

Abstract
Clathrin-mediated endocytosis dynamically regulates cell membrane abundance of CFTR and plays an essential role in CFTR-dependent Cl- conductance in fluid-transporting epithelia. It requires two closely related, but distinct processes: assembly of the clathrin coat and recruitment of cargo proteins for endocytosis. The assembly polypeptide-2 complex (AP-2) is the prototypical endocytic adaptor responsible for optimal clathrin coat formation. Disabled-2 (Dab2) is a clathrin associated sorting protein (CLASP) that also mediates clathrin assembly and cargo selection. Both of these complexes have clearly been shown to play roles in CFTR endocytosis in cells that endogenously express the channel. However, their precise functions exhibit cell-specific differences. While Dab2 appears to play a central role in CFTR recruitment to the clathrin coat in airway epithelial cells, it does not play a direct role in CFTR endocytosis in intestinal epithelial cells. Here, we review our current understanding of the role of Dab2 in CFTR endocytosis in different tissues. Next, we present new data demonstrating the role of Dab2 in endocytosis of the most commonly mutated CFTR gene product, ∆F508-CFTR, in human airwy epithelial cells. Finally we discuss the potential therapeutic implications of targeting the functional interaction between ∆F508-CFTR and Dab2.
Keywords: CFTR, Cystic fibrosis, Dab2, Endocytosis, clathrin coated vesicles, human airway epithelial cells, ∆F508 mutation
Tissue Specificity of CFTR Trafficking
The cystic fibrosis transmembrane conductance regulator (CFTR) belongs to the family of ATP binding cassette (ABC) transporters, but forms a cAMP-activated Cl- channel that mediates transepithelial Cl- secretion in various fluid-transporting epithelia.1-3 CFTR is mutated in patients with cystic fibrosis (CF). In the airway, CFTR plays an essential role in maintaining mucociliary clearance by regulating the airway surface liquid.4,5 A central aspect of this regulation involves control of Cl- secretion across polarized epithelial cells, which is modulated at the level of both CFTR channel activity and cell membrane abundance (reviewed in6,7). The plasma membrane abundance of CFTR in turn depends on its biosynthetic processing and post-maturational trafficking (reviewed in8). The long plasma membrane stability of CFTR stands in contrast to its inefficient biosynthetic processing and primarily reflects efficient recycling, which is required to compensate for rapid endocytosis in clathrin-coated vesicles (CCVs).9-11
Complex protein networks control the endocytic uptake of CFTR.7 While several proteins involved in the uptake process have been identified,12 their roles are incompletely defined, and there is evidence for tissue- and cell-specific differences. Nevertheless, it is clear that clathrin-mediated endocytosis requires two closely related, but distinct processes: (a) the assembly of the clathrin coat to form clathrin coated pits (CCPs) and CCVs; and (b) recruitment of cargo proteins for incorporation into CCPs and endocytosis in CCVs. Several endocytic adaptors are involved in both processes.
This is true in particular for central players in the process, such as the assembly polypeptide-2 (AP-2). AP-2 is a heterotetrameric complex of the α, β2, and σ2 adaptins, which are responsible for optimal clathrin coat formation, and the µ2 adaptin, which binds directly to YxxΦ motifs in transmembrane cargo proteins (reviewed in13). It is also true for the Disabled-2 (Dab2) protein, a clathrin-associated sorting protein (CLASP) with a complex, multidomain structure. The DAB homology (DH) domain binds to transmembrane cargo proteins containing the NPxY motif while the NPF sequence repeats assist in clathrin assembly. Dab2 can also promote endocytosis by binding directly via its DPF domain to the α adaptin and working in concert with AP-2 (Fig. 1). While individual functional domains are covalently linked within the Dab2 protein, there is a corresponding interdependence of function for the AP-2 adaptins: loss of any one disrupts the function of the entire complex.14-16
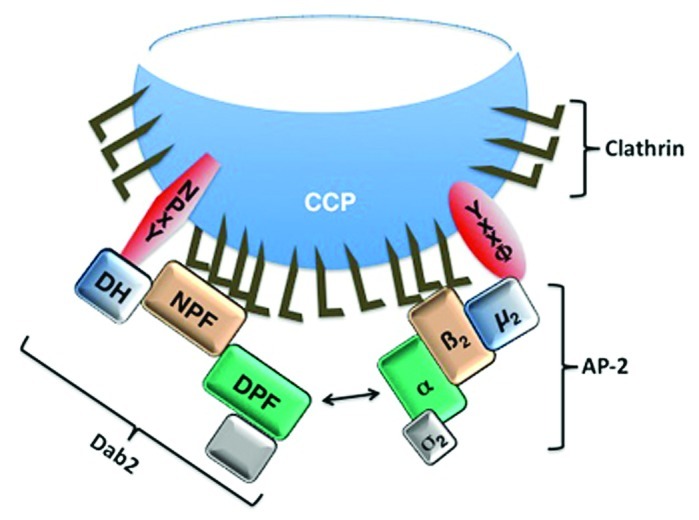
Figure 1.Schematic illustration of the Dab2 and AP-2 interactions during clathrin-mediated endocytosis. Dab2 and AP-2 mediate assembly of the clathrin coat and recruitment of transmembrane cargo proteins for incorporation into clathrin coated pits (CCPs). The Dab2 DH domain interacts with the NPxY motif-containing cargo proteins and the µ2 adaptin of AP-2 interacts with the YxxΦ motif-containing cargo proteins. Our recent data demonstrate that the DH domain may recruit cargo, such as CFTR that does not have a canonical NPxY motif (20). The Dab2 NPF repeats and the β2 adaptin of AP-2 bind clathrin. Moreover, Dab2 and AP-2 interact with each other by binding of the Dab2 DPF motif to the α adaptin of AP-2.
Depletion of either Dab2 or individual AP-2 subunits thus has the potential to disrupt both the CCV formation and cargo recruitment. However, in some cases, it is possible to distinguish these processes, since robust recruitment can sustain endocytosis even after a substantial fall of the CCV formation. For example, depletion of AP-2 by more than 90% results in a 10-fold reduction of CCV number.16 This profoundly inhibits the recruitment and endocytic uptake of cargo proteins that rely entirely on the YxxΦ motif, such as the transferrin receptor (TfR). In contrast, it has relatively modest effects on the uptake of the low-density lipoprotein receptor (LDLR), which undergoes privileged cargo recruitment independent of AP-2.16
Both of Dab2 and AP-2 have clearly been shown to play roles in CFTR endocytosis in cells that endogenously express the channel.17-20 In human airway epithelial cells, the knockdown of µ2 adaptin by more than 90% resulted in only a 2-fold reduction in CFTR endocytosis,19 compared with the 10-fold reduction that would be expected for purely YxxΦ-mediated uptake.16 This suggested that CFTR experiences AP-2 independent, privileged recruitment to a diminished population of CCVs. Consistent with this interpretation, a more limited (~60%) knockdown of µ2 adaptin caused no difference in either CFTR endocytosis or CFTR content in CCVs.20 By contrast, a similar depletion of Dab2 led to markedly decreased CFTR endocytosis and CFTR content in CCVs (20). Thus, in airway epithelial cells, Dab2 appears to play a central role in CFTR recruitment to the clathrin coat.20 However, in intestinal epithelial cells, Dab2 does not play a direct role in CFTR endocytosis. Instead it may promote CFTR endocytosis by working in concert with AP-2.17
In non-epithelial cells, even larger functional differences are seen. In HEK293 cells, AP-2 is strictly required for efficient CFTR endocytosis, and the µ2 adaptin interacts directly with the CFTR YDSI motif in vitro.10,21,22 Thus, while a ~60% knockdown of µ2 adaptin caused no change in airway epithelial cells, a comparable 64% knockdown of α adaptin in HEK293 cells caused a 2-fold reduction in the endocytic uptake of CFTR.17 To achieve an equivalent 2-fold CFTR reduction in airway cells, a 90% knockdown of µ2 adaptin in epithelial cells was required.19 Thus, in HEK293 cells, AP-2 appears to play a more critical role in CFTR endocytosis. Together, these results underscore the importance of studying protein trafficking processes in cells most representative of the appropriate physiological context.
Therapeutic Implications
Having identified the importance of Dab2-mediated recruitment in CFTR endocytosis in airway epithelial cells, we were interested in the therapeutic potential of Dab2 inhibition. Both biosynthetic processing and post-maturational trafficking are critically affected by the most common disease-associated mutation in the CFTR gene. In 70% of patient alleles, loss of Phe508 (∆F508) leads to a temperature-sensitive processing defect in the CFTR protein (reviewed in ref. 8). Low temperature and chemical chaperones increase biosynthetic processing and rescue the cell membrane abundance of ∆F508-CFTR, and disease-modifying therapies based on correction of the biosynthetic processing of ∆F508-CFTR have been highly anticipated (23,24). Rescued ∆F508-CFTR (r∆F508-CFTR) is partially functional as a Cl- channel but is unstable in the plasma membrane due to altered endocytic trafficking.11,25 Hence, addressing the post-maturational trafficking defect of r∆F508-CFTR represents a therapeutic approach complementary to biosynthetic rescue.
Specifically, proteins involved in either endocytic uptake or lysosomal degradation should be the targets for CFTR stabilization at the apical plasma membrane. Indeed, the plasma membrane half-life, cell-membrane abundance, and net chloride-channel activity of ∆F508-CFTR can be increased by depletion or inhibition of the CFTR-associated ligand CAL, which facilitates CFTR post-endocytic degradation.26-29 Similarly, depletion of proteins involved in CFTR endocytic uptake increases its apical membrane abundance.18-20 However, these studies have focused on WT-CFTR, and our earlier work clearly showed that WT- and r∆F508-CFTR experience differential rates of endocytic uptake in airway epithelial cells.11 We therefore directly tested the impact of Dab2 depletion on r∆F508-CFTR. Given the sensitive dependence of these processes on cell type, we studied paired CFBE41o- cell lines stably expressing either WT- or r∆F508-CFTR. Following siRNA treatment, similar levels of Dab2 depletion (~70%) were observed compared with cells treated with control siRNA (Fig. 2A&B). In both cells lines, this Dab2 knockdown was associated with similar (~50%) increases in the steady-state plasma membrane abundance of CFTR compared with control cells (Fig. 2A&C). Thus, despite differences between the endocytic uptake rates of WT- and ∆F508-CFTR, the uptake of both proteins is controlled to a similar extent by Dab2.
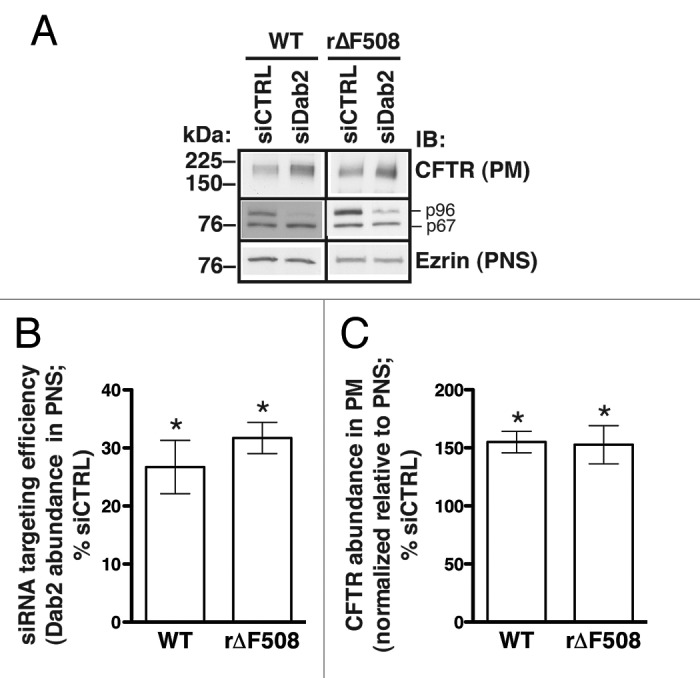
Figure 2. Experiments performed to determine the effects of Dab2 on the cell membrane abundance of WT-CFTR and r∆F508-CFTR in human airway epithelial cells (CFBE41o-). Cells were transfected with siRNA specific for the human Dab2 gene (siDab2) or with a non-silencing siRNA control (siCTRL). The Dab2 gene is alternatively spliced to produce two proteins, p96 and p67 (30). Because the Dab2 p96 functions as an endocytic adaptor, it was specifically targeted by the siRNA. CFBE41o- cells expressing ∆F508-CFTR were cultured at 27°C for 36 h before experiments to increase biosynthetic processing and thus enhance the cell membrane abundance of rescued ∆F508-CFTR (r∆F508-CFTR). Representative western blots (A, middle row) and summary of experiments (B) demonstrate that siDab2 decreased the Dab2 p96 protein abundance in postnuclear supernatants (PNS) by ~70%. Silencing Dab2 increased the steady-state plasma membrane (PM) abundance of WT-CFTR and r∆F508-CFTR (A, top row; C). PM CFTR was isolated by cell membrane biotinylation. siDab2 did not alter ezrin expression in PNS. *, p < 0.05 vs. siCTRL. Summary data reflect 4 experiments (WT-CFTR) or 6 experiments (r∆F508-CFTR). Error bars, SE.
Based on these results, targeting of Dab2 may offer an approach to investigate and control the limited plasma membrane stability of r∆F508-CFTR. CFTR does not contain a recognized Dab2 binding motif and the stereochemical basis of the functional interaction between Dab2 and CFTR remains unclear. Nevertheless, our data have previously shown that a peptide that binds to the Dab2 DH peptide-binding pocket can regulate CFTR abundance in the plasma membrane.20 Such compounds do not target the AP-2 mediated interactions and thus should not decrease the number of CCVs nor globally inhibit protein endocytosis. As a result, targeting of this interaction may provide future pharmacological approaches to correct the post-maturational trafficking defects of ∆F508-CFTR.
Methods
CFBE41o- cells stably expressing WT-CFTR or ∆F508-CFTR proteins were plated on collagen-coated tissue culture plates and incubated with the optimized transfection mixture containing 10 nM siRNA against human Dab2 gene (siDab2; Hs_Dab2_6 siRNA) or with a non-silencing siRNA control (siCTRL; Qiagen, Valencia, CA), using Lipofectamine 2000 (Invitrogen, Grand Island, NY), as previously described (20). The transfection medium was changed after 24 h and cells were cultured for 72 h to establish a monolayer. CFBE41o- cells expressing ∆F508-CFTR were cultured at 27°C for 36 h before experiments to increase the cell-membrane abundance of r∆F508-CFTR (11). Silencing the Dab2 gene under these conditions resulted in the corresponding protein depletion by ~70%. This level of silencing allowed sufficient clathrin coat formation to better differentiate the role of Dab2 in clathrin-coat formation vs. cargo recruitment and to prevent off-target effects that may occur with more complete gene silencing.20 The biochemical quantification of plasma membrane CFTR was performed by surface biotinylation, streptavidin bead capture, western blotting, and chemiluminescence, as previously described.20 The following mouse monoclonal antibodies were used: anti-human CFTR C-term (Millipore, Billerica, MA), anti-Dab2 p96 and anti-ezrin (BD Biosciences, Sparks, MD).
Statistical analysis of the data was performed using GraphPad Prism version 4.0 for Macintosh (GraphPad Software Inc., San Diego, CA). The means were compared by a two-tailed t-test. A p value < 0.05 was considered significant. Data are expressed as mean ± SE.
Acknowledgments
We would like to thank Karyn Hansen from the Geisel School of Medicine at Dartmouth for her technical assistance with the experiments. This study was supported by National Institutes of Health Grants R01HL090767 and R01HL090767–02S1 (to A.S.-U.) and R01-DK075309 (to D.R.M.). This work was also supported by the Pennsylvania Department of Health, Health Research Formula Fund to the Children's Hospital of Pittsburgh of the UPMC Health System (to A.S.-U.).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/cib/article/21375
References
- 1.Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 1989;245:1066–73. doi: 10.1126/science.2475911. [DOI] [PubMed] [Google Scholar]
- 2.Rommens JM, Iannuzzi MC, Kerem B, Drumm ML, Melmer G, Dean M, et al. Identification of the cystic fibrosis gene: chromosome walking and jumping. Science. 1989;245:1059–65. doi: 10.1126/science.2772657. [DOI] [PubMed] [Google Scholar]
- 3.Howard M, Jiang X, Stolz DB, Hill WG, Johnson JA, Watkins SC, et al. Forskolin-induced apical membrane insertion of virally expressed, epitope-tagged CFTR in polarized MDCK cells. Am J Physiol Cell Physiol. 2000;279:C375–82. doi: 10.1152/ajpcell.2000.279.2.C375. [DOI] [PubMed] [Google Scholar]
- 4.Boucher RC. New concepts of the pathogenesis of cystic fibrosis lung disease. Eur Respir J. 2004;23:146–58. doi: 10.1183/09031936.03.00057003. [DOI] [PubMed] [Google Scholar]
- 5.Tarran R, Button B, Boucher RC. Regulation of normal and cystic fibrosis airway surface liquid volume by phasic shear stress. Annu Rev Physiol. 2006;68:543–61. doi: 10.1146/annurev.physiol.68.072304.112754. [DOI] [PubMed] [Google Scholar]
- 6.Bertrand CA, Frizzell RA. The role of regulated CFTR trafficking in epithelial secretion. Am J Physiol Cell Physiol. 2003;285:C1–18. doi: 10.1152/ajpcell.00554.2002. [DOI] [PubMed] [Google Scholar]
- 7.Guggino WB, Stanton BA. New insights into cystic fibrosis: molecular switches that regulate CFTR. Nat Rev Mol Cell Biol. 2006;7:426–36. doi: 10.1038/nrm1949. [DOI] [PubMed] [Google Scholar]
- 8.Riordan JR. CFTR function and prospects for therapy. Annu Rev Biochem. 2008;77:701–26. doi: 10.1146/annurev.biochem.75.103004.142532. [DOI] [PubMed] [Google Scholar]
- 9.Lukacs GL, Segal G, Kartner N, Grinstein S, Zhang F. Constitutive internalization of cystic fibrosis transmembrane conductance regulator occurs via clathrin-dependent endocytosis and is regulated by protein phosphorylation. Biochem J. 1997;328:353–61. doi: 10.1042/bj3280353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Prince LS, Peter K, Hatton SR, Zaliauskiene L, Cotlin LF, Clancy JP, et al. Efficient endocytosis of the cystic fibrosis transmembrane conductance regulator requires a tyrosine-based signal. J Biol Chem. 1999;274:3602–9. doi: 10.1074/jbc.274.6.3602. [DOI] [PubMed] [Google Scholar]
- 11.Swiatecka-Urban A, Brown A, Moreau-Marquis S, Renuka J, Coutermarsh B, Barnaby R, et al. The short apical membrane half-life of rescued uF508-CFTR results from accelerated endocytosis uF508-CFTR in polarized human airway epithelial cells. J Biol Chem. 2005;280:36762–72. doi: 10.1074/jbc.M508944200. [DOI] [PubMed] [Google Scholar]
- 12.Okiyoneda T, Lukacs GL. Cell surface dynamics of CFTR: the ins and outs. Biochim Biophys Acta 2007; 1773:476-9. [DOI] [PubMed]
- 13.Traub LM. Sorting it out: AP-2 and alternate clathrin adaptors in endocytic cargo selection. J Cell Biol. 2003;163:203–8. doi: 10.1083/jcb.200309175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Meyer C, Zizioli D, Lausmann S, Eskelinen EL, Hamann J, Saftig P, et al. mu1A-adaptin-deficient mice: lethality, loss of AP-1 binding and rerouting of mannose 6-phosphate receptors. EMBO J. 2000;19:2193–203. doi: 10.1093/emboj/19.10.2193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Peden AA, Rudge RE, Lui WW, Robinson MS. Assembly and function of AP-3 complexes in cells expressing mutant subunits. J Cell Biol. 2002;156:327–36. doi: 10.1083/jcb.200107140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Motley A, Bright NA, Seaman MN, Robinson MS. Clathrin-mediated endocytosis in AP-2-depleted cells. J Cell Biol. 2003;162:909–18. doi: 10.1083/jcb.200305145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Collaco A, Jakab R, Hegan P, Mooseker M, Ameen N. Alpha-AP-2 directs myosin VI-dependent endocytosis of cystic fibrosis transmembrane conductance regulator chloride channels in the intestine. J Biol Chem. 2010;285:17177–87. doi: 10.1074/jbc.M110.127613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ye S, Cihil K, Stolz DB, Pilewski JM, Stanton BA, Swiatecka-Urban A. c-Cbl facilitates endocytosis and lysosomal degradation of cystic fibrosis transmembrane conductance regulator in human airway epithelial cells. J Biol Chem. 2010;285:27008–18. doi: 10.1074/jbc.M110.139881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fu L, Rab A, Tang LP, Rowe SM, Bebok Z, Collawn JF. Dab2 is a key regulator of endocytosis and post-endocytic trafficking of the cystic fibrosis transmembrane conductance regulator. Biochem J. 2012;441:633–43. doi: 10.1042/BJ20111566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cihil KM, Ellinger P, Fellows A, Stolz DB, Madden DR, Swiatecka-Urban A. Disabled-2 protein facilitates assembly polypeptide-2-independent recruitment of cystic fibrosis transmembrane conductance regulator to endocytic vesicles in polarized human airway epithelial cells. J Biol Chem. 2012;287:15087–99. doi: 10.1074/jbc.M112.341875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Weixel KM, Bradbury NA. The carboxyl terminus of the cystic fibrosis transmembrane conductance regulator binds to AP-2 clathrin adaptors. J Biol Chem. 2000;275:3655–60. doi: 10.1074/jbc.275.5.3655. [DOI] [PubMed] [Google Scholar]
- 22.Weixel KM, Bradbury NA. micro2 binding directs the cystic fibrosis transmembrane conductance regulator to the clathrin-mediated endocytic pathway. J Biol Chem. 2002;276:46251–9. doi: 10.1074/jbc.M104545200. [DOI] [PubMed] [Google Scholar]
- 23.Van Goor F, Hadida S, Grootenhuis PD, Burton B, Stack JH, Straley KS, et al. Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc Natl Acad Sci U S A. 2011;108:18843–8. doi: 10.1073/pnas.1105787108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lubamba B, Dhooghe B, Noel S, Leal T. Cystic fibrosis: Insight into CFTR pathophysiology and pharmacotherapy. Clin Biochem. 2012 doi: 10.1016/j.clinbiochem.2012.05.034. [accepted June 12; Epub ahead of print] In press. [DOI] [PubMed] [Google Scholar]
- 25.Sharma M, Pampinella F, Nemes C, Benharouga M, So J, Du K, et al. Misfolding diverts CFTR from recycling to degradation: quality control at early endosomes. J Cell Biol. 2004;164:923–33. doi: 10.1083/jcb.200312018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cheng J, Moyer BD, Milewski M, Loffing J, Ikeda M, Mickle JE, et al. A Golgi-associated PDZ domain protein modulates cystic fibrosis transmembrane regulator plasma membrane expression. J Biol Chem. 2002;277:3520–9. doi: 10.1074/jbc.M110177200. [DOI] [PubMed] [Google Scholar]
- 27.Wolde M, Fellows A, Cheng J, Kivenson A, Coutermarsh B, Talebian L, et al. Targeting CAL as a negative regulator of DeltaF508-CFTR cell-surface expression: an RNA interference and structure-based mutagenetic approach. J Biol Chem. 2007;282:8099–109. doi: 10.1074/jbc.M611049200. [DOI] [PubMed] [Google Scholar]
- 28.Cushing PR, Vouilleme L, Pellegrini M, Boisguerin P, Madden DR. A stabilizing influence: CAL PDZ inhibition extends the half-life of ΔF508-CFTR. Angew Chem Int Ed Engl. 2010;49:9907–11. doi: 10.1002/anie.201005585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Roberts KE, Cushing PR, Boisguerin P, Madden DR, Donald BR. Computational design of a PDZ domain peptide inhibitor that rescues CFTR activity. PLoS Comput Biol. 2012;8:e1002477. doi: 10.1371/journal.pcbi.1002477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xu XX, Yang W, Jackowski S, Rock CO. Cloning of a novel phosphoprotein regulated by colony-stimulating factor 1 shares a domain with the Drosophila disabled gene product. J Biol Chem. 1995;270:14184–91. doi: 10.1074/jbc.270.23.14184. [DOI] [PubMed] [Google Scholar]