

Abstract
The c-Met proto-oncogene is a multifunctional receptor tyrosine kinase that is stimulated by its ligand, hepatocyte growth factor (HGF), to induce cell growth, motility and morphogenesis. Dysregulation of c-Met function, through mutational activation or overexpression, has been observed in many types of cancer and is thought to contribute to tumor growth and metastasis by affecting mitogenesis, invasion, and angiogenesis. We identified human monoclonal antibodies that bind to the extracellular domain of c-Met and inhibit tumor growth by interfering with ligand-dependent c-Met activation. We identified antibodies representing four independent epitope classes that inhibited both ligand binding and ligand-dependent activation of c-Met in A549 cells. In cells, the antibodies antagonized c-Met function by blocking receptor activation and by subsequently inducing downregulation of the receptor, translating to phenotypic effects in soft agar growth and tubular morphogenesis assays. Further characterization of the antibodies in vivo revealed significant inhibition of c-Met activity (≥ 80% lasting for 72–96 h) in excised tumors corresponded to tumor growth inhibition in multiple xenograft tumor models. Several of the antibodies identified inhibited the growth of tumors engineered to overexpress human HGF and human c-Met (S114 NIH 3T3) when grown subcutaneously in athymic mice. Furthermore, lead candidate antibody CE-355621 inhibited the growth of U87MG human glioblastoma and GTL-16 gastric xenografts by up to 98%. The findings support published pre-clinical and clinical data indicating that targeting c-Met with human monoclonal antibodies is a promising therapeutic approach for the treatment of cancer.
Keywords: HGF, c-Met, antibody, xenograft, XenoMouse, antitumor
Introduction
The c-Met proto-oncogene is a multifunctional receptor tyrosine kinase that is stimulated by its high-affinity ligand hepatocyte growth factor (HGF). Both c-Met and HGF are expressed in a wide variety of tissues; however, the c-Met receptor is predominantly expressed in tissues of epithelial origin while HGF expression is normally confined to tissues of mesenchymal origin. 1, 2 Binding of HGF to c-Met activates multiple downstream signaling pathways resulting in cell proliferation, motility, migration, angiogenesis, morphogenic differentiation and cell survival during normal development and tissue repair. 3–6
c-Met mediated signaling is strictly regulated during normal mammalian development and tissue homeostasis. Dysregulation of c-Met function, through mutational activation or amplification and overexpression, has been observed in many types of cancer and is thought to contribute to tumor growth and metastasis by affecting mitogenesis, invasion, and angiogenesis. 3, 6–8 In fact, a correlation has been observed between overexpression of c-Met or HGF and elevated levels of circulating HGF and tumor stage or poor patient prognosis. 3, 6, 9, 10
Numerous pre-clinical studies suggest that c-Met is an attractive target for cancer drug discovery because blocking c-Met activation and inhibiting c-Met activity have been shown to circumvent tumor growth and metastasis. 11–14 Several different agents are being evaluated in cancer patients to assess the effects of c-Met/HGF signaling modulation, such as a human monoclonal antibody (mAb) that selectively binds and neutralizes HGF, antibodies targeting c-Met, including a single-armed humanized monovalent anti-c-Met antibody, and c-Met selective and multi-targeted small-molecule tyrosine kinase inhibitors. 6, 15–24
We describe here the isolation of four lead mAbs that bind the extracellular domain of the c-Met receptor and antagonize HGF binding, thereby preventing receptor activation and inducing receptor downregulation. These effects result in neutralization of c-Met function in cellular phenotypic assays. Further, we describe the in vitro and in vivo pharmacological attributes of lead candidate antibody CE-355621, which was previously reported to inhibit 18F-FDG accumulation in U87MG human glioblastoma mouse xenografts. 25 CE-355621 dramatically inhibited c-Met activity in vivo, corresponding to tumor growth inhibition of S114, U87MG, and GTL-16 tumor xenografts. These findings, along with other reports in the literature, suggest that targeting c-Met with human monoclonal antibody therapy is a promising therapeutic approach for the treatment of c-Met driven cancers.
Results
Binding properties of lead anti-c-Met antibodies
Antibodies specific for c-Met were generated by immunizing XenoMouse™ 26 transgenic mice with a recombinant fusion protein encoding the extracellular domain of human c-Met linked to the Fc domain of human IgG1 (Met ECD-Fc) or with NIH3T3 cells overexpressing human c-Met protein. Hybridomas were generated from immunized mice and screened for production of c-Met-specific antibodies in an ELISA that measured binding of antibody to immobilized Met ECD-Fc and in an FMAT assay that assessed binding of antibody to c-Met expressed on the surface of A549 cells.27 Positive hybridomas expressing human IgG2 antibody were identified and subcloned. Small-scale antibody purification was conducted and the binding of numerous lead antibodies to human c-Met ECD-Fc in vitro was assessed. Saturable binding and similar EC50 values for each of the four top leads were observed (Fig. S1; Table 1).
Table 1. Binding properties and neutralizing activity of human c-Met antibodies.
| Assay | CE-310393 | CE-310397 | CE-310400 | CE-310083 |
|---|---|---|---|---|
| ECD binding EC50* |
185 ± 12 pM10 |
377 ± 25 pM6 |
347 ± 89 pM6 |
281 ± 28 pM4 |
| Epitope class ** (Biacore) |
1 |
1 |
1 |
2 |
| Biacore** KD |
220 pM |
530 pM |
330 pM |
820 pM |
| Biacore** koff (1/s) |
1.5 × 10−4 |
1.5 × 10−4 |
6.1 × 10−5 |
6.8 × 10−5 |
| flow cytometry EC50# |
39 ± 7 pM4 |
26 ± 4 pM3 |
41 pM1 |
169 ± 46 pM3 |
| Ligand Binding IC50 (pM)## |
279 ± 21 pM10 |
652 ± 67 pM7 |
582 ± 113 pM6 |
304 ± 48 pM4 |
| Cell pMet IC50 (pM)^ |
150 ± 13 pM10 |
336 ± 61 pM7 |
666 ± 183 pM6 |
155 ± 39 pM4 |
| Soft Agar Growth IC50 (nM)^^ | 57 nM1 | 68 ± 2 nM2 | 167 nM1 | 47 ± 12 nM3 |
*Binding of lead antibodies to recombinant human c-Met ECD-Fc protein was evaluated by ELISA. **The binding kinetics of the four lead antibodies were determined using surface plasmon resonance (Biacore), and epitope class was determined in cross competition studies using Biacore. #The binding affinity (EC50) to native c-Met expressed on A549 cells was estimated using flow cytometry. ##Antibodies were assessed for their ability to inhibit binding of recombinant HGF to immobilized human c-Met ECD-Fc by ELISA. Antibodies were preincubated for 4 h followed by addition of HGF (100 ng/ml) for 15 min. HGF binding was detected with biotinylated anti-HGF antibodies and streptavidin-HRP. ^Antibodies inhibit HGF-dependent activation of c-Met in A549 cells. A549 cells were preincubated with varying concentrations of c-Met antibodies for 4 h then stimulated with 200 ng/ml HGF for 15 min. The level of c-Met autophosphorylation was measured by capture ELISA using PY20-HRP for detection. ^^Soft agar growth of S114 cells, which are NIH-3T3 cells engineered to overexpress human HGF and human c-Met, was evaluated 7–10 d after plating cells with varying concentrations of c-Met antibodies. Colony number and size of p-iodonitrotetrazolium violet stained colonies were determined. Values for the ECD binding, flow cytometry, ligand binding, cellular pMet, and soft agar growth assays represent means ± SEM of multiple dose titration experiments. IC50 values were calculated using GraphPad Prism software. The number of experimental replicates is noted in superscript.
We then characterized the binding of the lead antibodies to the target in greater detail. The kinetics of binding to recombinant Met ECD-Fc was determined using surface plasmon resonance (Biacore). The subnanomolar affinities for each antibody agreed well with the EC50 values seen in the ECD ELISA (Table 1). Cross competition experiments were performed using Biacore to assess the number of epitope classes represented among all the isolated antibodies. In these experiments, a saturating concentration of antibody was allowed to bind to c-Met ECD-Fc coupled to the chip and increasing concentrations of a second antibody were added. Competition is seen when no additional binding is observed upon presentation of the second antibody. Most antibodies fell into a single class (class 1), whereas CE-310083 was the sole antibody to define a second epitope class. In all, four epitope classes were identified from among the entire set of c-Met antibodies isolated and characterized.
The binding affinity of each of the leads for native c-Met expressed on A549 cells was estimated using flow cytometry. Antibody binding reached equilibrium within 6–8 h at RT, as determined with trace amounts of biotinylated antibody. Antibodies were incubated for 6 h at RT to allow equilibrium to be reached, and half maximal binding (EC50) was determined (Table 1). The binding affinities of the three leads from epitope class 1 were below 100 pM, ranging between 26 and 41 pM, whereas the affinity of CE-310083 was slightly lower (169 pM). The binding affinity of antibodies from epitope class 1 to native c-Met appeared to be somewhat higher (~5–15 fold) than those observed for binding to recombinant c-Met ECD protein used in the ECD binding ELISA and in SPR assays (Table 1), suggesting presentation of this binding epitope on native c-Met expressed on the cell surface and recombinant c-Met ECD protein may differ slightly.
Antibodies targeting c-Met block HGF binding and prevent ligand-dependent receptor activation
We focused on characterizing the neutralizing activity of lead antibodies on molecular events involved in c-Met activation, i.e., ligand binding and receptor autophosphorylation. First, lead antibodies were evaluated in a ligand binding assay in which the binding of recombinant HGF to immobilized c-Met ECD-Fc was inhibited by the addition of purified antibodies. Each of the leads was able to completely block ligand binding and showed subnanomolar potencies (Fig. 1A; Table 1) that correlated well with the binding activity observed in the ECD binding ELISA and affinity measurements using Biacore (Table 1). The correspondence of the binding measurements with the potency in the ligand binding assay is likely related to the use of recombinant c-Met ECD as the binding target in each of these assays.
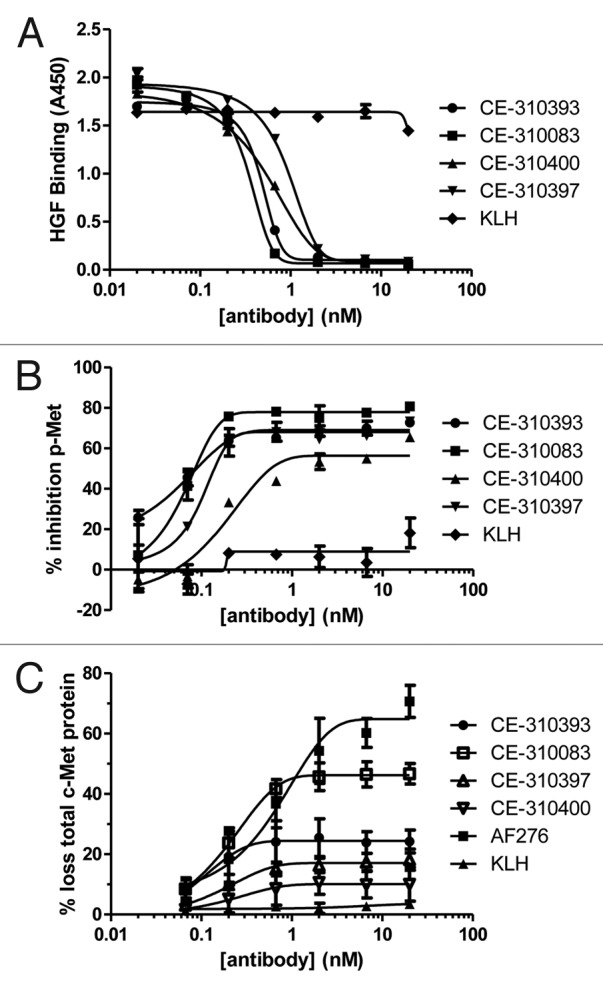
Figure 1. In vitro neutralizing activity of lead c-Met antibodies. (A) c-Met antibodies inhibit binding of HGF to c-Met ECD-Fc in vitro. Lead c-Met antibodies or anti-KLH antibodies were serially diluted and preincubated for 4 h with Met ECD-Fc (500 ng) immobilized in microtiter plates. Recombinant HGF was incubated for 15 min and its binding was determined. (B) c-Met antibodies inhibit HGF-dependent receptor activation in A549 cells. A549 cells were preincubated for 4 h with serially diluted antibodies then challenged with HGF (40 ng/well) for 15 min. Levels of pY-Met were determined in a capture ELISA using PY20 as the detection antibody. (C) Induction of c-Met degradation by c-Met antibodies. A549 cells were treated with varying concentrations of each lead antibody or anti-KLH for 4 h and the level of total c-Met was determined by ELISA.
Subsequently, neutralizing activity was demonstrated in a more native cellular setting using A549 cells. Normally, the basal level of c-Met kinase activity in A549 cells is low. Recombinant HGF is able to bind and activate c-Met on the surface of A549 cells, 27 resulting in receptor autophosphorylation on tyrosine, which can be measured in a sandwich ELISA. Each lead antibody effectively inhibited c-Met activation in this cellular assay (Fig. 1B), with CE-310393 and CE-310083 exhibiting the best potencies in the range of 150 pM (Table 1). The control antibody directed against KLH had no significant effect on ligand-dependent c-Met activation. Maximal inhibition of receptor activation varied among the leads between 60 and 80%. Incomplete inhibition may be the result of receptor-ligand internalization into endosomes and sequestration from antibody, 28 thereby preventing quantitative receptor blockade.
To better understand the mechanisms by which the c-Met antibodies inhibit receptor activation, we investigated the effect of antibody treatment on the steady-state levels of total c-Met protein. The stability of total c-Met protein following treatment with the lead antibodies was evaluated in both western blot and capture ELISA formats. Only minor loss (~25%) of total c-Met protein was seen following incubation of A549 cells with each of the leads from epitope class 1 (Fig. 1C); however, CE-310083, which falls into epitope class 2, induced 40–50% loss of c-Met protein at concentrations as low as 667 pM (Fig. 1C). The positive control polyclonal antibody AF276 decreased total c-Met protein levels approximately 70% at the highest antibody concentrations used (20 nM). Each mAb had a more dramatic effect on phospho-Met levels than total c-Met levels following stimulation of A549 cells with HGF (compare Fig. 1B and 1C). The data led us to conclude that the primary mechanism of inhibition of c-Met activation in cells is through blockade of ligand binding and that the effect of the antibody on total c-Met was only a minor contributor to the antibodies’ overall activity.
Phenotypic effects of c-Met antibodies
Autocrine signaling between HGF and c-Met can induce cellular transformation 3, 29 and promote tumor formation, malignant growth and dissemination of metastases. 3 Overexpression of human HGF and human c-Met in NIH3T3 cells induced cellular transformation yielding cells capable of forming colonies in soft agar and growing as aggressive xenograft tumors in athymic mice. 30, 31 Serial passage of the xenograft tumors in vivo resulted in a model, S114, that overexpressed human HGF and human Met. 30, 31 We used S114 cells in a soft agar colony formation assay to evaluate the functional activity of our four lead antibodies. Antibodies from epitope classes 1 and 2, CE-310393 and CE-310083, respectively, inhibited c-Met activity, seen as decreased levels of pY1234/1235-Met, in cultured S114 cells (Fig. 2A), suggesting they would interfere with colony formation and growth. The antibodies decreased the number of S114 colonies in soft agar in a dose-dependent manner (Fig. 2B). The IC50 values for each of the top four leads ranged from 47–167 nM (Table 1). The higher concentrations of antibody required to inhibit colony formation of S114 cells compared with the potency for the antibody in the A549 autophosphorylation assay is likely due to the difficulty for antibodies to diffuse and engage the target in soft agar and to the high level of c-Met receptor expression on the S114 cells.

Figure 2. Epitope class 1 and 2 antibodies inhibit c-Met activity in S114 cells and colony formation in soft agar. (A) S114 cells were treated with 10 µg/ml of CE-310393 (class 1), CE-310083 (class 2), or AF276 (R&D Systems) for between 15 min and 24 h. Cells were lysed and western blots were performed using an antibody to Y1234/1235 in the kinase domain to measure pY-Met levels and sc-10 to assess total Met protein. (B) Inhibition of S114 growth in soft agar. S114 cells were plated in soft agar with varying concentrations of CE-310393, CE-310083 or 10 μg/ml control anti-KLH antibodies and grown for 10 d before colony number was determined. Values represent means ± SEM. Results of Student’s t-test are indicated: *p < 0.05, **p < 0.01, ***p < 0.001
HepG2 cells are hepatocellular tumor cells that express c-Met 32 and respond to exogenous HGF by exhibiting elevated cell motility and matrigel invasion, 33, 34 which coincides with suppression of cell proliferation 33 and formation of tubular extensions in matrigel. We developed an assay measuring HGF-induced tubule formation in matrigel and employed this assay to assess the effects of our antibodies in this functional setting. The extent of tubule formation was quantified after staining with p-iodonitrotetrazolium violet using the ArrayScan system (Cellomics) and ImagePro software. In the analysis, the number and length of newly formed tubules were quantified with respect to colony size. Four days of treatment with 50 ng/ml HGF in matrigel stimulated significant tubule formation (Fig. 3A), whereas none of the lead antibodies alone showed significant tubulogenesis activity (Fig. 3A, CE-310083). However, coadministration of 50 ng/ml HGF with 1–50 μg/ml of anti-c-Met antibodies was able to inhibit tubulogenesis of HepG2 cells by 56–98% (Fig. 3A and B). We concluded from these functional studies that lead antibodies from both epitope classes were able to effectively inhibit multiple functions of c-Met that promote malignant tumor growth.
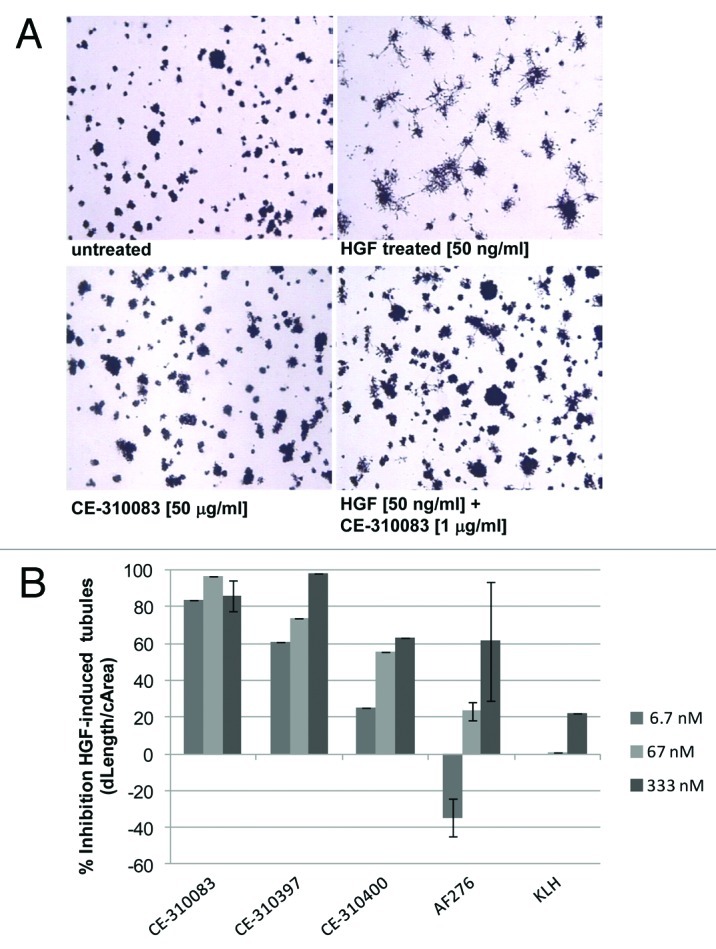
Figure 3. c-Met antibodies inhibit HGF-dependent tubulogenesis in HepG2 cells. (A) HGF induces tubulogenesis of HepG2 cells that is inhibited by c-Met antibodies. HepG2 cells grown in matrigel for 4 d form colonies lacking tubular extensions (top left panel). Plating cells with 50 ng/ml HGF induces tubulogenesis (top right panel), and plating cells with 50 ng/ml HGF and 1 μg/ml CE-310083 dramatically inhibited tubule formation (bottom right panel). Treatment of cells with antibody alone (50 µg/ml CE-3100833, bottom left panel) did not induce tubule formation. (B) c-Met antibodies antagonize tubule formation in a dose-dependent manner. Various concentrations of c-Met or KLH antibodies were plated with HGF (50 ng/ml) and after 4 d tubulogenesis of HepG2 cells was quantitated by image analysis of tubule length normalized to colony size (dLength/cArea).
c-Met antibodies exhibit variable degrees of agonism depending on epitope class
One of the concerns with developing therapeutic antagonist antibodies to receptor tyrosine kinases in general, and to c-Met in particular, is that the antibodies may possess agonist activity that counteracts the desired neutralizing activity of the therapeutic agent or induces ectopic pathway activation that could lead to side effects. Bivalent antibodies to receptor tyrosine kinases often mimic the mechanism of activation employed by natural ligands, such as HGF, by inducing receptor dimerization. 35–38 The lead antibodies were evaluated for the ability to activate c-Met in A549 cells. HGF induced maximal stimulation of c-Met activity in A549 cells within 15 min (data not shown). Cells were treated with various concentrations of each antibody for 15 min and c-Met activation was measured in the phosphoMet ELISA. Each of the epitope class 1 antibodies showed minimal stimulation of c-Met activity (Fig. 4A). Interestingly, CE-310083 from epitope class 2 behaved as a partial agonist, demonstrating ~50% of the maximal activity of HGF, similar to AF276. Both CE-310083 and AF276 were less potent than HGF, requiring higher concentrations to achieve their maximal effect. Maximal c-Met stimulation induced by CE-310083 and AF276 was reached within 15 min followed by decay of phosphoMet levels to basal levels within 1 h of treatment (data not shown).
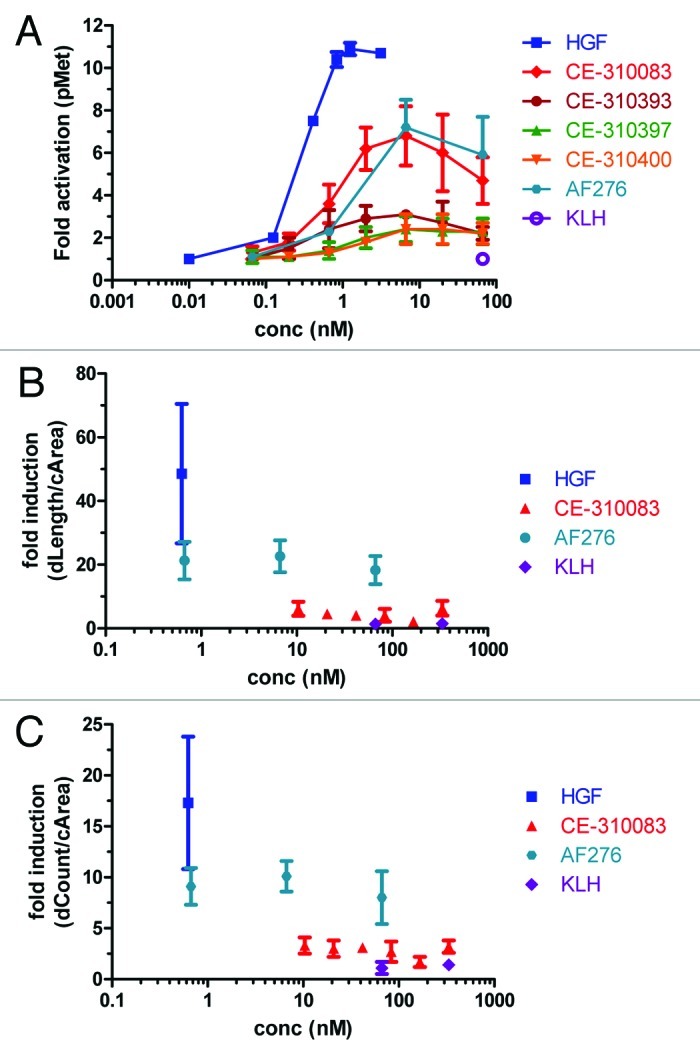
Figure 4. Evaluating agonist activity of c-Met antibodies. (A) Epitope class 2 but not epitope class 1 antibodies display partial agonist activity in the cellular c-Met activation assay. Various concentrations of HGF, c-Met antibodies (including those from epitope classes 1 and 2 and AF276) or KLH antibodies were added to A549 cells for 15 min and induction of phosphoMet was determined by capture ELISA with PY20 as the detection antibody. Each group represents triplicate samples, and values are means ± SD (B and C) CE-310083 does not promote c-Met-dependent tubular morphogenesis of HepG2 cells. HepG2 cells were treated with HGF, epitope class 2 antibody CE-310083, AF276 or KLH antibodies for 4 d and tubule length (B, dLength/cArea) and number (C, dCount/cArea) were determined. Values are means ± SD of 5 sample groups per treatment condition.
We evaluated whether the partial agonist activity of CE-310083 and AF276 translates to a functional response in an assay sensitive to stimulation with HGF, the HepG2 tubular morphogenesis assay. HGF efficiently promoted tubule formation, exhibited by ~17 fold increase in tubule number and ~50 fold increase in tubule length (Fig. 4B and C). The biochemical activity demonstrated by AF276 in A549 cells translated to partial agonism in HepG2 tubulogenesis. However, CE-310083 only weakly induced tubule formation, even though it showed similar activity to AF276 in the c-Met activation assay in A549 cells (Fig. 4A). Thus, even though CE-310083 stimulated c-Met activity, it did not translate to a robust functional response. Nonetheless, the lead c-Met antibodies, including CE-310083, effectively inhibited HGF-dependent tubulogenesis (Fig. 3), indicating the lead c-Met antibodies act as antagonists in the presence of HGF.
Differential anti-tumor activity of c-Met antibodies against S114 tumor xenografts
The four lead antibodies were tested for the ability to inhibit growth of xenograft tumors grown subcutaneously in athymic mice. Initially, we used the S114 tumor model because of its dependence on autocrine HGF/c-Met signaling for tumor growth. Tumor-bearing mice received a single i.p. bolus 200 μg dose (~10 mg/kg) on day 5 after implantation. Remarkably, there were clear differences in the growth delay caused by each antibody. Each antibody effectively inhibited tumor growth over the first 5 d of treatment (d5–10), at which point some of the tumors began to increase in size (Fig. 5). CE-310393 displayed the most robust anti-tumor activity, causing a growth delay of approximately 10 d. Based on the relative in vitro and in vivo activity of CE-310393, it underwent sequence optimization and its derivative CE-355621 was advanced to more detailed profiling in vivo.
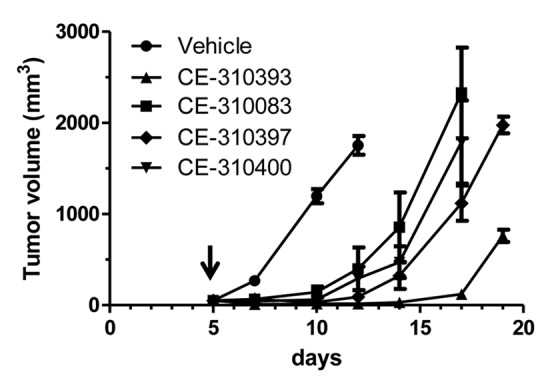
Figure 5. Lead c-Met antibodies show differential antitumor activity against S114 tumors. S114 tumors were injected subcutaneously into athymic mice and after tumors were established for five days, each mouse received a single 200 μg bolus dose (~10 mg/kg) of a lead c-Met antibody (arrow). Caliper measurements were made every 2–3 d. Values are means ± SEM.
Generation of CE-355621
One of the challenges associated with protein therapeutics such as antibodies is the development of an immune response against the therapeutic agent. Immunologic responses in the patient can result in the production of anti-drug antibodies that limit the effectiveness of the therapeutic antibodies by neutralizing antibody function, accelerating antibody clearance, and inducing toxic side effects. 39, 40 The emergence of fully human antibodies has limited the risk of immunogenicity seen with mouse, chimeric, and humanized therapeutic antibodies. Yet potential sources of immunogenicity in human antibodies remain, including deviations in amino acid sequences from germline. Sequence analysis of CE-310393 indicated there were two differences from germline in framework regions of VH chain gene 1–18/DP-14 (E42 and S97) and one difference in framework region 3 of Vκ gene L5/DPK5 (A91). The divergent sequences were restored to germline by site-directed mutagenesis, yielding CE-355621 (VH: E42K, S97T; Vκ:A91T). None of the differences from germline sequence found in CDRs of CE-310393 were altered in CE-355621. Sequence alignment of CE-310393 and CE-355621 with germline is shown in Fig. S2A. The activity of CE-355621 was virtually identical to that of CE-310393 in terms of binding affinity, activity in the in vitro ligand binding and ligand-dependent c-Met activation assays, and in an in vivo efficacy study with the U87MG model (Fig S2B–D; Table S1), which was used previously to demonstrate the antitumor activity of CE-355621.25
Pharmacodynamic effects of CE-355621
We focused on characterizing the pharmacodynamic (PD) effects of CE-355621 directly on c-Met activity and protein levels in vivo in the U87MG glioblastoma model reported to express HGF and c-Met and to be sensitive to c-Met inhibitors in vivo, including CE-355621. 17, 21, 25, 41 CE-355621 effectively inhibited c-Met activation by exogenous HGF in U87MG cells (Fig. S3), but, in contrast to Martens et al.’s 21 report that U87MG cells were sensitive to treatment with the single-arm c-Met antibody OA-5D5, CE-355621 had no effect on U87MG cell proliferation in culture in the presence or absence of exogenous HGF (data not shown). Treatment of athymic mice bearing U87MG tumors with increasing concentrations of CE-355621 resulted in significant inhibition of Met activity as measured by phosphoMet (pMet) in tumors extracted 24 h after dosing (Fig. 6A). Less dramatic effects on total Met protein levels were observed (Fig. 6A; Fig. S4), consistent with the modest effects on total Met protein levels seen with CE-310393 in cultured A549 cells (Fig. 1C) and CE-355621 in U87MG cells in culture (Fig. S5). Plasma was also collected from sacrificed animals and the levels of CE-355621 were determined. The pharmacokinetic (PK)/PD analysis indicated maximal inhibition at 24 h was ~75–90% at doses of 50 μg and above, which corresponded to plasma levels of CE-355621 of ≥ 10 μg/ml, and half maximal inhibition was seen at ~5 μg/ml (Fig. 6B).
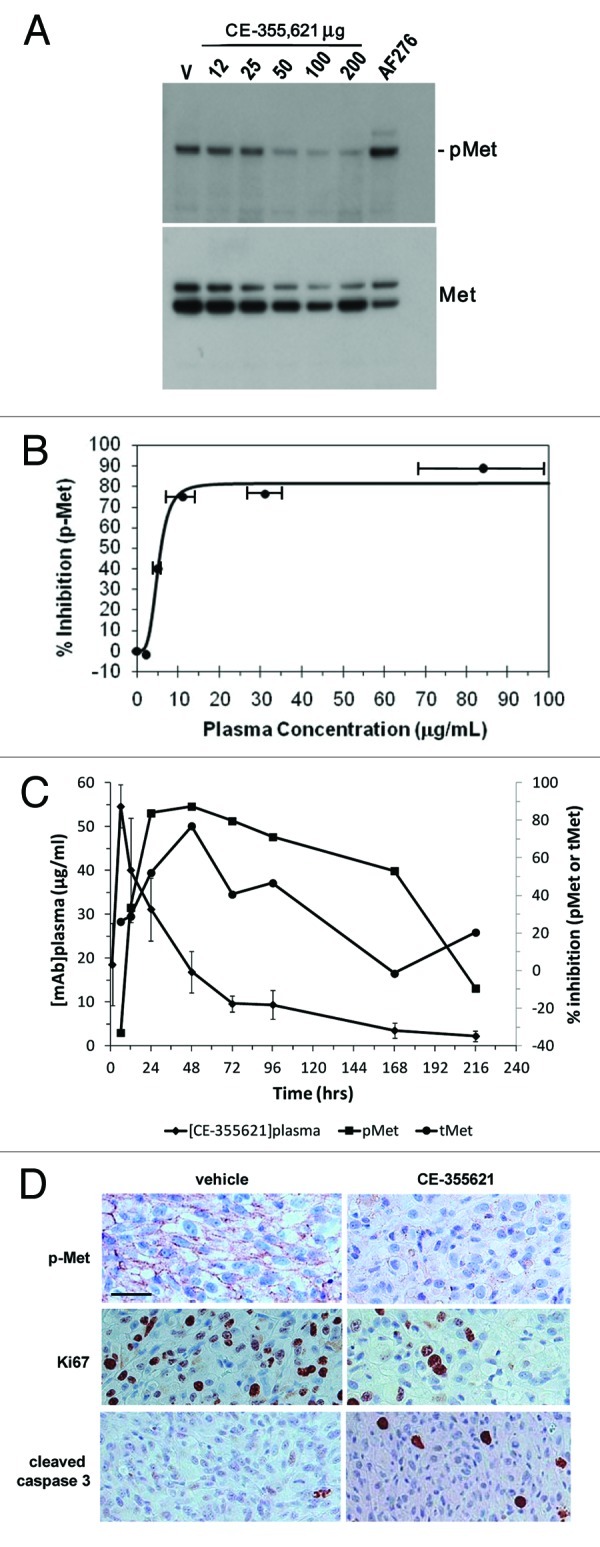
Figure 6. Pharmacodynamic modulation of phosphoMet levels in U87 xenograft tumors by CE-355621 . (A) Groups of four athymic female mice bearing U87MG human glioblastoma xenografts were treated with various amounts (12–200 μg) of CE-355621 as a single intraperitoneal bolus dose and tumors were excised 24 h later. Tumors were homogenized and phosphoMet and total c-Met protein levels were determined from pooled lysates via western blot analysis following immunoprecipitation of c-Met protein. (B). Graphical representation of PD modulation of phosphoMet by CE-355621 quantitated from the western blot in (A) as a function of the plasma concentration of circulating functional CE-355621, which was determined using a capture ELISA specific for human IgG competent to bind immobilized c-Met ECD-Fc. PK values represent means ± SD (C) PK/PD relationship of CE-355621 in athymic mice bearing U87MG xenograft tumors. U87MG tumors were grown to a size of ~300 mm3 in size and treated with vehicle or CE-355621 (100 µg intraperitoneal, single dose). Tumors were excised at various times after dose administration. Tumors were homogenized and phosphoMet and total c-Met levels and plasma levels of CE-355621 were determined as in (B). (D) CE-355621 decreases phosphoMet staining intensity in tumor cells, inhibits tumor cell proliferation and induces apoptosis in U87MG xenograft tumors. Established U87MG tumors were treated with vehicle or a single i.p. 200 µg dose of CE-355621, excised 72 h later, formalin-fixed then paraffin-embedded. Immunohistochemistry with a phosphoMet antibody against pY1234/1235 was used to assess the effect of CE-355621 on c-Met phosphorylation status. Staining for Ki67 and cleaved caspase 3 was used to evaluate effects on tumor cell proliferation and induction of apoptosis, respectively. Effects of CE-355621 on each of these endpoints were assessed by microscopic examination by a pathologist. All the images were acquired at the same magnification with a 40X objective and the scale bar = 10 µm.
The PK/PD relationship over time was determined in subsequent experiments performed in athymic mice bearing U87MG xenograft tumors. Mice were dosed with 100 μg CE-355621 and followed over a 9 d period. Maximal inhibition of pMet levels (~80% inhibition) was reached at 24 h post dosing (Fig. 6C), consistent with the results from the dose titration experiment done at 24 h (Fig. 6B). This level of inhibition was maintained between 24 and 96 h. Decreases in total c-Met protein levels were also observed, but, as seen in the dose titration experiment done at 24 h (Fig. 6A; Fig. S4), the magnitude of the effect on total Met was smaller than the effect on pMet (Fig. 6C). The PD effects of CE-355621 diminished as antibody levels decayed, and the effect on pMet levels was lost as antibody levels dropped below ~5 μg/ml by day 9, corroborating the results from the dose titration experiment (Fig. 6A and B), where the PD effect of CE-355621 was lost at plasma levels less than ~5 μg/ml. The dose titration experiment and the PK/PD time course experiment corresponded well with each other, indicating that a single dose of CE-355621 at or above 50 μg results in sufficient levels of circulating antibody (> 5 μg/ml) to effectively modulate c-Met activity. Finally, inhibition of phosphoMet in U87MG tumors by CE-355621 was corroborated by immunohistochemical staining of tumor sections (Fig. 6D). Antibody treatment also inhibited tumor cell proliferation and induced apoptosis, as evaluated by Ki67 and cleaved caspase 3 staining, respectively (Fig. 6D). Staining signal intensities were quantified by a pathologist who performed microscopic examination of sections from multiple tumors excised from both vehicle and antibody-treated groups, and scores (0–3+) are presented in Table S2.
Anti-tumor efficacy
The significant PD effects of CE-355621 on phosphoMet levels and tumor cell proliferation and apoptosis in U87MG xenografts suggested growth of this tumor model would be sensitive to treatment with the antibody. Indeed, CE-355621 did demonstrate dose-dependent efficacy against U87MG tumors following a single dose (Fig. 7A). In addition, increasing doses of antibody significantly extended the time for tumors in each treatment group to reach 1000 mm3 in size following a single bolus dose (Table 2). The results of these in vivo studies revealed the PK/PD/efficacy relationship for CE-355621 when treating U87MG tumors (Fig. 7B). As long as plasma levels of antibody remained above ~5 μg/ml, c-Met activity (pMet levels) was inhibited by ≥ 50% (percent inhibition of phosphoMet in excised tumor samples is noted in parentheses above the corresponding plotted value for CE-355621 plasma concentration in Fig. 7B) and tumor stasis was observed. When circulating levels of CE-355621 decayed significantly below the 5 μg/ml threshold, c-Met activity returned to pretreatment levels and tumor growth resumed. We concluded from this analysis that circulating levels of CE-355621 would need to be maintained at or above the 5 μg/ml threshold in patients to provide sufficient coverage for clinical efficacy.

Figure 7. Dose-dependent efficacy of CE-355621 against U87MG tumors reveals PK/PD/efficacy relationship. (A) CE-355621 inhibits the growth of U87MG xenograft tumors in a dose-dependent manner. U87MG cells were injected into athymic female mice, and mice bearing tumors of ~100 mm3 in size were randomized in four groups (6 animals/group). Mice were injected i.p. on day 6 with vehicle (circles) or 25 (squares), 100 (triangles) or 200 (diamonds) μg of CE-355621 and tumor growth was determined. Results are mean ± SEM plotted with day 0 corresponding to the day of dose administration. Results of Student’s t-test are indicated: *p < 0.05, **p < 0.01, ***p < 0.001. NB: The level of statistical significance for the 25 μg treatment on day 8 is p < 0.001 because the error bars are too small to be visible. The visible error bars on day 8 correspond to the 100 μg sample (p < 0.05). (B) Linking PK/PD of CE-355621 to tumor growth inhibition in U87MG xenograft tumors. The growth of control (circles) or CE-355621-treated (100 μg, triangles) tumors is plotted (right axis, mm3). The plasma concentration of CE-355621 and the degree of pMet inhibition determined in the PK/PD experiment depicted in Figure 5C is overlaid in this plot (PK is on the left axis [μg/ml]; PD is noted as % inhibition of pMet in parentheses above the corresponding PK symbol). Dosing of the efficacy study began on day 7 after implantation (arrow head). Data from the PD analysis displayed in Figure 5C indicated that there was ~50% decrease in phosphoMet signal corresponding to day 14 in this graph, or 7 d after dosing began. On day 16, 9 d after the start of dosing, the level of CE-355621 had decayed to ~3 μg/ml and there was no longer any detectable inhibition of phosphoMet. Tumor regrowth in mice treated with CE-355621 was first apparent on day 17 when CE-355621 serum levels had dropped below the minimal threshold required to maintain c-Met inhibition.
Table 2. Dose-dependent tumor growth delay with U87 tumors treated with CE-355621.
| Sample | Time to reach 1000 mm3 (days) |
|---|---|
| Vehicle |
6.2 ± 0.2 |
| 25 μg CE-355621 |
9.9 ± 0.2*** |
| 100 μg CE-355621 |
16.1 ± 1.1*** |
| 200 μg CE-355621 | 22.3 ± 0.4*** |
p < 0.001(by Student’s t-test). Increasing doses of CE-355621 induce progressively longer tumor growth delay in U87MG tumors. The size of each tumor in Figure 7A was measureon c-Met in GTL-16 xenograft tumorsd every 2–3 d and the time to reach 1000 mm3 (days) from the start of treatment was determined. Values are means ± SEM.
The MET locus is amplified ~10 fold in GTL-16 gastric tumor cells, 42 and even though they lack expression of HGF 42 (Hillerman and Michaud, unpublished observations), the c-Met pathway is constitutively activated in these cells. We evaluated whether CE-355621 could impact c-Met activity in GTL-16 cells in vivo and inhibit xenograft tumor growth. Surprisingly, the antibody exhibited robust activity against GTL-16 tumors, inhibiting growth by 85% following two 400 μg doses given on days 7 and 14 (Fig. 8A). Further, evaluation of the pharmacodynamics of CE-355621 in this model indicated it decreased phosphoMet levels by 48% at 24 h and 50% at 48 h after a single 400 μg dose and decreased total c-Met levels by 32 and 38%, respectively (Fig. 8B). Since pathway activation in GTL-16 cells occurs in a ligand-independent manner, the effects of c-Met antibodies appear to be mediated in part by inducing receptor turnover, as shown with CE-355621, though additional mechanisms may be involved. This may explain why more frequent administration of higher doses was required to detect pharmacodynamic and anti-tumor effects in GTL-16 tumors.

Figure 8. CE-355621 inhibits the growth of MET amplified, HGF-independent GTL-16 gastric cancer xenografts. (A) CE-355621 inhibits the growth of GTL-16 xenograft tumors. Tumor cells were injected subcutaneously and tumors were grown to about 100 mm3. 400 μg CE-355621 was administered i.p. on days 7 and 14 (arrow heads) into groups of 7 mice/group. Results are mean ± SEM *p < 0.001 (by Student’s t-test) (B) Pharmacodynamic effects of CE-355621 on c-Met in GTL-16 xenograft tumors. Mice bearing GTL-16 tumors were dosed i.p. with 400 μg CE-355621. Tumors were excised 24 or 48 h later and lysates were prepared. The levels phosphoMet and total Met were determined by ELISA, using PY99 and sc-10 as detection reagents, respectively, and % inhibition relative to the untreated group was calculated. Values are the means ± SD from 4 animals per group.
Discussion
Dysregulation of HGF/c-Met signaling has been described in numerous human tumors, and the involvement of HGF/c-Met function in tumor angiogenesis suggests targeting this signaling axis is an attractive therapeutic strategy. We describe here the isolation and characterization of several high affinity antibodies that specifically target c-Met and neutralize its function in vitro and in vivo. Each lead antibody described interfered with HGF binding and induced receptor downregulation, thereby preventing receptor activation. These effects translated to inhibition of c-Met function in soft agar growth and tubular morphogenesis assays. Furthermore, we determined the dose levels of CE-355621 required to maintain plasma levels of antibody sufficient to inhibit c-Met in vivo and found at sufficient doses that CE-355621 demonstrated antitumor activity against U87MG and GTL-16 xenograft tumors. The antitumor activity of CE-355621 is not likely the result of antibody-mediated cell cytoxicity (ADCC) in nude mice, as the antibody’s isotype is human IgG2, which has low affinity for Fc receptors and significantly reduced ability to induce ADCC. The broad activity of CE-355621 suggests it represents a viable option for the treatment of cancer patients with tumors exhibiting elevated levels of c-Met protein and pathway activation.
One advantage of therapeutic antibodies is their selectivity, and CE-355621 is exquisitely selective for c-Met. Extremely high concentrations of CE-355621 (up to 6000 μg/ml) did not inhibit the activation of two highly related receptor tyrosine kinases, c-Ron and IGF-1R, when each was stimulated by the addition of exogenous ligands MSP and IGF-1, respectively (data not shown). CE-355621 also did not bind or neutralize the mouse ortholog of c-Met, which hampered assessment of anti-angiogenic or other host activity in the mouse. As a result, evaluation of the in vivo efficacy of the lead molecules required either the use of a model in which an autocrine HGF/c-Met signaling loop existed or a model in which c-Met overexpression resulted in ligand-independent receptor autoactivation. Autocrine HGF/c-Met signaling has been reported in U87MG glioblastoma tumors, 17 and ligand-independent MET amplification in GTL-16 cells results in constitutive c-Met activity. 42 CE-355621 effectively inhibited growth of both models in vivo, suggesting the antitumor efficacy of CE-355621 demonstrated in these studies is due to neutralizing c-Met activity in tumor cells and that the overall activity of CE-355621 assessed with xenograft models in athymic mice is likely underestimated because any contribution to antitumor activity from inhibiting angiogenesis is not captured.
The apparent molecular mechanism by which each of the antibodies characterized in this study, including CE-355621 in particular inhibits c-Met function is primarily through blocking HGF binding. Each lead also caused a modest decrease in total c-Met levels in A549 cells (Fig. 1C), and CE-355621 had similar effects in U87MG cells (Fig. S5) and A549 cells (Fig. S6) grown in culture. To further ascertain the effect of CE-355621 on receptor levels at the cell surface, a flow cytometry assay was configured to detect c-Met on the surface of A549 cells following antibody treatment. In this assay, the polyclonal c-Met antibody BAF358 (R&D Systems) binds to c-Met and is not competed or displaced by CE-355621 or AF276. Following incubation of A549 cells with each lead antibody for various times up to 24 h, addition of sodium azide at 4°C or fixation with paraformaldehyde stabilized c-Met protein in the plasma membrane (Fig. S7). Similar results were seen with both methods. Minimal loss of c-Met from the surface was seen at 1 h, and maximal decreases were reached within 8 h. Receptor levels at the membrane decreased approximately 30–40% with CE-355621 treatment, whereas the control polyclonal antibody to c-Met, AF276 (R&D Systems), induced nearly 70% mobilization of c-Met from the cell surface. These observations suggest CE-355621 induces receptor internalization and degradation, providing a potential mechanistic explanation for the loss of total c-Met protein induced by antibody treatment.
Analysis of the effect of CE-355621 on total Met protein levels in xenografted U87MG tumors suggests the antibody had a slightly greater effect on Met protein levels in vivo compared with cultured cells. For example, CE-355621 induced a maximal decrease in c-Met protein levels of 60–75% in vivo (Figs. 6A and C; Fig. S4), whereas the maximal effects in cultured cells were 25–40% (Figs. 1C; Figs. S5–7). Nonetheless, the magnitude of the effect of the antibodies on phospho-Met levels was consistently greater than the effects on total c-Met in vitro, as seen when comparing Figure 1B and C, and in vivo, as seen when comparing effects on phospho- and total Met in Figure S4 and Figure 6C. In sum, these data support the conclusion that the primary mechanism by which CE-355621 neutralizes c-Met activation is by blocking ligand binding and that its effect on Met protein levels is secondary.
The molecular mechanism behind the efficacy exhibited by CE-355621 against GTL-16 tumors, however, is less clear. GTL-16 cells do not express HGF; the constitutive activation of c-Met results from amplification and overexpression of c-Met itself, leading to receptor dimerization and autoactivation in the absence of ligand. CE-355621 decreased phosphoMet levels in GTL-16 tumors by about 50% at 24–48 h following a 400 μg dose and receptor turnover diminished Met levels 32–38%, whereas doses of 50–200 μg inhibited c-Met activity by ~80% from 24–96 h in U87MG tumors. The absolute level of c-Met inhibition was lower in GTL-16 tumors than in U87MG tumors and higher doses were required to reach this level of inhibition, yet the degree of c-Met turnover was similar to what was observed in U87MG tumors. It appears that CE-355621 may affect the c-Met pathway by additional means. Alternative mechanisms to explain the sensitivity of GTL-16 cells to CE-355621 could include cleavage of c-Met within the extracellular domain, as reported for antibody DN-30, resulting in a decrease in the level of c-Met on the cell surface and shedding of a fragment of the ECD, 43 inhibition of receptor dimerization by antibody in the absence of ligand, or altering how accessory proteins, such as CD44v6 and α6β4 integrin, modulate c-Met function. 28 The observation that CE-355621 decreased c-Met activity in GTL-16 cells and inhibited GTL-16 tumor growth suggests that it may have antitumor activity against a broader range of tumors, including tumors in which c-Met activation is HGF-independent. Constitutive activation of the c-Met pathway is often genetically linked via amplification or mutation, rendering the tumors HGF-independent but addicted to Met function, potentially explaining the sensitivity of GTL-16 tumors to CE-355621 treatment in vivo. Further investigation into the mechanism by which CE-355621 inhibits c-Met function in tumors cells such as GTL-16 cells where ligand-independent pathway activation is genetically linked is warranted. Such tumors may represent an additional patient segment with the potential to respond to therapy with CE-355621.
It has been challenging historically to identify antibodies against c-Met that antagonize its function, as numerous bivalent c-Met antibodies have behaved as agonists presumably by inducing receptor dimerization in a manner similar to HGF. 16 In fact, onartuzumab (MetMab, Genentech/Roche) was engineered as a monovalent, single arm antibody to abrogate the agonist activity of the bivalent IgG parental antibody 5D5 that induced proliferative responses. 20 We describe several lead c-Met antibodies that exhibit minimal agonist activity in a cellular assay of c-Met activation, with the exception of CE-310083, which acted as a partial agonist of c-Met (Fig. 4A). Yet, CE-310083 was unable to effectively induce tubular morphogenesis in HepG2 cells (Fig. 4B and C). Further, none of the epitope class 1 antibodies stimulated HepG2 tubulogenesis. However, we cannot exclude the possibility that the mAbs we describe here may display some degree of functional agonism in a biological setting that we have not evaluated as part of these studies. Nonetheless, multiple lines of evidence support the conclusion that the lead antibodies, including CE-355621, act primarily as c-Met antagonists. None of the lead antibodies induce cellular responses that phenocopy HGF. The antibodies were unable to induce mitogenesis of A549 or U87MG cells. Each of the lead antibodies potently neutralized HGF function in multiple settings, including receptor autophosphorylation in A549 and U87 cells, soft agar growth with S114 cells, and tubulogenesis of HepG2 cells. Furthermore, the lead antibodies antagonized c-Met activity in xenograft tumor cells in a dose- and concentration-dependent manner resulting in tumor growth inhibition of multiple xenograft models. We, therefore, conclude that the lead c-Met antibodies possess the antagonist activity required of a therapeutic antibody.
c-Met is expressed on many cell types, including epithelial cells, in most tissues in the body, which could influence the clearance, exposure and distribution of CE-355621. It was not possible to assess the contribution of target-mediated clearance to its PK in rodent species because CE-355621 lacks cross-reactivity with murine c-Met. CE-355621 binds human and cynomolgus c-Met with similar affinity (Table S1), as demonstrated with Biacore and flow cytometry, and it potently inhibited HGF-mediated activation of c-Met in cynomolgus kidney cells (data not shown). The PK of CE-355621 was determined in cynomolgus primates following a bolus intravenous 5 mg/kg dose (Fig. S8). Beta phase clearance (12.96 ml/day/kg) resembled that of other human IgG2 mAbs, as did the half-life (5.9 d), suggesting any target mediated clearance of CE-355621 that may have been apparent at lower doses was at or near saturation at the 5 mg/kg dose. Simulations of the PK of CE-355621 in humans supported the feasibility of administering reasonable doses of the antibody and maintaining target coverage above the minimal threshold of 5 μg/ml required for efficacy in the U87MG xenograft model and postulated to be needed for clinical activity.
Important considerations for rapid assessment of clinical activity in patient populations most likely to respond to an antagonist c-Met antibody include the development of patient stratification and biomarker assays. We hypothesize that overexpression of c-Met coincident with pathway activation defines patients most likely to respond to CE-355621. Overexpression of c-Met has been reported in numerous tumor types, including gastric, non-small cell lung, breast, colon, prostate, pancreatic, ovarian, hepatocellular, and renal cell. 16 Immunohistochemical assessment of total Met in tumor micorarrays and has been reported, 44 and initial efforts to develop an immunohistochemical assay of phosphoMet in xenograft samples described here (Fig. 6D) suggests it may be feasible to assess Met activation and PD effects of CE-355621 in pre- and post-treatment tumor biopsies. In addition, preclinical evaluation of FDG-PET imaging as a means to assess effects of CE-355621 indicated the antibody dramatically reduced glucose uptake and utilization in U87MG tumors, coincident with inhibition of tumor growth. 25 FDG-PET represents an attractive approach to assess the clinical activity of CE-355621 in cancer patients as a non-invasive biomarker to discern proof of mechanism.
In recent years, several neutralizing antibodies targeting c-Met have been described, including DN-30, onartuzumab (MetMab, 5D5), 11E1, h224G11 and LMH 87. 6, 45–49 There is a broad spectrum of molecular mechanisms of action and biochemical activities represented by these antibodies, indicating neutralization of c-Met function can be accomplished by divergent mechanisms. Monovalent forms of 5D5 (MetMab, onartuzumab) and DN-30 have been engineered to eliminate agonist activity associated with the bivalent forms of those antibodies; 50, 51 whereas, 11E1, h224G11, and LMH 87 are described as bivalent, non-agonistic, neutralizing antibodies.46–48 Onartuzumab interacts with the Sema domain of c-Met and directly antagonizes HGF binding, but it does not appear to induce c-Met turnover. 20, 50 Though both DN-30 and LMH 87 do not antagonize HGF binding, each neutralizes c-Met function by inducing loss of c-Met protein by potentially different means. DN-30 causes receptor cleavage and shedding into the extracellular environment, 43, 51 and LMH 87 induces c-Met internalization. 46 11E1 and h224G11 differ from the above antibodies in that they both antagonize HGF binding and induce receptor turnover. 47, 48 Epitope class 1 and 2 antibodies described here, including CE-355621, neutralize c-Met activity primarily through inhibiting ligand binding and secondarily by inducing loss of receptor from the cell surface and protein degradation. Thus, the molecular and biochemical characteristics of CE-355621 and related antibodies appear to be most similar to those of 11E1 and h224G11. Nonetheless, head-to-head comparisons of these various antibodies would be required to accurately assess the relative effectiveness of each antibody as a potential therapeutic agent in the various settings of dysregulated Met function.
Proof of concept clinical activity related to targeting c-Met with a neutralizing antibody has been recently demonstrated with the monovalent single arm antibody onartuzumab (Genentech/Roche). Clinical benefit was demonstrated in NSCLC patients with tumors expressing elevated levels of Met protein (≥ 50% tumor cells having 2–3+ IHC staining) who were treated with onartuzumab and erlotinib, and a durable complete response was observed in a metastatic gastric cancer patient with high MET polysomy and Met protein expression detected by immunohistochemistry who was treated with onartuzumab. 52–55 CE-355621 appears to exhibit superior binding and neutralizing activity for its target in biochemical assays compared with onartuzumab. The affinity of bivalent CE-355621 for native c-Met is ~56 pM and for recombinant c-Met ECD is ~200 pM; whereas, the affinity of monovalent 5D5 (onartuzumab) for c-Met is ~4 nM. 14, 56 Further, CE-355621 displayed almost 20-fold better potency than monovalent 5D5 in an HGF displacement assay (~280 pM for CE-355621 compared with ~5 nM for 5D5). 56 The difference in binding activity is certainly related in part to the avidity of bivalent CE-355621 for its target, though the CE-355621 Fab retains significant potency of 740 pM in the HGF ligand displacement assay (data not shown), which is only a 2.7-fold shift compared with bivalent CE-355621. The pharmacokinetics of onartuzumab and CE-355621 do not differ appreciably in non-human primates, even though the isotypes of the antibodies differ. The clearance and half-life of 5 mg/kg CE-355621 (human IgG2) are 12.96 ml/day/kg and 5.9 d, respectively, and the clearance and T1/2 for 3–30 mg/kg onartuzumab (humanized IgG1) are 13 ml/day/kg and 3–6 d. 56, 57 The high affinity and enhanced potency of CE-355621 along with comparable pharmacokinetics to onartuzumab suggest CE-355621 could have considerable activity in the clinic.
In summary, we describe potent neutralizing c-Met antibodies with functional activity in vitro and in vivo. Further, lead candidate CE-355621 demonstrated concentration-dependent PD effects and robust in vivo efficacy. Thus, CE-355621 represents an attractive candidate therapeutic antibody to treat cancer patients suffering from disease in which dysregulation of HGF/c-Met signaling drives tumor growth and progression.
Materials and Methods
Ethics statement
All animal studies with mice and cynomolgus monkeys were conducted in accordance with animal care and use protocols according to the guidelines of the Association for the Assessment and Accreditation of Laboratory Animal Care and approved by the Institutional Animal Care and Use Committee (IACUC) of Pfizer Global Research and Development. The program of humane animal care and use at Pfizer Global Research and Development has been evaluated for its compliance with the US. Animal Welfare Act and The Guide for Care and Use of Laboratory Animals. 58
Cell lines
A549 human lung carcinoma cells, HepG2 human hepatocellular carcinoma cells, U87MG human glioblastoma, and GTL-16 gastric cells were obtained from American Type Culture Collection and grown according to culture conditions provided by ATCC. S114 cells 30, 31 were provided by Dr. George Vande Woude, Center for Cancer and Cell Biology, Van Andel Institute, Grand Rapids, MI.
In vitro antibody binding and HGF binding assays
Microtiter plate wells were coated with 500 ng c-Met ECD-Fc (R&D Systems) in PBS overnight. Plates were blocked with 3% BSA in TBS-T for 60 min at RT. Hybridoma supernatants or purified antibodies diluted in DMEM, 10% FBS were incubated for 4 h at room temperature, and antibody binding was detected with anti-human IgG2 antibodies (Zymed, 05-3500). The above assay was modified to evaluate inhibition of HGF binding to the c-Met ECD. Antibodies were pre-incubated for 4 h in wells coated with c-Met ECD-Fc prior to addition of HGF in serum-free DMEM to a concentration of 100 ng/ml. HGF was incubated for 15 min at RT, and HGF binding was detected with a biotinylated polyclonal anti-HGF antibody and streptavidin-HRP.
Affinity determination with surface plasmon resonance
The binding affinity of purified antibodies was determined using surface plasmon resonance. c-Met ECD-Fc (~220 RU) was immobilized on a B1 chip (BIACORE™) by standard direct amine coupling procedures and antibody samples were injected in duplicate at 5 μl/min flow rate for 4 min. Dissociation was monitored for 2000 sec. The data were fit globally to a simple 1:1 binding model using BIACORE™ Biavel software. In addition, to determine the koff independent of any potential error in the active concentration or fitting model, the dissociation data were fit globally and independently from association data to a simple dissociation model. In all cases, this method was used to obtain koff, and it was found that they compared well to data obtained from global fit of association and dissociation data.
Binding of lead antibodies to native c-Met expressed on A549 cells
The binding affinity of purified antibodies for c-Met expressed on the surface of human A549 lung carcinoma cells was determined by flow cytometry. Cells were lifted with 0.25% trypsin-EDTA, washed and suspended in PBS, 0.025% sodium azide, and 2% heat inactivated serum. Equilibrium binding of sub-saturating concentrations of each antibody was achieved within 6–8 h at RT, and half-maximal binding (EC50) of each antibody was determined. Each antibody was incubated with detached cells for 6–8 h and binding was detected with biotinylated anti-human IgG (Jackson Labs) and streptavidin R-phycoerythrin conjugate (Caltag).
Cellular c-Met phosphotyrosine and total c-Met assays
A549 cells were plated (1x105 cells/well) in DMEM, 10% FBS and incubated at 37°C. Anti-c-Met antibodies were diluted in DMEM, 10% FBS and typically incubated with cells for 4 h prior to stimulation with HGF (200 ng/ml) for 15 min at 37°C. Cells were disrupted in NP-40 lysis buffer (150 mM NaCl, 20 mM TRIS-HCl pH 8.0, 1% NP-40, 10 mM EDTA, 10% glycerol, 1 mM Na3VO4, and Roche Complete protease inhibitors). c-Met phosphotyrosine and protein levels were determined by ELISA or in some cases by western blot. For ELISAs, cell lysate was added to wells with 500 ng/well polyclonal c-Met capture antibody (Santa Cruz, sc-10) and incubated for 2 h at RT. c-Met phosphotyrosine levels were detected with PY20-HRP (Transduction Labs, P11625) and total c-Met was determined with c-Met ECD antibody UBI 05–237 antibody (clone DO24 Upstate Biotechnology, 21601).
Soft agar growth and tubulogenesis assays
S114 tumor cells, NIH-3T3 cells engineered to express human HGF and human c-Met, were plated (5000 cells/well) in 0.35% soft agar in DMEM, 10% calf serum with or without anti-c-Met antibodies and layered over 0.5% agar in growth medium. Colony number and size of p-iodonitrotetrazolium violet stained colonies were determined 7–10 d later with ROBOT automation (Ludel Electronics, Ltd.) using ETC3000 software (Engineering Technology Center).
HepG2 cells (40,000 cells/plate) were plated in 35 mm dishes in OptiMEM, 10% heat inactivated FBS, 2 mM L-glutamine, and 1X penicillin/streptomycin over a layer of solidified matrigel. HGF (final concentration 50 ng/ml) or c-Met antibodies (final concentration of 1–10 μg/ml) were added, and cells were grown for 4 d at 37°C. Tubulogenesis was determined from images of p-iodonitrotetrazolium violet stained cells using the image analysis macro within Image Pro Plus v4.1 (Media Cybernetics).
Animals
Athymic female mice (CD-1 nu/nu, 20 g) obtained from Charles River Laboratories were used for all of the in vivo studies. Mice were housed in specific pathogen-free conditions according to the guidelines of the Association for the Assessment and Accreditation of Laboratory Animal Care (Institute of Laboratory Animal Research, 1996) and all of the in vivo studies were performed under approved Pfizer Global Research and Development institutional experimental animal care and use protocols in accordance with Pfizer Institutional Animal Care and Use Committee (IACUC), State, and Federal guidelines for the humane treatment and care of laboratory animals. Animal handling was done in a laminar flow hood. Animals were provided pelleted food and water ad libitum and kept in a room conditioned at 70–75°F with 50–60% relative humidity. Sentinel mice were monitored at regular intervals by serologic assays and were found free of murine pathogens (murine hepatitis virus, Sendai virus, pneumonia virus of mice, minute virus of mice, mouse poliovirus type 3 reovirus, Mycoplasma pulmonis, mouse parvovirus, epizootic diarrhea of infant mice, lymphocytic choriomeningitis virus, mouse adenovirus, ectromelia, mouse pneumonitis, and polyomavirus). For all of the studies, mice were allowed to acclimate a minimum of 3 d after receipt of shipment and randomized before commencement of studies.
Pharmacokinetics and pharmacodynamic effects of c-Met antibodies
The effects of the anti-c-Met antibodies on the phosphorylation state and protein levels of c-Met in vivo were determined by western blot. U87MG human glioblastoma cells (5 × 106) were injected subcutaneously into athymic (nu/nu) mice. Mice harboring established tumors (~300 mm3) were treated via intraperitoneal (i.p.) injection of vehicle or c-Met antibodies. Tumors were excised and homogenized in NP-40 lysis buffer and c-Met was immunoprecipitated with 25 μl of sc-10 agarose beads (Santa Cruz) for 2 h at 4°C. Phospho- and total c-Met levels were determined in western blots using PY100 (Cell Signaling Technology) or sc-10-HRP (Invitrogen) antibodies, respectively. Results were quantified by measuring chemiluminescent ECL signals by LumiImager (Roche).
The level of circulating c-Met antibodies in athymic mice or cynomolgus monkeys was determined in plasma following intraperitoneal dosing of mice or intravenous (i.v.) dosing of nonhuman primates by ELISA. Microtiter plates (Costar) were coated with 300 ng human Met ECD-Fc (R&D Systems, 358-MT) in PBS and blocked with 0.5% BSA-TBST (Sigma, T9039). Plasma samples were diluted with PBS/1% Tween/10% BSA (PBSTB), added to 96-well microtiter plates for 3 h at room temperature (RT). After washing with TBS-T, anti-Human IgG2-HRP (Zymed, 05-3500, or Jackson ImmunoResearch, 709-035-098) diluted in PBST was incubated for 1 h at RT. Following incubation the plate was washed and TMB (KPL, 5076-18) was added and monitored for color development. A stop solution of 0.9 M H2sub>SO4 was added and read at 450 nm using a Wallac microplate reader.
Sample and QC preparation was performed by sample dilution in PBSTB and adjustment to an appropriate standard concentration range (0.0003 to 1 µg/mL) by serial titration. QCs were made by spiking in known amounts of clone into naïve monkey or mouse serum. QCs were treated like samples and brought into appropriate dilution range. Naïve monkey serum was spiked into PBST at the highest monkey serum concentration (10%) as a negative control.
Immunohistochemistry on U87MG Xenografts
Tumors excised humanely from euthanized athymic mice were fixed by immersion in neutral buffered formalin. Tumor samples were processed to paraffin blocks following routine histology procedures and 5 µm sections cut for immunohistochemical procedures.
To expose antigenic sites, tissue sections were heated in Citra Antigen Retrieval pH 6 (Biogenex) using a steamer at 96°C for 20 min. Immunostaining was performed by incubating tissues with Met pTyr 1234–1235 (Millipore) at a dilution of 1:500; Ki67 (clone MIB1, Dako) at a dilution of 1:100; or cleaved caspase-3 (Cell Signaling) at a dilution of 1:100. Antibody binding sites for pMet Tyr 1234–1235 or cleaved caspase-3 were detected by incubating tissues with a biotinylated secondary goat anti-rabbit antibody (Vector Laboratories) at a dilution of 1:150 followed by incubation with Elite® ABC (Vector Laboratories), or for Ki67 by using the Dako ARK™ Kit (Dako). Dako Liquid DAB+ was used to visualize antibody staining, while Mayer’s hematoxylin was used as a counterstain.
In vivo efficacy of CE-355621
S114, U87MG human glioblastoma and GTL-16 human gastric cancer cells were maintained in DMEM, 10% heat inactivated FBS, 2 mM L-Glutamine, and 10 units/ml penicillin, 10 μg/ml streptomycin at 37°C, 10% CO2. Tumors cells (1.0–5.0 × 106 cells) were inoculated subcutaneously in 0.2 ml Hank’s Buffered Saline Solution into athymic (nu/nu) mice. Once tumors had reached 100–200 mm3 in size, 200 μl of antibody solution diluted with sterile PBS or vehicle alone was injected i.p. into each experimental animal subject, and tumor sizes were measured in the mice using calipers every 2–3 d.
Supplementary Material
Acknowledgments
The manuscript is dedicated to the memory of Elisabeth “Lissie” Knauth, who was an important contributor to this work. In addition, the authors would like to thank David Cunningham and Simone Paillet for their contribution to this body of work.
Potential Conflicts of Interest
Neil R. Michaud, Jitesh P. Jani, Stephen Hillerman, Konstantinos E. Tsaparikos, Elsa G. Barbacci-Tobin, Elisabeth Knauth, Henry Putz Jr, Mary Campbell, George A. Karam, Boris Chrunyk, David F. Gebhard, Jinghai J. Xu, Margaret C. Dunn, Tim M. Coskran, Jean-Martin Lapointe, Bruce D. Cohen, Kevin G. Coleman, Vahe Bedian, Patrick Vincent, Shama Kajiji, Stefan J. Steyn, Gary V. Borzillo, and Gerrit Los are/were employed by Pfizer who provided funding, equipment and supplies for this study. Some of the authors have personal financial interests by owning shares in the company (Pfizer). At the time of the study Larry Green was employed by Abgenix, Inc.
Footnotes
Previously published online: www.landesbioscience.com/journals/mabs/article/22160
References
- 1.Michalopoulos GK, DeFrances MC. Liver regeneration. Science. 1997;276:60–6. doi: 10.1126/science.276.5309.60. [DOI] [PubMed] [Google Scholar]
- 2.Di Renzo MF, Narsimhan RP, Olivero M, Bretti S, Giordano S, Medico E, et al. Expression of the Met/HGF receptor in normal and neoplastic human tissues. Oncogene. 1991;6:1997–2003. [PubMed] [Google Scholar]
- 3.Birchmeier C, Birchmeier W, Gherardi E, Vande Woude GF. Met, metastasis, motility and more. Nat Rev Mol Cell Biol. 2003;4:915–25. doi: 10.1038/nrm1261. [DOI] [PubMed] [Google Scholar]
- 4.Bladt F, Riethmacher D, Isenmann S, Aguzzi A, Birchmeier C. Essential role for the c-met receptor in the migration of myogenic precursor cells into the limb bud. Nature. 1995;376:768–71. doi: 10.1038/376768a0. [DOI] [PubMed] [Google Scholar]
- 5.Schmidt C, Bladt F, Goedecke S, Brinkmann V, Zschiesche W, Sharpe M, et al. Scatter factor/hepatocyte growth factor is essential for liver development. Nature. 1995;373:699–702. doi: 10.1038/373699a0. [DOI] [PubMed] [Google Scholar]
- 6.Gherardi E, Birchmeier W, Birchmeier C, Vande Woude G. Targeting MET in cancer: Rationale and progress. Nat Rev Cancer 2012; 12:89-103; PMID: 22270953; DOI: 10.1038/nrc3205; 10.1038/nrc3205. [DOI] [PubMed]
- 7.Maulik G, Shrikhande A, Kijima T, Ma PC, Morrison PT, Salgia R. Role of the hepatocyte growth factor receptor, c-Met, in oncogenesis and potential for therapeutic inhibition. Cytokine Growth Factor Rev. 2002;13:41–59. doi: 10.1016/S1359-6101(01)00029-6. [DOI] [PubMed] [Google Scholar]
- 8.Gao CF, Vande Woude GF. HGF/SF-Met signaling in tumor progression. Cell Res. 2005;15:49–51. doi: 10.1038/sj.cr.7290264. [DOI] [PubMed] [Google Scholar]
- 9.Ichimura E, Maeshima A, Nakajima T, Nakamura T. Expression of c-met/HGF receptor in human non-small cell lung carcinomas in vitro and in vivo and its prognostic significance. Jpn J Cancer Res. 1996;87:1063–9. doi: 10.1111/j.1349-7006.1996.tb03111.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Han SU, Lee JH, Kim WH, Cho YK, Kim MW. Significant correlation between serum level of hepatocyte growth factor and progression of gastric carcinoma. World J Surg. 1999;23:1176–80. doi: 10.1007/s002689900642. [DOI] [PubMed] [Google Scholar]
- 11.Kuba K, Matsumoto K, Date K, Shimura H, Tanaka M, Nakamura T. HGF/NK4, a four-kringle antagonist of hepatocyte growth factor, is an angiogenesis inhibitor that suppresses tumor growth and metastasis in mice. Cancer Res. 2000;60:6737–43. [PubMed] [Google Scholar]
- 12.Tomioka D, Maehara N, Kuba K, Mizumoto K, Tanaka M, Matsumoto K, et al. Inhibition of growth, invasion, and metastasis of human pancreatic carcinoma cells by NK4 in an orthotopic mouse model. Cancer Res. 2001;61:7518–24. [PubMed] [Google Scholar]
- 13.Martin TA, Parr C, Davies G, Watkins G, Lane J, Matsumoto K, et al. Growth and angiogenesis of human breast cancer in a nude mouse tumour model is reduced by NK4, a HGF/SF antagonist. Carcinogenesis. 2003;24:1317–23. doi: 10.1093/carcin/bgg072. [DOI] [PubMed] [Google Scholar]
- 14.Christensen JG, Burrows J, Salgia R. c-Met as a target for human cancer and characterization of inhibitors for therapeutic intervention. Cancer Lett. 2005;225:1–26. doi: 10.1016/j.canlet.2004.09.044. [DOI] [PubMed] [Google Scholar]
- 15.Cao B, Su Y, Oskarsson M, Zhao P, Kort EJ, Fisher RJ, et al. Neutralizing monoclonal antibodies to hepatocyte growth factor/scatter factor (HGF/SF) display antitumor activity in animal models. Proc Natl Acad Sci U S A. 2001;98:7443–8. doi: 10.1073/pnas.131200498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Comoglio PM, Giordano S, Trusolino L. Drug development of MET inhibitors: targeting oncogene addiction and expedience. Nat Rev Drug Discov. 2008;7:504–16. doi: 10.1038/nrd2530. [DOI] [PubMed] [Google Scholar]
- 17.Zou HY, Li Q, Lee JH, Arango ME, McDonnell SR, Yamazaki S, et al. An orally available small-molecule inhibitor of c-Met, PF-2341066, exhibits cytoreductive antitumor efficacy through antiproliferative and antiangiogenic mechanisms. Cancer Res. 2007;67:4408–17. doi: 10.1158/0008-5472.CAN-06-4443. [DOI] [PubMed] [Google Scholar]
- 18.Tu WH, Zhu C, Clark C, Christensen JG, Sun Z. Efficacy of c-Met inhibitor for advanced prostate cancer. BMC Cancer. 2010;10:556. doi: 10.1186/1471-2407-10-556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ma PC, Schaefer E, Christensen JG, Salgia R. A selective small molecule c-MET Inhibitor, PHA665752, cooperates with rapamycin. Clin Cancer Res. 2005;11:2312–9. doi: 10.1158/1078-0432.CCR-04-1708. [DOI] [PubMed] [Google Scholar]
- 20.Jin H, Yang R, Zheng Z, Romero M, Ross J, Bou-Reslan H, et al. MetMAb, the one-armed 5D5 anti-c-Met antibody, inhibits orthotopic pancreatic tumor growth and improves survival. Cancer Res. 2008;68:4360–8. doi: 10.1158/0008-5472.CAN-07-5960. [DOI] [PubMed] [Google Scholar]
- 21.Martens T, Schmidt NO, Eckerich C, Fillbrandt R, Merchant M, Schwall R, et al. A novel one-armed anti-c-Met antibody inhibits glioblastoma growth in vivo. Clin Cancer Res. 2006;12:6144–52. doi: 10.1158/1078-0432.CCR-05-1418. [DOI] [PubMed] [Google Scholar]
- 22.van der Horst EH, Chinn L, Wang M, Velilla T, Tran H, Madrona Y, et al. Discovery of fully human anti-MET monoclonal antibodies with antitumor activity against colon cancer tumor models in vivo. Neoplasia. 2009;11:355–64. doi: 10.1593/neo.81536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stabile LP, Rothstein ME, Keohavong P, Jin J, Yin J, Land SR, et al. Therapeutic targeting of human hepatocyte growth factor with a single neutralizing monoclonal antibody reduces lung tumorigenesis. Mol Cancer Ther. 2008;7:1913–22. doi: 10.1158/1535-7163.MCT-07-2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Buchanan SG, Hendle J, Lee PS, Smith CR, Bounaud PY, Jessen KA, et al. SGX523 is an exquisitely selective, ATP-competitive inhibitor of the MET receptor tyrosine kinase with antitumor activity in vivo. Mol Cancer Ther. 2009;8:3181–90. doi: 10.1158/1535-7163.MCT-09-0477. [DOI] [PubMed] [Google Scholar]
- 25.Tseng JR, Kang KW, Dandekar M, Yaghoubi S, Lee JH, Christensen JG, et al. Preclinical efficacy of the c-Met inhibitor CE-355621 in a U87 MG mouse xenograft model evaluated by 18F-FDG small-animal PET. J Nucl Med. 2008;49:129–34. doi: 10.2967/jnumed.106.038836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Green LL. Antibody engineering via genetic engineering of the mouse: XenoMouse strains are a vehicle for the facile generation of therapeutic human monoclonal antibodies. J Immunol Methods. 1999;231:11–23. doi: 10.1016/S0022-1759(99)00137-4. [DOI] [PubMed] [Google Scholar]
- 27.Graziani A, Gramaglia D, Cantley LC, Comoglio PM. The tyrosine-phosphorylated hepatocyte growth factor/scatter factor receptor associates with phosphatidylinositol 3-kinase. J Biol Chem. 1991;266:22087–90. [PubMed] [Google Scholar]
- 28.Trusolino L, Bertotti A, Comoglio PM. MET signalling: principles and functions in development, organ regeneration and cancer. Nat Rev Mol Cell Biol. 2010;11:834–48. doi: 10.1038/nrm3012. [DOI] [PubMed] [Google Scholar]
- 29.Jeffers M, Rong S, Anver M, Vande Woude GF. Autocrine hepatocyte growth factor/scatter factor-Met signaling induces transformation and the invasive/metastastic phenotype in C127 cells. Oncogene. 1996;13:853–6. [PubMed] [Google Scholar]
- 30.Rong S, Bodescot M, Blair D, Dunn J, Nakamura T, Mizuno K, et al. Tumorigenicity of the met proto-oncogene and the gene for hepatocyte growth factor. Mol Cell Biol. 1992;12:5152–8. doi: 10.1128/mcb.12.11.5152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rong S, Oskarsson M, Faletto D, Tsarfaty I, Resau JH, Nakamura T, et al. Tumorigenesis induced by coexpression of human hepatocyte growth factor and the human met protooncogene leads to high levels of expression of the ligand and receptor. Cell Growth Differ. 1993;4:563–9. [PubMed] [Google Scholar]
- 32.Tajima H, Matsumoto K, Nakamura T. Regulation of cell growth and motility by hepatocyte growth factor and receptor expression in various cell species. Exp Cell Res. 1992;202:423–31. doi: 10.1016/0014-4827(92)90095-P. [DOI] [PubMed] [Google Scholar]
- 33.Okano Y, Mizuno K, Osada S, Nakamura T, Nozawa Y. Tyrosine phosphorylation of phospholipase C gamma in c-met/HGF receptor-stimulated hepatocytes: comparison with HepG2 hepatocarcinoma cells. Biochem Biophys Res Commun. 1993;190:842–8. doi: 10.1006/bbrc.1993.1125. [DOI] [PubMed] [Google Scholar]
- 34.Neaud V, Faouzi S, Guirouilh J, Le Bail B, Balabaud C, Bioulac-Sage P, et al. Human hepatic myofibroblasts increase invasiveness of hepatocellular carcinoma cells: evidence for a role of hepatocyte growth factor. Hepatology. 1997;26:1458–66. doi: 10.1002/hep.510260612. [DOI] [PubMed] [Google Scholar]
- 35.Gherardi E, Sandin S, Petoukhov MV, Finch J, Youles ME, Ofverstedt LG, et al. Structural basis of hepatocyte growth factor/scatter factor and MET signalling. Proc Natl Acad Sci U S A. 2006;103:4046–51. doi: 10.1073/pnas.0509040103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tolbert WD, Daugherty J, Gao C, Xie Q, Miranti C, Gherardi E, et al. A mechanistic basis for converting a receptor tyrosine kinase agonist to an antagonist. Proc Natl Acad Sci U S A. 2007;104:14592–7. doi: 10.1073/pnas.0704290104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tolbert WD, Daugherty-Holtrop J, Gherardi E, Vande Woude G, Xu HE. Structural basis for agonism and antagonism of hepatocyte growth factor. Proc Natl Acad Sci U S A. 2010;107:13264–9. doi: 10.1073/pnas.1005183107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ohashi K, Marion PL, Nakai H, Meuse L, Cullen JM, Bordier BB, et al. Sustained survival of human hepatocytes in mice: A model for in vivo infection with human hepatitis B and hepatitis delta viruses. Nat Med. 2000;6:327–31. doi: 10.1038/73187. [DOI] [PubMed] [Google Scholar]
- 39.Carter PJ. Potent antibody therapeutics by design. Nat Rev Immunol. 2006;6:343–57. doi: 10.1038/nri1837. [DOI] [PubMed] [Google Scholar]
- 40.Tangri S, Mothé BR, Eisenbraun J, Sidney J, Southwood S, Briggs K, et al. Rationally engineered therapeutic proteins with reduced immunogenicity. J Immunol. 2005;174:3187–96. doi: 10.4049/jimmunol.174.6.3187. [DOI] [PubMed] [Google Scholar]
- 41.Abounader R, Ranganathan S, Lal B, Fielding K, Book A, Dietz H, et al. Reversion of human glioblastoma malignancy by U1 small nuclear RNA/ribozyme targeting of scatter factor/hepatocyte growth factor and c-met expression. J Natl Cancer Inst. 1999;91:1548–56. doi: 10.1093/jnci/91.18.1548. [DOI] [PubMed] [Google Scholar]
- 42.Smolen GA, Sordella R, Muir B, Mohapatra G, Barmettler A, Archibald H, et al. Amplification of MET may identify a subset of cancers with extreme sensitivity to the selective tyrosine kinase inhibitor PHA-665752. Proc Natl Acad Sci U S A. 2006;103:2316–21. doi: 10.1073/pnas.0508776103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Petrelli A, Circosta P, Granziero L, Mazzone M, Pisacane A, Fenoglio S, et al. Ab-induced ectodomain shedding mediates hepatocyte growth factor receptor down-regulation and hampers biological activity. Proc Natl Acad Sci U S A. 2006;103:5090–5. doi: 10.1073/pnas.0508156103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tolgay Ocal I, Dolled-Filhart M, D’Aquila TG, Camp RL, Rimm DL. Tissue microarray-based studies of patients with lymph node negative breast carcinoma show that met expression is associated with worse outcome but is not correlated with epidermal growth factor family receptors. Cancer. 2003;97:1841–8. doi: 10.1002/cncr.11335. [DOI] [PubMed] [Google Scholar]
- 45.Beck A, Reichert JM, Wurch T. 5th european antibody congress 2009: November 30-december 2, 2009, geneva, switzerland. MAbs. 2010;2:108–28. doi: 10.4161/mabs.2.2.11302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Greenall SA, Gherardi E, Liu Z, Donoghue JF, Vitali AA, Li Q, et al. Non-agonistic bivalent antibodies that promote c-MET degradation and inhibit tumor growth and others specific for tumor related c-MET. PLoS One. 2012;7:e34658. doi: 10.1371/journal.pone.0034658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Goetsch L. Antibodies inhibiting c-met dimerization, and therapeutic (anticancer) and diagnostic uses thereof. PCT Int Appl 2009; 2008-EP59026; 2007-301231:175.
- 48.Goetsch L, Wurch T, Bes C. Anti-c-met antibodies for cancer therapy. PCT Int Appl 2011; 2011-EP59139; 2010-791681:119.
- 49.Prat M, Crepaldi T, Pennacchietti S, Bussolino F, Comoglio PM. Agonistic monoclonal antibodies against the Met receptor dissect the biological responses to HGF. J Cell Sci. 1998;111:237–47. doi: 10.1242/jcs.111.2.237. [DOI] [PubMed] [Google Scholar]
- 50.Kong-Beltran M, Stamos J, Wickramasinghe D. The Sema domain of Met is necessary for receptor dimerization and activation. Cancer Cell. 2004;6:75–84. doi: 10.1016/j.ccr.2004.06.013. [DOI] [PubMed] [Google Scholar]
- 51.Pacchiana G, Chiriaco C, Stella MC, Petronzelli F, De Santis R, Galluzzo M, et al. Monovalency unleashes the full therapeutic potential of the DN-30 anti-Met antibody. J Biol Chem. 2010;285:36149–57. doi: 10.1074/jbc.M110.134031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sharma N, Adjei AA. In the clinic: ongoing clinical trials evaluating c-MET-inhibiting drugs. Ther Adv Med Oncol. 2011;3(Suppl):S37–50. doi: 10.1177/1758834011423403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Surati M, Patel P, Peterson A, Salgia R. Role of MetMAb (OA-5D5) in c-MET active lung malignancies. Expert Opin Biol Ther. 2011;11:1655–62. doi: 10.1517/14712598.2011.626762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Belalcazar A, Azaña D, Perez CA, Raez LE, Santos ES. Targeting the Met pathway in lung cancer. Expert Rev Anticancer Ther. 2012;12:519–28. doi: 10.1586/era.12.16. [DOI] [PubMed] [Google Scholar]
- 55.Catenacci DV, Henderson L, Xiao SY, Patel P, Yauch RL, Hegde P, et al. Durable complete response of metastatic gastric cancer with anti-Met therapy followed by resistance at recurrence. Cancer Discov. 2011;1:573–9. doi: 10.1158/2159-8290.CD-11-0175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schwall RH, Adams CW, Zheng Z, Romero M, Mai E, Moffat B, et al. Inhibition of cMet activation by a one-armed antibody. AACR Meeting Abstracts 2004; 2004:327-b-. [Google Scholar]
- 57.Reyes AE, II, Xiang H, Bender B, Haughney P, Oldendorp A, Nijem I, et al. Pharmacokinetics of a novel one-armed antibody to C-met in mice, rats, and monkeys. AAPS PharmSci. 2008;10(S2):587. [Google Scholar]
- 58.Institute of Laboratory Animal Resources (U.S.). Guide for the Care and use of Laboratory Animals. Washington, D.C.; Great Britain: National Academy Press, 1996. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.