

Abstract
Background
Interleukin-4 (IL-4) and STAT6 play an important role in progression of allergic airway disease (AAD) or asthma. IL-4 and STAT6 mediate T helper 2 (Th2) responses in T cells, and immunoglobulin class switching to IgE in B cells. Both, Th2 responses and IgE promote the asthmatic condition. We have previously demonstrated that PARP-14, a member of the poly ADP-ribose polymerase (PARP) family of proteins regulates the transcription function of STAT6. However, the role of PARP-14 in AAD is not known.
Objective
Here we investigate the role of PARP-14 and the enzyme activity associated with it in AAD dependent on airway hyper-responsiveness (AHR) and lung inflammation. We also elucidate the mechanism by which PARP-14 regulates AAD.
Methods
The role of PARP-14 and its enzyme activity in AAD and Th2 differentiation were examined using a mouse model of AAD and in vitro T helper cell differentiation.
Results
PARP-14 deficient animals when compared to controls show reduced lung pathology and IgE. Treating mice with a pharmacological inhibitor for PARP activity reduced the severity of AHR and lung inflammation. Mechanistically, our data indicate that PARP-14 and its enzyme activity aid in the differentiation of T cells towards a Th2 phenotype by regulating the binding of STAT6 to the Gata3 promoter.
Conclusion
PARP-14 and the catalytic activity associated with it promote Th2 differentiation and AAD in a murine model, and targeting PARP-14 may be a potential new therapy for allergic asthma.
Keywords: Interleukin-4, STAT6, PARP-14, Th2 cells, Gata3, lung inflammation, airway hyper-responsiveness, PARP inhibitor, allergic airway disease
INTRODUCTION
Asthma is a chronic inflammatory airway disease, which affects more than 300 million people worldwide 1. The cardinal features of asthma are airway hyper-responsiveness (AHR), severe tissue inflammation and goblet cell metaplasia. Traditionally, drugs used to treat asthma promote relaxation of airway smooth muscle (bronchodilators) or suppress airway inflammation (corticosteroids) 1. These drugs are directed towards relieving the symptoms of the asthmatic response not the underlying cause. Dysregulated Th2 immune responses play a key role in the pathogenesis of asthma 2. Therefore, a superior approach for therapy for asthma may be to target Th2 immunity. IL-4 activated STAT6 is involved in Th2 responses 3 and participates in progression of asthma 4–7. Thus, STAT6 is an attractive therapeutic target for asthma.
Previously we have shown that PARP-14, a member of the poly ADP ribose polymerase (PARP) superfamily, acts as a transcriptional switch for IL-4 dependent STAT6 mediated transcription 8. PARP activity contained in PARP-14, is a reversible post-translational modification and is characterized by the addition of ADP-ribose moieties onto acceptor proteins 9. PARP-1 is well studied in DNA damage repair mechanisms and has been observed to play a role in tumor progression. A number of PARP inhibitors are available and are being tested as cancer drugs 10. We have shown that ablation of PARP-14 expression and pharmacological inhibition of PARP activity both results in reduced expression of STAT6 dependent gene expression 8, 11. Based on these observations we hypothesize that PARP-14 promotes Th2 cell differentiation and targeting the catalytic activity of PARP-14 may attenuate a Th2 response resulting in the alleviation of AAD.
In this report, we evaluate the role of PARP-14 on AAD and T cell differentiation. In mice lacking PARP-14 expression, allergen induced airway disease is attenuated and correlates with reduced Th2 cytokines and IgE secretion when compared to controls. Importantly, administration of PJ34, a PARP inhibitor alleviates AAD. We find that PARP-14 modulates the binding of STAT6 to the Gata3 promoter. Taken together, our results provide strong evidence for the involvement of PARP-14 in AAD and our data indicate that inhibiting its activity may be a potential therapy for allergic asthma.
METHODS
Mice
6–8 week old Parp14 −/− mice on a C57BL/6 background and wild-type littermates were used for AAD studies, or BALB/c mice were employed in the PARP inhibitor AAD experiments. Mice were maintained in a pathogen free condition and all studies were approved by Indiana University School of Medicine and the IACUC and adhere to the ARRIVE (Animal Research: Reporting on In Vivo Experiments) guidelines.
In vitro T cultures
Naïve T cells (CD4+CD62high) were isolated from spleens using the MACS system and 1X106 cells were cultured in IMDM-10% FBS under Th1 and Th2 conditions as described previously 12. Differentiated cells were restimulated overnight with anti-CD3/CD28, and cytokines in cell-free supernatants were analyzed by ELISA.
RNAi for PARP-14
Two approaches for acutely inhibiting PARP-14 expression were used, siRNA and Short Hairpin transduction. For detailed method see Online Repository Material.
Induction of AAD and drug administration
Mice were sensitized by intra-peritoneal injections on days 0 and 7 with OVA/alum (20 μg OVA (BALB/c) and 50 μg OVA (C56BL/6) per 2 mg alum). On day 14, mice were challenged with OVA (100 μg) intra-nasally. The progression of AAD was determined by evaluating AHR and analysis of BAL fluid. Mice were administered PJ34, a PARP inhibitor. For detailed methods see online repository material.
Gene expression analysis
Total RNA was isolated using the TRIzol reagent and cDNA was synthesized using the reverse transcriptase. To evaluate gene expression of various cytokines and chemokines quantitative PCR was performed using commercially available primers.
Chromatin immunoprecipitation
ChIP assays were performed on purified CD4 cells essentially as described before 8. The immunoprecipitated DNA was analyzed for S4 and S7 gene fragments from the Gata3 promoter 14 by SYBR green qPCR and expressed as a percentage of the total chromatin in the sample.
ChIP-Seq analysis
Naïve CD4+ T cells were cultured and restimulated essentially as described by Wei et al 15. ChIP-Seq was performed by Active Motif using the TranscriptionPath service on the chromatin isolated from these cells.
RESULTS
PARP-14 and the catalytic activity associated with it participate in Th2 differentiation
It is well established that STAT6 activated by IL-4 plays an important role in the Th2 differentiation 16–18. As we have shown that PARP-14 enhances STAT6 dependent transcription 19, we hypothesized that PARP-14 also participates in Th2 differentiation. Thus, naïve T cells (CD4+CD62L+) were isolated from the spleens of Parp14 −/− mice and WT controls, and cultured under either Th1 or Th2 conditions. The phenotype of the differentiated cells was determined by evaluating the cytokines secreted after secondary stimulation. While, Th2-primed cells with wild-type expression of PARP-14 secreted high levels of IL-4, IL-5 and IL-13, PARP-14-deficient Th2 cells secreted significantly reduced amounts of Th2 cytokines (Figure 1a). However, PARP-14-deficient Th1 cells produced similar amount of IFN-γ as compared to wild-type Th1 cells (Figure 1b).
Figure 1. PARP-14 and its activity are required for optimal Th2 differentiation.
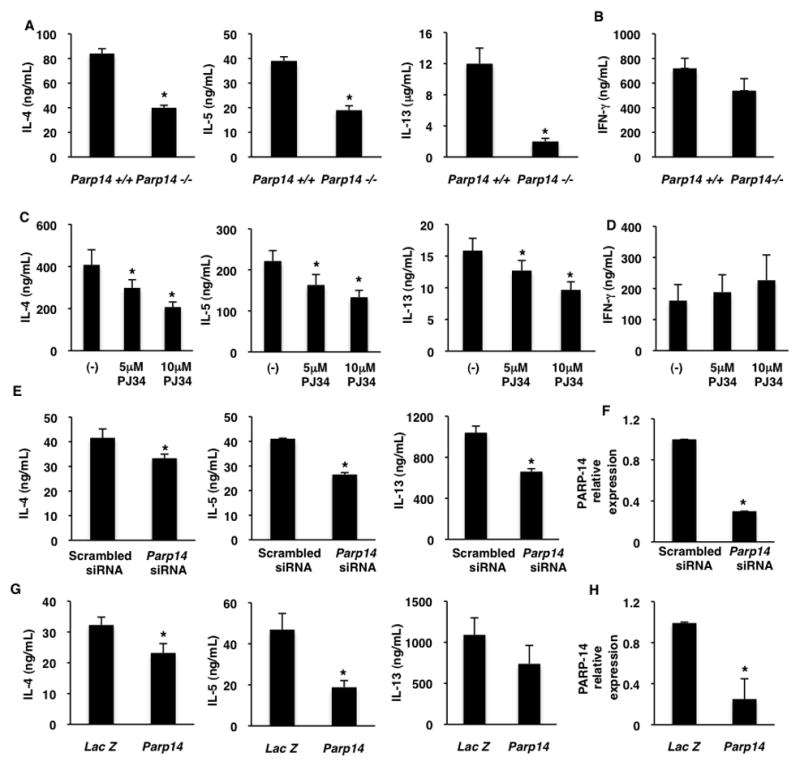
(A–B) Naïve T cell from Parp14−/− and WT mice were cultured under Th2 (A) and Th1 (B) conditions. differentiated cells were re-stimulated and the indicated cytokines were measured by ELISA. (C–D) CD4+CD62Lhigh T cells were cultured under Th2 (C) and Th1 (D) skewing conditions in the presence or absence of PJ34 for 7 days. The indicated cytokines were measured. (E–F) Naïve T cells were nucleofected with the indicated siRNA and then cultured under Th2 conditions for 5 days. Cytokines and PARP-14 expression were measured as indicated. (G–H) Short Hairpins specific for PARP-14 or LacZ were retrovirally transduced into activated T cells and cultured under Th2 conditions. Cytokines and PARP-14 expression were measured as indicated. The results are mean (± SEM) of three different experiments. An asterisk on the graphs indicates a p value of ≤ 0.05 when compared to controls.
To determine if inhibiting PARP enzyme activity would abrogate Th2 differentiation specifically, WT naïve CD4+ T cells were cultured under Th1 or Th2 conditions in the presence or absence of PJ34, a PARP inhibitor. A dose dependent decrease in IL-4, IL-5 and IL-13 was observed in cells treated with PJ34 compared to untreated control samples (Figure 1c). However, similar amounts of IFN-γ were secreted from Th1 skewed cells treated with or without PJ34 (Figure 1d). These results indicate that PARP-14 and the enzyme activity associated with it participate in the differentiation of T helper cells towards a Th2 but not Th1 phenotype.
To determine the effect of acute disruption of PARP-14 expression on Th2 differentiation, Naïve T cells were nucleofected with PARP-14 specific or scrambled siRNA and cultured under Th2 conditions and the phenotype was evaluated after secondary stimulation. Lower levels of Th2 cytokines were observed in the presence of siRNA specific for PARP-14 (Figure 1e) and correspondingly there was lower expression of PARP-14 (Figure 1f and Online Repository Figure E1). A second approach of retrovirally transducing a Short Hairpin (Sh) against PARP-14 into activated T cells was used to specifically reduce PARP-14 expression and determine its effect on Th2 differentiation. Consistent with our previous data significantly lower Th2 cytokines and PARP-14 were observed with a Sh against PARP-14 as compared to LacZ controls (Figure 1g and h). Previously it has been demonstrated that the Sh used to target PARP-14 significantly reduces the protein levels of PARP-14 as well 19. Taken together these data indicate that acute disruption of PARP-14 expression specifically impacts Th2 differentiation.
Parp14 −/− mice are protected from AAD
As the expression of PARP-14 increases in the lungs of allergen sensitized and challenged mice (Online Repository Figure E2). We next determined the exact role of PARP-14 in AAD, wild-type (WT) and PARP-14 deficient mice were sensitized and challenged with OVA. The lung function in mice was assessed by measuring their lung resistance dependent on increasing doses of inhaled methacholine. An increase in lung resistance was observed with increasing concentrations of methacholine in SC WT mice (Figure 2a). Importantly, the magnitude of lung resistance in SC Parp14 −/− mice was significantly decreased compared to WT and similar to control NSNC mice (Figure 2a). We next investigated the inflammation in the lung. The BAL fluid of SC Parp14 −/− mice showed significantly lower numbers of total inflammatory cells compared to Parp14 +/+ mice (Figure 2b). The numbers of eosinophils, T cells, neutrophils and macrophages were significantly reduced in PARP-14 deficient animals as compared to WT (Figure 2b). No significant differences in B cells in the BAL were found between WT and Parp14 −/− mice (Figure 2b). PARP-14 deficient mice displayed reduced peribronchial inflammation when compared to mice with intact PARP-14 expression as observed with H&E staining of lung sections (Online Repository Figure E3). Taken together, these data indicate that PARP-14 plays an important role in allergen induced AHR and inflammation.
Figure 2. PARP-14 plays a role in the development of allergic airway disease.

(A–E) Parp14+/+ and Parp14−/− mice were induced for AAD. (A) Lung Resistance (RL) in the indicated mice sensitized and challenged (SC) with OVA or PBS (NSNC) was measured by invasive Resistance and Compliance (RC) system, B – baseline and S – saline. (B) Cells recovered from the BAL fluid from the experimental animals were counted (left graph) and stained with lineage specific markers (middle and right graphs). mRNA levels of Th2 cytokines (C) and chemokines (D) were measured in the lung homogenates of above mice. (E) IgE ELISA was performed on the BAL fluid. (F) Total and OVA specific IgE levels were measured in the serum collected from the experimental animals. (G) Splenocytes isolated from the mice were re-stimulated and the indicated cytokines were measured. The results plotted are mean values (± SEM) from three independent experiments and * indicates a p value of ≤ 0.05 when compared to WT controls.
Consistent with AHR and inflammation data the expression of IL-4, IL-5 and IL-13 was diminished in lungs of PARP-14 deficient mice in comparison to control mice (Figure 2c). The transcripts of chemokines CCL17/TARC, CCL11/eotaxin-1 and CCL24/eotaxin-2 that are required for the recruitment of T cells and eosinophils respectively were lower in Parp14 −/− mice as compared to WT littermates (Figure 2d). The levels of IgE in BAL fluid were reduced by 70% in the lungs of Parp14 −/− mice as compared to WT mice (Figure 2e). Airways within lung sections of SC Parp14 +/+ showed a significant increase in goblet cell hyperplasia as indicated by PAS positivity, whereas very few cells positive for PAS were detected in PARP-14 deficient mice (Online Repository Figure E3). Taken together, all of the above data demonstrate that PARP-14 plays a role in AAD by modulating inflammatory mediators and AHR.
To investigate if PARP-14 is required for systemic Th2 immune responses, the amount of total and OVA-specific IgE in serum were measured, and were found to be greater in Parp14 +/+ than in Parp14 −/− samples (Figure 2f). Also, restimulated splenocytes isolated from SC mice showed significant reduction in IL-4, IL-5 and IL-13 but not IFN-γ secretion with PARP-14 deficiency (Figure 2g). Thus, ablation of PARP-14 lead to reduced systemic responses and these data indicate a pro-allergic role of PARP-14.
Inhibition of PARP activity during antigen sensitization and challenge reduces allergic airway disease
As PARP-14 contains the PARP catalytic domain, and as we have shown previously that the PARP activity of PARP-14 regulates the transcription function of STAT6 8, 11, we next tested if inhibiting the PARP catalytic activity would alleviate AAD. Mice were sensitized and challenged with OVA as before and a PARP inhibitor (PJ34), which has previously been shown to inhibit PARP-14 8, was administered via three different routes during induction of AAD. PJ34 was delivered during allergen challenge (PJ34 C), during both sensitization and challenge (PJ34 SC) and was injected multiple times during the sensitization phase (mPJ34 S) (Online Repository Figure E4). These groups of mice were analyzed in a similar manner as that of the WT and Parp14 −/− experiment. All of the PJ34 treated experimental groups were compared to the NSNC and SC controls. For some of the graphs we have not shown the NSNC controls as the values for this group were negligible. SC mice showed a robust increase of inflammatory cells in the BAL fluid, whereas all the three PJ34 treatment protocols showed a decrease in cellular infiltration (Figure 3a). The numbers of eosinophils, T cells, B cells, macrophages and neutrophils mimicked the total BAL cell count data (Figure 3b). These data were confirmed by histological analysis of lung sections of mice stained with H&E (Online Repository Figure E5). Consistent with the BAL data, mice in which PJ34 was given intra-nasally during challenge showed very modest reductions in inflammation (Online Repository Figure E5). The expression of IL-4, IL-5 and IL-13 in lung tissue was reduced with PJ34 treatment, the most profound effect was observed in mice treated with PJ34 during both sensitization and challenge (Figure 3c). The expression levels of CCL11, CCL17 and CCL24 followed a similar pattern as that of the number of eosinophils and T cells recruited to the lung (Figure 3c). IgE levels in BAL fluid in mice treated with PJ34 showed a significant reduction from the untreated controls (Figure 3d). A decrease in AHR to methacholine was also observed in mice treated with PJ34 compared to untreated control animals (Figure 3e). Histological analysis revealed the attenuation of mucous hyper-secretion by goblet cells in PJ34 treated mice as compared to the control mice (Online Repository Figure E5). The above data indicates the requirement of PARP activity in the pathogenesis of lung inflammatory response and lung reactivity against OVA antigen.
Figure 3. Inhibition of PARP enzymatic activity during establishment of AAD reduces OVA induced lung disease.

(A–G) BALB/c mice were immunized and challenged with OVA and administered with PJ34 as described in online repository Figure E4. BAL fluid was collected from the experimental mice and cells were counted (A), and stained for specific cell populations (B). (C) Relative expression of the indicated cytokines and chemokines was determined. (D) Amount of IgE was measured in BAL fluid. (E) Airway reactivity in each group of mice was determined as lung resistance (RL) to increasing concentrations of methacholine dissolved in saline (S). (F) Splenocytes were re-stimulated and levels of the indicated cytokines was measured. (G) Total and OVA specific IgE were measured in serum collected. Each of the above graphs is means (± SEM) of three independent experiments with each experiment having 5 mice per group. An asterisk on the graphs indicates a p value of ≤ 0.05 when compared to controls.
Splenocytes isolated from the same group of mice were stimulated with 100μg of OVA for 72 hrs and the secreted cytokines were measured. IL-4, IL-5 and IL-13 were all reduced in PJ34 treated mice, with the maximum decrease seen when PJ34 was given during sensitization and challenge (Figure 3f). Conversely, levels of IFN-γ, a classical Th1 cytokine, were increased in two of the PJ34 treatment protocols as compared to control samples (Figure 3f). Lastly, we evaluated IgE levels in the serum, and administration of PJ34 attenuated OVA induced IgE (Figure 3g). Taken together, our data indicate that inhibition of PARP enzymatic activity during induction of AAD alleviates allergen induced airway disease in mice. All three routes of PJ34 administration were effective, but the maximum effect was observed when PJ34 was given during sensitization and challenge. As PJ34 is a general PARP inhibitor, from the above data it cannot be inferred that PJ34 is inhibiting the PARP activity of PARP-14 to alleviate AAD. To address this we treated WT and PARP-14 deficient animals with PJ34 and observed no further decrease in the AAD phenotype when Parp14 −/− mice were treated with PJ34 as compared to WT mice treated with inhibitor (Online Repository Figure E6). These data indicate that PJ34 treatment is primarily targeting PARP-14 in this model.
Administration of a PARP inhibitor to mice with established AAD alleviates the disease pathology
Our data indicate that inhibiting PARP activity during the induction of AAD reduces the disease phenotype. We next tested if administration of a PARP inhibitor to mice with established AAD would also alleviate the pathology. Mice were sensitized with OVA like before and then to establish AAD mice were challenged with intranasal OVA for 10 days. On the 5th day of OVA challenge a group of mice were sacrificed and the AAD phenotype was evaluated like before. We confirmed that the mice at this time point had full blown AAD by evaluating recruitment of inflammatory cells, airways resistance, cytokine and IgE production (Online Repository Figure E7&8). To test if PJ34 would lessen the disease phenotype, the OVA challenges were continued for an additional five days, and during this time mice were either left untreated (SC (−)), or treated with PJ34 (SC PJ34) (Online Repository Figure E7). The inflammation in the lung was reduced significantly in mice that were treated with PJ34 as evaluated by BAL cell count and H&E staining of lung sections (Figure 4a and Online Repository Figure E9). The lung function was also significantly improved in mice that were treated with PJ34 (Figure 4b). The levels of IL-4, IL-5, IL-13 and IgE were all reduced in mice that were administered with a PARP inhibitor with the exception of IFN-γ (Figure 4c–e). Inflammation around airways and goblet cell hyperplasia were consistent with this data (Online Repository Figure E9). Taken together, our data indicate that inhibiting PARP activity during the induction of AAD, or during an ongoing allergen challenge can be effective to alleviate the disease.
Figure 4. Administration of PJ34 reduces lung disease in an established AAD model.
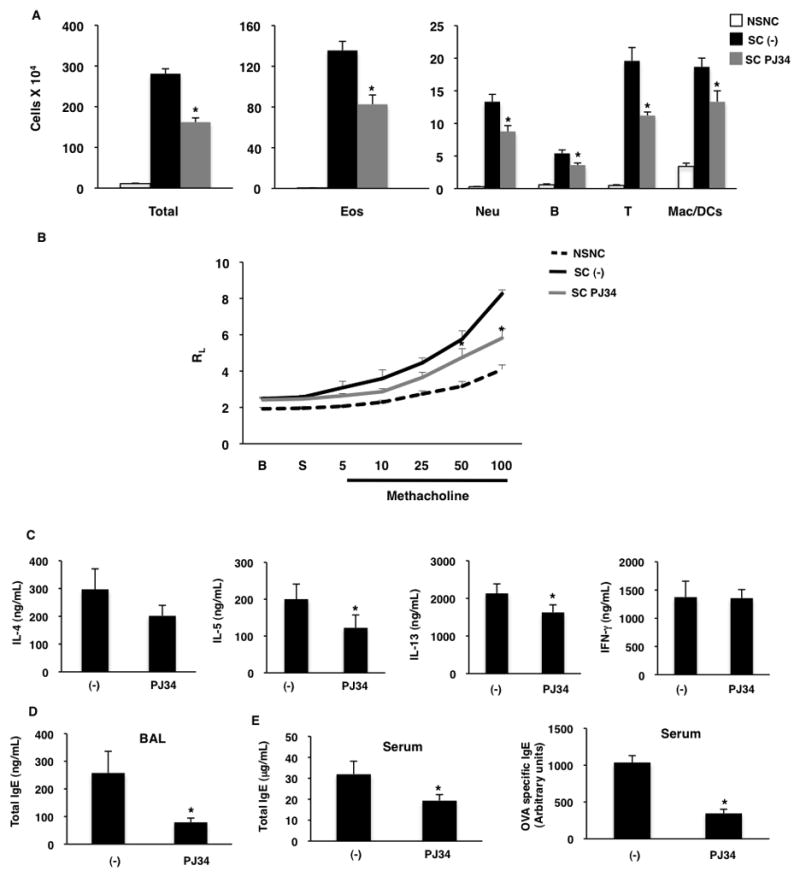
(A–E)AAD was induced in mice and treated with PJ34 as described in online repository Figure E7. (A) Inflammatory cells in the BAL fluid were counted (left graph) and stained for lineage specific markers (middle and right graph). (B) Lung resistance (RL) in response to increasing doses of bronchoconstrictor was measured. (C) Splenocytes were re-stimulated and cytokines were measured. (D) IgE levels were measured in the BAL fluid. (E) Amount of total and OVA specific IgE in the serum of these mice was also determined. The above results are a mean (± SEM) of two independent experiments with 4–5 mice in each group for each experiment. A p value of ≤ 0.05 is indicated by * symbol.
PARP-14 and its enzyme activity regulate expression of GATA3 by modulating the binding of STAT6 to its promoter
To define the mechanism by which PARP-14 participates in Th2 cell differentiation and hence impact AAD, we compared the global expression profile of genes between WT and Parp14 −/− Th2 cells. Naïve T cells were isolated from WT and Parp14 −/− mice and differentiated under Th2 conditions. These cells were then used for ChIP-seq analysis with an antibody directed against the active form of RNA polymerase II. Using this approach we were able to capture active transcription profile of genes on a genomic scale in Th2 cells. We evaluated the peak profiles and average peak values of the Th2 cytokine genes Il4, Il5 and Il13 in WT and Parp14 −/− Th2 cells (Figure 5a and Table 1). Consistent with our ELISA data the peak intensity of all three cytokines was markedly reduced with PARP-14 deficiency (Figure 1a, 5a and Table 1). We next screened for the RNA pol II binding profile of Batf, Gfi1, Irf4, Gata3 and Maf genes that are implicated in Th2 differentiation 20–24. The peak profile and average peak value of only Gata3 was reduced due to PARP-14 deficiency, the other transcription factor genes had comparable values in WT and Parp14 −/− samples (Figure 5b and Table 1). These data suggest that PARP-14 may impact Th2 differentiation by regulating the expression of GATA3.
Figure 5. PARP-14 and activity associated with it is required for ambient GATA-3 expression.

Naïve CD4+ T cells from Parp14+/+ and Parp14−/− mice were differentiated under Th2 conditions and then re-stimulated in the presence of IL-4. ChIP-Seq was performed on these cells using an antibody directed towards the active form of RNA Polymerase II antibody. Screen capture views of ChIP-seq signal profile maps for the indicated cytokines (A) and transcriptional factors (B) from the genome browser are shown. (C) CD4+ cells isolated from Parp14+/+ and Parp14−/− mice were cultured for 5 days under Th2 conditions and Gata3 mRNA levels was measured on each day. (D) CD4+ cells were activated in the presence of IL-4 for 1 hr. ChIP was performed using indicated antibodies. The immunoprecipitated DNA was evaluated for Gata3 promoter fragments, S4 (left graph) and S7 (right graph). (E) CD4+ cells were cultured under Th2 conditions with or without 10μM PJ34. Transcripts of Gata3 were quantified using qPCR. (F) CD4+ T cells isolated from WT mice were stimulated with IL-4 in the presence or absence of 10μM PJ34 for 1 hr. ChIP experiments were performed as in (D). The results are mean (± SEM) of three independent experiments. The asterisk indicates a p value of ≤ 0.05 when compared to controls.
Table 1.
The values indicated are the average peak intensities of the ChIP-seq signal for each cytokine gene.
| Gene | Parp14 +/+ | Parp14 −/− |
|---|---|---|
| IL-4 | 19.065 | 7.469 |
| IL-5 | 5.224 | 0.732 |
| IL-13 | 10.730 | 3.569 |
| IFN-γ | 2.972 | 2.933 |
| Batf | 5.904 | 6.560 |
| Gfi1 | 6.088 | 5.662 |
| Irf4 | 19.016 | 18.963 |
| Gata3 | 18.697 | 14.529 |
| Maf | 5.413 | 4.745 |
To confirm if GATA3 expression is regulated by PARP-14, we cultured CD4 positive T cells from WT and Parp14 −/− mice under Th2 conditions and determined the level of Gata3 expression by qPCR during the differentiation process. The expression of Gata3 was significantly reduced at the later time points during Th2 differentiation in the absence of PARP-14 expression (Figure 5c). Consistent to these data the silencing of PARP-14 by ShRNA targeting PARP-14 resulted in reduced expression of Gata3 (Online Repository Figure E10). To determine if PARP-14 impacted the binding of STAT6 to the Gata3 promoter so as to regulate its expression, we determined the binding ability of STAT6 to two binding sites within the Gata3 regulatory region 14 with and without PARP-14 expression. STAT6 ChIP experiments were performed on T cells from WT and Parp14 −/− and the level of the Gata3 promoter fragments was quantified by qPCR. Consistent with the Gata3 expression data, the binding of STAT6 to both promoter regions was significantly reduced in PARP-14 deficient T cells (Figure 5d). Next we determined if PARP enzymatic would also regulate Gata3 expression. Inhibiting PARP activity had a profound effect on Gata3 expression throughout the differentiation process (Figure 5e). Consistent with this data, inhibition of PARP activity resulted in decreased STAT6 binding to the Gata3 promoter (Figure 5f). Taken together, these data indicate that PARP-14 and the enzyme activity associated with it participate in Th2 differentiation by regulating the binding ability of STAT6 to the Gata3 promoter.
DISCUSSION
In this study we have provided evidence that a PARP family member, PARP-14 regulates the pathogenesis of AAD in a mouse model. Our data demonstrate that with PARP-14 deficiency and inhibiting the catalytic activity associated with PARP-14, the severity of AHR and airway inflammation induced by an allergen are markedly reduced. We have found that PARP-14 through its catalytic activity modulates Th2 differentiation. Mechanistically, we demonstrate that PARP-14 regulates binding of STAT6 to the Gata3 promoter. Our data indicate that there is residual responsiveness to allergen in the absence of PARP-14, indicating that PARP-14 may provide part of the transcription activation potential to STAT6. The other cofactors that aid STAT6 are still available in the absence of PARP-14 to mediate the allergic response. Based on these studies we propose that PARP-14 is an important mediator of Th2 mediated AAD and targeting its catalytic activity may be a potential therapy for allergen induced asthma.
The current therapies of asthma target the symptoms of asthma rather than the underlying cause. Th2 cells activated by innocuous antigens are the initiators of the allergic asthmatic condition 2. Therefore, targeting Th2 cells via PARP-14 may be a more effective therapy for this form of asthma. Here we show that administration of the PARP inhibitor during ongoing allergen exposure alleviates AAD in mice, suggesting that administering PARP inhibitors to a patient undergoing allergen challenge may be effective in alleviating the disease. Moreover, when the PARP inhibitor is administered during the sensitization phase of our AAD protocol, the allergic disease in the mice is also reduced. Our data indicate that there is a reduction of Th2 responses and this is in turn is responsible for the alleviation of the allergic disease. Taken together, our data provide a proof of principle that inhibiting PARP catalytic activity can affect both local and systemic Th2 responses, and this may be an effective therapy for allergic asthma.
The PARP inhibitor, PJ34 used in these studies is a general PARP inhibitor and does not discriminate amongst different PARP enzymes. Therefore, our data with the PJ34 itself does not prove that it is targeting PARP-14 specifically to reduce the allergic response. However, we provide evidence that when PJ34 is used in Parp14 −/− mice there is no further reduction in AAD, suggesting that PJ34 targets predominantly PARP-14 (Online Repository Figure E6). Interestingly, the expression of IL-5 in in vitro differentiated Parp14 −/− Th2 cells is significantly reduced in the presence of PJ34 as compared to without the inhibitor. These data suggest that PJ34 may be inhibiting another PARP enzyme besides PARP-14 to decrease the expression of IL-5. Indeed, PARP-1 has been shown to regulate IL-5 expression by regulating the calpain-dependent degradation of STAT6 25. This study showed that when PARP-1 was absent there were reduced levels of STAT6 protein that impacted the expression of IL-5. The same group previously had shown that PARP-1 deficiency or inhibiting PARP activity with another PARP inhibitor (TIQ-A), resulted in reduced IL-5 production but not IL-4 in their OVA/Alum AAD model 26. There are several differences in this study and ours: First, our study uses 6–10 intranasal challenges with OVA which mimics a more severe model of allergen challenge as compared to one aerosolized administration of the antigen. Secondly, our data shows that PARP-14 and inhibition of PARP activity play a role in Th2 differentiation which impacts the expression of all Th2 cytokines not only IL-5. Third, the route of administration of PARP inhibitor in the other study was limited as it was given as a single i.p. injection, 2 hours prior to challenge with antigen. This would not attenuate the Th2 differentiation process that takes place during the sensitization phase. In this present study we have used several protocols for the administration of the PARP inhibitor, and show that PARP inhibition targets Th2 differentiation systemically and not only the inflammatory response that ensues right after local challenge with an antigen. Nevertheless, both, the previous work and the present study indicate that inhibition of PARP activity is an attractive therapy for asthma. Moreover, both these studies indicate that there is a need to identify specific inhibitors for the different PARP enzymes, as each enzyme may play a distinct biological role.
Our ex vivo data demonstrate that PARP-14 deficiency and inhibiting PARP activity results in the attenuation of all signature Th2 cytokines. We have shown that amongst all of the transcription factors involved in Th2 differentiation, only the expression of Gata3 is dependent on PARP-14 and its catalytic activity (Figure 5). As the expression of GATA3 is dependent on IL-4 and STAT6 27 our data imply that PARP-14 may regulate the expression of Gata3 via STAT6. Indeed, we observe that the binding of STAT6 to two regions within the Gata3 promoter is diminished in the absence of PARP-14 or its catalytic activity (Figure 5). These data are consistent with our previous finding that PARP-14 and its enzymatic activity impact the expression of Iε and Fcer2a by modulating the binding of STAT6 to the promoter elements of these genes 8. Hence, we can speculate that the reduced expression of IgE in our in vivo model may be due to the B cell specific intrinsic role of PARP-14 in immunoglobulin class switching to IgE. From this current study we cannot rule out the possibility that the reduction in IgE is due to reduced help from the T cell. Further studies will be needed to address the exact role of PARP-14 in immunoglobulin class switching.
Here we have provided strong evidence that PARP-14 and the catalytic activity associated with it plays a role in AAD dependent on IL-4 and STAT6. There are several other allergic conditions such as, atopic dermatitis and eosinophillic esophagitis whose pathologies are associated with IL-4 and STAT6. It will be important to study if PARP-14 plays a role in the pathogenesis of these other diseases, and if targeting PARP activity could be a potential therapy for these conditions as well.
Key Messages.
PARP-14, a cofactor for STAT6, participates in allergic airway disease by impacting allergen induced airway hyper-responsiveness and inflammation in the lung.
Inhibiting the catalytic activity associated with PARP-14 alleviates the pathogenesis of allergic airway disease in mice implicating this as a potential new therapy for asthma.
PARP-14 and the enzyme activity associated with it participate in AAD by promoting Th2 differentiation and regulating Gata3 expression by modulating the DNA binding of STAT6 to the Gata3 promoter.
Acknowledgments
Funding: National Institutes of Health Grant (HL093105) and the Showalter Foundation Grant
We thank Drs. Mark Kaplan, Rebecca Shilling and Robert Tepper for their critical review of this manuscript.
Abbreviations
- IL-4
Interleukin-4
- STAT6
Signal transducer activator of transcription 6
- PARP-14
poly ADP-ribose polymerase-14
- Th2
T helper cell 2
- AAD
Allergic airway disease
- AHR
airway hyper-responsiveness
- NSNC
non-sensitized non-challenged
- SC
sensitized challenged
- OVA
ovalbumin
- BAL
bronchoalveolar lavage
- H&E
hematoxylin and eosin
- PAS
periodic acid-Schiff
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Fanta CH. Asthma. N Engl J Med. 2009;360:1002–14. doi: 10.1056/NEJMra0804579. [DOI] [PubMed] [Google Scholar]
- 2.Broide DH, Finkelman F, Bochner BS, Rothenberg ME. Advances in mechanisms of asthma, allergy, and immunology in 2010. J Allergy Clin Immunol. 2011;127:689–95. doi: 10.1016/j.jaci.2011.01.027. [DOI] [PubMed] [Google Scholar]
- 3.Goenka S, Kaplan MH. Transcriptional regulation by STAT6. Immunol Res. 2011;50:87–96. doi: 10.1007/s12026-011-8205-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kuperman DA, Schleimer RP. Interleukin-4, interleukin-13, signal transducer and activator of transcription factor 6, and allergic asthma. Curr Mol Med. 2008;8:384–92. doi: 10.2174/156652408785161032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sehra S, Bruns HA, Ahyi AN, Nguyen ET, Schmidt NW, Michels EG, et al. IL-4 is a critical determinant in the generation of allergic inflammation initiated by a constitutively active Stat6. J Immunol. 2008;180:3551–9. doi: 10.4049/jimmunol.180.5.3551. [DOI] [PubMed] [Google Scholar]
- 6.Kuperman D, Schofield B, Wills-Karp M, Grusby MJ. Signal transducer and activator of transcription factor 6 (Stat6)-deficient mice are protected from antigen-induced airway hyperresponsiveness and mucus production. J Exp Med. 1998;187:939–48. doi: 10.1084/jem.187.6.939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Akimoto T, Numata F, Tamura M, Takata Y, Higashida N, Takashi T, et al. Abrogation of bronchial eosinophilic inflammation and airway hyperreactivity in signal transducers and activators of transcription (STAT)6-deficient mice. J Exp Med. 1998;187:1537–42. doi: 10.1084/jem.187.9.1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mehrotra P, Riley JP, Patel R, Li F, Voss L, Goenka S. PARP-14 functions as a transcriptional switch for Stat6-dependent gene activation. J Biol Chem. 2011;286:1767–76. doi: 10.1074/jbc.M110.157768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schreiber V, Dantzer F, Ame JC, de Murcia G. Poly(ADP-ribose): novel functions for an old molecule. Nat Rev Mol Cell Biol. 2006;7:517–28. doi: 10.1038/nrm1963. [DOI] [PubMed] [Google Scholar]
- 10.Virag L, Szabo C. The therapeutic potential of poly(ADP-ribose) polymerase inhibitors. Pharmacol Rev. 2002;54:375–429. doi: 10.1124/pr.54.3.375. [DOI] [PubMed] [Google Scholar]
- 11.Goenka S, Cho SH, Boothby M. Collaborator of Stat6 (CoaSt6)-associated poly(ADP-ribose) polymerase activity modulates Stat6-dependent gene transcription. J Biol Chem. 2007;282:18732–9. doi: 10.1074/jbc.M611283200. [DOI] [PubMed] [Google Scholar]
- 12.Stritesky GL, Muthukrishnan R, Sehra S, Goswami R, Pham D, Travers J, et al. The transcription factor STAT3 is required for T helper 2 cell development. Immunity. 2011;34:39–49. doi: 10.1016/j.immuni.2010.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.van Rijt LS, Kuipers H, Vos N, Hijdra D, Hoogsteden HC, Lambrecht BN. A rapid flow cytometric method for determining the cellular composition of bronchoalveolar lavage fluid cells in mouse models of asthma. J Immunol Methods. 2004;288:111–21. doi: 10.1016/j.jim.2004.03.004. [DOI] [PubMed] [Google Scholar]
- 14.Onodera A, Yamashita M, Endo Y, Kuwahara M, Tofukuji S, Hosokawa H, et al. STAT6-mediated displacement of polycomb by trithorax complex establishes long-term maintenance of GATA3 expression in T helper type 2 cells. J Exp Med. 2010;207:2493–506. doi: 10.1084/jem.20100760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wei L, Vahedi G, Sun HW, Watford WT, Takatori H, Ramos HL, et al. Discrete roles of STAT4 and STAT6 transcription factors in tuning epigenetic modifications and transcription during T helper cell differentiation. Immunity. 2010;32:840–51. doi: 10.1016/j.immuni.2010.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kaplan MH, Schindler U, Smiley ST, Grusby MJ. Stat6 is required for mediating responses to IL-4 and for development of Th2 cells. Immunity. 1996;4:313–9. doi: 10.1016/s1074-7613(00)80439-2. [DOI] [PubMed] [Google Scholar]
- 17.Shimoda K, van Deursen J, Sangster MY, Sarawar SR, Carson RT, Tripp RA, et al. Lack of IL-4-induced Th2 response and IgE class switching in mice with disrupted Stat6 gene. Nature. 1996;380:630–3. doi: 10.1038/380630a0. [DOI] [PubMed] [Google Scholar]
- 18.Takeda K, Tanaka T, Shi W, Matsumoto M, Minami M, Kashiwamura S, et al. Essential role of Stat6 in IL-4 signalling. Nature. 1996;380:627–30. doi: 10.1038/380627a0. [DOI] [PubMed] [Google Scholar]
- 19.Goenka S, Boothby M. Selective potentiation of Stat-dependent gene expression by collaborator of Stat6 (CoaSt6), a transcriptional cofactor. Proc Natl Acad Sci U S A. 2006;103:4210–5. doi: 10.1073/pnas.0506981103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Betz BC, Jordan-Williams KL, Wang C, Kang SG, Liao J, Logan MR, et al. Batf coordinates multiple aspects of B and T cell function required for normal antibody responses. J Exp Med. 2010;207:933–42. doi: 10.1084/jem.20091548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhu J, Guo L, Min B, Watson CJ, Hu-Li J, Young HA, et al. Growth factor independent-1 induced by IL-4 regulates Th2 cell proliferation. Immunity. 2002;16:733–44. doi: 10.1016/s1074-7613(02)00317-5. [DOI] [PubMed] [Google Scholar]
- 22.Ahyi AN, Chang HC, Dent AL, Nutt SL, Kaplan MH. IFN regulatory factor 4 regulates the expression of a subset of Th2 cytokines. J Immunol. 2009;183:1598–606. doi: 10.4049/jimmunol.0803302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zheng W, Flavell RA. The transcription factor GATA-3 is necessary and sufficient for Th2 cytokine gene expression in CD4 T cells. Cell. 1997;89:587–96. doi: 10.1016/s0092-8674(00)80240-8. [DOI] [PubMed] [Google Scholar]
- 24.Ho IC, Hodge MR, Rooney JW, Glimcher LH. The proto-oncogene c-maf is responsible for tissue-specific expression of interleukin-4. Cell. 1996;85:973–83. doi: 10.1016/s0092-8674(00)81299-4. [DOI] [PubMed] [Google Scholar]
- 25.Datta R, Naura AS, Zerfaoui M, Errami Y, Oumouna M, Kim H, et al. PARP-1 deficiency blocks IL-5 expression through calpain-dependent degradation of STAT-6 in a murine asthma model. Allergy. 2011;66:853–61. doi: 10.1111/j.1398-9995.2011.02549.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Oumouna M, Datta R, Oumouna-Benachour K, Suzuki Y, Hans C, Matthews K, et al. Poly(ADP-ribose) polymerase-1 inhibition prevents eosinophil recruitment by modulating Th2 cytokines in a murine model of allergic airway inflammation: a potential specific effect on IL-5. J Immunol. 2006;177:6489–96. doi: 10.4049/jimmunol.177.9.6489. [DOI] [PubMed] [Google Scholar]
- 27.Paul WE. What determines Th2 differentiation, in vitro and in vivo? Immunol Cell Biol. 2010;88:236–9. doi: 10.1038/icb.2010.2. [DOI] [PubMed] [Google Scholar]