116:3015–3025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vandanmagsar B, Youm YH, Ravussin A, et al. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat Med 2011;17:179–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li Z, Potts-Kant EN, Garantziotis S, Foster WM, Hollingsworth JW. Hyaluronan signaling during ozone-induced lung injury requires tlr4, myd88, and tirap. PLoS ONE 2011;6:e27137. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 38.Takeda K, Akira S. Toll-like receptors. Curr Protoc Immunol 2007;Chapter 14:Unit 14.12. [DOI] [PubMed] [Google Scholar]
- 39.Dostert C, Pétrilli V, Van Bruggen R, Steele C, Mossman BT, Tschopp J. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science 2008;320:674–677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sun L, Yu Z, Ye X, et al. A marker of endotoxemia is associated with obesity and related metabolic disorders in apparently healthy Chinese. Diabetes Care 2010;33:1925–1932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lepper PM, Schumann C, Triantafilou K, et al. Association of lipopolysaccharide-binding protein and coronary artery disease in men. J Am Coll Cardiol 2007;50:25–31 [DOI] [PubMed] [Google Scholar]
- 42.Imai Y, Kuba K, Neely GG, et al. Identification of oxidative stress and Toll-like receptor 4 signaling as a key pathway of acute lung injury. Cell 2008;133:235–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Deiuliis JA, Kampfrath T, Zhong J, et al. Pulmonary T cell activation in response to chronic particulate air pollution. Am J Physiol Lung Cell Mol Physiol 2012;302:L399-L409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Goto Y, Ishii H, Hogg JC, et al. Particulate matter air pollution stimulates monocyte release from the bone marrow. Am J Respir Crit Care Med 2004;170:891–897 [DOI] [PubMed] [Google Scholar]
- 45.Laroux FS, Romero X, Wetzler L, Engel P, Terhorst C. Cutting edge: MyD88 controls phagocyte NADPH oxidase function and killing of gram-negative bacteria. J Immunol 2005;175:5596–5600 [DOI] [PubMed] [Google Scholar]
- 46.Dominici F, Peng RD, Ebisu K, Zeger SL, Samet JM, Bell ML. Does the effect of PM10 on mortality depend on PM nickel and vanadium content? A reanalysis of the NMMAPS data. Environ Health Perspect 2007;115:1701–1703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liberda EN, Cuevas AK, Gillespie PA, Grunig G, Qu Q, Chen LC. Exposure to inhaled nickel nanoparticles causes a reduction in number and function of bone marrow endothelial progenitor cells. Inhal Toxicol 2010;22(Suppl. 2):95–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gardai SJ, Xiao YQ, Dickinson M, et al. By binding SIRPalpha or calreticulin/CD91, lung collectins act as dual function surveillance molecules to suppress or enhance inflammation. Cell 2003;115:13–23 [DOI] [PubMed] [Google Scholar]
- 49.Davis IC, Zhu S, Sampson JB, Crow JP, Matalon S. Inhibition of human surfactant protein A function by oxidation intermediates of nitrite. Free Radic Biol Med 2002;33:1703–1713 [DOI] [PubMed] [Google Scholar]
- 50.Crouch EC, Hirche TO, Shao B, et al. Myeloperoxidase-dependent inactivation of surfactant protein D in vitro and in vivo. J Biol Chem 2010;285:16757–16770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fernández-Real JM, Valdés S, Manco M, et al. Surfactant protein d, a marker of lung innate immunity, is positively associated with insulin sensitivity. Diabetes Care 2010;33:847–853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hill J, Heslop C, Man SF, et al. Circulating surfactant protein-D and the risk of cardiovascular morbidity and mortality. Eur Heart J 2011;32:1918–1925 [DOI] [PubMed] [Google Scholar]
- 53.Nguyen AB, Rohatgi A, Garcia CK, et al. Interactions between smoking, pulmonary surfactant protein B, and atherosclerosis in the general population: the Dallas Heart Study. Arterioscler Thromb Vasc Biol 2011;31:2136–2143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Brook RD, Jerrett M, Brook JR, Bard RL, Finkelstein MM. The relationship between diabetes mellitus and traffic-related air pollution. J Occup Environ Med 2008;50:32–38 [DOI] [PubMed] [Google Scholar]
- 55.Krämer U, Herder C, Sugiri D, et al. Traffic-related air pollution and incident type 2 diabetes: results from the SALIA cohort study. Environ Health Perspect 2010;118:1273–1279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pearson JF, Bachireddy C, Shyamprasad S, Goldfine AB, Brownstein JS. Association between fine particulate matter and diabetes prevalence in the U.S. Diabetes Care 2010;33:2196–2201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Puett RC, Hart JE, Schwartz J, Hu FB, Liese AD, Laden F. Are particulate matter exposures associated with risk of type 2 diabetes? Environ Health Perspect 2011;119:384–389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Andersen ZJ, Raaschou-Nielsen O, Ketzel M, et al. Diabetes incidence and long-term exposure to air pollution: a cohort study. Diabetes Care 2012;35:92–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Coogan PF, White LF, Jerrett M, et al. Air pollution and incidence of hypertension and diabetes mellitus in black women living in Los Angeles. Circulation 2012;125:767–772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kelishadi R, Mirghaffari N, Poursafa P, Gidding SS. Lifestyle and environmental factors associated with inflammation, oxidative stress and insulin resistance in children. Atherosclerosis 2009;203:311–319 [DOI] [PubMed] [Google Scholar]
- 61.Chuang KJ, Yan YH, Chiu SY, Cheng TJ. Long-term air pollution exposure and risk factors for cardiovascular diseases among the elderly in Taiwan. Occup Environ Med 2011;68:64–68 [DOI] [PubMed] [Google Scholar]