

Abstract
The synthetic triterpenoid CDDO-Me has been shown to directly inhibit the growth of myeloid leukemias and lends itself to a wide array of therapeutic indications, including inflammatory conditions, due to its inhibition of NFκB. We have previously demonstrated protection from acute graft-versus-host disease (GVHD) after CDDO-Me administration in an allogeneic bone marrow transplant (BMT) model (Li, et al. BBMT, 2010). In the current study, we observed that CDDO-Me promoted myelopoiesis in both naïve and transplanted mice. This effect was dose-dependent as high doses of CDDO-Me inhibited myeloid growth in vitro. All lineages (CFU-GM, BFU-E) were promoted by CDDO-Me. We then compared the effects with G-CSF, a known inducer of myeloid expansion and mobilization from the bone marrow. Whereas both drugs induced terminal myeloid expansion in the spleen, peripheral blood and bone marrow, G-CSF only induced CFU-GM precursors in the spleen while CDDO-Me increased these precursors in the spleen and bone marrow. After sublethal total body irradiation, mice pretreated with CDDO-me further displayed an accelerated recovery of myeloid progenitors and total nucleated cells in the spleen. A similar expansion of myeloid and myeloid progenitors was noted with CDDO-Me treatment after syngeneic BMT. Combined, these data suggest that CDDO-Me may be of use post-transplant to accelerate myeloid recovery in addition to the prevention of GVHD.
INTRODUCTION
Hematopoietic stem cell transplantation (HSCT) is frequently used in the treatment of various hematological malignancies, immune deficiencies, and anemias. The recovery period after HSCT is characterized by prolonged neutropenia, thus increasing susceptibility to opportunistic infections and related mortalities (1, 2). Granulocyte colony-stimulating factor (G-CSF, filgrastim) is used clinically to accelerate donor myeloid recovery and minimize the duration of neutropenia after HSCT. Administration of G-CSF after HSCT has been shown to decrease the time required for absolute neutrophil counts to reach above 500/μL, a commonly examined clinical threshold, and is regularly used in the clinic (3).
CDDO-Me (methyl-2-cyano-3, 12-dioxooleana-1, 9-diene-28-oate; bardoxolone methyl) is a synthetic triterpenoid which has been previously shown to induce apoptosis and inhibit growth in a number of hematological and non-hematological malignancies (4-7) as well as to improve renal function in patients with chronic kidney disease (8). CDDO-Me induces the Keap1-Nrf2 pathway, thereby upregulating many antioxidant and cytoprotective genes (8, 9). CDDO-Me has been shown to protect against oxidative stress caused by inducible nitric oxide synthase (iNOS). An elevation of NAD(P)H-quinone oxidoreductase and heme oxygenase, both of which protect cells from redox cycling and oxidative stress, are believed to mediate the antioxidant properties of CDDO-Me (10). However, growth and survival inhibition have also been demonstrated by CDDO-Me, most likely mediated by the blockade of NF-κB through the inhibition of IκBα kinase β (IKKβ)(11). Based on the antioxidant role of CDDO-Me, a recent study found that CDDO-Me can inhibit the suppressive activity of myeloid-derived suppressor cells, which utilize reactive oxygen species as a mechanism of suppression, and thus improve immune function and anti-tumor responses (12). Studies in our laboratory have additionally demonstrated that CDDO-Me inhibits the severity and development of acute murine graft-versus-host disease (GVHD) after allogeneic HSCT in mice and was shown to induce cell death in proliferating lymphocytes in a mixed lymphocyte reaction (13). To our surprise in these studies, mice which received CDDO-Me were not only protected from GVHD, but also displayed an expansion of myeloid progenitor CFU-GM cells within the spleen and bone marrow.
In the current study, we examined the effects of CDDO-Me on hematopoiesis and compared administration of CDDO-Me with G-CSF in terms of their respective abilities to induce myelopoiesis and expand myeloid cells in resting and syngeneic HSCT mice. We found that both CDDO-Me and G-CSF expanded CD11b+ Gr-1+ myeloid cells in the bone marrow and spleen. However, CDDO-Me administration also increased the amount of CFU-GM and BFU-e colonies in the spleen, bone marrow and blood while G-CSF administration only increased CFU-GM and BFU-e colonies in the spleen and blood. Additionally, the number of CFU-GM and hematopoietic progenitor cells (HPCs) in the spleen and bone marrow showed a significant increase in mice which received CDDO-Me. In vitro studies showed that CDDO-Me was sufficient to stimulate colony growth of naïve bone marrow cells in suboptimal colony-stimulating cytokine conditions. Given the broad effects of CDDO-Me, the myeloid expansion we observed may offer advantages in HSCT outcome.
MATERIALS AND METHODS
Animals
Female C57BL/6 (B6) mice were obtained from the Animal Production Area of the National Cancer Institute (Frederick, MD). Mice were kept in specific pathogen-free conditions at the University of Nevada, Reno (Reno, NV) or at the University of California Davis Medical Center (Sacramento, CA). All mice were between 8 and 12 weeks of age at the start of the experiments. Animal protocols were approved by the Institutional Animal Care and Use Committees at the respective institutions in which studies were performed.
Cells and Reagents
CDDO-Me was manufactured through the NIH RAID Program and provided by Reata Pharmaceuticals, Inc. (Irving, TX). CDDO-Me was prepared in a vehicle solution of 10% DMSO (Sigma, St. Louis, MO), 10% Cremophor EL (Sigma), and 80% DPBS. A vehicle control consisting of 10% DMSO, 10% Cremophor EL and 80% DPBS was used as a control in all experiments. G-CSF (Neupogen®, Amgen, Thousand Oaks, CA) was diluted to the appropriate concentration in DPBS. Except where indicated, mice received a treatment schedule of CDDO-Me (120μg/dose, twice daily i.p.) or vehicle control for 7 days or G-CSF (2.5μg/dose, twice daily s.q.) for 5 days. In naïve mice, a treatment schedule of 120μg per injection for 7 days was chosen because it resulted in the greatest increase in colony formation and was well tolerated.
Flow Cytometry
Single cell suspensions (1 × 106) were prepared from the spleen and bone marrow as described previously (14) and labeled with fluorochrome-conjugated anti-mouse antibodies, and nonspecific binding was corrected with isotype-matched controls. FITC anti-CD11b (M1/70), PE-Cy5 anti-Gr-1 (RB6-8C5), PE-Sca-1 (D7), FITC c-kit (2B8) and 7-AAD were purchased from BD Biosciences. Lineage positive cells were excluded by simultaneous staining with PE-Cy5-labeled anti-CD3ε, (145-2C11), anti-Gr-1(RB6-8C5), anti- CD11b (M1/70), anti-CD45R/B220 (RA3-6B2) and anti-(TER-119), all purchased from BioLegend (San Diego, CA). Flow cytometric acquisition was performed on a BD LSR Fortessa (Becton Dickinson, San Jose, CA) or Stratedigm S1400 (San Jose, CA). Data sets were analyzed using FlowJo software (Tree Star, Ashland, OR).
Colony Assays
CFU-GM, BFU-e and CFU-HPP colony assays were performed as previously described (14, 15). For CFU-GM and BFU-e assays, 5 × 104 bone marrow cells or 5 × 105 splenocytes were plated in 35mm Petri dishes with methylcellulose-containing medium. Colony formation was stimulated with 10 ng/mL recombinant murine GM-CSF, 10 ng/mL recombinant murine interleukin (IL)-3, and 5 U/mL erythropoietin. Plates were incubated 7 days at 37°C at 5% CO2. Colonies were defined as aggregates of 50 or more cells which contained only red cells (BFU-e) or only white cells (CFU-GM). In assays with suboptimal cytokines, colony growth was stimulated with 300pg/mL GM-CSF (without IL-3 or erythropoietin) and CDDO-Me or vehicle control was added to dishes at the indicated concentrations with the amount of vehicle in each dish remaining constant. In CFU-HPP assays, bone marrow cells or spleen cells were plated in 60-mm Petri dishes at, 5 × 104 or 5 × 105 cells per plate, respectively. Colony formation was stimulated with 20 ng/mL rmIL-6, 20 ng/mL rmIL-3 and 50 ng/mL stem cell factor (PeproTech, Rocky Hill, NJ). Plates were incubated for 11 days at 37° C after which time colonies (CFU-HPP) with a diameter of >2 mm were counted.
Total Body Irradiation (TBI) and Bone Marrow Transplantation (BMT)
For radioprotection studies, mice received 120μg CDDO-Me on days -7 through -1 before sublethal irradiation with 500 cGy from a 137Cs irradiator on day 0. Mice were harvested on day 8 to determine radioprotection through CFU assays and total cell counts through flow cytometry. For BMT studies. C57BL/6 (Ly5.1) mice received a myeloablative dose (950 cGy) followed by infusion of 107 congeneic bone marrow cells (BMCs) from C57BL/6 (Ly5.2) donors into the tail veins of recipient mice. Recipients received 60μg CDDO-Me or vehicle control daily on days 1-13 after BMT. Mice were sacrificed on day 14 and spleens and bone marrow were harvested.
RESULTS
CDDO-Me stimulates CFU-GM colony growth in vitro
We first studied the effects of CDDO-Me on myeloid progenitor differentiation and expansion ex vivo by culturing naïve C57BL/6 bone marrow cells in the presence of CDDO-Me at varying concentrations. Colony formation was stimulated by using a suboptimal amount of colony-stimulating cytokines. We chose 300pg/mL GM-CSF to stimulate myeloid progenitor differentiation in these assays as we found this dose to induce 50% as many colonies as with an optimal dose of cytokines (10ng/mL rmGM-CSF, 10ng/mL rmIL-3; data not shown). These data could only be reproduced under conditions of suboptimal cytokines, as higher concentrations of colony-promoting cytokines most likely masked any direct effects of CDDO-Me. As expected given the effects of CDDO-Me on inhibiting NFκB, high concentrations of CDDO-Me above 10nM significantly decreased the number of colonies formed in a seven-day CFU-GM assay (Figure 1). However, in contrast, a low dose of 0.5nM CDDO-Me significantly stimulated colony growth ex vivo, suggesting direct myelopoietic effects of the CDDO-Me on bone marrow cells. The amount of the vehicle present in each dish was kept constant in these studies due to the potentially toxic effects of the vehicle necessary to achieve solubility of CDDO-Me: a solution of 0.9% saline containing 10% DMSO, 10% Cremophor EL. This vehicle control (VC) was used as a comparison in all subsequent experiments. Thus, CDDO-Me appears to be able, at low concentrations, to directly promote myelopoiesis.
Figure 1. CDDO-Me stimulates CFU-GM formation at low concentrations, but is toxic at higher concentrations.
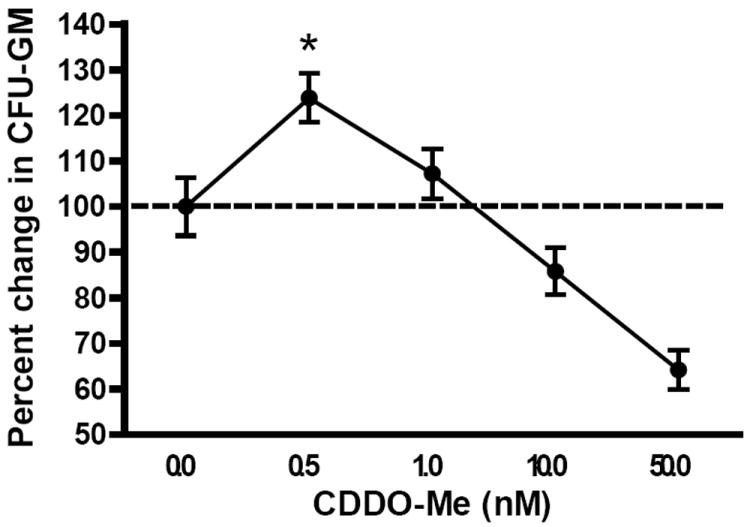
Naïve mouse splenocytes were plated in a CFU-GM with a suboptimal amount of colony-stimulating cytokines (0.3ng/mL GM-CSF). CDDO-Me was added the Petric dishes at the indicated concentrations while keeping the amount of vehicle in each sample normalized. Plates were incubated for 7 days at which time colonies of greater that 50 cells were counted. Colony counts from individual experiments were normalized to the vehicle control in each experiment and pooled. *p<0.05 by Dunnett’s multiple comparison test.
CDDO-Me expands myeloid CD11b+ Gr-1+ cells in the spleen and bone marrow
We next evaluated the ability of CDDO-Me to stimulate myeloid expansion in vivo in naïve mice. C57BL/6 mice were treated with CDDO-Me or vehicle control (120μg/dose, twice daily) for 7 days or G-CSF (2.5μg/dose, twice daily) for 5 days. Spleens and femurs were collected, processed, and stained for flow cytometric analysis of CD11b+ Gr-1+ myeloid cells (Fig. 2A-C). Both mouse monocytes and neutrophils were phenotyped by CD11b and Gr-1 (Ly6G/C) expression: monocytes exist as CD11b+/Gr-1hi, Gr-1int or Gr-1low populations whereas neutrophils are also Gr-1hi (16). Although these populations can be further differentiated by flow cytometric panels to identify the phenotype and maturation state of each population, we gated total CD11b+ Gr-1+ events in the spleen and bone marrow as a broad indicator of myelopoietic activity. In the spleen, CDDO-Me increased both the percentage and the number of total myeloid cells (3.18×106 ± 1.06×106) 2.5-fold compared with vehicle control (1.27×106 ± 0.415×106), though not to the extent of G-CSF (1.41×107 ± 2.46×106), a positive-control for myeloid expansion in the periphery (Figure 2). Similarly, CDDO-Me expanded CD11b+ Gr-1+ myeloid cells in the bone marrow 2-fold, where a much larger population of myeloid cells exists in naïve mice. This myeloid expansion in the bone marrow was similar to the expansion observed with the administration of G-CSF (Figure 2). We did not observe a change in either the number or percentage of CD11b+ Gr-1- macrophages within the spleen or bone marrow. Thus, in vivo treatment of mice with CDDO-Me promoted myeloid cell expansion to an extent comparable with G-CSF.
Figure 2. CDDO-Me administration causes a myeloid expansion in the spleen and bone marrow.

C57BL/6 mice were treated for 7 days with vehicle control (VC) or CDDO-Me, or 5 days with G-CSF after which mice were sacrificed and spleens and bone marrow were harvested. Single cell suspensions were stained with anti-CD11b and anti-Gr-1. A, Representative flow cytometry panels showing percentages of cells in either organ. B and C, total CD11b+Gr-1+ cell numbers in the spleen and bone marrow were enumerated. P values were determined by Dunnett’s multiple comparison test.
CDDO-Me administration results in an increase in CFU-GM and BFU-E in the bone marrow, spleen and blood
Given the increased total myeloid cells observed in CDDO-Me treated mice, we performed CFU-GM and BFU-e colony assays to determine if CDDO-Me treatment acted on terminal stages of myeloid differentiation or in early hematopoietic development and what lineages were specifically enhanced. Mice were treated as before and sacrificed on day 7. Splenocytes and bone marrow cells were plated for CFU-GM and BFU-e colony assays to detect myeloid or erythroid progenitors, respectively, with optimal concentrations of growth-stimulating cytokines (10ng/mL rmGM-CSF,10ng/mL rmIL-3 and 5 U/mL erythropoietin). Colonies were allowed to incubate for 7 days before CFU-GM or BFU-e colonies were counted. CDDO-Me-treated mice displayed an expansion of both CFU-GM and BFU-e progenitor cells in the bone marrow, spleen and blood (Figure 3). As expected, G-CSF treatment resulted in an increase in CFU-GM and BFU-e in both the spleen and blood, but not the bone marrow consistent with clinical observations of stem cell mobilization after G-CSF treatment. The retention of myeloid progenitors in the bone marrow after CDDO-Me but not G-CSF treatment implicates a discrete mechanism for CDDO-Me-induced myelopoiesis. Similarly, high proliferative potential (CFU-HPP) progenitors in the spleen and bone marrow were enumerated after CDDO-Me treatment. CDDO-Me treatment increased the number of CFU-HPP significantly in the spleen and non-significantly in the bone marrow (Figure 4). G-CSF caused a pronounced increase in splenic CFU-HPP, resulting in colony formation too numerous to count, but showed no effect in the bone marrow.
Figure 3. CDDO-Me administration results in a significant increase of CFU-GM and BFU-e in the BM and periphery.
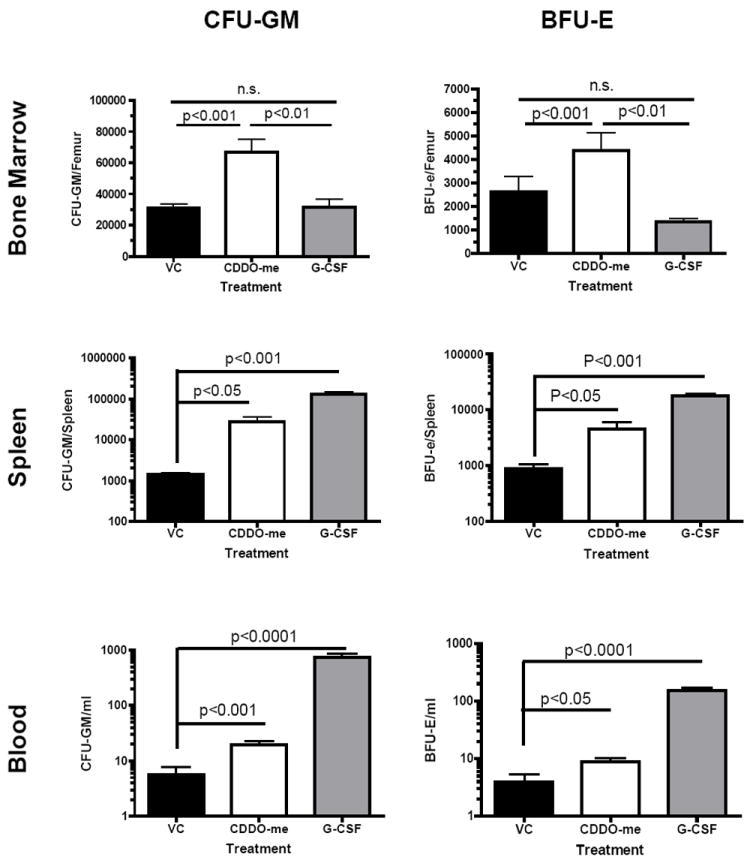
Mice were treated with vehicle control (VC), CDDO-me or G-CSF as before. CFU-GM and BFU-e colony assays were performed on bone marrow, spleens, and peripheral blood of treated animals. The data is representative of three independent experiments with three mice per group per experiment. Statistical differences were determined by one-way ANOVA with Tukey’s post-test and a p-value<0.05 was considered significant.
Figure 4. CDDO-Me administration results in increased CFU-HPP in the spleen and BM.
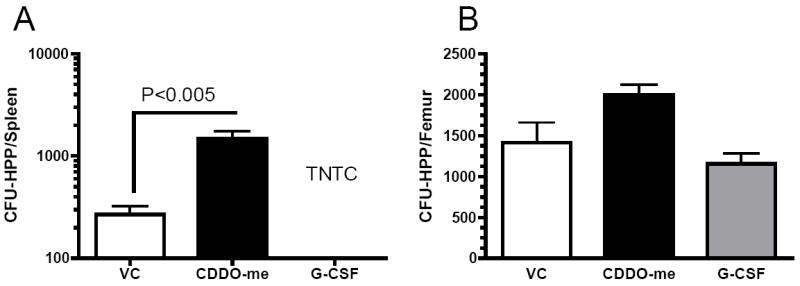
Mice were treated with vehicle control or CDDO-me (120 μg/dose BID) a day intraperitoneally (i.p) for 7 days. Bone marrow and spleen were collected at day 7, processed into single cell suspensions and used for colony assays. Recombinant hG-CSF (2.5 μg/dose BID) was used as a control. The number of CFU-HPP colonies were enumerated in both the spleen (A) and femurs (B). The data is representative of three independent experiments with three mice per group per experiment. Statistical differences were determined by One-way ANOVA (Tukey) and p-value<0.05 was considered significant. TNTC = too numerous to count
CDDO-Me expands hematopoietic progenitor cells in the spleen and bone marrow
We next determined whether the increased CFU-GM and BFU-e colony-forming cells observed in the spleen and bone marrow after CDDO-Me treatment would correlate with increased numbers of multipotent hematopoietic progenitor cells (HPCs) in each organ. HPCs in the spleen and bone marrow were assessed by staining single-cell suspensions of CDDO-Me or G-CSF-treated mice with lineage markers, c-kit (stem cell factor receptor), Sca-1(stem cell antigen-1) and 7-AAD to exclude dead cells. We observed an increase in the number of lin- c-kit+ Sca-1+ HPCs in both the bone marrow (3.9-fold) and spleen (4.5-fold) of CDDO-Me-treated mice (Fig 5A-C). The increase in HPCs in the spleen after CDDO-Me treatment was less pronounced than observed with G-CSF treatment (8.9-fold). Interestingly, treatments with G-CSF or CDDO-Me equally increased the number of HPCs in the bone marrow, suggesting that higher numbers of HPCs in G-CSF treated mice did not lead to increased CFU-GM or BFU-e as observed in Figure 3. These results demonstrate that CDDO-Me administration can affect early hematopoietic progenitors and HPC expansion in vivo in a manner distinct from mobilization via G-CSF.
Figure 5. CDDO-me administration increases the number of hematopoietic progenitor cells in the bone marrow and spleen.
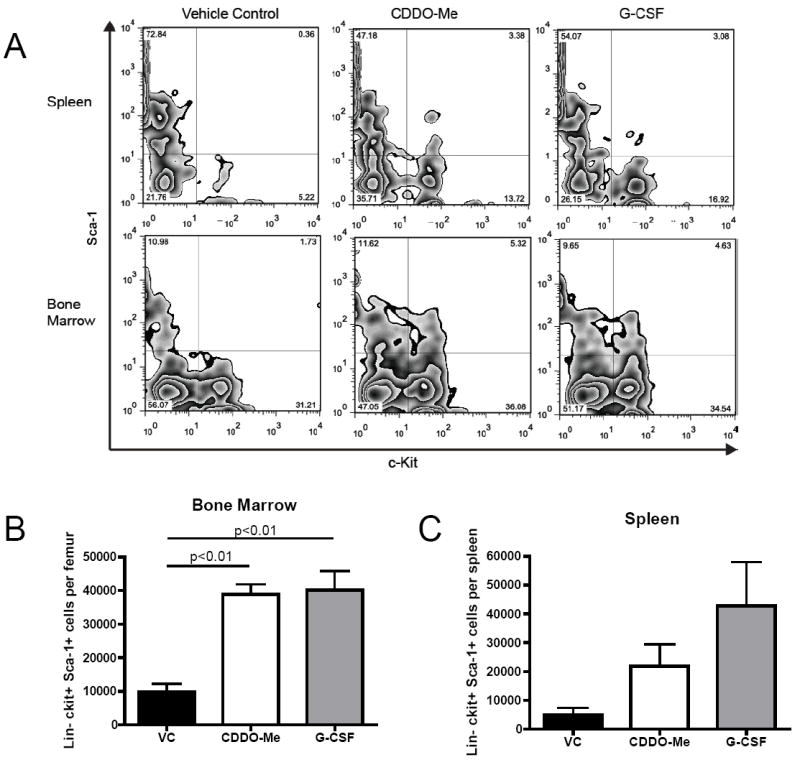
Mouse bone marrow and spleens were collected after the previously described treatment schedule and stained for lineage markers, c-kit, Sca-1 and 7-AAD to exclude dead cells. 7-AAD emits in the same fluorescent channel as the lineage markers used in this study (PE-Cy-5); thus dead cells were eliminated from gating during lineage exclusion. A, Representative flow plots of spleens and bone marrow HPCs are displayed. B and C, total bone marrow and spleen hematopoietic progenitor cells were determined. Statistical differences were determined by one-way ANOVA with Tukey’s post-test and a p-value<0.05 was considered significant.
CDDO-Me Improves Hematopoietic Recovery When Administered Prior to Sublethal Total Body Irradiation
We next assessed whether CDDO-Me administration before sublethal total body irradiation (TBI) would accelerate myeloid recovery. C57BL/6 mice were myelosuppressed with 500 cGy of TBI after administration of CDDO-Me or vehicle control. Mice received 120μg CDDO-Me or vehicle control twice daily on days -7 through -1 before irradiation on day 0. Mice were sacrificed on day 8 to determine the extent of hematopoietic recovery. Spleens were harvested and counted for all treatment groups (Figure 6). Spleens of mice pre-treated with CDDO-Me were significantly larger than control spleens (1.82×107 ± 0.19 vs. 7.37×107 ± 2.44; 4.1-fold) and displayed a significant increase in neutrophils, although other cell types were not significantly increased. Importantly, CDDO-Me pretreatment increased CFU-GM colony numbers 14-fold in the spleen to levels similar to those seen in naïve, unirradiated mice. Interestingly, no effects on peripheral blood composition recovery were observed suggesting that CDDO-Me is relatively restricted to early lineage recovery.
Figure 6. CDDO-Me is radioprotective when administered before sublethal TBI.
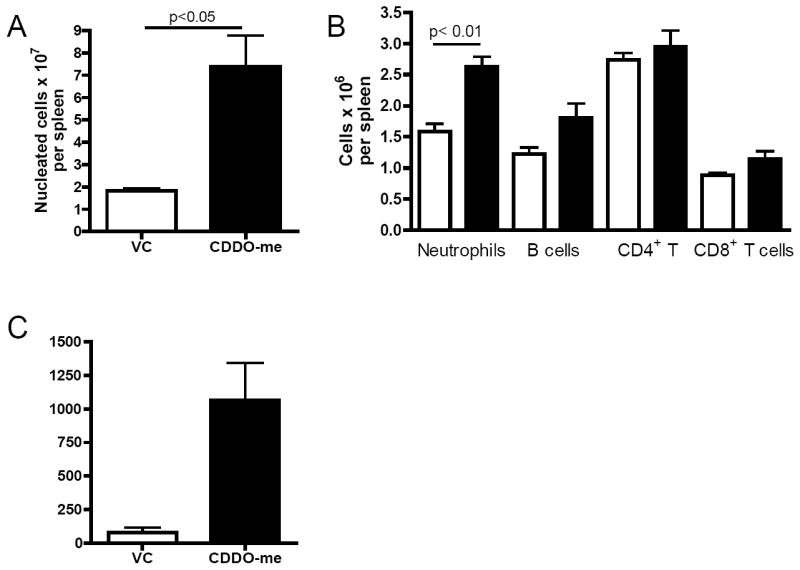
Mice were treated with CDDO-Me or vehicle control on days -7 through -1 before sublethal total body irradiation. Mice were harvested on day 8 after irradiaiton. A, Total nucleated cell counts in the spleen were quantified. B, leukocyte cell types in the spleen were quantified by flow cytometry for vehicle control (white bars) and CDDO-Me (black bars) treted mice. C, The total number of CFU-GM cells per spleen were quantified. Statistical differences were determined by one-way ANOVA with Tukey’s post-test and a p-value<0.05 was considered significant.
CDDO-Me Administration After Congenic HSCT Improves Myeloid Recovery
Due to the expansion of myeloid progenitors seen in naïve mice after CDDO-Me administration, we hypothesized that CDDO-Me could accelerate myeloid recovery after HSCT. C57BL/6 mice were lethally irradiated with 950 cGy and intravenously injected with 107 Ly5.2 congenic bone marrow cells. We performed a dose titration of CDDO-Me after HSCT and identified a dose of 60μg per day as the maximum tolerated dose; the dose used in naïve mice (120μg BID) was lethal in 100% of mice by day 7 after HSCT primarily due to gut toxicity (data not shown). Therefore, we administered 60μg CDDO-Me or vehicle control daily on days 1-13 after congenic HSCT. Mice were sacrificed on day 14 after HSCT and analyzed for myeloid and erythroid progenitors as well as donor (CD45.1) myeloid recovery (Figure 7). CDDO-Me administration resulted in significant increases in BFU-e progenitors as well as donor CD45.1+ CD11b+ Gr-1+ cells in the spleen compared with vehicle control. However, CDDO-Me did not induce progenitor differentiation or myeloid expansion in the femur and CBC were not significantly different consistent with the early lineage effects observed.
Figure 7. CDDO-Me administration after HSCT accelerates myeloid recovery.
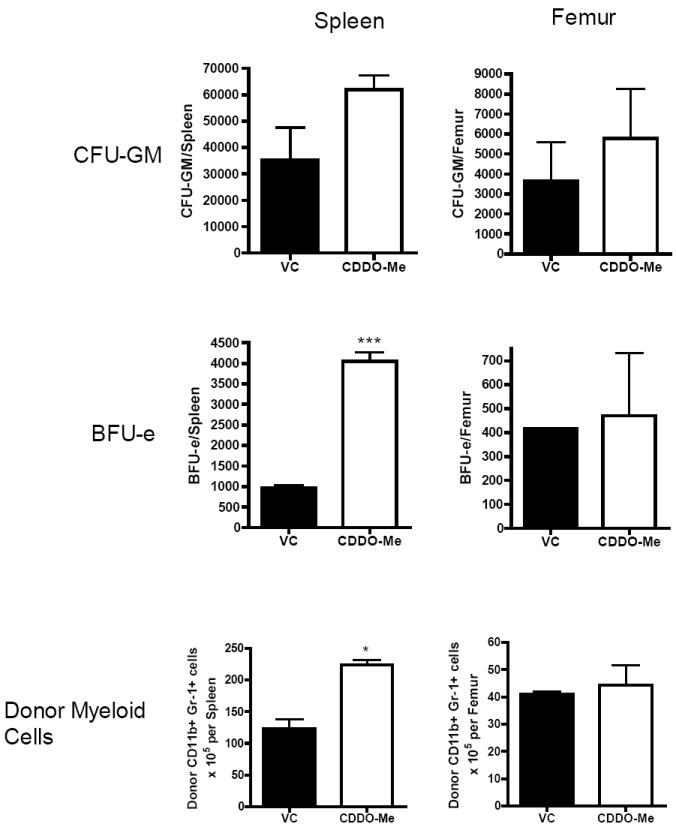
C57BL/6 (CD45.2) mice underwent a congenic BMT with 107 BMC from C57BL/6 Ly5.2 (CD45.1) donors. Mice received 60μg CDDO-Me daily on days 1 through 13 after BMT. Mouse spleens and femurs were collected on day 14 and were assessed for CFU-GM and BFU-e activity. Donor myeloid cells (CD45.1+/CD11b+/Gr-1+) were also assessed by flow cytometry. Statistical differences were determined by Student’s t-test and a p-value<0.05 was considered significant.
DISCUSSION
We have shown that the synthetic triterpenoid, CDDO-Me, is a potent inducer of myeloid progenitor expansion and terminal differentiation both in vitro and in vivo. Adding CDDO-Me directly to naïve bone marrow cells in a CFU-GM assay significantly increased colony counts when cultured in suboptimal colony-stimulating cytokines. Since the number of myeloid progenitor cells capable of producing CFU-GM colonies is fixed in these assays, increased colony counts suggest that CDDO-Me enhances the activity of GM-CSF to induce terminal differentiation of these progenitors. However, in vivo treatments with CDDO-Me not only increased CD11b+/Gr-1+ myeloid cells, but also myeloid progenitor cells. Splenic CD11b+/Gr-1+ cells have been described as myeloid-derived suppressor cells (MDSCs) which regulate suppressive activity on a broad variety of immune cells. In this regard, an expansion of CD11b+/Gr-1+ cells would be expected to suppress anti-tumor activity; however, Nagaraj et al. recently described that treatment of MDSCs with CDDO-Me eliminated the suppressive ability of these cells (12). CDDO-Me-treated mice displayed increased colony counts, indicative of a higher amount of myeloid and erythroid progenitors present in each organ. Interestingly, CDDO-Me showed similar effects in both the periphery and the bone marrow. Treatment of mice with G-CSF caused an increase of differentiated CD11b+/Gr-1+ cells in both the spleen and bone marrow similar to CDDO-Me. However, G-CSF failed to induce CFU-GM or BFU-e colony formation in the bone marrow. The mobilization of hematopoietic stem cells into the peripheral blood by G-CSF has been well characterized due to its importance in hematopoietic stem cell transplantation (17, 18). G-CSF disturbs interactions between the chemokine SDF-1 and its receptor, CXCR4, allowing HSCs to escape into the periphery. Similarly, a CXCR4 agonist (AMD3100, plerixafor) was FDA approved in 2008 for mobilizing peripheral blood in conjunction with G-CSF in patients undergoing autologous HSC transplants for multiple myeloma and non-Hodgkin’s lymphoma (19). G-CSF also increased HPCs in the bone marrow similar to CDDO-Me, an interesting observation given the use of G-CSF as a HSC mobilizer. Although bone marrow HSCs are infrequently examined in studies concerning G-CSF in naïve mice, this observation has been previously demonstrated either alone or in combination with cyclophosphamide (20, 21). However, the increased HPCs in the bone marrow of G-CSF-treated mice failed to induce CFU-GM colony formation. This observation may give insight into the differential mobilization of various subsets in the bone marrow. Whereas LT- or ST-HSC may require a higher threshold to become mobilized, more differentiated myeloid cells may have a lower threshold due to increased responsiveness to G-CSF. Kim, et al. demonstrated that CXCR4 expression was highly responsive to incubation with G-CSF in human CD33+ bone marrow cells whereas CD33- cells showed no change in CXCR4 expression (17). While G-CSF treatment may increase HSCs in the bone marrow with some of these cells becoming mobilized and some remaining, as these cells differentiate toward the myeloid lineage they may mobilize more rapidly due to decreased CXCR4 expression. This differential mobilization may account for the lack of any differences in CFU-GM or BFU-e colonies between mice treated with G-CSF or vehicle control. In contrast, CDDO-Me appears to act not as a bone marrow mobilizer, but as a broad stimulator of stem cell expansion and differentiation down the myeloid lineage. CDDO-Me induced a robust expansion of progenitor cells on three levels: HPCs, CFU-HPP, and CFU-GM/BFU-e. Interestingly, the magnitude of the effect induced by CDDO-Me seemed to decrease with increasing “primitiveness” of the precursor cells examined.
We were unable to determine the mechanism of action of CDDO-Me-induced myelopoiesis. The sera of treated mice were tested for colony-stimulating cytokines by ELISA as well as mRNA transcripts of cytokines and chemokines and chemokine receptors associated with hematopoieisis and mobilization on spleen and bone marrow samples but were inconclusive in CDDO-Me-treated animals. Due to the discrete mechanism of action of CDDO-Me versus G-CSF, it is unlikely that CDDO-Me induces a similar profile of surface receptor modulation and cytokine secretion as with G-CSF. However, the ability of CDDO-Me to induce differentiation of naïve bone marrow cells into CFU-GM colonies suggests a direct cellular mechanism of action. Mice lacking IκBα have been reported to show sharply increased CFU-GEMM, suggesting a role for NFκB in myelopoiesis. Given that CDDO-Me blocks IκBα kinase β (IKKβ) and thus prevents IκBα from becoming phosphorylated, the mechanism of action of CDDO-Me may lie in the NKκB pathway.
The most important application for CDDO-Me may be in the context of hematopoietic stem cell transplantation. When administered before sublethal TBI, CDDO-Me improves hematopoietic recovery, most significantly in neutrophils. This finding may be due to either general antioxidant and anti-inflammatory properties of CDDO-Me which have been demonstrated to be radioprotective (23) or the increase in hematopoietic progenitor cells. Primitive hematopoietic cells display decreased sensitivity to irradiation-induced damage, most likely due to their quiescent state (24). By increasing the number of these precursor cells, CDDO-Me helps to maintain hematopoietic function post-TBI when more differentiated cells are ablated. CDDO-Me administration post-transplantation also increased CFU-GM and BFU-e recovery. Importantly, CDDO-Me accelerated the recovery of neutrophils and all CD11b+/Gr-1+ cells which are clinical indicators of hematopoietic recovery post transplant. Furthermore, we have previously demonstrated that CDDO-Me and its parent compound, CDDO, delay graft-versus-host disease while still maintaining graft-versus-leukemia effects (13, 25) in addition to direct anti-tumor effects of CDDO-Me. In this previous report, we also examined effects on hematopoietic recovery in an allogeneic model and observed increased engraftment and HSCT recovery data in that study but could not determine if the effects were direct or affecting host resistance mechanisms. The current study demonstrates that direct effects can indeed occur. Clinically, CDDO-Me may potentially be used to supplement G-CSF both in the setting of HSC mobilization and HSCT recovery. Given the distinct mechanisms of action and sites of myeloid expansion seen in CDDO-Me and G-CSF, these agents may show synergistic effects when given in concert. Combined, these data suggest multiple advantageous effects of CDDO-Me when administered before or after allogeneic or autologous transplants.
Acknowledgments
The authors thank Weihong Ma, Monja Metcalf and Megan Whitaker for their excellent technical assistance. This work was supported by NIH grants RO1 CA102282 and PO1 CA055164.
Footnotes
Financial disclosure: William J. Murphy is a consultant for Reata Pharmaceuticals, Inc. Colin J. Meyer is an employee of Reata Pharmaceuticals and receives stock/stock options.
The remaining authors declare no competing financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Einsele H, Bertz H, Beyer J, et al. Infectious complications after allogeneic stem cell transplantation: epidemiology and interventional therapy strategies--guidelines of the Infectious Diseases Working Party (AGIHO) of the German Society of Hematology and Oncology (DGHO) Annals of hematology. 2003;82(Suppl 2):S175–185. doi: 10.1007/s00277-003-0772-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Weaver CH, Schwartzberg LS, Hainsworth J, et al. Treatment-related mortality in 1000 consecutive patients receiving high-dose chemotherapy and peripheral blood progenitor cell transplantation in community cancer centers. Bone marrow transplantation. 1997;19:671–678. doi: 10.1038/sj.bmt.1700713. [DOI] [PubMed] [Google Scholar]
- 3.Trivedi M, Martinez S, Corringham S, Medley K, Ball ED. Optimal use of G-CSF administration after hematopoietic SCT. Bone marrow transplantation. 2009;43:895–908. doi: 10.1038/bmt.2009.75. [DOI] [PubMed] [Google Scholar]
- 4.Ryu K, Susa M, Choy E, et al. Oleanane triterpenoid CDDO-Me induces apoptosis in multidrug resistant osteosarcoma cells through inhibition of Stat3 pathway. BMC cancer. 2010;10:187. doi: 10.1186/1471-2407-10-187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gao X, Deeb D, Hao J, et al. Synthetic triterpenoids inhibit growth, induce apoptosis and suppress pro-survival Akt, mTOR and NF-{kappa}B signaling proteins in colorectal cancer cells. Anticancer research. 2010;30:785–792. [PMC free article] [PubMed] [Google Scholar]
- 6.Shishodia S, Sethi G, Konopleva M, Andreeff M, Aggarwal BB. A synthetic triterpenoid, CDDO-Me, inhibits IkappaBalpha kinase and enhances apoptosis induced by TNF and chemotherapeutic agents through down-regulation of expression of nuclear factor kappaB-regulated gene products in human leukemic cells. Clin Cancer Res. 2006;12:1828–1838. doi: 10.1158/1078-0432.CCR-05-2044. [DOI] [PubMed] [Google Scholar]
- 7.Gao X, Deeb D, Jiang H, Liu Y, Dulchavsky SA, Gautam SC. Synthetic triterpenoids inhibit growth and induce apoptosis in human glioblastoma and neuroblastoma cells through inhibition of prosurvival Akt, NF-kappaB and Notch1 signaling. Journal of neuro-oncology. 2007;84:147–157. doi: 10.1007/s11060-007-9364-9. [DOI] [PubMed] [Google Scholar]
- 8.Pergola PE, Raskin P, Toto RD, et al. Bardoxolone methyl and kidney function in CKD with type 2 diabetes. N Engl J Med. 2011;365:327–336. doi: 10.1056/NEJMoa1105351. [DOI] [PubMed] [Google Scholar]
- 9.Yates MS, Tauchi M, Katsuoka F, et al. Pharmacodynamic characterization of chemopreventive triterpenoids as exceptionally potent inducers of Nrf2-regulated genes. Mol Cancer Ther. 2007;6:154–162. doi: 10.1158/1535-7163.MCT-06-0516. [DOI] [PubMed] [Google Scholar]
- 10.Dinkova-Kostova AT, Liby KT, Stephenson KK, et al. Extremely potent triterpenoid inducers of the phase 2 response: correlations of protection against oxidant and inflammatory stress. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:4584–4589. doi: 10.1073/pnas.0500815102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ahmad R, Raina D, Meyer C, Kharbanda S, Kufe D. Triterpenoid CDDO-Me blocks the NF-kappaB pathway by direct inhibition of IKKbeta on Cys-179. The Journal of biological chemistry. 2006;281:35764–35769. doi: 10.1074/jbc.M607160200. [DOI] [PubMed] [Google Scholar]
- 12.Nagaraj S, Youn JI, Weber H, et al. Anti-inflammatory triterpenoid blocks immune suppressive function of MDSCs and improves immune response in cancer. Clin Cancer Res. 2010;16:1812–1823. doi: 10.1158/1078-0432.CCR-09-3272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li M, Sun K, Redelman D, Welniak LA, Murphy WJ. The triterpenoid CDDO-Me delays murine acute graft-versus-host disease with the preservation of graft-versus-tumor effects after allogeneic bone marrow transplantation. Biol Blood Marrow Transplant. 2010;16:739–750. doi: 10.1016/j.bbmt.2010.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Welniak LA, Karas M, Yakar S, Anver MR, Murphy WJ, LeRoith D. Effects of organ-specific loss of insulin-like growth factor-I production on murine hematopoiesis. Biol Blood Marrow Transplant. 2004;10:32–39. doi: 10.1016/j.bbmt.2003.09.008. [DOI] [PubMed] [Google Scholar]
- 15.Woody MA, Welniak LA, Sun R, et al. Prolactin exerts hematopoietic growth-promoting effects in vivo and partially counteracts myelosuppression by azidothymidine. Experimental hematology. 1999;27:811–816. doi: 10.1016/s0301-472x(99)00019-3. [DOI] [PubMed] [Google Scholar]
- 16.Tacke F, Randolph GJ. Migratory fate and differentiation of blood monocyte subsets. Immunobiology. 2006;211:609–618. doi: 10.1016/j.imbio.2006.05.025. [DOI] [PubMed] [Google Scholar]
- 17.Kim HK, De La Luz Sierra M, Williams CK, Gulino AV, Tosato G. G-CSF down-regulation of CXCR4 expression identified as a mechanism for mobilization of myeloid cells. Blood. 2006;108:812–820. doi: 10.1182/blood-2005-10-4162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Petit I, Szyper-Kravitz M, Nagler A, et al. G-CSF induces stem cell mobilization by decreasing bone marrow SDF-1 and up-regulating CXCR4. Nature immunology. 2002;3:687–694. doi: 10.1038/ni813. [DOI] [PubMed] [Google Scholar]
- 19.Attolico I, Pavone V, Ostuni A, et al. Plerixafor Added to Chemotherapy Plus G-CSF Is Safe and Allows Adequate PBSC Collection in Predicted Poor Mobilizer Patients with Multiple Myeloma or Lymphoma. Biol Blood Marrow Transplant. 2011 doi: 10.1016/j.bbmt.2011.07.014. [DOI] [PubMed] [Google Scholar]
- 20.Neipp M, Zorina T, Domenick MA, Exner BG, Ildstad ST. Effect of FLT3 ligand and granulocyte colony-stimulating factor on expansion and mobilization of facilitating cells and hematopoietic stem cells in mice: kinetics and repopulating potential. Blood. 1998;92:3177–3188. [PubMed] [Google Scholar]
- 21.Morrison SJ, Wright DE, Weissman IL. Cyclophosphamide/granulocyte colony-stimulating factor induces hematopoietic stem cells to proliferate prior to mobilization. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:1908–1913. doi: 10.1073/pnas.94.5.1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rupec RA, Jundt F, Rebholz B, et al. Stroma-mediated dysregulation of myelopoiesis in mice lacking I kappa B alpha. Immunity. 2005;22:479–491. doi: 10.1016/j.immuni.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 23.Eskiocak U, Kim SB, Roig AI, et al. CDDO-Me protects against space radiation-induced transformation of human colon epithelial cells. Radiation research. 2010;174:27–36. doi: 10.1667/RR2155.1. [DOI] [PubMed] [Google Scholar]
- 24.Lacorazza HD, Yamada T, Liu Y, et al. The transcription factor MEF/ELF4 regulates the quiescence of primitive hematopoietic cells. Cancer cell. 2006;9:175–187. doi: 10.1016/j.ccr.2006.02.017. [DOI] [PubMed] [Google Scholar]
- 25.Sun K, Li M, Konopleva M, et al. The synthetic triterpenoid, CDDO, suppresses alloreactive T cell responses and reduces murine early acute graft-versus-host disease mortality. Biol Blood Marrow Transplant. 2007;13:521–529. doi: 10.1016/j.bbmt.2006.12.453. [DOI] [PMC free article] [PubMed] [Google Scholar]