

Abstract
Birth asphyxia is often associated with a high seizure burden that is predictive of poor neurodevelopmental outcome. The mechanisms underlying birth asphyxia seizures are unknown. Using an animal model of birth asphyxia based on 6-day-old rat pups, we have recently shown that the seizure burden is linked to an increase in brain extracellular pH that consists of the recovery from the asphyxia-induced acidosis, and of a subsequent plateau level well above normal extracellular pH. In the present study, two-photon imaging of intracellular pH in neocortical neurons in vivo showed that pH changes also underwent a biphasic acid–alkaline response, resulting in an alkaline plateau level. The mean alkaline overshoot was strongly suppressed by a graded restoration of normocapnia after asphyxia. The parallel post-asphyxia increase in extra- and intracellular pH levels indicated a net loss of acid equivalents from brain tissue that was not attributable to a disruption of the blood–brain barrier, as demonstrated by a lack of increased sodium fluorescein extravasation into the brain, and by the electrophysiological characteristics of the blood–brain barrier. Indeed, electrode recordings of pH in the brain and trunk demonstrated a net efflux of acid equivalents from the brain across the blood–brain barrier, which was abolished by the Na/H exchange inhibitor, N-methyl-isobutyl amiloride. Pharmacological inhibition of Na/H exchange also suppressed the seizure activity associated with the brain-specific alkalosis. Our findings show that the post-asphyxia seizures are attributable to an enhanced Na/H exchange-dependent net extrusion of acid equivalents across the blood–brain barrier and to consequent brain alkalosis. These results suggest targeting of blood–brain barrier-mediated pH regulation as a novel approach in the prevention and therapy of neonatal seizures.
Keywords: birth asphyxia, neonatal seizures, resuscitation, pH, Na/H exchange
Introduction
Seizures are common in the newborn baby and most frequently occur as a result of birth asphyxia. Increased morbidity and mortality, brain injury and poor neurodevelopmental outcome have been associated with a higher seizure burden (McBride et al., 2000; Lingwood et al., 2003; Haynes et al., 2005; Silverstein and Jensen, 2007; Lindstrom et al., 2008; Bjorkman et al., 2010; Stolp et al., 2012). Directly addressing neonatal seizures is therefore a priority, but current drugs are largely ineffective and may have serious side effects on the neonatal brain (Rennie and Boylan, 2003; Evans et al., 2007; Rennie and Boylan, 2007). The molecular and cellular mechanisms underlying birth-asphyxia seizures are unknown, but it is obvious that information on the seizure-triggering mechanisms is of crucial importance in the design of novel therapeutic strategies.
A profound acidosis (blood pH 7.00 or lower) at the time of birth is an essential criterion in the diagnosis of perinatal asphyxia (Poland and Freeman, 2002). However, birth asphyxia seizures do not coincide with the maximal blood acidosis but are typically first observed during the recovery period after a delay that usually ranges in human babies from 2 to 16 h (Shalak and Perlman, 2004). A wide range of observations have shown that changes in extra- and intracellular pH exert a strong modulatory effect on brain excitability under normal and pathophysiological conditions, whereby an alkalosis enhances excitability while an acidosis has an opposite effect (Siesjö, 1985; Chesler, 1990; Kaku et al., 1993; Kaila, 1994; Casey et al., 2010; Enyedi and Czirjak, 2010; Ruusuvuori et al., 2010). This underscores the crucial role of brain pH regulation in seizure generation (Schuchmann et al., 2006, 2011; Ziemann et al., 2008; Tolner et al., 2011).
Using an animal model of birth asphyxia based on 6-day-old rat pups, we recently showed that the post-asphyxia seizure burden was tightly linked to an increase in brain extracellular pH during the recovery from the asphyxia-induced acidosis and its subsequent alkaline overshoot, which reached values that were well above the normal brain pH level (Helmy et al., 2011). Several lines of evidence showed that this alkaline shift had a direct, causal role in seizure generation. Moreover, the post-asphyxia alkalosis was dramatically reduced by restoring the CO2 levels down to normal in a slow, controlled manner. Accordingly, this ‘graded restoration of normocapnia’ virtually abolished the seizures, suggesting a novel therapeutic manoeuvre for suppression of birth-asphyxia seizures.
It is not known how post-asphyxia brain alkalosis is caused. In the present study we used two-photon imaging of intracellular pH in neocortical neurons in vivo, which revealed an intraneuronal alkalosis after asphyxia, with a time course and sensitivity to graded restoration of normocapnia that were similar to what we described before for extracellular alkalosis (Helmy et al., 2011). The post-asphyxia brain alkalosis is generated in the absence of a rise of pH in the trunk and blood. Thus, the rise in both extracellular and intracellular pH recorded in the brain implies a net loss of acid equivalents and is attributable to net extrusion of acid across the blood–brain barrier. This conclusion gained direct support from measurements of the pH gradient across the blood–brain barrier. The post-asphyxia increase in blood–brain barrier acid extrusion was inhibited by systemically administered pharmacological blockers of Na/H exchange, which notably, also abolished post-asphyxia seizures.
Materials and methods
In vivo measurement of intraneuronal pH
Male Sprague-Dawley rat pups, postnatal Day 6–7 (where Day 0 refers to the day of birth) were used. All experiments were approved by the University of Maryland Institutional Animal Care and Use Committee. Animals were anaesthetized during surgery with isoflurane. A small, bloodless craniotomy (3 mm in diameter) was made above parietal cortex. A small tear was made in the dura, and the exposed brain was covered with physiological saline, described in Stosiek et al.. (2003). For in vivo imaging, anaesthesia was maintained with 0.7–0.9% isoflurane in air or in special gas mixtures (as described later) applied through a funnel loosely placed on the muzzle. Body temperature was maintained by a heating pad at 35°C throughout the experiments. The pups were first exposed for 60 min to 20% CO2 and 9% O2 (asphyxia). In one of the two groups, the pups breathed air after asphyxia. In the other group, the experimental asphyxia was followed by an immediate restoration of normoxia, but with a graded re-establishment of normocapnia (Helmy et al., 2011), by lowering the CO2 levels first from 20 to 10% (with appropriate adjustments of N2) for 30 min, then to 5% for a further 30 min and finally to virtually zero or room air (graded restoration of normocapnia).
Acetoxymethyl ester dye solution was prepared by dissolving 50 μg of 2′,7′-bis-(2-carboxyethyl)-5-(and-6-)-carboxyfluorescein (BCECF-AM) in 4 μl of 20% pluronic acid in dimethyl sulphoxide (DMSO, Invitrogen) and diluted (1:8) with physiological saline (Stosiek et al., 2003). Glass pipettes (2–4 μm tip diameter) were pulled (Sutter P2000) and loaded with dye solution. For bulk loading of cortical neurons, pipettes were gradually advanced to a depth of ∼500 μm into the parietal cortex under two-photon scanning mode, and the dye solution was pressure injected as described by Bandyopadhyay et al.. (2010). After loading, the pipette was withdrawn from the cortex, and the craniotomy was covered with warm agarose and a glass coverslip. Imaging was done using a two-photon laser-scanning microscope (Ultima IV, Prairie Technologies) with a Spectra Physics Mai Tai Deep See Ti-Sapphire mode-locked femto-second laser. Layer 2/3 cells were imaged using a ×20 water-immersion objective lens (LUMPlanFI/IR Olympus 0.95 NA) at a depth of 250–350 µm from the surface of the cortex. Twelve frames per minute were acquired and averaged; data were collected over the course of 4 min, with excitation at 825 nm (F825), and over 1 min at 770 nm (F770) at a higher light intensity to maximize signal-to-noise ratio. Images were acquired using a 570 nm dichroic filter. Averaged frames were analysed using a custom software written in MATLAB. The averaged data collected at 825 nm excitation was divided by the averaged data collected at 770 nm to give the ratiometric intraneuronal pH measurement of one 5 min period (F825/F770).
Calibration of the BCECF signal was done in slice preparation. Sprague-Dawley rat pups were anesthetized with isoflurane and decapitated. Equipment and solutions used to prepare 200-µm thick brain slices were described in Zhao et al.. (2009). All physiological solutions were equilibrated with carbogen (5% CO2, 95% O2) and kept at 35°C. BCECF-AM (50 μg) was dissolved in 4 μl of 20% pluronic acid in DMSO and then diluted in physiological solution to a final concentration of 5 µM. Slices were placed into the dye solution and allowed to load for 20 min. A slice was placed under the two-photon microscope and first perfused with physiological solution, followed by high K+/nigericin (Eisner et al., 1989) solutions with a of pH 7.4, 7.2, 7.0 and 6.8, equilibrated with carbogen. Superficial cells were imaged using the paradigm described earlier. The calibration curve was linear over the pH range tested. Although the linear slope is expected to translate faithfully from the in vitro calibration to in vivo measurements, this is not necessarily true for absolute pH values. Therefore, the data on intracellular pH have been given as changes from the baseline (see Fig. 1).
Figure 1.

An intraneuronal alkalosis is triggered after asphyxia and suppressed by graded restoration of normocapnia. The experimental changes in inhaled CO2 and O2 are schematically shown above the recordings. (A) Two-photon in vivo measurements of intracellular pH changes (mean ΔpH indicated by black line) in 40 layer 2/3 pyramidal neurons shown in colour from five Day 6–7 rat pups. Inset shows BCECF-loaded neurons. Scale bar = 20 µm. (B) Intracellular ΔpH in 41 neurons from five rat pups in the graded restoration of normocapnia paradigm.
pH at the end of the two experimental paradigms was compared using Student's two-tailed unpaired t-test. Data are presented as mean ± SD.
In vivo measurement of body and brain pH and blood–brain barrier potential
In these and subsequent experiments, male Wistar rat pups (Day 6) were used with approval by the National Animal Ethics Committee in Finland. The in vivo intracranial pH measurements with H+-sensitive microelectrodes were done as described previously (Helmy et al., 2011). For simultaneous body pH measurements, small incisions were made in the loose skin of the pup’s dorsum. Two 4 cm lengths of polyvinyl chloride tube (outer diameter 0.6 mm, inner diameter 0.2 mm) were prepared as pH-sensitive and reference electrodes and inserted into the incisions. The pH-sensitive body electrodes had the same polyvinyl chloride-gelled membrane and backfilling solutions as the intracranial pH microelectrodes (Voipio and Kaila, 1993), with a H+-sensitive membrane column length of ∼1 mm. The reference electrode was filled with 0.5% agar in 0.9% NaCl.
Brain extracellular pH and body pH were measured in asphyxic conditions (20% CO2, 9% O2 for 1 h) and for 2 h afterwards. N-methyl-isobutyl-amiloride (MIA; Sigma-Aldrich) was given 2.5 mg/kg bodyweight intraperitoneally 30 min before asphyxia (Kendall et al., 2006; Helmy et al., 2011). The potential difference across the blood–brain barrier (VBBB) was obtained as the difference of the reference electrodes in the brain and the body (Held et al., 1964; Woody et al., 1970). Values of pH or VBBB in the two experimental paradigms were compared using the Mann-Whitney U-test (two tailed). Data are shown as mean ± SD.
Assessment of blood–brain barrier permeability using sodium fluorescein
For the asphyxia group, 30 min after termination of asphyxia, 4 ml/kg body weight of 10% sodium fluorescein (Sigma-Aldrich) was injected intraperitoneally (Ferrari et al., 2010). Thirty minutes after sodium fluorescein administration (Farkas et al., 1998), pups were anaesthetized with isoflurane and then perfused with ice-cold 0.9% NaCl through the left ventricle, with a cut in the right auricle for 25 min, a period of time that allows for the outflowing fluid to run clear. Whole brains were dissected out, weighed and frozen in liquid nitrogen, and then kept at −80°C till further processing. For the negative control group (with intact blood–brain barrier), rat pups were removed from the nest and kept at 35°C for 90 min, injected with sodium fluorescein and, 30 min later, processed as described earlier. For the positive control group (with mannitol-induced damage of the blood–brain barrier), rat pups were injected with sodium fluorescein; 30 min later, under isoflurane anaesthesia and on a heating pad set to 35°C, a small thoracotomy was made to expose the tip of the left ventricle. Over a period of 2 min, 30 µl of 24% d-mannitol (w/w) in 0.9% NaCl (Hooper et al., 1995) was infused into the left ventricle. The pups were left under anaesthesia for a further 20 min before being processed, as described earlier.
Brain content of sodium fluorescein was measured as described before (Lenzser et al., 2005) with some modifications as follows. Brains were homogenized in 2.5 ml of 50% trichloroacetic acid and centrifuged for 15 min at 10 000g. The supernatant was diluted with 5 M NaOH, 1:0.8. Fluorometry was performed with a Victor2 (Perkin-Elmer) in plates with black frames and clear wells (IsoPlate 96, Perkin-Elmer).
Brain content of sodium fluorescein was compared between the experimental paradigms using Mann-Whitney U-test (two-tailed). Data are shown as mean ± SD.
Behavioural assessment of seizure burden
Seizure burden was quantified during the 2 h post-asphyxia period from video recordings, based on the number of loss of righting reflex in freely moving animals as described before (Helmy et al., 2011). MIA was administered intraperitoneally 30 min before the onset of experimental asphyxia (Kendall et al., 2006). In some experiments, amiloride (amiloride hydrochloride hydrate; Sigma-Aldrich), the blood–brain barrier-impermeable parent drug of MIA (Sipos and Brem, 2000; Fisher, 2002; Liu et al., 2010), was administered at an identical concentration. Each incidence of loss of righting reflex was given a score of one. The scores for every pup were added to give the total individual seizure burden. Seizure quantifications based on loss of righting reflex were done in uninstrumented animals. Total seizure burden was compared between the experimental paradigms using the Mann-Whitney U-test (two-tailed). Data are shown as mean ± SD. Control experiments with vehicle alone (1.3 mM acetic acid in 0.9% NaCl, 100 ml) did not have any effect.
Effect of N-methyl-isobutyl-amiloride on acid extrusion in cortical pyramidal neurons
To examine the effects of MIA on acid extrusion in neocortical pyramidal neurons, measurements of intracellular pH based on BCECF were done on acute slices in CO2/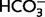 -buffered and CO2/
-buffered and CO2/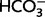 -free HEPES (20 mM) buffered solutions. For further details on methods and data, see Supplementary material and Ruusuvuori et al. (2010).
-free HEPES (20 mM) buffered solutions. For further details on methods and data, see Supplementary material and Ruusuvuori et al. (2010).
Results
The intracellular pH of parietal cortex layer 2/3 neurons was measured in Day 6 rat pups using in vivo two-photon microscopy and the pH-sensitive dye BCECF (inset in Fig. 1A). When pups were exposed to asphyxia (20% CO2 and 9% O2), a conspicuous fall in fluorescence took place that levelled off by the end of the 1-h exposure period. This decrease in fluorescence corresponded to an acidification of 0.30 ± 0.08 pH units below the baseline (n = 40 neurons from five pups; Fig. 1A). During the 2-h post-asphyxia period, intracellular pH not only recovered from acidosis but also showed a slower alkalosis with a plateau level of 0.27 ± 0.12 pH units above baseline pH. Importantly, this is a time point when blood pH has already recovered from the post-asphyxia acidosis and, moreover, blood pH does not become alkaline at any point of time under the present experimental conditions (Helmy et al., 2011). Hence, the above data and our previous observations on brain extracellular pH (see also Fig. 2A) clearly indicate that the post-asphyxia alkalosis is confined to brain tissue.
Figure 2.

Enhanced net acid extrusion across the blood–brain barrier takes place after asphyxia and is suppressed by the Na/H exchange blocker, MIA. (A) From top to bottom: the experimental paradigm; sample recording of extracellular pH changes in the brain (red) and in the body (black); sample recording of trans-blood–brain barrier potential difference (VBBB, calculated as Vbrain–Vbody); mean values ± SD in extracellular brain ΔpH (red triangles) and body ΔpH (black squares) in the asphyxia paradigm from six rats; mean values ± SD of VBBB. (B) Data from experiments on five rats where MIA was injected 30 min before the experimental asphyxia. Note strict dependence of steady-state values of VBBB on the pH changes across the blood–brain barrier. (C) Blood–brain barrier permeability is not disrupted by experimental birth asphyxia, as indicated with sodium fluorescein (NaF) extravasation into the brain (n = 4 rat pups in each paradigm). For further details see text.
We have previously shown that graded restoration of normocapnia brought about by gradually reducing CO2 in inhaled air during recovery from asphyxia (Helmy et al., 2011) suppresses the post-asphyxia alkalosis of brain extracellular pH, but it is not known how graded restoration of normocapnia affects intracellular pH. Therefore, we exposed rats to asphyxia, which again resulted in a decrease in intracellular pH similar to that described above (0.31 ± 0.08 pH units, n = 41 neurons from five pups; Fig. 1B). Importantly, during graded restoration of normocapnia, intracellular pH gradually recovered but with a marked suppression of the overshoot of intracellular pH (cf. Fig. 1A), which was only ∼0.03 ± 0.10 pH units above baseline at 2-h (P < 0.0001, immediate versus graded restoration of normocapnia) closely resembling the suppressing action of graded restoration of normocapnia on the overshoot of extracellular pH (Helmy et al., 2011).
As concluded earlier, the fact that brain extracellular and intracellular pH undergo a post-asphyxia alkalosis indicates that there is a net loss of acid equivalents from both compartments. Thus, the most likely explanation that could account for the parallel increase in extracellular and intracellular pH is an increase in net extrusion of acid equivalents across the blood–brain barrier. We directly tested this idea by simultaneous measurements of brain extracellular pH and body pH (Fig. 2) using one pH-sensitive electrode plus its reference in the brain parenchyma and a similar pair of electrodes in body subcutaneous tissue (Voipio and Kaila, 1993). To manipulate acid transport across the blood–brain barrier (Pedersen et al., 2006), we used the Na/H exchange blocker, MIA.
The brain extracellular pH changes during and after asphyxia were similar to previous measurements (Helmy et al., 2011), with a baseline level of 7.20 ± 0.01, peak acidosis at 6.80 ± 0.05 and plateau alkalosis at 7.57 ± 0.04 (n = 6 pups). Body pH during asphyxia decreased from 7.42 ± 0.03 to 7.19 ± 0.03 and recovered with no alkaline overshoot, at any instant of time, to a level that was identical to the pre-exposure baseline (7.42 ± 0.01). The recordings in Fig. 2A show that the time course of pH changes in response to asphyxia is strikingly different in the two compartments, with the brain extracellular pH changes being larger and faster than those in the body. Unlike brain extracellular pH, body pH does not show a post-asphyxia alkaline overshoot. This observation is in full agreement with the blood acid–base data obtained in our previous study (Helmy et al., 2011). A finding of crucial importance here is that the application of MIA completely suppressed the post-asphyxia increase in brain extracellular pH (P = 0.004, n = 5; Fig. 2B). Clearly, brain extracellular pH and body pH behave as distinct compartments in response to asphyxia and during the post-asphyxia period.
The only structure that could be responsible for the above pH compartmentalization is the blood–brain barrier. The blood–brain barrier in the Day 6 rats is likely tight enough with regard to ionic movements to maintain such a compartmentalization (Butt et al., 1990), which we found was the case. A trans-blood–brain barrier potential difference (Woody et al., 1970; Voipio et al., 2003), which was sensitive to the extracellular pH changes in a manner similar to the mature human brain (Voipio et al., 2003), was clearly seen in the electrophysiological recordings (Fig. 2A). The average peak shift during the asphyxia-induced acidosis was similar in the absence (+4.08 ± 0.24 mV, n = 6) and presence (+4.05 ± 0.22 mV, n = 5) of MIA (P = 0.9). At the end of the 2-h post-asphyxia period, the blood–brain barrier potential shift was −3.08 ± 0.37 mV and 0 ± 0.1 mV in the asphyxia and asphyxia plus MIA-treated groups, respectively (P = 0.004; Fig. 2B), indicating that the steady negative shift in trans-blood–brain barrier potential was abolished in parallel with the extracellular pH overshoot in the presence of MIA.
To gain further information on blood–brain barrier permeability, we used sodium fluorescein as an indicator (Fig. 2C). No significant difference was found in brain fluorescein extravasation after asphyxia when compared with control (128.1 ± 38.7 and 129.9 ± 33.1 ng sodium fluorescein/mg brain tissue, respectively, n = 4, P = 0.9). In contrast, when the blood–brain barrier was disrupted by injecting 30 µl of 24% mannitol in saline into the left ventricle, a large increase in sodium fluorescein extravasation was observed (549.8 ± 99.1 ng sodium fluorescein/mg brain tissue, n = 5, P = 0.02). These data provide further evidence that blood–brain barrier permeability is not altered under the present experimental conditions.
If the increase in brain pH is caused by an enhanced efflux of acid equivalents from the brain across the blood–brain barrier, one might assume that it is large enough to cause a detectable transient in the body pH measurements. Indeed, we found that, during the time window of maximum increase in brain extracellular pH, a small deflection to the acid direction in body pH could be observed in four out of six recordings, including the one shown in Fig. 2A.
Consistent with a key role of the blood–brain barrier in the functional compartmentalization of the brain pH during and after asphyxia was that, when plotting brain extracellular pH against body pH, a profound hysteresis was observed (Fig. 3A). Blocking the blood–brain barrier Na/H exchange using the inhibitor MIA completely inhibited the post-asphyxia overshoot alkalosis of brain extracellular pH (Fig. 2B), which abolished the hysteresis and, under these conditions, brain extracellular pH was related to body pH in an almost linear manner. Thus, the trajectories of body pH and brain extracellular pH in the presence and absence of MIA in Fig. 3A directly illustrate the role of blood–brain barrier Na/H exchange in dissociating brain extracellular pH regulation from body pH during the post-asphyxia period. MIA had no effect on either body or brain extracellular pH under control conditions during 3.5 h post-injection, which covers the duration of the present experiments (n = 3).
Figure 3.

Brain extracellular pH and body pH are functionally compartmentalized in an Na/H exchanger-dependent manner as indicated by the hysteresis in response to experimental asphyxia and by the MIA sensitive seizures. (A) The change in brain extracellular pH plotted against the change in body pH. Pups exposed to asphyxia only are shown as blue circles; pups to which MIA was administered are shown as green diamonds. Data taken from Fig. 2. (B) Total seizure burden [quantified as the number of loss of righting reflex (LRR) per pup] after asphyxia and asphyxia with administration of MIA or amiloride, at equal doses, from eight rat pups in each paradigm.
In line with the above and previous results (Kendall et al., 2006; Helmy et al., 2011), we found that MIA reduced seizure burden by 88% (P < 0.0002, n = 8; Fig. 3B). We repeated the behavioural tests with the Na/H exchange inhibitor amiloride, because amiloride penetration across the blood–brain barrier is known to be poor (Sipos and Brem, 2000; Fisher, 2002; Liu et al., 2010). Notably, the effects of amiloride and MIA were similar (Fig. 3B), providing further support for the conclusion that activation of Na/H exchange in the blood–brain barrier leads to brain alkalosis and consequent seizures.
All the above evidence points to an action of the blockers of Na/H exchange that is not mediated by changes in neuronal regulation of intracellular pH. Moreover, intracellular pH regulation in hippocampal pyramidal neurons is insensitive to amiloride and its derivatives (Raley-Susman et al., 1991) but whether this is true for neocortical neurons is not known (Mellergard et al., 1993; Ou-yang et al., 1993; Chesler, 2003). Hence, we examined the effects of MIA on layer 2/3 pyramidal neurons in neocortical slices (Supplementary material). Consistent with previous results on hippocampal pyramidal neurons (Schwiening and Boron, 1994; Baxter and Church, 1996; Bevensee et al., 1996), acid extrusion following an ammonium prepulse (Boron and De Weer, 1976) was much more efficient (by a factor of ∼10) in the presence of 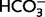 than in the HEPES-buffered solution, indicating a minor role for any
than in the HEPES-buffered solution, indicating a minor role for any 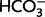 -independent acid extruder, such as Na/H exchange (Supplementary Fig. 1). Moreover, application of 10 µM MIA (Vinnikova et al., 2004) did not have any detectable effect on steady-state intracellular pH or acid extrusion either in the presence (n = 8 cells) or absence (n = 10 cells) of CO2/
-independent acid extruder, such as Na/H exchange (Supplementary Fig. 1). Moreover, application of 10 µM MIA (Vinnikova et al., 2004) did not have any detectable effect on steady-state intracellular pH or acid extrusion either in the presence (n = 8 cells) or absence (n = 10 cells) of CO2/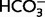 (Supplementary material and Supplementary Fig. 1). Taken together, these data do not support the view that the actions of MIA and amiloride on brain extracellular pH changes caused by asphyxia are mediated by a block in neuronal Na/H exchange. Here, it is also worth emphasizing that a cell-autonomous increase in neuronal pH caused by acid extrusion during the post-asphyxia period would impose an acid shift in extracellular pH (Siesjö, 1985; Chesler and Kaila, 1992; Chesler, 2003), exactly the opposite of what was seen in our present and previous (Helmy et al., 2011) study.
(Supplementary material and Supplementary Fig. 1). Taken together, these data do not support the view that the actions of MIA and amiloride on brain extracellular pH changes caused by asphyxia are mediated by a block in neuronal Na/H exchange. Here, it is also worth emphasizing that a cell-autonomous increase in neuronal pH caused by acid extrusion during the post-asphyxia period would impose an acid shift in extracellular pH (Siesjö, 1985; Chesler and Kaila, 1992; Chesler, 2003), exactly the opposite of what was seen in our present and previous (Helmy et al., 2011) study.
Discussion
The present work shows that post-asphyxia alkalosis in the brain and the consequent seizures are attributable to an enhanced efflux of acid equivalents from the brain that is suppressed by blocking Na/H exchange located in the blood–brain barrier (Pedersen et al., 2006; Lam et al., 2009). This conclusion is based on (i) the post-asphyxia increase and alkaline overshoot of both brain intracellular pH and extracellular pH, which indicates a net loss of acid equivalents from brain tissue; (ii) direct measurements of the extracellular pH levels and dynamics in the brain and the rest of the body, which indicated a strict compartmentalization of brain extracellular pH by the blood–brain barrier under normal conditions and in response to asphyxia in the Day 6 rats; (iii) the specific block of the post-asphyxia brain alkalosis by MIA, an inhibitor of Na/H exchange (Kendall et al., 2006; Pedersen et al., 2006) and (iv) the equally strong inhibitory action on seizures by MIA and amiloride, whereof the latter is an Na/H exchange inhibitor that has a very low permeability across the blood–brain barrier (Sipos and Brem, 2000; Fisher, 2002; Liu et al., 2010).
An increase in seizure burden after birth has an incremental, deleterious effect on neurological outcome and therefore it is of utmost importance to identify the mechanisms underlying the pathophysiological post-asphyxia brain alkalosis. It is a well-established fact that a rise in both intracellular and extracellular pH enhances neuronal excitability (Chesler, 1990; Kaku et al., 1993; Kaila, 1994; Casey et al., 2010; Enyedi and Czirjak, 2010; Ruusuvuori et al., 2010). The immature brain appears to be particularly sensitive to changes in pH. Recent experiments on neonatal hippocampal slices have shown that a change of 0.05 pH units has a profound effect on endogenous network activity (Ruusuvuori et al., 2010). A robust cause–effect between brain alkalosis and seizure induction in the immature rat brain was shown in our work on experimental febrile seizures, where tonic–clonic seizures were triggered following an increase in extracellular pH of ∼0.2 pH units induced by hyperthermia-evoked hyperventilation (Schuchmann et al., 2006, 2011). Moreover, an intraperitoneal injection of sodium bicarbonate that produced an increase in brain extracellular pH of ∼0.2 pH units triggered seizures in rat pups (Schuchmann et al., 2006).
Consistent with the present results, magnetic resonance spectroscopy following severe human birth asphyxia has shown an abnormally high brain intracellular pH (Robertson et al., 2002). A high magnitude and prolonged duration of alkalosis are predictive of poor neurodevelopmental outcome including cerebral atrophy (Robertson et al., 1999). The elevated intracellular and extracellular pH in our animal model (Helmy et al., 2011) as well as the elevated intracellular pH in the newborn human brain after birth asphyxia (Robertson et al., 2002) persist beyond the time of normalization of the post-asphyxia acid–base parameters in the blood (Brouillette and Waxman, 1997).
We hypothesized that among the acid–base transporters in the blood–brain barrier (Pedersen et al., 2006), the Na/H exchanger is the most relevant one under our experimental conditions. In full agreement with this idea, we found that MIA abolished the brain-specific post-asphyxia alkalosis and the associated seizures, while having no effects on the changes in body pH or its basal level under control conditions.
It is unlikely that neuronal Na/H exchange contributes to the effects of asphyxia or to the actions of MIA and amiloride seen in the present study. It has been shown that hippocampal neurons are resistant to amiloride and its derivatives (Raley-Susman et al., 1991; Chesler, 2003), and our present findings with MIA indicate that this is also true with regard to neocortical neurons. Furthermore, if the post-asphyxia increase in intracellular pH would be caused by enhanced neuronal Na/H exchange (or by any acid extruder), this would lead to a simultaneous decrease in extracellular pH (Siesjö, 1985; Chesler and Kaila, 1992; Chesler, 2003) while the opposite was observed. Indeed, that an extracellular alkalosis leads to an intracellular alkalosis has been demonstrated in various kinds of mammalian neurons (Roos and Boron, 1981; Ou-yang et al., 1993; Kaila and Ransom, 1998; Ritucci et al., 1998; Bouyer et al., 2004). The most parsimonious explanation for the near-parallel increase in intracellular and extracellular pH is that the blood–brain barrier Na/H exchanger-dependent rise in extracellular pH causes a rise in neuronal pH.
Na/H exchange in the blood–brain barrier is prominently sensitive to amiloride and MIA (Murphy and Johanson, 1990; Sipos et al., 2005; Pedersen et al., 2006) and a high binding capacity of MIA has been demonstrated in cerebral microvessels (Kalaria et al., 1998). The endothelial isoforms of Na/H exchangers are located on the luminal side of endothelial cells (Crone, 1986; Goldstein et al., 1986; Redzic, 2011; Benarroch, 2012), and therefore, they are easily accessible to systemically applied Na/H exchange inhibitors, regardless of the drug permeability across the blood–brain barrier. Amiloride is poorly permeable in the blood–brain barrier (Sipos and Brem, 2000; Fisher, 2002; Liu et al., 2010), and notably, our present study shows that amiloride is as effective as MIA in abolishing post-asphyxia seizures.
The persistent activation of Na/H exchange in the blood–brain barrier most likely involves a change in the kinetic set point (Roos and Boron, 1981). This set point renders Na/H exchangers functionally silent at normal intracellular pH and active when intracellular pH becomes acidic (Casey et al., 2010). Notably, an alkaline shift in the set point of intracellular pH occurs when Na/H exchangers are persistently stimulated by hormones and other neuroactive factors (Sardet et al., 1991; Orlowski and Grinstein, 2004; Pedersen et al., 2006). It is well known that various signalling molecules, including hormones, cytokines and growth factors, are released following birth asphyxia and related states (Lagercrantz and Slotkin, 1986; Yoon et al., 1996; Nordstrom and Arulkumaran, 1998; Savman et al., 1998; Silveira and Procianoy, 2003; Fliegel and Karmazyn, 2004; Gazzolo et al., 2009), and an important question for future studies is to identify the factor(s) that act on the Na/H exchanger in the blood–brain barrier.
Interestingly, block of Na/H exchange by MIA administered systemically before hypoxia/ischaemia insult has been shown to have a neuroprotective effect (Kendall et al., 2006) that may be at least partly attributable to the drug-induced block of seizures. Another, mutually non-exclusive possibility is that the post-asphyxia recovery of acidosis and the subsequent alkalosis trigger cell damage by activating enzymes, such as proteases and phospholipases (Currin et al., 1991). Analysing the contributions of these mechanisms to neuronal damage following birth asphyxia is an important topic for future studies.
An interesting hypothesis based on the enhanced efflux of acid across the blood–brain barrier is that in human neonates suffering from severe birth asphyxia, the net acid efflux from the brain during the initial alkalosis might cause a slowing down of the early recovery of the systemic acidosis as monitored using blood samples. The total-body volume fraction of the newborn human brain is higher than that of a Day 6 rat, and even in the rat pups, we found evidence that the maximum rate of recovery of brain extracellular pH after asphyxia was paralleled by a small acid transient in body pH (Fig. 2A). These findings predict that an increase in the severity of birth asphyxia in human newborns is associated with a progressively slower recovery of blood pH that might even include a transient acid shift. This hypothesis can be readily tested in future clinical work.
In conclusion, following neonatal asphyxia, extrusion of acid equivalents across the blood–brain barrier generates a brain alkalosis that promotes seizures. Our results raise the important and worrying question whether resuscitation paradigms where normocapnic conditions established in a fast manner will, in fact, lead to the promotion of birth-asphyxia seizures. Graded restoration of normocapnia and/or drugs targeting the Na/H exchange in the blood–brain barrier offer an effective and straightforward means to functionally suppress seizures and ameliorate other neurological sequela after birth asphyxia (Rakhade and Jensen, 2009; Bonifacio et al., 2011), and can be readily used in conjunction with other treatment modalities, such as hypothermia (Johnston et al., 2011) and optimization of blood oxygenation levels (Saugstad, 2010).
Funding
This work was supported by grants from the Letten Foundation, the Academy of Finland, the Sigrid Juselius Foundation, and the Jane and Aatos Erkko Foundation. K.K. is a member of the Finnish Center of Excellence in Molecular and Integrative Neuroscience Research. P.O.K. is supported by National Institutes of Health [R01DC009607], and the Alfred P. Sloan Foundation. P.V.W is supported by National Institutes of Health [NIH T32DC000046].
Supplementary material
Supplementary material is available at Brain online.
Glossary
Abbreviations
- BCECF
2′,7′-bis-(2-carboxyethyl)-5-(and-6-)-carboxyfluorescein
- MIA
N-methyl-isobutyl-amiloride
- VBBB
blood-brain barrier potential
References
- Bandyopadhyay S, Shamma SA, Kanold PO. Dichotomy of functional organization in the mouse auditory cortex. Nat Neurosci. 2010;13:361–8. doi: 10.1038/nn.2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baxter KA, Church J. Characterization of acid extrusion mechanisms in cultured fetal rat hippocampal neurones. J Physiol. 1996;493:457–70. doi: 10.1113/jphysiol.1996.sp021396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benarroch EE. Blood-brain barrier: recent developments and clinical correlations. Neurology. 2012;78:1268–76. doi: 10.1212/WNL.0b013e318250d8bc. [DOI] [PubMed] [Google Scholar]
- Bevensee MO, Cummins TR, Haddad GG, Boron WF, Boyarsky G. pH regulation in single CA1 neurons acutely isolated from the hippocampi of immature and mature rats. J Physiol. 1996;494:315–28. doi: 10.1113/jphysiol.1996.sp021494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjorkman ST, Miller SM, Rose SE, Burke C, Colditz PB. Seizures are associated with brain injury severity in a neonatal model of hypoxia-ischemia. Neuroscience. 2010;166:157–67. doi: 10.1016/j.neuroscience.2009.11.067. [DOI] [PubMed] [Google Scholar]
- Bonifacio SL, Glass HC, Peloquin S, Ferriero DM. A new neurological focus in neonatal intensive care. Nat Rev Neurol. 2011;7:485–94. doi: 10.1038/nrneurol.2011.119. [DOI] [PubMed] [Google Scholar]
- Boron WF, De Weer P. Intracellular pH transients in squid giant axons caused by CO2, NH3, and metabolic inhibitors. J Gen Physiol. 1976;67:91–112. doi: 10.1085/jgp.67.1.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouyer P, Bradley SR, Zhao J, Wang W, Richerson GB, Boron WF. Effect of extracellular acid-base disturbances on the intracellular pH of neurones cultured from rat medullary raphe or hippocampus. J Physiol. 2004;559:85–101. doi: 10.1113/jphysiol.2004.067793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouillette RT, Waxman DH. Evaluation of the newborn's blood gas status. Clin Chem. 1997;43:215–21. [PubMed] [Google Scholar]
- Butt AM, Jones HC, Abbott NJ. Electrical resistance across the blood-brain barrier in anaesthetized rats: a developmental study. J Physiol. 1990;429:47–62. doi: 10.1113/jphysiol.1990.sp018243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casey JR, Grinstein S, Orlowski J. Sensors and regulators of intracellular pH. Nat Rev Mol Cell Biol. 2010;11:50–61. doi: 10.1038/nrm2820. [DOI] [PubMed] [Google Scholar]
- Chesler M. Regulation and modulation of pH in the brain. Physiol Rev. 2003;83:1183–221. doi: 10.1152/physrev.00010.2003. [DOI] [PubMed] [Google Scholar]
- Chesler M. The regulation and modulation of pH in the nervous system. Prog Neurobiol. 1990;34:401–27. doi: 10.1016/0301-0082(90)90034-e. [DOI] [PubMed] [Google Scholar]
- Chesler M, Kaila K. Modulation of pH by neuronal activity. Trends Neurosci. 1992;15:396–402. doi: 10.1016/0166-2236(92)90191-a. [DOI] [PubMed] [Google Scholar]
- Crone C. The blood-brain barrier as a tight epithelium: where is information lacking? Ann NY Acad Sci. 1986;481:174–85. doi: 10.1111/j.1749-6632.1986.tb27149.x. [DOI] [PubMed] [Google Scholar]
- Currin RT, Gores GJ, Thurman RG, Lemasters JJ. Protection by acidotic pH against anoxic cell killing in perfused rat liver: evidence for a pH paradox. FASEB J. 1991;5:207–10. doi: 10.1096/fasebj.5.2.2004664. [DOI] [PubMed] [Google Scholar]
- Eisner DA, Kenning NA, O’Neill SC, Pocock G, Richards CD, Valdeolmillos M. A novel method for absolute calibration of intracellular pH indicators. Pflugers Arch. 1989;413:553–8. doi: 10.1007/BF00594188. [DOI] [PubMed] [Google Scholar]
- Enyedi P, Czirjak G. Molecular background of leak K+ currents: two-pore domain potassium channels. Physiol Rev. 2010;90:559–605. doi: 10.1152/physrev.00029.2009. [DOI] [PubMed] [Google Scholar]
- Evans DJ, Levene MI, Tsakmakis M. Anticonvulsants for preventing mortality and morbidity in full term newborns with perinatal asphyxia. Cochrane Database Syst Rev. 2007:CD001240. doi: 10.1002/14651858.CD001240.pub2. [DOI] [PubMed] [Google Scholar]
- Farkas G, Marton J, Nagy Z, Mandi Y, Takacs T, Deli MA, et al. Experimental acute pancreatitis results in increased blood-brain barrier permeability in the rat: a potential role for tumor necrosis factor and interleukin 6. Neurosci Lett. 1998;242:147–50. doi: 10.1016/s0304-3940(98)00060-3. [DOI] [PubMed] [Google Scholar]
- Ferrari DC, Nesic OB, Perez-Polo JR. Oxygen resuscitation does not ameliorate neonatal hypoxia/ischemia-induced cerebral edema. J Neurosci Res. 2010;88:2056–65. doi: 10.1002/jnr.22358. [DOI] [PubMed] [Google Scholar]
- Fisher JL. Amiloride inhibition of gamma-aminobutyric acid(A) receptors depends upon the alpha subunit subtype. Mol Pharmacol. 2002;61:1322–8. doi: 10.1124/mol.61.6.1322. [DOI] [PubMed] [Google Scholar]
- Fliegel L, Karmazyn M. The cardiac Na-H exchanger: a key downstream mediator for the cellular hypertrophic effects of paracrine, autocrine and hormonal factors. Biochem Cell Biol. 2004;82:626–35. doi: 10.1139/o04-129. [DOI] [PubMed] [Google Scholar]
- Gazzolo D, Abella R, Marinoni E, Di Iorio R, Li Volti G, Galvano F, et al. Circulating biochemical markers of brain damage in infants complicated by ischemia reperfusion injury. Cardiovasc Hematol Agents Med Chem. 2009;7:108–26. doi: 10.2174/187152509787847119. [DOI] [PubMed] [Google Scholar]
- Goldstein GW, Betz AL, Bowman PD, Dorovini-Zis K. In vitro studies of the blood-brain barrier using isolated brain capillaries and cultured endothelial cells. Ann NY Acad Sci. 1986;481:202–13. doi: 10.1111/j.1749-6632.1986.tb27151.x. [DOI] [PubMed] [Google Scholar]
- Haynes RL, Baud O, Li J, Kinney HC, Volpe JJ, Folkerth DR. Oxidative and nitrative injury in periventricular leukomalacia: a review. Brain Pathol. 2005;15:225–33. doi: 10.1111/j.1750-3639.2005.tb00525.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Held D, Fencl V, Pappenheimer JR. Electrical potential of cerebrospinal fluid. J Neurophysiol. 1964;27:942–59. doi: 10.1152/jn.1964.27.5.942. [DOI] [PubMed] [Google Scholar]
- Helmy MM, Tolner EA, Vanhatalo S, Voipio J, Kaila K. Brain alkalosis causes birth asphyxia seizures, suggesting therapeutic strategy. Ann Neurol. 2011;69:493–500. doi: 10.1002/ana.22223. [DOI] [PubMed] [Google Scholar]
- Hooper DC, Ohnishi ST, Kean R, Numagami Y, Dietzschold B, Koprowski H. Local nitric oxide production in viral and autoimmune diseases of the central nervous system. Proc Natl Acad Sci USA. 1995;92:5312–6. doi: 10.1073/pnas.92.12.5312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston MV, Fatemi A, Wilson MA, Northington F. Treatment advances in neonatal neuroprotection and neurointensive care. Lancet Neurol. 2011;10:372–82. doi: 10.1016/S1474-4422(11)70016-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaila K. Ionic basis of GABAA receptor channel function in the nervous system. Prog Neurobiol. 1994;42:489–537. doi: 10.1016/0301-0082(94)90049-3. [DOI] [PubMed] [Google Scholar]
- Kaila K, Ransom BR. pH and brain function. Wiley-Liss; 1998. [Google Scholar]
- Kaku DA, Giffard RG, Choi DW. Neuroprotective effects of glutamate antagonists and extracellular acidity. Science. 1993;260:1516–8. doi: 10.1126/science.8389056. [DOI] [PubMed] [Google Scholar]
- Kalaria RN, Premkumar DR, Lin CW, Kroon SN, Bae JY, Sayre LM, et al. Identification and expression of the Na+/H+ exchanger in mammalian cerebrovascular and choroidal tissues: characterization by amiloride-sensitive [3H]MIA binding and RT-PCR analysis. Brain Res Mol Brain Res. 1998;58:178–87. doi: 10.1016/s0169-328x(98)00108-9. [DOI] [PubMed] [Google Scholar]
- Kendall GS, Robertson NJ, Iwata O, Peebles D, Raivich G. N-Methyl-isobutyl-amiloride ameliorates brain injury when commenced before hypoxia ischemia in neonatal mice. Pediatr Res. 2006;59:227–31. doi: 10.1203/01.pdr.0000196805.68082.22. [DOI] [PubMed] [Google Scholar]
- Lagercrantz H, Slotkin TA. The “stress” of being born. Sci Am. 1986;254:100–7. doi: 10.1038/scientificamerican0486-100. [DOI] [PubMed] [Google Scholar]
- Lam TI, Wise PM, O'Donnell ME. Cerebral microvascular endothelial cell Na/H exchange: evidence for the presence of NHE1 and NHE2 isoforms and regulation by arginine vasopressin. Am J Physiol Cell Physiol. 2009;297:C278–89. doi: 10.1152/ajpcell.00093.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenzser G, Kis B, Bari F, Busija DW. Diazoxide preconditioning attenuates global cerebral ischemia-induced blood-brain barrier permeability. Brain Res. 2005;1051:72–80. doi: 10.1016/j.brainres.2005.05.064. [DOI] [PubMed] [Google Scholar]
- Lindstrom K, Hallberg B, Blennow M, Wolff K, Fernell E, Westgren M. Moderate neonatal encephalopathy: pre- and perinatal risk factors and long-term outcome. Acta Obstet Gynecol Scand. 2008;87:503–9. doi: 10.1080/00016340801996622. [DOI] [PubMed] [Google Scholar]
- Lingwood BE, Dunster KR, Healy GN, Ward LC, Colditz PB. Cerebral impedance and neurological outcome following a mild or severe hypoxic/ischemic episode in neonatal piglets. Brain Res. 2003;969:160–7. doi: 10.1016/s0006-8993(03)02295-9. [DOI] [PubMed] [Google Scholar]
- Liu F, Zhang M, Tang ZQ, Lu YG, Chen L. Inhibitory effects of amiloride on the current mediated by native GABA(A) receptors in cultured neurons of rat inferior colliculus. Clin Exp Pharmacol Physiol. 2010;37:435–40. doi: 10.1111/j.1440-1681.2009.05325.x. [DOI] [PubMed] [Google Scholar]
- McBride MC, Laroia N, Guillet R. Electrographic seizures in neonates correlate with poor neurodevelopmental outcome. Neurology. 2000;55:506–13. doi: 10.1212/wnl.55.4.506. [DOI] [PubMed] [Google Scholar]
- Mellergard P, Ouyang YB, Siesjo BK. Intracellular pH regulation in cultured rat astrocytes in CO2/HCO3(-)-containing media. Exp Brain Res. 1993;95:371–80. doi: 10.1007/BF00227129. [DOI] [PubMed] [Google Scholar]
- Murphy VA, Johanson CE. Na+/H+ exchange in choroid plexus and CSF in acute metabolic acidosis or alkalosis. Am J Physiol. 1990;258:F1528–37. doi: 10.1152/ajprenal.1990.258.6.F1528. [DOI] [PubMed] [Google Scholar]
- Nordstrom L, Arulkumaran S. Intrapartum fetal hypoxia and biochemical markers: a review. Obstet Gynecol Surv. 1998;53:645–57. doi: 10.1097/00006254-199810000-00023. [DOI] [PubMed] [Google Scholar]
- Orlowski J, Grinstein S. Diversity of the mammalian sodium/proton exchanger SLC9 gene family. Pflugers Arch. 2004;447:549–65. doi: 10.1007/s00424-003-1110-3. [DOI] [PubMed] [Google Scholar]
- Ou-yang Y, Mellergard P, Siesjo BK. Regulation of intracellular pH in single rat cortical neurons in vitro: a microspectrofluorometric study. J Cereb Blood Flow Metab. 1993;13:827–40. doi: 10.1038/jcbfm.1993.105. [DOI] [PubMed] [Google Scholar]
- Pedersen SF, O'Donnell ME, Anderson SE, Cala PM. Physiology and pathophysiology of Na+/H+ exchange and Na+ -K+ -2Cl- cotransport in the heart, brain, and blood. Am J Physiol Regul Integr Comp Physiol. 2006;291:R1–25. doi: 10.1152/ajpregu.00782.2005. [DOI] [PubMed] [Google Scholar]
- Poland R, Freeman R. In: Guidelines for perinatal care. Elk Grove Village, IL: American Academy of Pediatrics, and American College of Obstetricians and Gynecologists; 2002. Relationship between perinatal factors and neurologic outcome. [Google Scholar]
- Rakhade SN, Jensen FE. Epileptogenesis in the immature brain: emerging mechanisms. Nat Rev Neurol. 2009;5:380–91. doi: 10.1038/nrneurol.2009.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raley-Susman KM, Cragoe EJ, Jr, Sapolsky RM, Kopito RR. Regulation of intracellular pH in cultured hippocampal neurons by an amiloride-insensitive Na+/H+ exchanger. J Biol Chem. 1991;266:2739–45. [PubMed] [Google Scholar]
- Redzic Z. Molecular biology of the blood-brain and the blood-cerebrospinal fluid barriers: similarities and differences. Fluids Barriers CNS. 2011;8:3. doi: 10.1186/2045-8118-8-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rennie J, Boylan G. Treatment of neonatal seizures. Arch Dis Childhood-Fetal Neonatal Edn. 2007;92:F148–50. doi: 10.1136/adc.2004.068551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rennie JM, Boylan GB. Neonatal seizures and their treatment. Curr Opin Neurol. 2003;16:177–81. doi: 10.1097/01.wco.0000063768.15877.23. [DOI] [PubMed] [Google Scholar]
- Ritucci NA, Chambers-Kersh L, Dean JB, Putnam RW. Intracellular pH regulation in neurons from chemosensitive and nonchemosensitive areas of the medulla. Am J Physiol. 1998;275:R1152–63. doi: 10.1152/ajpregu.1998.275.4.R1152. [DOI] [PubMed] [Google Scholar]
- Robertson NJ, Cowan FM, Cox IJ, Edwards AD. Brain alkaline intracellular pH after neonatal encephalopathy. Ann Neurol. 2002;52:732–42. doi: 10.1002/ana.10365. [DOI] [PubMed] [Google Scholar]
- Robertson NJ, Cox IJ, Cowan FM, Counsell SJ, Azzopardi D, Edwards AD. Cerebral intracellular lactic alkalosis persisting months after neonatal encephalopathy measured by magnetic resonance spectroscopy. Pediatr Res. 1999;46:287–96. doi: 10.1203/00006450-199909000-00007. [DOI] [PubMed] [Google Scholar]
- Roos A, Boron WF. Intracellular pH. Physiol Rev. 1981;61:296–434. doi: 10.1152/physrev.1981.61.2.296. [DOI] [PubMed] [Google Scholar]
- Ruusuvuori E, Kirilkin I, Pandya N, Kaila K. Spontaneous network events driven by depolarizing GABA action in neonatal hippocampal slices are not attributable to deficient mitochondrial energy metabolism. J Neurosci. 2010;30:15638–42. doi: 10.1523/JNEUROSCI.3355-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sardet C, Fafournoux P, Pouyssegur J. Alpha-thrombin, epidermal growth factor, and okadaic acid activate the Na+/H+ exchanger, NHE-1, by phosphorylating a set of common sites. J Biol Chem. 1991;266:19166–71. [PubMed] [Google Scholar]
- Saugstad OD. New guidelines for newborn resuscitation—a critical evaluation. Acta Paediatr. 2010;100:1058–62. doi: 10.1111/j.1651-2227.2011.02301.x. [DOI] [PubMed] [Google Scholar]
- Savman K, Blennow M, Gustafson K, Tarkowski E, Hagberg H. Cytokine response in cerebrospinal fluid after birth asphyxia. Pediatr Res. 1998;43:746–51. doi: 10.1203/00006450-199806000-00006. [DOI] [PubMed] [Google Scholar]
- Schuchmann S, Hauck S, Henning S, Gruters-Kieslich A, Vanhatalo S, Schmitz D, et al. Respiratory alkalosis in children with febrile seizures. Epilepsia. 2011;52:1949–55. doi: 10.1111/j.1528-1167.2011.03259.x. [DOI] [PubMed] [Google Scholar]
- Schuchmann S, Schmitz D, Rivera C, Vanhatalo S, Salmen B, Mackie K, et al. Experimental febrile seizures are precipitated by a hyperthermia-induced respiratory alkalosis. Nat Med. 2006;12:817–23. doi: 10.1038/nm1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwiening CJ, Boron WF. Regulation of intracellular pH in pyramidal neurones from the rat hippocampus by Na(+)-dependent Cl(-)-HCO3- exchange. J Physiol. 1994;475:59–67. doi: 10.1113/jphysiol.1994.sp020049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalak L, Perlman JM. Hypoxic-ischemic brain injury in the term infant-current concepts. Early Hum Dev. 2004;80:125–41. doi: 10.1016/j.earlhumdev.2004.06.003. [DOI] [PubMed] [Google Scholar]
- Siesjö BK. Acid-base homeostasis in the brain: physiology, chemistry, and neurochemical pathology. Prog Brain Res. 1985;63:121–54. doi: 10.1016/S0079-6123(08)61980-9. [DOI] [PubMed] [Google Scholar]
- Silveira RC, Procianoy RS. Interleukin-6 and tumor necrosis factor-alpha levels in plasma and cerebrospinal fluid of term newborn infants with hypoxic-ischemic encephalopathy. J Pediatr. 2003;143:625–9. doi: 10.1067/S0022-3476(03)00531-6. [DOI] [PubMed] [Google Scholar]
- Silverstein FS, Jensen FE. Neonatal seizures. Ann Neurol. 2007;62:112–20. doi: 10.1002/ana.21167. [DOI] [PubMed] [Google Scholar]
- Sipos EP, Brem H. Local anti-angiogenic brain tumor therapies. J Neurooncol. 2000;50:181–8. doi: 10.1023/a:1006482120049. [DOI] [PubMed] [Google Scholar]
- Sipos H, Torocsik B, Tretter L, Adam-Vizi V. Impaired regulation of pH homeostasis by oxidative stress in rat brain capillary endothelial cells. Cell Mol Neurobiol. 2005;25:141–51. doi: 10.1007/s10571-004-1379-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolp H, Neuhaus A, Sundramoorthi R, Molnar Z. The long and the short of it: gene and environment interactions during early cortical development and consequences for long-term neurological disease. Front Psychiatry. 2012;3:50. doi: 10.3389/fpsyt.2012.00050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stosiek C, Garaschuk O, Holthoff K, Konnerth A. In vivo two-photon calcium imaging of neuronal networks. Proc Natl Acad Sci USA. 2003;100:7319–24. doi: 10.1073/pnas.1232232100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolner EA, Hochman DW, Hassinen P, Otahal J, Gaily E, Haglund MM, et al. Five percent CO2 is a potent, fast-acting inhalation anticonvulsant. Epilepsia. 2011;52:104–14. doi: 10.1111/j.1528-1167.2010.02731.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinnikova AK, Alam RI, Malik SA, Ereso GL, Feldman GM, McCarty JM, et al. Na+-H+ exchange activity in taste receptor cells. J Neurophysiol. 2004;91:1297–313. doi: 10.1152/jn.00809.2003. [DOI] [PubMed] [Google Scholar]
- Voipio J, Kaila K. Interstitial PCO2 and pH in rat hippocampal slices measured by means of a novel fast CO2/H-sensitive microelectrode based on a PVC-gelled membrane. Pflugers Arch. 1993;423:193–201. doi: 10.1007/BF00374394. [DOI] [PubMed] [Google Scholar]
- Voipio J, Tallgren P, Heinonen E, Vanhatalo S, Kaila K. Millivolt-scale DC shifts in the human scalp EEG: evidence for a nonneuronal generator. J Neurophysiol. 2003;89:2208–14. doi: 10.1152/jn.00915.2002. [DOI] [PubMed] [Google Scholar]
- Woody CD, Marshall WH, Besson JM, Thompson HK, Aleonard P, Albefess D. Brain potential shift with respiratory acidosis in cat and monkey. Am J Phys. 1970;218:275–83. doi: 10.1152/ajplegacy.1970.218.1.275. [DOI] [PubMed] [Google Scholar]
- Yoon BH, Romero R, Yang SH, Jun JK, Kim IO, Choi JH, et al. Interleukin-6 concentrations in umbilical cord plasma are elevated in neonates with white matter lesions associated with periventricular leukomalacia. Am J Obstet Gynecol. 1996;174:1433–40. doi: 10.1016/s0002-9378(96)70585-9. [DOI] [PubMed] [Google Scholar]
- Zhao C, Kao JP, Kanold PO. Functional excitatory microcircuits in neonatal cortex connect thalamus and layer 4. J Neurosci. 2009;29:15479–88. doi: 10.1523/JNEUROSCI.4471-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziemann AE, Schnizler MK, Albert GW, Severson MA, Howard MA, III, Welsh MJ, et al. Seizure termination by acidosis depends on ASIC1a. Nat Neurosci. 2008;11:816–22. doi: 10.1038/nn.2132. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}