

Abstract
Quorum sensing in Burkholderia cenocepacia H111 involves two signalling systems that depend on different signal molecules, namely N-acyl homoserine lactones (AHLs) and the diffusible signal factor cis-2-dodecenoic acid (BDSF). Previous studies have shown that AHLs and BDSF control similar phenotypic traits, including biofilm formation, proteolytic activity and pathogenicity. In this study we mapped the BDSF stimulon by RNA-Seq and shotgun proteomics analysis. We demonstrate that a set of the identified BDSF-regulated genes or proteins are also controlled by AHLs, suggesting that the two regulons partially overlap. The detailed analysis of two mutually regulated operons, one encoding three lectins and the other one encoding the large surface protein BapA and its type I secretion machinery, revealed that both AHLs and BDSF are required for full expression, suggesting that the two signalling systems operate in parallel. In accordance with this, we show that both AHLs and BDSF are required for biofilm formation and protease production.
Introduction
Many bacteria are capable of coordinating gene expression in a cell density-dependent manner, a phenomenon commonly referred to as quorum sensing (QS) [1]. QS systems rely on the production and release of small signal molecules into the environment. Bacteria respond to these signals when their concentration has reached a certain threshold (and thus the bacterial population has attained a critical density), upon which expression of target genes is activated or repressed. Among the various QS signal molecules identified to date, the two most thoroughly investigated classes are the N-acyl-homoserine lactones (AHLs), which are produced by many Gram-negative bacteria, and small peptides, which are produced by many Gram-positive species [2], [3].
Burkholderia cenocepacia is a Gram-negative opportunistic pathogen belonging to the Burkholderia cepacia complex (Bcc), a group of 17 closely related bacterial species [4]. B. cenocepacia can cause airway infections in susceptible individuals, particularly in persons suffering from cystic fibrosis [5]. All members of the Bcc investigated so far utilize the AHL-dependent CepIR QS system [6]. CepI was shown to catalyze the synthesis of N-octanoyl homoserine lactone (C8-HSL) along with minor amounts of N-hexanoyl homoserine lactone (C6-HSL) [7]. At quorate population densities C8-HSL binds to its cognate transcriptional regulator CepR and in the AHL-bound form CepR binds to specific DNA sequences (so-called cep boxes) in the promoter region of target genes, thereby inducing or repressing gene expression [8]. Previous work has shown that the CepIR system regulates multiple functions, including virulence, biofilm formation, swarming motility, and the production of proteases, siderophores and antifungal compounds (reviewed in [9]). The CepR regulons of two B. cenocepacia, K56-2 and H111, have previously been determined using functional genomics approaches [8], [10], [11]. These investigations not only identified many genes encoding virulence factors [12] but has also shown that in strain H111 AHL-dependent expression of a large surface protein (bapA, BCAM2143) is critical for biofilm formation on abiotic surfaces [11].
Recent work has identified an additional QS system in B. cenocepacia that relies on BDSF (Burkholderia diffusible signal factor, cis-2-dodecenoic acid), which belongs to a rapidly growing family of fatty acid signal molecules [13], [14]. The biosynthesis of BDSF is driven by the product of rpfF Bc (BCAM0581), an enoyl CoA hydratase [15]. RpfFBc is the first protein described to possess both dehydratase and thioesterase activity, which enables the direct conversion of the acyl carrier protein thioester of 3-hydroxydodecanoic acid into cis-2-dodecenoic acid [15]. More recently it has been demonstrated that the gene adjacent to rpfF Bc encodes the BDSF receptor protein RpfR, which contains PAS-GGDEF-EAL domains [16]. It has been shown that upon binding of BDSF to RpfR the c-di-GMP phosphodiesterase activity of the protein is stimulated and as a consequence the intracellular c-di-GMP level is lowered. Hence, RpfR is the first example of a c-di-GMP metabolic enzyme that is directly activated by a cell-cell communication signal [16]. Disruption of either rpfR or rpfFBc was shown to result in reduced motility, impaired biofilm formation, lowered proteolytic activity, and attenuated virulence [16]. All these phenotypes are also known to be AHL-regulated and we were therefore interested to investigate whether the two regulatory circuits regulate the same set of genes and whether they are interconnected or operate independently of each other.
In this study the BDSF stimulon of B. cenocepacia H111 was defined both at the transcript and protein level using RNA-Seq and shotgun proteomics. To determine the overlap of the AHL- and BDSF-dependent QS systems we compared the BDSF stimulon to the previously published CepR regulon [11]. In addition, we constructed a cepI rpfF Bc double mutant and used it to assess the influence of the two signal molecules individually and in combination on biofilm formation and the production of proteases and on transcription of QS-regulated target genes. Our data demonstrate that, in spite of the observed decrease in AHL production in the rpfF Bc mutant, the two QS systems regulate the tested phenotypes and genes independently, suggesting that they are not hierarchically arranged but operate in parallel under the experimental conditions used in this study.
Results
Mapping of the B. cenocepacia H111 BDSF Stimulon
To analyse the mode of action of the signalling molecule BDSF in strain H111, we conducted RNA-Seq genome-wide transcriptome analyses comparing the expression levels of a mutant in rpfF Bc with those of the wild type and of the rpfF Bc mutant supplemented by exogenous addition of BDSF. The sum of mapped reads matching to mRNA transcripts varied between 296’000–634’000 reads per sample. This yield of uniquely mapping reads is in the range of what Yoder-Himes and colleagues [17] have reported for RNA-Seq analyses of two B. cenocepacia strains [17]. Using the software DESeq [18], we focused on the 150 top ranked differentially expressed genes (Figure 1A), and noted that most of them (112 genes) were down-regulated in the rpfF Bc mutant. For the majority of the down-regulated genes (85%), gene expression could be restored to wild type levels by supplementing the medium with 10 µM BDSF (Figure 1B). Among the 150 differentially expressed genes (Table S1), 38 showed increased expression in the rpfF Bc mutant, suggesting that BDSF also represses a set of genes. For a functional analysis we transferred the classifications that were made by the EggNOG software [19] for B. cenocepacia strain J2315 to the respective orthologs in B. cenocepacia strain H111. Using the EggNOG resource, a large number of protein-coding genes could be functionally classified in 26 proNOG categories (5270, 73%, see Material and Methods). Many of the genes that showed decreased expression in the rpfF Bc mutant were previously shown to be controlled by the CepI/CepR QS system, including genes encoding the sugar-binding lectins BclACB (BCAM0186-84), the large surface protein BapA (BCAM2143) and the protease ZmpB (BCAM2307). Additionally, two gene clusters coding for the exopolysaccharide cepacian (BCAM1004-10 and BCAM0854-0864) were identified. We next compared the rpfF Bc regulon of strain H111 with the recently published BDSF regulon of strain J2315 [20]. Surprisingly, from the set of 150 top ranked genes identified as BDSF-regulated in our study only three genes overlapped with the BDSF regulon determined for strain J2315, namely a dioxygenase (BCAM0811), a component of a type I secretion system located downstream of bapA (BCAM2142) and the permease of an ABC efflux pump (BCAM2225). This discrepancy may be due to differences between the strains and culture conditions.
Figure 1. Mapping of the BDSF stimulon.

(A) MA plot showing the fold change in transcript expression of all B. cenocepacia H111 genes versus the mean of identified reads in an rpfF Bc mutant and wild type. The 112 down-regulated transcripts/proteins in the rpfF Bc mutant are indicated in red, the 38 up-regulated genes in green. (B) Box plot demonstrating that supplementing the medium with 10 µM BDSF rescues the gene expression defects in the rpfF Bc mutant.
Comparative Proteome Analysis of the B. cenocepacia H111 Wild Type and its Isogenic rpfFBc Mutant Derivative
In order to obtain a comprehensive view of the RpfFBc regulon, we also analyzed the protein expression profiles of the wild type, the rpfFBc mutant and the rpfFBc mutant supplemented with BDSF. For this purpose, proteins and total RNA (used for the RNA-Seq study, see above) were extracted from each sample in parallel, allowing for the best possible comparison of these datasets. Using stringent criteria (see Material & Methods for details), a total of 2′565, 2′420, and 2′685 proteins were detected in wild type, rpfFBc mutant and supplemented rpfFBc mutant, respectively. Therefore, approximately 35% of the in silico predicted B. cenocepacia H111 proteins were identified. Based on the target-decoy database search results, we could estimate that the overall peptide and protein false discovery rate (FDR) amounted to 0.8% and 2.7%, respectively. Similar to reads from RNA-Seq, the individual peptide spectrum matches (PSMs) also represent count data that can be analyzed with the DESeq software to identify differentially expressed proteins. In total, 116 proteins displayed significant changes in the rpfFBc mutant (Table S2). Out of these, 81 proteins were significantly down-regulated in the mutant. When the medium was supplemented with BDSF, protein expression was restored to wild type levels in 91.5% of the cases. A correlation between proteomic and transcriptomic data showed that 29 genes/proteins are shared among the top ranked candidates when combining the results from both approaches (Table 1).
Table 1. List of genes and proteins differentially expressed using RNA-Seq and proteomics in the rpfFBc mutant compared to the wild type.
| RNA Seq | Proteomics | ||||||
| Locus ID CCEa | Orthologues J2315b | Descriptionc | Gene name | wt vs rpfF d | wt vs rpfF comple | wt vs rpfF f | wt vs rpfF complg |
| CCE49245 | BCAL0111 | O-linked N-acetylglucosamine transferase | 3.0 | 2.1 | na | 2.8 | |
| CCE46674 | BCAL0249 | 50S ribosomal protein L6 | 0.4 | 0.9 | 1.6 | 1.5 | |
| CCE48190 | BCAL0524 | Flagellar motor switch protein | 5.1 | 3.2 | 3.9 | 1.4 | |
| CCE48189 | BCAL0525 | Flagellar MS-ring protein | 4.2 | 6.5 | 2.9 | 1.6 | |
| CCE53354 | BCAL0762 | Methyl-Apting chemotaxis protein | 4.6 | 1.4 | 2.7 | 0.9 | |
| CCE53285 | BCAL0831 | Phasin family protein | 2.9 | 0.7 | 3.2 | 1.4 | |
| CCE51557 | BCAL1059 | Bifunctional N-succinyldiaminopimelate-aminotransferase/acetylornithine transaminase protein | 2.8 | 1.6 | 0.5 | 0.8 | |
| CCE50475 | BCAL2352 | Carbonic anhydrase | 4.3 | 1.0 | 7.0 | 1.1 | |
| CCE49840 | BCAL3041 | Protein involved in carbohydrate transport | 4.5 | 2.0 | 0.5 | 1.6 | |
| CCE51244 | BCAL3285 | Nitric oxide dioxygenase; flavohemoprotein | 6.7 | 0.6 | na | 0.8 | |
| CCE46722 | BCAM0184 | Fucose-Binding lectin protein | bclB | 183.0 | 0.5 | na | 0.8 |
| CCE46720 | BCAM0186 | Fucose-Binding lectin protein | bclA | 280.2 | 0.5 | na | 1.1 |
| CCE46715 | BCAM0191 | Non-ribosomal peptide synthetase | 5.2 | 0.9 | na | 0.7 | |
| CCE46713 | BCAM0192 | 9.4 | 1.5 | na | 1.2 | ||
| CCE46711 | BCAM0194 | 7.9 | 1.6 | na | 0.4 | ||
| CCE46710 | BCAM0195 | Non-ribosomal peptide synthetase | 6.6 | 1.4 | na | 0.8 | |
| CCE46709 | BCAM0196 | 5.9 | 0.9 | 14.0 | 0.9 | ||
| CCE50899 | BCAM0853 | Transposase | 3.1 | 0.8 | 10.0 | 0.7 | |
| CCE50898 | BCAM0854 | Mannose-6-phosphate isomerase | 8.3 | 0.4 | na | 0.4 | |
| CCE50892 | BCAM0859 | Protein involved in capsule organization | 31.8 | 0.4 | na | 0.7 | |
| CCE48736 | BCAM1004 | Gdp-Mannose 4,6-dehydratase | 4.3 | 0.3 | na | 0.5 | |
| CCE47169 | BCAM1572 | Methyl-Apting chemotaxis protein | 3.0 | 1.2 | 3.0 | 1.5 | |
| CCE46959 | BCAM1745 | Atpase | 0.01 | 0.8 | 0.2 | 2.2 | |
| CCE53120 | BCAM2140 | Type I secretion membrane fusion protein | 15.8 | 1.0 | na | 1.4 | |
| CCE53119 | BCAM2141 | ABC transporter protein | 16.0 | 0.9 | na | 1.0 | |
| CCE53117 | BCAM2143 | Calcium ion binding protein | bapA | 12.2 | 0.8 | 41.5 | 0.7 |
| CCE53024 | BCAM2224 | Protein involved in siderophore transport | 31.5 | 28.0 | na | na | |
| CCE53019 | BCAM2230 | Non-ribosomal peptide synthetase | 11.8 | 7.9 | na | na | |
| CCE46201 | BCAM2627 | Protein involved in iron ion transport | 6.2 | 11.0 | 10.0 | 10.0 | |
Nomenclature according to GenBank file CAFQ01000001.1.
Orthologs were identified as described in the Material and Methods section.
Description according to the EggNOG classification.
Fold change (FC) of transcript expression, comparing wild type strain with rpfF mutant grown in LB medium until an OD of 2.
Fold change (FC) of transcript expression, comparing wild type strain with complemented rpfF mutant grown in LB medium until an OD of 2.
Fold change (FC) of protein expression, comparing wild type strain with rpfF mutant grown in LB medium until an OD of 2.
Fold change (FC) of protein expression, comparing wild type strain with complemented rpfF mutant grown in LB medium until an OD of 2.
na, not applicable because the read number or spectral counts in the mutant or complemented strains is equal 0.
Comparison of the B. cenocepacia H111 RpfFBc and CepR Regulons
A recent transcriptomic study using a custom B. cenocepacia oligonucleotide microarray showed that expression of 103 genes was decreased at least two-fold in a cepR mutant derived from B. cenocepacia strain H111 [11]. When those data were compared to the RNA-Seq data obtained in the present study, a total of 31 overlapping genes were found to be differentially expressed in both the cepR and the rpfF Bc mutant (Figure S1, Table 2). Among the co-regulated genes was the bclACB lectin operon and genes coding for the large surface protein BapA and enzymes for the biosynthesis of the exopolysaccharide cepacian (Table 2). Interestingly, expression of aidA and cepI, which were previously demonstrated to be directly controlled by CepR through binding of the CepR/C8-HSL complex to the promoter regions of these target genes [21], was only marginally affected by BDSF. The expression and regulation of selected target genes (bclA, cepI, aidA) was validated by quantitative real time PCR using RNA from an independent biological replicate (Table 3). The expression profile of a cepR or a cepI mutant did not reveal any influence on rpfFBc expression by the AHL-dependent QS system, neither at the transcript (Table 3) nor at the protein level [11]. Moreover, cepR transcription was not affected by the BDSF signalling molecule. However, qPCR analysis and shotgun proteomics indicated a significant decrease of cepI and CepI levels in an rpfFBc mutant (Table 3 and Table S2). In agreement with these results we observed that the production of AHL signal molecules was reduced in the rpfFBc mutant when compared to the wild type (see below).
Table 2. List of genes differentially expressed in the rpfFBc and cepR mutant, using the wild type as baseline.
| Locus ID CCEa | Orthologues J2315b | Descriptionc | Gene name | wt vs rpfF d | wt vs rpfF compl e | wt vs cepR |
| CCE49231 | BCAL0124 | Transcriptional activator FlhD | 3.7 | 1.5 | 0.5 | |
| CCE53285 | BCAL0831 | Phasin family protein | 2.8 | 0.7 | 3.2 | |
| CCE52825 | BCAL0833 | Acetoacetyl-Coa reductase | 4.3 | 1.1 | 3.0 | |
| CCE51553 | BCAL1063 | Succinylarginine dihydrolase | 2.8 | 1.4 | 0.5 | |
| CCE50476 | BCAL2353 | Sulfate permease | 18.4 | 0.9 | 2.2 | |
| CCE51244 | BCAL3285 | Nitric oxide dioxygenase; flavohemoprotein | 6.5 | 0.6 | 25.8 | |
| CCE46722 | BCAM0184 | Fucose-Binding lectin protein | bclB | 181.0 | 0.5 | 5.1 |
| CCE46721 | BCAM0185 | Fucose-Binding lectin protein | bclC | 73.5 | 0.7 | 3.3 |
| CCE46720 | BCAM0186 | Fucose-Binding lectin protein | bclA | 274.4 | 0.5 | 9.0 |
| CCE46715 | BCAM0191 | Non-ribosomal peptide synthetase | 5.3 | 0.9 | 12.8 | |
| CCE46713 | BCAM0192 | 9.2 | 1.5 | 46.8 | ||
| CCE46712 | BCAM0193 | 17.1 | 2.3 | 47.7 | ||
| CCE46711 | BCAM0194 | 8.0 | 1.6 | 52.2 | ||
| CCE46710 | BCAM0195 | Non-ribosomal peptide synthetase | 6.5 | 1.4 | 34.0 | |
| CCE46709 | BCAM0196 | 6.1 | 0.9 | 37.7 | ||
| CCE50917 | BCAM0835 | Transcriptional regulator, AraC family protein | 4.9 | 1.2 | 4.3 | |
| CCE48735 | BCAM1005 | Acyltransferase | 4.6 | 0.6 | 2.2 | |
| CCE48728 | BCAM1010 | Utp–Glucose-1-Phosphate uridylyltransferase | 39.4 | 0.5 | 2.4 | |
| CCE46959 | BCAM1745 | Atpase | 0.01 | 0.8 | 0.4 | |
| CCE50321 | BCAM1871 | 3-Hydroxy-3-Methylglutaryl-Coenzyme A reductase | 3.5 | 0.8 | 32.5 | |
| CCE53256 | BCAM2060 | Natural resistance-associated macrophage protein | 0.1 | 0.8 | 0.4 | |
| CCE53120 | BCAM2140 | Type I secretion membrane fusion protein | 16.0 | 0.9 | 3.1 | |
| CCE53119 | BCAM2141 | ABC transporter protein | 16.0 | 0.9 | 4.4 | |
| CCE53118 | BCAM2142 | 14.9 | 1.0 | 5.3 | ||
| CCE53117 | BCAM2143 | Calcium ion binding protein | 12.1 | 0.8 | 5.1 | |
| CCE53089 | BCAM2169 | Outer membrane autotransporter protein | 2.6 | 0.9 | 2.1 | |
| CCE53021 | BCAM2227 | Pyochelin biosynthetic protein | na | na | 2.2 | |
| CCE52940 | BCAM2307 | Metalloendopeptidase | zmpB | 3.7 | 1.4 | 8.7 |
| CCE52939 | BCAM2308 | Aminopeptidase | 4.0 | 1.5 | 5.3 | |
| CCE52108 | BCAS0292 | aidA' | 3.2 | 0.9 | 138.1 | |
| CCE52109 | BCAS0293 | aidA | 4.0 | 0.8 | 167.2 |
Nomenclature according to GenBank file CAFQ01000001.1.
Orthologs were identified as described in the Material and Methods section.
Description according to EggNOG categories.
Fold change (FC) of expression, comparing wild type strain with rpfF mutant grown in LB medium until an OD of 2.
Fold change (FC) of expression, comparing wild type strain with complemented rpfF mutant grown in LB medium until an OD of 2.
Fold change (FC) of expression comparing wild type strain with cepR mutant grown in LB medium until an OD of 2 [11].
na, not applicable because the read number in the mutant or complemented strains is equal 0.
Table 3. Validation of RNA-Seq results using quantitative PCR analysis.
| Locus ID CCEa | Orthologues J2315b | Descriptionc | gene name | wt vs rpfF d | wt vs rpfF comple | wt vs cepR f | wt vs cepR complg |
| CCE46720 | BCAM0186 | Fucose-Binding lectin protein | bclA | 74 | 1.2 | 4.1 | 0.4 |
| CCE48446 | BCAM0581 | Enoyl-CoA hydratase/carnithine racemase | rpfF | 0.6 | 0.3 | 0.7 | 0.8 |
| CCE48447 | BCAM0580 | EAL/GGDEF domain protein | rpfR | 1 | 0.6 | 0.8 | 0.5 |
| CCE50322 | BCAM1870 | N-acyl-L-homoserine lactone synthetase | cepI | 3.2 | 1.6 | 65.0 | 0.8 |
| CCE50324 | BCAM1868 | Transcriptional regulator protein | cepR | 1.7 | 1.8 | 0.6 | 0.3 |
| CCE50476 | BCAL2353 | Sulfate permease | 6 | 1.8 | 4.2 | 1.1 | |
| CCE50898 | BCAM0854 | Mannose-6-phosphate isomerase | 11.3 | 1 | 6.3 | 0.6 | |
| CCE52109 | BCAS0293 | AidA | aidA | 6 | 1.8 | 685.0 | 0.5 |
Nomenclature according to GenBank file CAFQ01000001.1.
Orthologs were identified as described in the Material and Methods section.
Description according to EggNOG classification.
Fold change (FC) of expression, comparing wild type strain with rpfF mutant grown in LB medium until an OD of 2.
Fold change (FC) of expression, comparing wild type strain with complemented rpfF mutant grown in LB medium until an OD of 2.
Fold change (FC) of expression comparing wild type strain with cepR mutant grown in LB medium until an OD of 2.
Fold change (FC) of expression, comparing wild type strain with complemented cepR mutant grown in LB medium until an OD of 2.
Both Signal Molecules are Required for Maximal Expression of bapA and bclACB
In order to understand how the two QS systems co-regulate gene expression, we assessed the impact of AHL and of BDSF on the expression of selected target genes. To this end, we constructed a cepI rpfFBc double mutant and used this strain as genetic background to test the effect of the two signal molecules individually and in combination on transcription of target genes. Specifically, we generated lacZ transcriptional fusions to the promoter regions of genes that were regulated either by both AHL and BDSF (bapA), primarily by BDSF (bclACB), or primarly by AHLs (aidA) (Table 1, Table 3).
In agreement with the global analyses, which showed that bapA expression is regulated by both AHLs and BDSF (Table 2), the PbapA-lacZ fusion showed greatly decreased activity in the cepI rpfF Bc double mutant compared to the wild type (Figure 2A). Interestingly, the activity of the bapA promoter was restored to wild type levels only when the medium was supplemented with both C8-HSL and BDSF (Figure 2A). Addition of either C8-HSL or BDSF resulted in only partial restoration of bapA promoter activity. Likewise, activity of the PbclA-lacZ fusion was only restored to the level of the wild type when both signalling molecules were added to the growth medium (Figure 2B). In contrast to the bapA promoter the addition of C8-HSL alone did not stimulate transcription of bclACB while the addition of BDSF resulted in a partial restoration of bclACB promoter activity. To confirm these results we performed a Western Blot analysis using antibodies directed against the lectin BclB (BCAM0184, which is co-transcribed with blcA [11]) (Figure 2D). We observed that BclB could only be detected in the double mutant ΔcepI rpfF Bc when it was grown in the presence of BDSF. These data further support the idea that expression of BclB is primarily driven by BDSF. We also assessed the promoter activity of aidA, a gene already known to be very stringently AHL-regulated [11], [22], in the cepI rpfF Bc mutant background. The activity of this promoter fusion was negligible in a double cepI rpfF Bc mutant background and could be fully restored after supplementing the medium with AHLs, whereas the addition of BDSF did not show any effect (Figure 2C). In agreement with these results, AidA levels could only be restored after supplementing the strain with AHLs (Figure 2D). In conclusion, our data suggest that the expression of several genes is controlled by both QS systems and that the contribution of each of the systems is variable.
Figure 2. The role of the two QS systems in the regulation of selected genes.

The bapA (A), bclA (B), and aidA (C) promoter activities were assessed by means of transcriptional lacZ fusions in the H111 wild type strain and in the mutant defective in AHL and BDSF synthesis (ΔcepI rpfF Bc). The strains were grown to late exponential growth phase in LB Lennox broth in the absence or presence of signal molecules (200 nM C8-HSL; 10 µM BDSF) as indicated by+and - below each bar. Error bars indicate SEM, n = 3. * P<0.05, ** P<0.01, *** P<0.001 (t-test, two-tailed) compared to ΔcepI rpfF Bc without signalling molecule (ns, not significant) (D) Expression of BclB and AidA in the H111 wild type and the double mutant ΔcepI rpfFBc as assessed by Western Blot analysis. The strains were grown on plates in the presence or absence of signal molecules as indicated by+and - below each band.
The BDSF-dependent QS System Affects AHL Levels
The gene cluster coding for BapA and its export machinery was recently shown to be under the control of CepI/CepR [11]. In the present study we observed that expression of bapA in the rpfF Bc mutant background was reduced (Table 1, Figure 2A). Given that our data also showed a reduced expression of cepI in this mutant background (Table 3, Table S2), we hypothesised that the BDSF-dependent QS system could positively regulate the production of AHLs. To test this hypothesis, we quantified the amount of AHLs produced by the rpfF Bc mutant by the aid of the biosensor P. putida F117/pAS-C8 [23]. We found that the AHL-levels were 50% lower in the rpfF Bc mutant relative to the wild type (Figure 3). The AHL levels were fully rescued when cepI was expressed from plasmid pBBRcepI in the BDSF-deficient mutant (Figure 3). These findings were in agreement with our qPCR and proteomics data, which showed a down-regulation of cepI in an rpfF Bc mutant (Table 3 and Table S2). We also observed that the activity of a PcepI-lacZ promoter fusion was reduced in the rpfFBc mutant (Figure S2) and the promoter activity was fully restored after supplementing the medium with either BDSF or C8-HSL. Interestingly, when the fusion was tested in a cepI mutant background the promoter activity could not be restored by the addition of BDSF (Figure S2), suggesting that the reduced transcription of cepI is a direct consequence of the lowered amounts of AHLs produced by the BDSF mutant.
Figure 3. AHL-levels are reduced in an rpfFBc mutant.
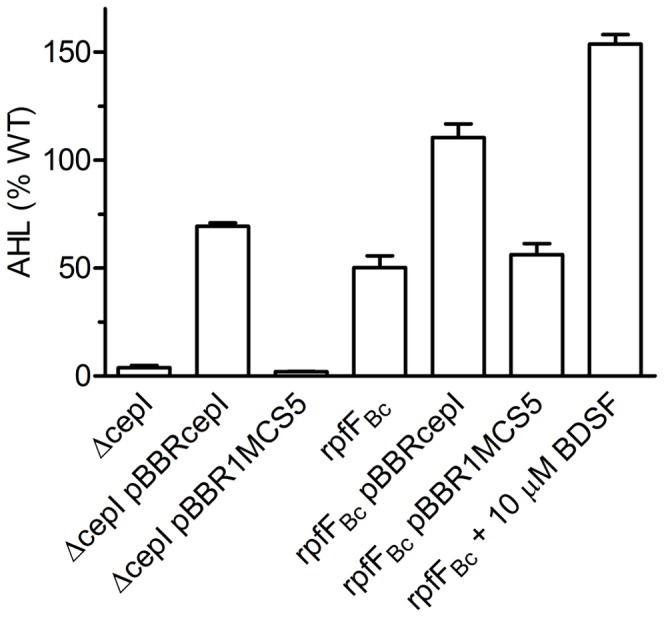
The amount of AHLs produced by the cepI and the rpfFBc mutant with cepI constitutively expressed from plasmid pBBRcepI or with the empty plasmid control pBBR1MCS5 was quantified by the aid of the biosensor P. putida/pAS-C8. Error bars indicate SEM, n≥3.
In agreement with our global analyses, we observed that inactivation of cepI or cepR did not affect the amount of BDSF produced (Figure S3).
BDSF and AHLs co-regulate Biofilm Formation and Production of Proteolytic Activity
We next examined whether the reduced AHL level of the rpfF Bc mutant affected known AHL-controlled phenotypes. To this end, we quantified the biofilms formed by the cepI rpfF Bc double mutant grown with or without exogenous addition of C8-HSL or BDSF. As shown in Figure 4A, the double mutant was defective in biofilm formation relative to the wild type and this defect could only be restored when both signalling molecules were added to the growth medium. In agreement with this observation, neither overexpression of CepI nor the addition of C8-HSL or BDSF to the growth medium lead to full restoration of the biofilm defect in the rpfF Bc mutant (Figure S4).
Figure 4. Biofilm formation and protease activity are co-regulated by AHL and BDSF.
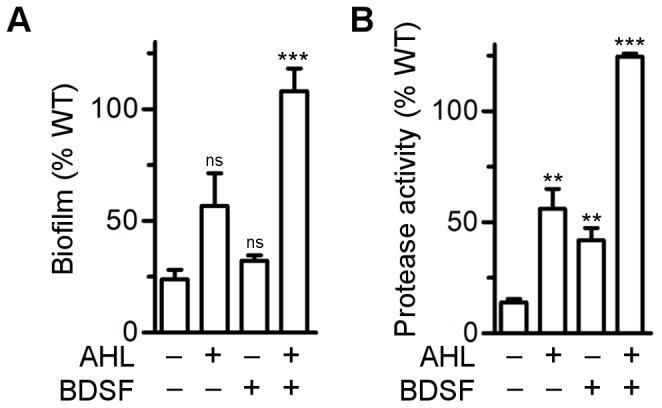
(A) Biofilm formation in ABC minimal. (B) Protease activity in NYG medium. The strains tested are the wild type H111 and the cepI rpfF Bc double mutant. Strains were grown in the presence of absence of signal molecules (200 nM C8-HSL; 10 µM BDSF) as indicated by+and - below each bar. Error bars indicate SEM, n≥3. ** P<0.01, *** P<0.001 (t-test, two-tailed) compared to ΔcepI rpfF Bc without signalling molecule (ns, not significant).
Previous studies have shown that the proteolytic activity of B. cenocepacia H111 is regulated by both AHLs and BDSF [11], [24]. Consistently, we found that transcription of a zinc-metalloprotease (zmpB, BCAM2307) was down-regulated in the rpfFBc mutant (Table 2). We therefore tested the influence of BDSF and C8-HSL on the production of extracellular proteases of the cepI rpfF Bc double mutant. We observed that, similar to biofilm formation, both signalling molecules were needed to fully restore proteolytic activity (Figure 4B).
Discussion
Our combined RNA-Seq and proteome analysis revealed that the set of genes regulated by BDSF in B. cenocepacia H111 shows a substantial overlap with the set of genes recently shown to be CepR-regulated (Figure S1). However, we also identified some genes that were almost exclusively regulated by one of the two QS systems. For example, the gene aidA, which encodes a protein required for killing of the nematode Caenorhabditis elegans [22], is stringently regulated by C8-HSL (>100-fold at the transcript level, Table 3, Figure 2C and D) whereas the effect of BDSF is marginal (4-fold to 6-fold at the transcript level, Tables 2 and 3, Figure 2C and D). It is important to note that the aidA promoter region contains a cep box, i.e. a CepR binding site, which is required for AHL-dependent transcriptional activation of this gene [22]. Likewise, we observed that genes containing a bona fide cep box in their upstream regions are more strongly affected by the CepI/R than the RpfR/F system (Table S3). At the other extreme, expression of the lectin BclB, which is encoded by the last gene of the bclACB operon [11], in the cepI rpfFBc double mutant was found to be strongly dependent on BDSF while C8-HSL showed little effect (Figure 2D, Table 2). We have previously shown that in a cepI mutant strain transcription of the bclACB operon is approximately 6-fold down-regulated relative to the wild type and that this defect is reversed by the addition of C8-HSL to the medium [11]. The evidence presented in this study suggests that AHL-dependent regulation of the bclACB operon only occurs when the BDSF-dependent QS system is intact (Figure 2D, Table 3). In summary, these data suggests that the CepIR system synergistically enhances BDSF-dependent activation of bclACB expression. The underlying molecular mechanism of how the two QS systems interact in the expression of the lectin operon remains to be elucidated.
It has not escaped our attention that the rpfFBc mutant produces significantly reduced amounts of AHL signal molecules, most likely due to lowered transcription of cepI (Table 3, Figure S2). This result suggests a hierarchical arrangement of the two QS systems with the RpfF/RpfR system being on top of the CepI/CepR system. However, several of our results do not support this conclusion. Expression of some of the well-characterized AHL-regulated genes, including aidA, was only marginally affected in an rpfFBc mutant background. In the case of bapA, which was shown to be regulated by both systems, addition of AHLs to the BDSF-deficient mutant did not restore expression of this gene to the level of the wild type, which would have been expected if the AHL-dependent circuitry operated downstream of the BDSF system. Likewise, biofilm formation and proteolytic activity was dependent on both signal molecules (Figure 4) and could not be rescued to wild type levels when the BDSF mutant was grown in the presence of AHLs (Figures S4 and S5). Only in the case of proteolytic activity a partial complementation was observed in the presence of AHLs (Figure S5) and this effect may be attributed to the lowered level of the signal molecule. Extracellular proteolytic activity of B. cenocepacia is mainly conferred by two metalloproteases, ZmpA and ZmpB [25], [26]. The expression of zmpA in a rpfFBc mutant of B. cenocepacia J2315 was previously shown to be restored when the medium was supplemented with either AHLs or BSDF [27], which may in part explain the slight increase in proteolytic activity when the cepI rpfFBc double mutant was grown in the presence of C8-HSL. Importantly, it has been demonstrated that the ZmpB protease has greater activity against casein [26] and thus our measurements mainly reflect the activity of this protease, the expression of which appears to be dependent on both QS systems (Table 2). In conclusion, our data suggest that the reduced AHL levels of the BDSF-deficient mutant are not, at least not solely, responsible for the observed phenotypic defects.
It is noteworthy that the amount of C8-HSL produced by different B. cenocepacia strains varies dramatically, with concentrations ranging from 1 nM to 0.2 µM [7]. The CF isolate used in this study, strain H111 [7], [28] produces very high levels (0.2 µM) of C8-HSL. Although the BDSF mutant produces less (50%) AHLs relative to the H111 wild type it still produces much higher amounts of the signal molecule than many other B. cenocepacia wild type strains, including the frequently studied strains K56-2 and J2315 [7]. We therefore cannot exclude the possibility that in strains producing very low amounts of C8-HSL inactivation of rpfFBc has a more pronounced effect on expression of AHL-regulated functions. In a recent study evidence was presented that inactivation of rpfFBc in B. cenocepacia J2315 also resulted in a lowered AHL level, albeit this reduction was found to be insignificant when the AHL concentration was normalized against the cell density [27].Given that the amount of AHLs produced by strain J2315 is very low, a further reduction is difficult to quantify with standard methods and this may be the reason for the statistically insignificant results. In addition, the difference between the studies may be due to different growth conditions (Anwar minimal medium versus LB) or the fact that B. cenocepacia J2315 harbors an additional QS system [29].
In conclusion, our data support a model in which the two QS systems operate in parallel to control specific as well as overlapping sets of genes (Figure 5). This model is also in accordance with the finding that the AHL and BDSF stimulons do not completely overlap and some genes are almost exclusively regulated by just one of the QS systems. It has recently been shown that binding of BDSF to its cognate receptor RpfR activates the c-di-GMP phosphodiesterase activity of this protein, which leads to a lowered intracellular c-di-GMP level [16]. At present it is unknown how this change in c-di-GMP level affects transcription of target genes.
Figure 5. Schematic presentation of the two B. cenocepacia H111 QS circuitries.
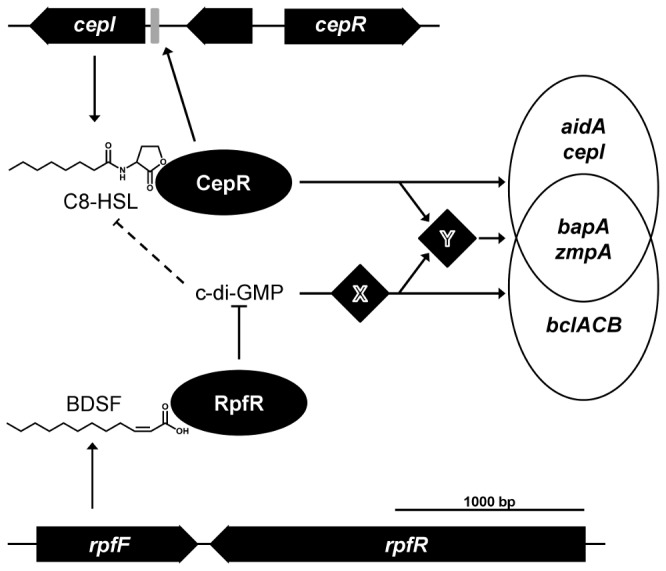
The CepI/CepR system consists of the AHL synthase CepI directing the synthesis of C8-HSL, and of the transcriptional regulator CepR. The RpfF/RpfR system consists of RpfF which directs the synthesis of BDSF, and of its cognate receptor RpfR. Upon binding of BDSF to RpfR the c-di-GMP phosphodiesterase activity of the protein is stimulated and as a consequence the intracellular c-di-GMP level is lowered. The two QS systems operate in parallel to control specific as well as overlapping sets of genes. Our working model assumes an unknown c-di-GMP receptor protein × that stimulates transcription of target genes. Alternatively, the two QS cascades converge and control the expression or the activity status of an unknown common regulator Y, which in turn regulates expression of target genes. C-di-GMP has a negative regulatory effect on AHL levels via an unknown mechanism (depicted by the dashed line).
In our working model we assume an unknown c-di-GMP receptor protein × that activates transcription of target genes either directly or via a regulatory cascade. Given that most genes that were found to be regulated by both QS systems are not directly regulated by CepR/C8-HSL, it is also possible that the two QS cascades converge and control the expression or the activity status of an unknown common regulator Y, which in turn regulates expression of target genes (Figure 5). Work currently under way aims at distinguishing between the two possibilities and at identifying the c-di-GMP effector.
Materials and Methods
Bacterial Strains, Plasmids and Growth Conditions
Strains and plasmids used in this study are listed in Table S4. Unless otherwise stated, strains were grown aerobically at 37°C in LB Lennox broth (Difco). Complementation assays were performed with 10 µM BDSF (Sigma) and/or 200 nM C8-HSL (Sigma). Antibiotics were used at the concentrations (in µg/ml) indicated in parentheses: for E. coli, ampicillin (100), kanamycin (30), and tetracycline (10); and for B. cenocepacia, kanamycin (50), gentamicin (10), trimethoprim (100), and chloramphenicol (60). Growth was monitored spectrophotometrically by measurement of optical density at 600 nm (Ultrospec Pro 2100, GE Healthcare, Switzerland).
Construction of B. cenocepacia H111 Mutants
A deletion mutant of B. cenocepacia H111 cepI was created using a modified version of the Gateway method described by Choi and Schweizer [30] and modified by Carlier et al. [31]. Briefly, the flanking regions of the cepI gene were amplified by PCR using oligonucleotide primers cepIDnkan, cepIDnGW, cepIUpGW and cepIUpkan listed in Table S5. A kanamycin-resistance cassette derived from plasmid pKD4 was inserted between the flanking regions of the gene of interest using a PCR overlap technique with primer GW-attB1 and GW-attB2, listed in Table S5. The resulting PCR product containing the Km-resistance cassette flanked by gene-specific DNA was cloned into the Gateway Entry vector pDONR221 using the BP clonase II kit (Invitrogen). The constructs were then genetically transferred into the suicide vector pAUC40 using the LR clonase kit II (Invitrogen), resulting in plasmid pAUC51. The plasmid was introduced into E. coli strain S17-1 and conjugally transferred into the wild type strain B. cenocepacia H111. Allelic replacement events were selected based on resistance to kanamycin and sensitivity to streptomycin and verified by PCR. Marker-free strain BcepX2 was created by conjugally transferring plasmid pBBR1MCS5::FLP into strain BcepX1. Colonies sensitive to kanamycin were selected and pBBR1MCS-5::FLP was cured after passaging on sucrose-containing medium and selecting colonies sensitive to gentamycin. The loss of the kanR cassette was verified by PCR. To generate an rpfF Bc insertion mutant, an internal fragment of rpfF Bc was amplified by PCR using the primer pair rpfFBc-fw/rpfFBc-rev (Table S5) and inserted as a BamHI/HindIII fragment into the respective sites of plasmid pSHAFT2, generating pSHAFT-rpfFBc. The plasmid was transferred by triparental conjugation to B. cenocepacia H111 wild type to generate a BDSF deficient single mutant rpfF Bc or to B. cenocepacia H111 ΔcepI to generate the AHL and BDSF deficient double mutant ΔcepI rpfF Bc. The integrity of the insertion was verified by PCR using oligonucleotides rpfFBccheck and pSHAFTcheck (Table S5). For constitutive expression of cepI, the gene was amplified with primers cepI_orf_fw and cepI_orf_rev (Table S5), digested with EcoRI and XbaI and cloned into pBBR1MCS-5 cut with the same enzymes.
RNA Isolation and cDNA Synthesis
Total RNA from wild type and mutant strains grown aerobically in complex LB medium until an OD of 2 was extracted using a modified hot acid phenol protocol [32], further purified (RNeasy kit, Qiagen), and its quality checked using RNA Nano Chips (Agilent 2100 Bioanalyzer; RIN>8). The removal of genomic DNA by DNAseI (Promega) treatment was controlled by a PCR (which targets aidA) with 40 cycles. Ten micrograms from each total RNA sample were annealed with random primers (kindly provided by C. Majerczyk and P. Greenberg, Seattle) and cDNA synthesized using MLV reverse transcriptase (Promega). Second strand synthesis was carried out according to Yoder-Himes and colleagues [17] using nick translation. Double strand cDNA was purified with MinElute columns (Qiagen).
Illumina Sequencing and Data Analysis
Libraries were prepared for sequencing according to the manufacturer’s instructions (Illumina). Single-end 51 nucleotide sequence reads were obtained using the Illumina HiSeq2000 system at GATC (Konstanz, Germany), processed with Casava version 1.8 and provided to us in fastq format. Sequencing reads were mapped to the Bcc H111 genome using CLC Genomics Workbench v4.9 (CLCbio) allowing up to 2 mismatches per read. RNA-Seq count data were subsequently analyzed with the DESeq software [18], which ranks the differentially expressed genes according to statistical significance. We chose to rely on the top 150 differentially expressed genes (DESeq p-value <0.1) for our further analyses. The RNA-Seq raw data files are accessible through the GEO Series accession number GSE41244.
Verification of RNA-Seq Data by Real Time PCR
The expression of H111 orthologs of J2315 genes BCAL2353, BCAM0186, BCAM0580, BCAM0581, BCAM0854, BCAM1868, BCAM1870 and BCAS0293 was analyzed by qPCR using Brilliant III Ultra-Fast SYBR® Green QPCR Master Mix (Agilent, Switzerland) and a Mx3000P instrument (Agilent, Switzerland). As a template, cDNA prepared from biological replicates was used. Each PCR reaction contained 12.5 µl 2× Brilliant III Ultra-Fast SYBR® Green QPCR Master Mix, 0.7 µM of individual primers and 3 dilutions of cDNA in a total volume of 25 µl. Reactions were run in triplicates. Melting curves were generated to verify the specificity of the respective amplification. Gene expression fold-changes were calculated as described elsewhere [33]. Expression of the primary sigma factor rpoD (BCAM0918) which, based on the RNA-Seq data, was found to be unchanged under different conditions was used as a reference for normalization. The primers used are listed in Table S5.
Preparation of Protein Samples for Shotgun Proteomics
Cellular proteins and extracellular proteins were prepared as described previously [34], [35]. Cells were lysed by two consecutive passes through a French Press homogenizer (Hypramag/Aminco). Unbroken cells and cell debris were removed by 15 min centrifugation at 4000 g. Total cell membranes were then harvested by ultracentrifugation for 1 h at 80000 g, 4°C. The pellet containing total membrane proteins was finally dissolved in 100 mM Tris-HCl, pH 7.5, 2% SDS by incubation at 50°C for 1 h. The cell lysate supernatant containing soluble intracellular proteins was extracted with 6 volumes of ice-cold acetone at −20°C overnight. The precipitated proteins were harvested by centrifugation at 20000 g and dissolved in 100 mM Tris-HCl, pH 7.5, 0.1% SDS. Total protein concentration was determined according to Bradford using the Coomassie Plus™ Protein Assay (Pierce) with BSA as a standard. Approximately 15 µg total protein for each extracellular, intracellular and membrane fractions were separated by 1D SDS-PAGE on 12.5% polyacrylamide gels. Gels were stained with colloidal Coomassie Blue (Serva). Individual protein lanes were cut into ten slices and immediately subjected to in-gel tryptic digestion.
Protein Identification and Further Analysis
Peptides were separated by RP-HPLC and analyzed by a hybrid LTQ-Orbitrap XL mass spectrometer (Thermo Fisher Scientific, Waltham, MA, USA) interfaced with a nanoelectrospray source. Mass spectrometric detection was performed in data-dependent mode. Precursor mass spectra were acquired at the Orbitrap mass analyzer with a scan range from m/z 300.0 to 1,600.0 using real-time internal calibration on polydimethylcyclosiloxane (PCM) background ions. Resolution was set to 60,000 at m/z 400. Mass spectra processing was performed with Xcalibur 2.0.7 (Thermo Fisher Scientific). Peak list generation for database searches was done with Mascot Distiller 2.1.1.0 (Matrix Science, London, UK). The protein search database was built by combining 7,258 B. cenocepacia H111 proteins (downloaded from NCBI link: http://www.ncbi.nlm.nih.gov/nuccore/CAFQ00000000.1) with 260 common contaminants (e.g. human keratin, trypsin). All experimental fragment ion spectra were searched with Mascot 2.3 (Matrix Science, London, UK) against a target and randomized decoy database. The following search parameters were applied: fixed modification, cysteine carbamidomethylation; variable modification, methionine oxidation; enzyme, trypsin; peptide tolerance, ±10 ppm; MS/MS tolerance, ±0.5 Da; maximum number of missed cleavages, 2. PSMs were post-processed with Percolator [36] and stringently filtered such that the final FDR at the PSM level was below 0,1%. All PSMs identified at this stringent FDR were subjected to a PeptideClassifier analysis [37]. Only peptides that unambiguously identify one protein (either class 1a or 3a) were used to generate a minimal list of protein identifications, while shared peptides of class 3b that cannot distinguish between proteins encoded by different gene models were not considered. Furthermore, we did not consider proteins identified by a single spectrum (e.g. those supported by transcriptomics evidence [38]), but required at least 2 independent spectra in order to include true low abundant expressed proteins, while at the same time keeping the FDR rates low without going into manual validation of spectra [39]. Total spectral counts were used for the proteins in each experiment as a basis for DESeq analysis. Due to the lower number of counts compared to sequenced reads, we chose a more lenient cut-off of p<0.2 to select the 116 top-ranked differentially expressed proteins for further analysis. The data associated with this manuscript can be downloaded from the ProteomeCommons.org Tranche network using the following hash: t+/Mpgxtm8V3LTW8MYd5cgth39TXezCbwVchze0oCrAzIF07N18bZSOdSCWF8MhYDqAa27VYeUR+Un7R+kM41GyBUV4AAAAAAABluw = = .
Ortholog Mapping and Functional Classification
Throughout the manuscript, we rely on the gene nomenclature of strain J2315 orthologs of H111 genes. Amino acid sequences, including the predicted protein sequences from H111, were clustered using the OrthoMCL v1.4 program [40], with the following parameters: -e 1.0e-6 -v 1000–b 1000, an identity cut-off of 50% and a match cut-off of 50%. We also transferred the functional annotations made by the EggNOG software for the well-studied strain J2315 to H111. The EggNOG resource contains non-supervised orthologous groups that were constructed from 1133 organisms (including J2315 but not H111) [19]. It contains extensive functional annotations, whose depth and coverage differ depending on the evolutionary level that is selected, a feature that is based on the underlying principle of the last common ancestor. Therefore, a greater percentage of annotation is given for more focused groups, e.g. the level proteobacteria (proNOG) has more annotations than the level bacteria. When choosing proteobacteria, we could assign functional predictions to 5270 of 7258 H111 protein coding genes (72.6%).
Construction and Assessment of Transcriptional lacZ Fusions
A modified derivative of pSU11 [41] was constructed by amplifying the dhfr cassette from pRN3 with primer pair dhfr-fw/dhfr-rev listed in Table S5 and subsequent cloning of the sphI fragment into the respective site of pSU11, giving rise to pSU11Tp. The pPbapA-lacZ promoter-probe vector was then cloned using pSU11Tp as described in [11]. Plasmid pPcepI-lacZ was constructed as pPbapA-lacZ with primer pair PcepI-fw and PcepI-rev. For determination of the ß-galactosidase activity, the strains were grown overnight in LB broth, then subcultured in LB medium complemented with AHL and/or BDSF as indicated and grown to late exponential growth phase. ß-galactosidase quantification was performed as described by Stachel et al. [42] with some modifications. Briefly, 50–200 µl of cells were harvested and resuspended in 1 ml Z-buffer. After addition of 25 µl of CHCl3 and 25 µl of 0.05% SDS, the cells were vortexed for 10 seconds and then incubated at 30°C for 15 min. The reaction was started by adding 200 µl of ONPG (4 mg/ml) and incubated at 30°C. The reaction was stopped by the addition of 500 µl of 1 M Na2CO3. Cell debris was removed by centrifugation and absorbance at 442 nm was recorded. β-galactosidase activity was graphed as Miller Units, using the formula Miller Units = (1000*OD420)/(time[min]*V[ml]*OD600). Data are based on three independent biological replicates (n = 3).
Phenotypic Assays
AHL levels were quantified with P. putida F117 pAS-C8 as biosensor. 100 µl of supernatant of an overnight culture was mixed in a black 96-well plate with 100 µl of the biosensor strain grown to exponential growth phase. After 16 hours of incubation at 30°C, fluorescence was recorded using a microtiter plate reader (Synergy HT; Bio-Tek, Germany) with excitation at 485 nm and detection of emission at 528 nm. Background fluorescence (sensor strain with LB medium) was subtracted to give relative fluorescence units (RFU). AHL levels (calculated as RFU/OD600) were plotted as percentage of wild type. Data are based on at least 3 independent experiments with 6 technical replicates each.
For quantification of BDSF, one-liter cultures of Bcc strains were grown in YEB medium [43] to an OD600 of about 3.5 and centrifuged. The supernatants were acidified to a pH of 4.0 with diluted HCl and extracted with ethyl acetate (1.0, vol/vol) twice. Following evaporation of the ethyl acetate, the residue was dissolved in methanol, subjected to flash chromatography for high-performance liquid chromatography (HPLC) profiling analysis on a reverse-phase column (Phenomenex Luna, 5 M C18, 250 by 4.60 mm) and eluted with 80% methanol in H2O at a flow rate of 1 ml min−1. Peaks were monitored with a UV detector (210 and 254 nm). Synthetic BDSF was used as control.
Biofilm formation was quantified in a microtiter dish assay as described by Huber et al. [44]. Briefly, overnight cultures were diluted to an OD600 of 0.01 in AB minimal medium [45] supplemented with 10 mM citrate and 100 µl of this suspension were added per well to a 96-well-plate. After 48 h of static incubation at 30°C, the planktonic cells were removed and 100 µl of a 1% (w/v) aqueous solution of crystal violet were added. Following 20 min incubation at room temperature, the wells were washed thoroughly with distilled water. For quantification, the dye was solubilized by the addition of 120 µl DMSO and absorbance was determined at 570 nm. Biofilm levels were plotted as percentage of the wild type. Data are based on at least 3 independent experiments with 8 technical replicates each.
Proteolytic activity was quantified based on the method described by Safarik et al. [46], with some modifications: Bacteria were grown in NYG medium (0.5% peptone, 0.3% yeast extract, 2% glycerol) at 37°C to late exponential growth phase and OD600 was recorded. Cells were centrifuged in a microcentrifuge and 100 µl of cell free supernatant was incubated with an equal volume of azocasein (5 mg/ml, in 50 mM Tris-Cl pH 8) for 60 min at 37°C. The proteins were then precipitated by adding 400 µl of 10% TCA in ddH2O and removed by centrifugation. The supernatant was incubated with 750 µl of 525 mM NaOH and the absorbance at 442 nm was recorded. Protease activity was calculated as OD442/OD600 and expressed as percentage of the wild type activity. Data are based on at least 3 independent experiments.
Western Blot
Bacterial strains were incubated on NB plates (3 g/L Bacto Peptone, 5 g/L meat extract, 1.5% agar), supplemented with 10 µM BDSF or 200 nM C8-HSL or both, for 24 h at 37°C. The cells were scraped of, resuspended in 0.9% NaCl and the OD600 was adjusted to 4.0. From this cell suspension, 13 µl were loaded on a 12% SDS-PAGE gel and transferred to a polyvinylidene difluoride (PVDF) membrane (Amersham HybondTM-P, GE Healthcare, Munich, Germany). Membranes were incubated with antibodies against BclB [11] or AidA [22]. Detection was performed as described in Inhülsen et al. [11].
Supporting Information
Overlap between the RpfFBc stimulon and CepR regulon. Venn diagram of the RpfFBc stimulon (light grey circle) and CepR regulon (dark grey circle) as determined by RNA Seq and microarray analysis, respectively. The number of genes with decreased expression in the rpfFBc mutant is shown in brackets.
(TIF)
Transcription of cepI is reduced in an rpfFBc mutant background. The activity of a cepI-lacZ transcriptional fusion was determined in the wild type, the rpfFBc and the cepI mutant strain. Exogenous addition of 200 nm C8-HSL (AHL) restored cepI promoter activity in both mutant backgrounds, whereas the addition of 10 µM BDSF only rescued activity of the transcriptional fusion in the rpfFBc mutant background. Error bars indicate SEM, n = 3.
(TIF)
BDSF levels are not influenced by the CepI/R system. BDSF was extracted with ethyl acetate from culture supernatant and quantified by high-performance liquid chromatography (HPLC) as described in the Material and Methods section.
(TIF)
Neither exogenous addition of AHLs nor in trans expression of cepI rescues the biofilm formation defect of an rpfFBc mutant. Biofilm formation of the cepI and the rpfFBc mutant in the presence or absence of 200 nM C8-HSL (AHL) or with cepI constitutively expressed from plasmid pBBRcepI (empty plasmid control pBBR1MCS5) using the microtiter plate assay. Error bars indicate SEM, n≥3.
(TIF)
Biofilm formation and protease activity cannot be rescued to wild type levels when the BDSF mutant is grown in the presence of AHLs. (A) Biofilm formation and (B) protease activity in the rpfFBc mutant. The growth medium was supplemented with 200 nM C8-HSL (AHL), with 10 µM BDSF or both signalling molecules as indicated by+and - below each bar. Error bars indicate SEM, n≥3.
(TIF)
Classification of 150 B. cenocepacia H111 genes that showed differential expression in a rpfFBc mutant strain compared to the wild-type (DE-Seq analysis, p-value <0.1).
(XLSX)
Classification of 116 B. cenocepacia H111 proteins that showed differential expression in a rpfF mutant strain compared to the wild-type (DE-Seq analysis, p-value <0.2).
(XLSX)
Comparison of AHL and BDSF dependent transcriptional regulation of genes with an experimentally verified cep box.
(DOCX)
Bacterial strains and plasmids used in this study.
(DOCX)
Oligonucleotides used in this study.
(DOCX)
Acknowledgments
We thank Peter Gehrig, Claudia Fortes, Simon Barkow and Jonas Grossman for their expert help with mass spectrometry and data processing at the Functional Genomics Center Zurich. We are grateful to Amy Schäfer and Peter Greenberg for sharing the specific primers used for cDNA synthesis. Furthermore we thank Susanne Uehlinger for construction of pBBRcepI and pPcepI-lacZ and Stefanie Heller for technical assistance.
Funding Statement
This work was financially supported by the Swiss National Science Foundation (Project 31003A_122013) to LE, the Swiss SystemsX.ch initiative (grant IPP 2011/121) to CHA and LE and the Biomedical Research Council, the Agency of Science, Technology and Research (A*Star) to LHZ. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Fuqua WC, Winans SC, Greenberg EP (1994) Quorum sensing in bacteria: the LuxR-LuxI family of cell density-responsive transcriptional regulators. J Bacteriol 176: 269–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Fuqua C, Winans SC, Greenberg EP (1996) Census and consensus in bacterial ecosystems: the LuxR-LuxI family of quorum-sensing transcriptional regulators. Annu Rev Microbiol 50: 727–751 doi:10.1146/annurev.micro.50.1.727 [DOI] [PubMed] [Google Scholar]
- 3. Whitehead N (2001) Quorum-sensing in Gram-negative bacteria. FEMS Microbiology Reviews 25: 365–404 doi:10.1016/S0168-6445(01)00059-6 [DOI] [PubMed] [Google Scholar]
- 4. Vanlaere E, Lipuma JJ, Baldwin A, Henry D, De Brandt E, et al. (2008) Burkholderia latens sp. nov., Burkholderia diffusa sp. nov., Burkholderia arboris sp. nov., Burkholderia seminalis sp. nov. and Burkholderia metallica sp. nov., novel species within the Burkholderia cepacia comple. Int J Syst Evol Microbiol 58: 1580–1590 doi:10.1099/ijs.0.65634-0 [DOI] [PubMed] [Google Scholar]
- 5. Vandamme P, Holmes B, Vancanneyt M, Coenye T, Hoste B, et al. (1997) Occurrence of Multiple Genomovars of Burkholderia cepacia in Cystic Fibrosis Patients and Proposal of Burkholderia multivorans sp. nov. International Journal of Systematic Bacteriology 47: 1188–1200 doi:10.1099/00207713-47-4-1188 [DOI] [PubMed] [Google Scholar]
- 6. Lewenza S, Conway B, Greenberg EP, Sokol PA (1999) Quorum sensing in Burkholderia cepacia: identification of the LuxRI homologs CepRI. J Bacteriol 181: 748–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gotschlich A, Huber B, Geisenberger O, Tögl A, Steidle A, et al. (2001) Synthesis of multiple N-acylhomoserine lactones is wide-spread among the members of the Burkholderia cepacia complex. Syst Appl Microbiol 24: 1–14 doi:10.1078/0723-2020-00013 [DOI] [PubMed] [Google Scholar]
- 8. O’Grady EP, Viteri DF, Malott RJ, Sokol PA (2009) Reciprocal regulation by the CepIR and CciIR quorum sensing systems in Burkholderia cenocepacia . BMC Genomics 10: 441 doi:10.1186/1471-2164-10-441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Eberl L (2006) Quorum sensing in the genus Burkholderia . Int J Med Microbiol 296: 103–110 doi:10.1016/j.ijmm.2006.01.035 [DOI] [PubMed] [Google Scholar]
- 10. Riedel K, Arevalo-Ferro C, Reil G, Görg A, Lottspeich F, et al. (2003) Analysis of the quorum-sensing regulon of the opportunistic pathogen Burkholderia cepacia H111 by proteomics. Electrophoresis 24: 740–750 doi:10.1002/elps.200390089 [DOI] [PubMed] [Google Scholar]
- 11. Inhülsen S, Aguilar C, Schmid N, Suppiger A, Riedel K, et al. (2012) Identification of functions linking quorum sensing with biofilm formation in Burkholderia cenocepacia H111. MicrobiologyOpen 1: 225–242 doi:10.1002/mbo3.24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Uehlinger S, Schwager S, Bernier SP, Riedel K, Nguyen DT, et al. (2009) Identification of specific and universal virulence factors in Burkholderia cenocepacia strains by using multiple infection hosts. Infect Immun 77: 4102–4110 doi:10.1128/IAI.00398-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Boon C, Deng Y, Wang L-H, He Y, Xu J-L, et al. (2008) A novel DSF-like signal from Burkholderia cenocepacia interferes with Candida albicans morphological transition. ISME J 2: 27–36 doi:10.1038/ismej.2007.76 [DOI] [PubMed] [Google Scholar]
- 14. Deng Y, Wu J, Tao F, Zhang L-H (2011) Listening to a new language: DSF-based quorum sensing in Gram-negative bacteria. Chem Rev 111: 160–173 doi:10.1021/cr100354f [DOI] [PubMed] [Google Scholar]
- 15. Bi H, Christensen QH, Feng Y, Wang H, Cronan JE (2012) The Burkholderia cenocepacia BDSF quorum sensing fatty acid is synthesized by a bifunctional crotonase homologue having both dehydratase and thioesterase activities. Mol Microbiol 83: 840–855 doi:10.1111/j.1365-2958.2012.07968.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Deng Y, Schmid N, Wang C, Wang J, Pessi G, et al. (2012) Cis-2-dodecenoic acid receptor RpfR links quorum-sensing signal perception with regulation of virulence through cyclic dimeric guanosine monophosphate turnover. Proc Natl Acad Sci U S A 108: 15479–15484 doi:10.1073/pnas.1205037109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yoder-Himes DR, Chain PSG, Zhu Y, Wurtzel O, Rubin EM, et al. (2009) Mapping the Burkholderia cenocepacia niche response via high-throughput sequencing. Proc Natl Acad Sci U S A 106: 3976–3981 doi:10.1073/pnas.0813403106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Anders S, Huber W (2010) Differential expression analysis for sequence count data. Genome Biol 11: R106 doi:10.1186/gb-2010-11-10-r106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Powell S, Szklarczyk D, Trachana K, Roth A, Kuhn M, et al. (2012) eggNOG v3.0: orthologous groups covering 1133 organisms at 41 different taxonomic ranges. Nucleic Acids Res 40: D284–9 doi:10.1093/nar/gkr1060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. McCarthy Y, Yang L, Twomey KB, Sass A, Tolker-Nielsen T, et al. (2010) A sensor kinase recognizing the cell-cell signal BDSF (cis-2-dodecenoic acid) regulates virulence in Burkholderia cenocepacia . Mol Microbiol 77: 1220–1236 doi:10.1111/j.1365-2958.2010.07285.x [DOI] [PubMed] [Google Scholar]
- 21. Wei Y, Ryan GT, Flores-Mireles AL, Costa ED, Schneider DJ, et al. (2011) Saturation mutagenesis of a CepR binding site as a means to identify new quorum-regulated promoters in Burkholderia cenocepacia . Mol Microbiol 79: 616–632 doi:10.1111/j.1365-2958.2010.07469.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Huber B, Feldmann F, Köthe M, Vandamme P, Wopperer J, et al. (2004) Identification of a novel virulence factor in Burkholderia cenocepacia H111 required for efficient slow killing of Caenorhabditis elegans . Infect Immun 72: 7220–7230 doi:10.1128/IAI.72.12.7220-7230.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Riedel K, Hentzer M, Geisenberger O, Huber B, Steidle A, et al. (2001) N-acylhomoserine-lactone-mediated communication between Pseudomonas aeruginosa and Burkholderia cepacia in mixed biofilms. Microbiology 147: 3249–3262. [DOI] [PubMed] [Google Scholar]
- 24. Deng Y, Wu J, Eberl L, Zhang L-H (2010) Structural and functional characterization of diffusible signal factor family quorum-sensing signals produced by members of the Burkholderia cepacia complex. Appl Environ Microbiol 76: 4675–4683 doi:10.1128/AEM.00480-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Corbett CR, Burtnick MN, Kooi C, Woods DE, Sokol PA (2003) An extracellular zinc metalloprotease gene of Burkholderia cepacia . Microbiology 149: 2263–2271 doi:10.1099/mic.0.26243-0 [DOI] [PubMed] [Google Scholar]
- 26. Kooi C, Subsin B, Chen R, Pohorelic B, Sokol PA (2006) Burkholderia cenocepacia ZmpB is a broad-specificity zinc metalloprotease involved in virulence. Infect Immun 74: 4083–4093 doi:10.1128/IAI.00297-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Deng Y, Boon C, Eberl L, Zhang L-H (2009) Differential modulation of Burkholderia cenocepacia virulence and energy metabolism by the quorum-sensing signal BDSF and its synthase. J Bacteriol 191: 7270–7278 doi:10.1128/JB.00681-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Römling U, Wingender J, Müller H, Tümmler B (1994) A major Pseudomonas aeruginosa clone common to patients and aquatic habitats. Appl Environ Microbiol 60: 1734–1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Holden MTG, Seth-Smith HMB, Crossman LC, Sebaihia M, Bentley SD, et al. (2009) The genome of Burkholderia cenocepacia J2315, an epidemic pathogen of cystic fibrosis patients. J Bacteriol 191: 261–277 doi:10.1128/JB.01230-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Choi K-H, Schweizer HP (2005) An improved method for rapid generation of unmarked Pseudomonas aeruginosa deletion mutants. BMC Microbiol 5: 30 doi:10.1186/1471-2180-5-30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Carlier A, Burbank L, von Bodman SB (2009) Identification and characterization of three novel EsaI/EsaR quorum-sensing controlled stewartan exopolysaccharide biosynthetic genes in Pantoea stewartii ssp. stewartii . Mol Microbiol 74: 903–913 doi:10.1111/j.1365-2958.2009.06906.x [DOI] [PubMed] [Google Scholar]
- 32. Pessi G, Ahrens CH, Rehrauer H, Lindemann A, Hauser F, et al. (2007) Genome-wide transcript analysis of Bradyrhizobium japonicum bacteroids in soybean root nodules. Mol Plant Microbe Interact 20: 1353–1363 doi:10.1094/MPMI-20-11-1353 [DOI] [PubMed] [Google Scholar]
- 33. Pfaffl MW (2001) A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Research 29: 45e–45 doi:10.1093/nar/29.9.e45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Riedel K, Carranza P, Gehrig P, Potthast F, Eberl L (2006) Towards the proteome of Burkholderia cenocepacia H111: setting up a 2-DE reference map. Proteomics 6: 207–216 doi:10.1002/pmic.200500097 [DOI] [PubMed] [Google Scholar]
- 35. Carranza P, Grunau A, Schneider T, Hartmann I, Lehner A, et al. (2010) A gel-free quantitative proteomics approach to investigate temperature adaptation of the food-borne pathogen Cronobacter turicensis 3032. Proteomics 10: 3248–3261 doi:10.1002/pmic.200900460 [DOI] [PubMed] [Google Scholar]
- 36. Brosch M, Yu L, Hubbard T, Choudhary J (2009) Accurate and sensitive peptide identification with Mascot Percolator. J Proteome Res 8: 3176–3181 doi:10.1021/pr800982s [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Qeli E, Ahrens CH (2010) PeptideClassifier for protein inference and targeted quantitative proteomics. Nat Biotechnol 28: 647–650 doi:10.1038/nbt0710-647 [DOI] [PubMed] [Google Scholar]
- 38. Delmotte N, Ahrens CH, Knief C, Qeli E, Koch M, et al. (2010) An integrated proteomics and transcriptomics reference data set provides new insights into the Bradyrhizobium japonicum bacteroid metabolism in soybean root nodules. Proteomics 10: 1391–1400 doi:10.1002/pmic.200900710 [DOI] [PubMed] [Google Scholar]
- 39. Grobei MA, Qeli E, Brunner E, Rehrauer H, Zhang R, et al. (2009) Deterministic protein inference for shotgun proteomics data provides new insights into Arabidopsis pollen development and function. Genome Res 19: 1786–1800 doi:10.1101/gr.089060.108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Li L, Stoeckert CJ, Roos DS (2003) OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res 13: 2178–2189 doi:10.1101/gr.1224503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Malott RJ, O’Grady EP, Toller J, Inhülsen S, Eberl L, et al. (2009) A Burkholderia cenocepacia orphan LuxR homolog is involved in quorum-sensing regulation. J Bacteriol 191: 2447–2460 doi:10.1128/JB.01746-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Stachel SE, An G, Flores C, Nester EW (1985) A Tn3 lacZ transposon for the random generation of beta-galactosidase gene fusions: application to the analysis of gene expression in Agrobacterium . EMBO J 4: 891–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhang H-B, Wang L-H, Zhang L-H (2002) Genetic control of quorum-sensing signal turnover in Agrobacterium tumefaciens . Proc Natl Acad Sci U S A 99: 4638–4643 doi:10.1073/pnas.022056699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Huber B, Riedel K, Hentzer M, Heydorn A, Gotschlich A, et al. (2001) The cep quorum-sensing system of Burkholderia cepacia H111 controls biofilm formation and swarming motility. Microbiology 147: 2517–2528. [DOI] [PubMed] [Google Scholar]
- 45. Clark DJ, Maaløe O (1967) DNA replication and the division cycle in Escherichia coli . Journal of Molecular Biology 23: 99–112 doi:10.1016/S0022-2836(67)80070-6 [Google Scholar]
- 46. Safarík I (1987) Thermally modified azocasein–a new insoluble substrate for the determination of proteolytic activity. Biotechnol Appl Biochem 9: 323–324. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Overlap between the RpfFBc stimulon and CepR regulon. Venn diagram of the RpfFBc stimulon (light grey circle) and CepR regulon (dark grey circle) as determined by RNA Seq and microarray analysis, respectively. The number of genes with decreased expression in the rpfFBc mutant is shown in brackets.
(TIF)
Transcription of cepI is reduced in an rpfFBc mutant background. The activity of a cepI-lacZ transcriptional fusion was determined in the wild type, the rpfFBc and the cepI mutant strain. Exogenous addition of 200 nm C8-HSL (AHL) restored cepI promoter activity in both mutant backgrounds, whereas the addition of 10 µM BDSF only rescued activity of the transcriptional fusion in the rpfFBc mutant background. Error bars indicate SEM, n = 3.
(TIF)
BDSF levels are not influenced by the CepI/R system. BDSF was extracted with ethyl acetate from culture supernatant and quantified by high-performance liquid chromatography (HPLC) as described in the Material and Methods section.
(TIF)
Neither exogenous addition of AHLs nor in trans expression of cepI rescues the biofilm formation defect of an rpfFBc mutant. Biofilm formation of the cepI and the rpfFBc mutant in the presence or absence of 200 nM C8-HSL (AHL) or with cepI constitutively expressed from plasmid pBBRcepI (empty plasmid control pBBR1MCS5) using the microtiter plate assay. Error bars indicate SEM, n≥3.
(TIF)
Biofilm formation and protease activity cannot be rescued to wild type levels when the BDSF mutant is grown in the presence of AHLs. (A) Biofilm formation and (B) protease activity in the rpfFBc mutant. The growth medium was supplemented with 200 nM C8-HSL (AHL), with 10 µM BDSF or both signalling molecules as indicated by+and - below each bar. Error bars indicate SEM, n≥3.
(TIF)
Classification of 150 B. cenocepacia H111 genes that showed differential expression in a rpfFBc mutant strain compared to the wild-type (DE-Seq analysis, p-value <0.1).
(XLSX)
Classification of 116 B. cenocepacia H111 proteins that showed differential expression in a rpfF mutant strain compared to the wild-type (DE-Seq analysis, p-value <0.2).
(XLSX)
Comparison of AHL and BDSF dependent transcriptional regulation of genes with an experimentally verified cep box.
(DOCX)
Bacterial strains and plasmids used in this study.
(DOCX)
Oligonucleotides used in this study.
(DOCX)