

Abstract
The Na+/Ca2+ exchanger (NCX) is a bi-directional regulator of cytosolic Ca2+, causing Ca2+ efflux in forward-mode and Ca2+ influx in reverse-mode. We hypothesized that reverse-mode NCX is a means of Ca2+ entry in rat aorta (RA) and vena cava (RVC). NCX protein in RA and RVC was confirmed by immunoprecipitation. To assess NCX function, isometric contraction and intracellular Ca2+ was measured in RA and RVC rings in response to low extracellular Na+, endothelin-1 (ET-1), and KCl, in the presence or absence of the NCX antagonist KB-R7943. In RVC, low extracellular Na+ caused vasoconstriction and an increase in intracellular Ca2+ that was attenuated by 10 μM KB-R7943. KB-R7943 (10 μM) attenuated maximal contraction to ET-1 in RVC (53±9% of control), but not RA (91±1% of control). KB-R7943 (10 μM) reduced the maximal contraction to KCl in RA (48±5%) and nearly abolished it in RVC (9±2%), suggesting that voltage-dependent Ca2+ influx may be inhibited by KB-R7943 as well. However, the L-type Ca2+ channel inhibitor nifedipine (1 μM) did not alter ET-1-induced contraction. Our findings suggest that reverse-mode NCX is an important mechanism of Ca2+ influx in RVC but not RA, especially during ET-1-induced contraction. Also, the effects of KB-R7943 on ET-1-induced contraction of RA and RVC are predominantly mediated by reverse-mode NCX inhibition and not due to off-target inhibition of Ca2+ channels.
Keywords: Na+/Ca2+ Exchanger, vasoconstriction, veins, calcium, endothelin-1
1. Introduction
The Na+/Ca2+ exchanger (NCX) is a bi-directional regulator of cytosolic Ca2+, capable of both Ca2+ influx and Ca2+ efflux [1]. In forward mode, the NCX transports Ca2+ out of cells; in reverse mode, NCX takes up extracellular Ca2+. As the NCX does not hydrolyze ATP to provide energy for ion transport, the direction of Ca2+ movement through the NCX depends on the net electrochemical gradients for Na+ and Ca2+ [1,2]. Ion transport by the NCX is itself electrogenic, with a stoichiometry of 3 Na+ ions exchanged for each Ca2+ ion [3]. The direction of transport is associated with changes in membrane potential, whereby membrane depolarization augments Ca2+ influx and reduces Ca2+ efflux through the NCX and vice versa [4]. Thus, both the function and regulation of the NCX are highly complex as they depend on the ionic concentration, membrane potential, and the electrogenic nature of the Na+/Ca2+ exchange.
Ca2+ regulation by the NCX is thought to be important in the maintenance of arterial tone and blood pressure [5]. Animals overexpressing smooth muscle NCX have elevated blood pressure and salt-sensitive hypertension [6]. Likewise, knockout of smooth muscle NCX decreases vasoconstriction and lowers blood pressure [7]. The relationship between increased NCX expression and increased arterial tone implies that Ca2+ influx through the reverse-mode NCX is an important determinant of arterial smooth muscle tone [8].
While a growing body of evidence suggests that venous tone contributes to blood pressure maintenance [9], little is known about the mechanisms regulating venous smooth muscle calcium handling and contraction. Two mathematical models, based upon research conducted using rabbit inferior vena cava, predict that Na+ influx and subsequent reverse-mode NCX activation are required for sarcoplasmic stores refilling during vascular smooth muscle contraction [10,11]. It remains unclear if the reverse-mode NCX is an important regulator of venous smooth muscle tone.
Pharmacological investigation of NCX function is hampered by the lack of commercially available NCX inhibitors. Only one NCX inhibitor, 2-(2-(4-(4-nitrobenzyloxy)phenyl)ethyl)-isothiourea methanesulfonate (KB-R7943), was both readily available and usable in our experiments. The compound SN-6, while also characterized as an NCX inhibitor, did not remain soluble for the duration of our experiments and thus could not be used as a comparator. Another NCX inhibitor, SEA-0400, is reported to have increased potency and selectivity as compared to KB-R7943. However, this compound currently is not commercially available. Thus, KB-R7943 is the only pharmacological tool currently available to assess NCX function. In this study, we used KB-R7943 to test the hypothesis that reverse-mode NCX is a means of Ca2+ entry in rat aorta (RA) and vena cava (RVC). Additional experiments to assess the specificity of KB-R7943 for the reverse-mode NCX in RA and RVC were performed because of possible off-target effects that may influence the interpretation of our results and the paucity of commercially-available NCX inhibitors.
2. Methods
2.1. Animal Care and Use
All procedures that involved animals were performed in accordance with the Institutional Animal Care and Use Committee and the Guide for the Care and Use of Laboratory Animals at Michigan State University. Normal male Sprague-Dawley rats (SD) (250–300 g) were used. Animals were euthanized with sodium pentobarbital (60 mg/kg i.p.).
2.2. Protein Isolation
Freshly dissected RA and RVC tissues were cleaned of adipose tissue in physiological salt solution (PSS) containing (mM): NaCl, 130; KCl, 4.7; KH2PO4, 1.18; MgSO4·7H2O, 1.17; NaHCO3, 14.8; dextrose, 5.5; Na2EDTA·2H2O, 0.03; CaCl2, 1.6; (pH=7.2). Tissues were then ground with mortar and pestle under liquid nitrogen in 1 ml of ice-cold homogenation buffer (125 mM Tris (pH 6.8), 4% SDS, 20% glycerol, 0.5 mM phenylmethylsulfonyl fluoride, 1 mM orthovanadate, 10 μg/ml aprotinin, 10 μg/ml leupeptin). Homogenate was vortexed, sonicated, transferred to a plastic centrifuge tube, and spun at 4 °C to pellet debris; the supernatant was kept. A Bicinchoninic Acid (BCA) assay was used to determine protein concentration.
2.3. Immunoprecipitation
NCX-1 antibody (2 μg, Swant, Switzerland) was added to 200 μg of RA and RVC tissue homogenate. Two hours after addition, protein A/G agarose beads (30 μl, Santa Cruz Biotechnology, USA) were then added to each sample and tumbled overnight at 4° C. Samples were then centrifuged (2500 rpm for 1 min), after which the supernatant was removed and replaced with fresh phosphate buffered saline (PBS) before re-suspension of precipitate. This process was repeated 3 times. After the final centrifugation, the supernatant was removed and replaced with 35 μl of denaturing sample buffer, boiled for 5 minutes, and centrifuged (2500 rpm for 1 min). The resulting supernatant was separated on a 7.5% SDS-polyacrylamide gel and wet-transferred to PVDF membrane for standard Western analysis using NCX-1 antibody (1:1000; Swant, Switzerland). Positive control for NCX-1 was rat heart homogenate (Santa Cruz Biotechnology, Santa Cruz, CA USA). Band density was quantified using ImageJ software (NIH, USA).
2.4. Isometric Contraction
RA and RVC were dissected and cleaned of outer adipose tissue in PSS. Endothelium-intact tissue rings were then mounted in warmed, aerated PSS (37°C; 95/5% O2/CO2) in isolated tissue baths (20 ml) for measurement of isometric contractile force using a 750 TOBS Tissue Organ Bath System (Danish Myo Technology, Aarhus, Denmark) and PowerLab for Windows (ADInstruments, Colorado Springs, CO, USA). Two rings of RA and two rings of RVC were placed under optimum resting tension (1g for RVC, 4g for RA) [12] and initially challenged with 10−5 M phenylephrine (PE) (RA) or norepinephrine (NE) (RVC) to test for tissue viability. Different agonists were used for the initial challenge because RVC do not respond to PE, and to remain consistent with previously published work [12–16]. Endothelium viability was confirmed by relaxation to 10−6 M acetylcholine after contraction by PE (RA) or NE (RVC). Tissues were washed every 15 minutes until they returned to resting tension. Rings were then incubated with either vehicle or antagonist for 1h prior to addition of agonists. Cumulative concentration response curves or responses to single concentrations of agonists were recorded. The specific agonists and antagonists (and corresponding solvents) were: KB-R7943 (dimethyl sulfoxide), nifedipine (ethanol), ET-1 (dH2O), NE (dH2O) and PE (dH2O).
2.5. Extracellular Calcium
Calcium influx during ET-1-induced contraction was measured as previously described [17]. Briefly, RA and RVC were first incubated in Ca2+-replete PSS and tested for tissue viability (as described above). After washout and upon return to resting tension, tissues were incubated for 30 min in Ca2+-free PSS with 1 mM EGTA. Tissues were then switched to nominally Ca2+-free PSS, without EGTA, for 10 minutes before the addition of a maximum concentration of ET-1 (100 nM). After plateau of any ET-1-induced contraction, cumulative concentration response curves to Ca2+ (1 μM – 3 mM CaCl2) were performed in the presence of ET-1.
2.6. Reverse-mode NCX Function
While no specific agonist of reverse-mode NCX is currently available, removal of extracellular Na+ will cause NCX-dependent Ca2+ influx and elicit a transient contraction of vascular smooth muscle [7]. Therefore, isometric contractility was used to determine if NCX could function in reverse-mode in RA and RVC to cause Ca2+ influx. RA and RVC were hung in a custom-fabricated wire myograph, and challenge with 10−5 M NE (RVC) or PE (RA) confirmed tissue viability. Endothelium viability was confirmed by relaxation to 10−6 M acetylcholine after contraction by PE (RA) or NE (RVC). Tissues were then exposed to vehicle (dimethyl sulfoxide, 0.1%) for 30 min before exposure to low-Na+ (14 mM) PSS for 10 min. To control for changes in osmolarity, N-methyl D-glucamine was substituted for NaCl in low-Na+ PSS. Tissues then recovered in normal-Na+ (145 mM) PSS for 30 minutes. After recovery, tissues were incubated with KB-R7943 (10 μM) for 30 min before a second exposure to low-Na+ PSS for 10 min.
2.7. Intracellular Calcium
Using a custom-fabricated wire myograph imaging chamber, RVC were loaded with the ratiometric Ca2+ indicator Fura 2-AM (Life Technologies, Grand Island, NY, USA) by bath incubation, after application of resting tension as described above. The dye solution was made in Ca2+-replete PSS and contained: 5 μM Fura 2-AM dye, 0.5% dimethyl sulfoxide, and 0.01% Pluronic (Life Technologies, USA). The dye solution was applied to RVC for 1 hour at room temperature, and exchanged once during that time. Before imaging, a 30-minute superfusion with aerated PSS (95/5% O2/CO2) was performed to wash any extracellular dye from the bath, allow for dye de-esterification, and gradual temperature increase to 37°C. Isometric contraction and fluorescence ratio were measured simultaneously during exposure to low-Na+ (14 mM) PSS for 10 min. Fura 2 fluorescence ratio (excitation: 340 and 380 nm; emission: 510 nm) was recorded using a DeltaRam-X multi-wavelength illuminator and a D-104 photomultiplier system (Photon Technologies Int’l (PTI), Birmingham NJ, USA) mounted on a Nikon TE-300 inverted microscope (Nikon Instruments, Melville NY, USA) equipped with a 40x (N.A. 0.75) Plan-Fluor long working-distance objective. To isolate changes in smooth muscle cell Ca2+ from changes in endothelial cell Ca2+, a focal plane was chosen in the cell layer immediately below the endothelium. FeliX software (PTI) was used to control the illuminator and photometer, and record all acquired data.
2.8. Statistical Analysis
All data from contractility experiments were normalized to the maximal tissue contraction during initial adrenergic (PE or NE) challenge [12]. Mean, standard error and variance were calculated from the normalized data. To control for variability between gels, the density of aorta and vena cava samples were normalized to the density of the corresponding positive control sample. Mean, standard error and variance were calculated from these data. For samples of equal variance, statistical significance between groups was established using two-tailed, unpaired Student’s t-tests (α=0.05). For samples of unequal variance, the Mann-Whitney U test was used (α=0.05). Calculations were performed using Microsoft Excel (Microsoft Corporation, USA) or GraphPad Prism (GraphPad Software Inc., USA).
3. Results
3.1. Presence of NCX-1 Protein
Immunoprecipitation, followed by Western blotting, was used to confirm the presence of NCX1 protein in equivalent amounts (200 μg) of RA and RVC whole-tissue homogenate. A distinct band was visible in all immunoprecipitates at ~110 kDa, which corresponds to the expected molecular weight of the NCX1 protein (fig. 1a). An identical band was also present in the positive control (rat heart lysate) and absent in samples from which NCX1 antibody was omitted. Densitometry was then used to quantify NCX1 protein expression in a total of 6 aorta and vena cava samples from two different experiments (fig. 1b). Data were normalized to the density of their respective positive control samples. Neither normalized band density nor raw band density were significantly different between aorta and vena cava immunoprecipitates, indicating that aorta and vena cava express NCX1 protein similarly.
Figure 1.
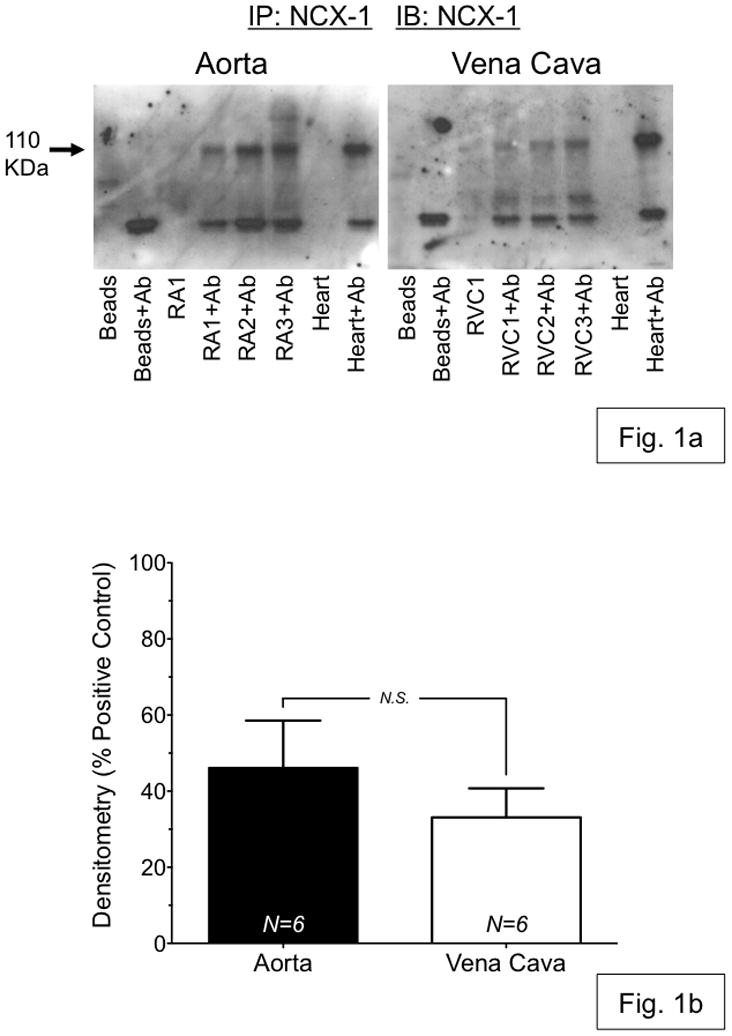
(a) Representative Western blot analysis of immunoprecipitation of NCX1 from 200 μg of whole-tissue protein homogenate from aorta (RA1-3) and vena cava (RVC1-3), isolated from Sprague-Dawley rats. NCX1 protein was immunoprecipitated using protein A/G beads (Santa Cruz Biotechnology, CA USA) and NCX1 antibody (Ab) (Swant, Switzerland). Blots were probed with antibody against NCX1. (b) Densitometry of NCX western blot analysis shows no significant difference in NCX expression between rat aorta (black) and vena cava (white). N.S. = p>0.05; N=6.
3.2. Reverse-mode NCX function
Having established the presence of NCX-1 protein, we proceeded to measure reverse-mode NCX function in RA and RVC using isometric contractility. Rapid reduction of extracellular Na+ (Na+O) is a common experimental test of NCX function, as NCX function is regulated by the Na+ electrochemical gradient [18]. In RA, exposure to low-Na+ PSS (14 mM) for 10 minutes caused a small relaxation followed by a small, transient contraction that was not inhibited by the reverse-mode NCX inhibitor KB-R7943 at either concentration tested (1 μM or 10 μM) (fig. 2a–e). However, low-Na+ PSS exposure caused a sustained contraction in RVC that was not attenuated by KB-R7943 (1 μM), but was significantly reduced by KB-R7943 (10 μM) (fig. 3a–e). Simultaneous measurement of contraction and Fura 2 fluorescence ratio showed that contraction during low-Na+ PSS exposure was accompanied by an increase in intracellular Ca2+, which was attenuated by KB-R7943 (10 μM) (fig. 4a–d). Thus, contraction of vena cava due to low-Na+ exposure is mediated, in part, by Ca2+ influx through KB-R7943-sensitive Ca2+ influx.
Figure 2.

(a–c) Representative tracings of rat aorta contraction, in response to rapid exposure to low-Na+ (~15 mM) physiological salt solution. Shown are responses from tissues incubated with vehicle (DMSO) (a), 1 μM KB-R7943 (b) and 10 μM KB-R7943 (c). Arrows indicate the baseline and maximal contraction used to measure the responses. (d,e) Summary graphs of low-Na+-induced contraction in aorta, in the presence or absence of KB-R7943 (1 μM and 10 μM). All bars represent mean ± SEM for the number of animals indicated. Black bars represent vehicle-exposed tissues (differences between vehicle contractions were not significant). White bars represent tissues exposed to 1 μM KB-R7943 (d) or 10 μM KB-R7943 (e). Results are shown as percentages of initial phenylephrine contraction (PE). N=3–5; * = p<0.05 versus vehicle.
Figure 3.
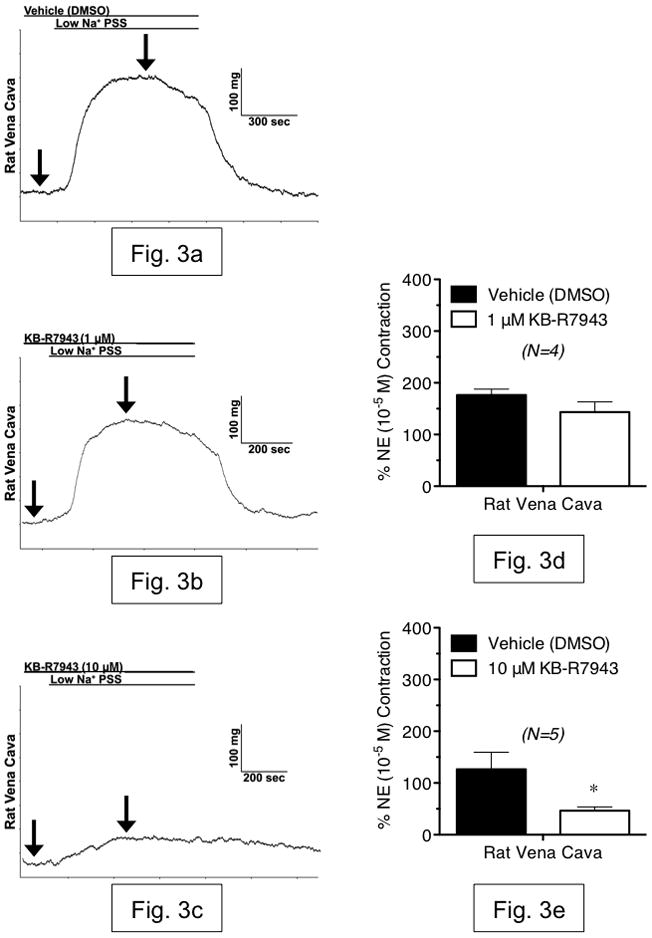
(a–c) Representative tracings of rat vena cava contraction, in response to rapid exposure to low-Na+ (~15 mM) physiological salt solution. Shown are responses from tissues incubated with vehicle (a), 1 μM KB-R7943 (b) and 10 μM KB-R7943 (c). Arrows indicate the baseline and maximal contraction used to measure the responses. (d,e) Summary graphs of low-Na+-induced contraction in vena cava, in the presence or absence of KB-R7943 (1 μM and 10 μM). All bars represent mean ± SEM for the number of animals indicated. Black bars represent vehicle-exposed tissues (differences between vehicle contractions were not significant). White bars represent tissues exposed to 1 μM KB-R7943 (d) or 10 μM KB-R7943 (e). Results are shown as percentages of initial norepinephrine contraction (NE). N=4–5; * = p<0.05 versus vehicle.
Figure 4.

(a,b) Representative measurements of Fura2-AM fluorescence ratio (dashed line, left axis) and contraction (solid line, right axis) in vena cava exposed to low Na+ PSS in the presence of vehicle (a) or 10 μM KB-R7943 (b). (c,d) Summary bar graphs indicating the maximum change in fluorescence ratio (c) and contraction (d) from these same experiments. Black bars represent vehicle-exposed tissues. White bars represent tissues exposed to 10 μM KB-R7943. N=5–6; * = p<0.05 versus vehicle.
3.3. The effects of KB-R7943 on agonist-induced contraction
To investigate the contribution of Ca2+ influx through reverse-mode NCX during vascular contraction, isometric contraction to ET-1 was measured in an isolated tissue bath in the presence or absence of KB-R7943. KB-R7943 (1 μM) had no effect on ET-1-induced contraction in either RA or RVC (fig. 5a,b). However, KB-R7943 (10 μM) significantly attenuated maximal contraction to ET-1 in vena cava (52.93 ± 9.22% of control), but not aorta (90.06 ± 1.04% of control) (fig. 6a,b). KB-R7943 significantly inhibited maximal contraction to ET-1 in RA as well as RVC upon inhibition of nitric oxide synthase and cyclooxygenase-1 and -2 by by LNNA (100 μM) and indomethacin (5 μM) (fig. 7a–b). However, inhibition of maximal contraction to ET-1 by KB-R7943 was still greater in vena cava (29.25 ± 3.84% of control) as compared to aorta (73.25 ± 0.98% of control).
Figure 5.
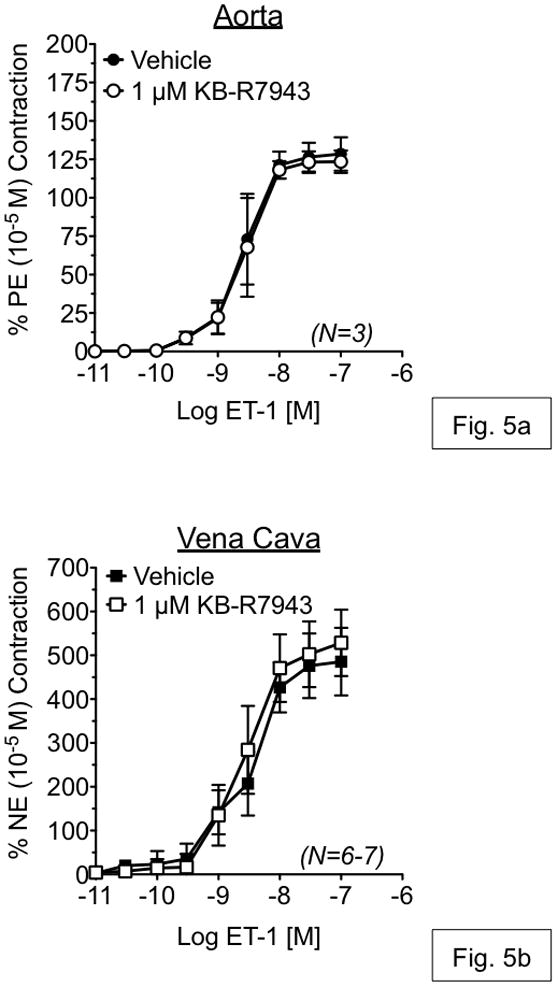
Measurement of endothelin-1-induced responses in aorta (a) and vena cava (b), exposed to vehicle or KB-R7943 (10 μM). Vehicle or antagonists were incubated with tissue for 1h prior to ET-1 exposure. Points represent mean ± SEM for the number of animals indicated in parentheses. * = p<0.05 versus vehicle.
Figure 6.
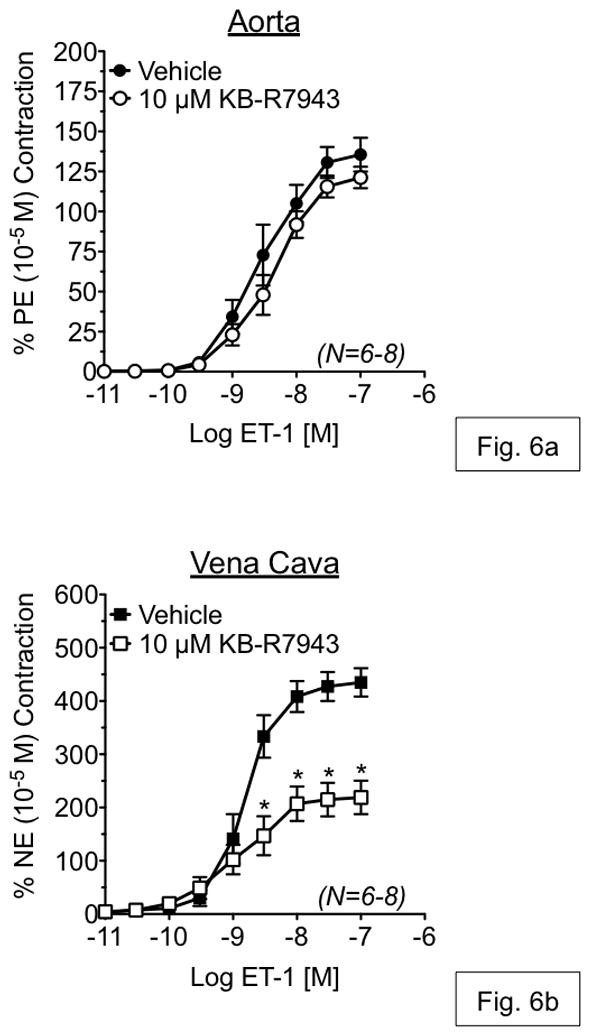
Measurement of endothelin-1-induced responses in aorta (a) and vena cava (b), exposed to vehicle or KB-R7943 (1 μM). Vehicle or antagonists were incubated with tissue for 1h prior to ET-1 exposure. Points represent mean ± SEM for the number of animals indicated in parentheses. * = p<0.05 versus vehicle.
Figure 7.
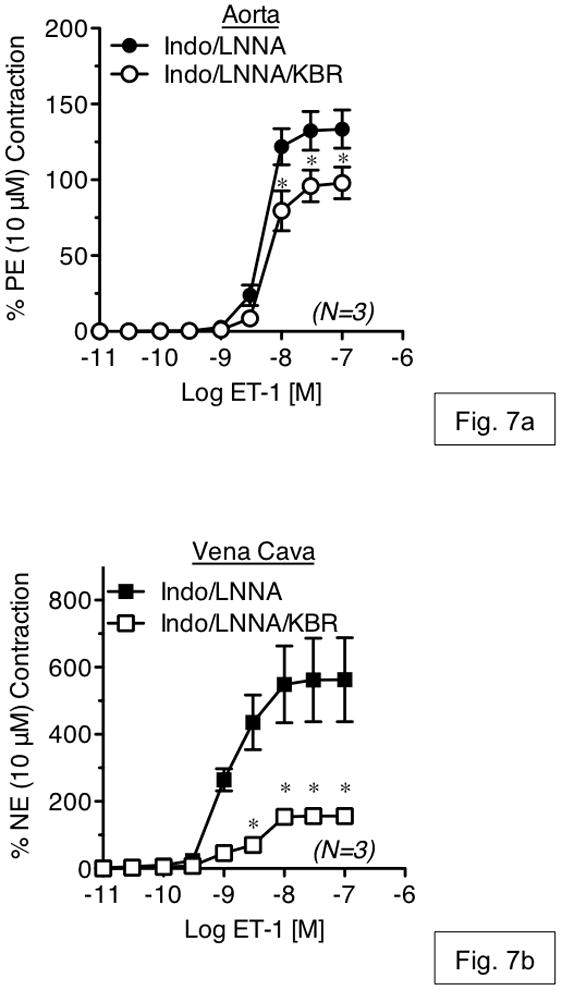
The effects of KB-R7943 (10 μM) on endothelin-1-induced contraction in aorta (a) and vena cava (b) exposed to L-NNA (100 μM) and indomethacin (5 μM). Vehicle or antagonists were incubated with tissue for 1h prior to ET-1 exposure. Points represent mean ± SEM for the number of animals indicated in parentheses. * = p<0.05 versus vehicle.
The effects of KB-R7943 on KCl-induced contraction were also tested. KB-R7943 (10 μM) attenuated maximal contraction to KCl in aorta (47.81±4.77%) and nearly abolished the response of vena cava (8.77±2.20%) (fig. 8a,b).
Figure 8.
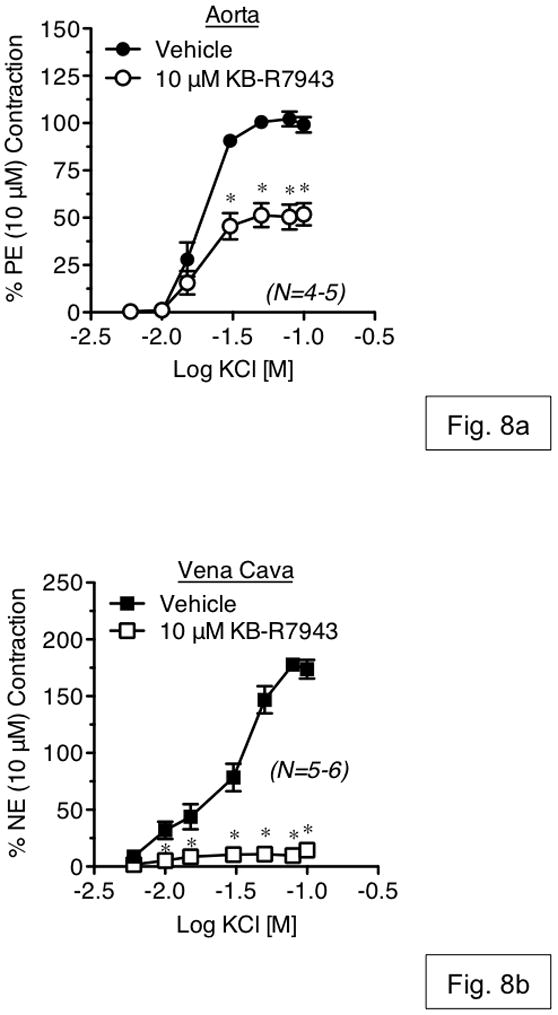
Measurement of KCl-induced contraction in aorta (a) and vena cava (b), exposed to vehicle or KB-R7943 (10 μM). Vehicle or antagonists were incubated with tissue for 1h prior to agonist exposure. Points represent mean ± SEM for the number of animals indicated in parentheses. * = p<0.05 versus vehicle.
3.4. Inhibition of Ca2+ influx by KB-R7943 during ET-1-induced contraction
To test if KB-R7943 inhibited extracellular Ca2+ influx during contraction to ET-1, contraction to increasing concentrations of CaCl2 was measured in the presence of ET-1 (100 nM) as described above (see Methods). As compared to vehicle, KB-R7943 (10 μM) caused a 7.1-fold rightward-shift in the contractile response to CaCl2 in aorta (EC50=37.33 μM vs. 264.50 μM) but had no effect on maximal contraction to CaCl2 (fig. 9a). In vena cava, KB-R7943 (10 μM) reduced the maximal contraction to CaCl2 in addition to causing a 10.4-fold rightward shift in the contractile response (EC50=55.90 μM vs. 580.00 μM) (fig. 9b). At a CaCl2 concentration equivalent to that of our physiological salt solution used in the previous experiments (1.6 mM), KB-R7943 (10 μM) had no effect on maximal contraction to CaCl2 in the presence of ET-1 in aorta, but significantly reduced the maximal contraction in vena cava (boxes, fig 9a–b), consistent with the data presented in Figure 6.
Figure 9.
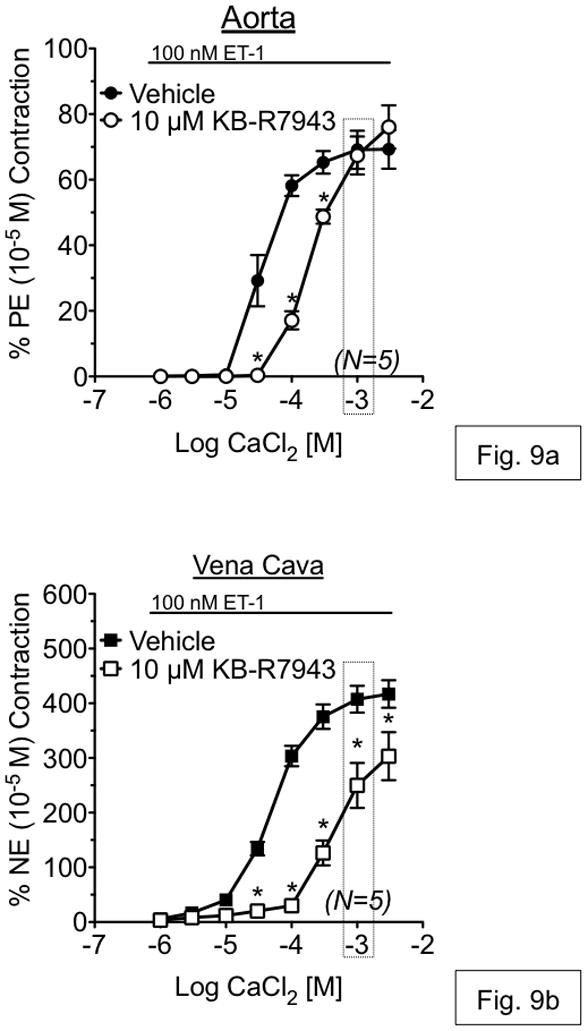
Measurement of CaCl2 concentration response curves, in the presence of ET-1 (100 nM), in aorta (a) and vena cava (b). Tissues were first incubated for 30 minutes in Ca2+-free buffer with 1 mM EGTA, then in Ca2+-free buffer (no EGTA) for 10 minutes before addition of 100nM ET-1. After plateau of any contraction to ET-1, tissues were incubated with vehicle (solid shapes) or 10 μM KB-R7943 (open shapes). Points represent mean ± SEM for the number of animals indicated in parentheses. Boxes represent the CaCl2 concentration that is equivalent to that of physiological salt solution used in all other experiments. * = p<0.05 versus vehicle.
3.5. Potential Secondary Actions of KB-R7943
Our finding that KB-R7943 attenuated KCl-induced contraction suggested that the effects of KB-R7943 on ET-1-induced contraction could be due to voltage-dependent Ca2+ influx inhibition and not inhibition of the reverse-mode NCX. To test this, we measured KCl- and ET-1-induced contraction in the presence or absence of the L-type Ca2+ channel antagonist nifedipine (1 μM), and compared these results to our previous experiments with KB-R7943. As with KB-R7943, nifedipine (1 μM) markedly attenuated the maximal contraction to KCl in both aorta (16.65±2.17%) and vena cava (69.55±15.88%) (fig. 10a,b). However, nifedipine had no effect on ET-1-induced contraction in either aorta or vena cava (fig. 10c,d). Thus, it was unlikely that the effects of KB-R7943 on ET-1-induced contraction resulted from off-target inhibition of voltage-gated Ca2+ channels.
Figure 10.

Top: Measurement of KCl-induced contraction in aorta (a) and vena cava (b), exposed to vehicle or nifedipine (1 μM). Bottom: Measurement of ET-1-induced contraction in aorta (c) and vena cava (d), exposed to vehicle or nifedipine (1 μM). In (ad), vehicle or antagonists were incubated with tissue for 1h prior to agonist exposure. Points represent mean ± SEM for the number of animals indicated in parentheses. * = p<0.05 versus vehicle.
4. Discussion
The principal and novel findings of this study are: (1) activation of the reverse-mode NCX by reducing extracellular Na+ causes significant contraction and increases intracellular Ca2+ in vena cava but not aorta; (2) the reverse-mode NCX inhibitor KB-R7943 selectively attenuates contraction to ET-1 in vena cava but not aorta; and (3) 10 μM KB-R7943 also inhibits KCl-induced contraction to a greater degree in vena cava than in aorta. These latter results suggest that KB-R7943 may also inhibit contraction by blocking voltage-gated Ca2+ channels. However, this does not account for the effects of KB-R7943 on ET-1-induced contraction, because the response to ET-1 appears to be independent of voltage-gated Ca2+ channels in both aorta and vena cava (fig. 9). The findings that KB-R7943 inhibits contraction and decreases intracellular Ca2+ during low Na+ exposure in vena cava suggest that the effects of KB-R7943 on venous contraction are due, at least in part, to Na+-dependent Ca2+ influx, possibly through reverse-mode NCX.
4.1. Reverse-mode NCX and Na+-dependent contraction
We found that removal of Na+, a test used to activate NCX without addition of pharmacological agents, caused only a small, transient response in aorta that was not affected by KB-R7943. These data suggest that stimulation of reverse-mode NCX does not, in and of itself, cause contraction of aorta. The lack of a response was also not due to lack of protein, since aorta robustly expressed NCX-1 protein. Similar published results show that low Na+ only caused aortic contraction when intracellular Ca2+ stores were depleted, suggesting that Ca2+ sequestration by the sarcoplasmic reticulum attenuated any contraction that could be caused by low Na+-dependent Ca2+ influx [18]. Unlike aorta, vena cava exhibited a prolonged and significant contraction and an increase in intracellular Ca2+ when exposed to low Na+ PSS. The contraction and rise in intracellular Ca2+ were attenuated by KB-R7943, suggesting that both depended upon Ca2+ influx through the reverse-mode NCX. As with rat aorta, previous studies using rabbit inferior vena cava suggest that the reverse-mode NCX is active after sarcoplasmic stores depletion [10]. Our findings that low Na+ PSS causes contraction and increases intracellular Ca2+ suggest that reverse-mode NCX contributes significantly to contraction in rat vena cava.
4.2. Reverse-mode NCX and agonist-induced contraction
We found that KB-R7943 (10 μM) attenuated ET-1-induced contraction only in vena cava, but attenuated KCl-induced contraction in both aorta and vena cava. However, the inhibitory effect of KB-R7943 on KCl-induced contraction was greater in vena cava than in aorta. KCl-induced contraction is primarily caused by membrane depolarization and subsequent influx of Ca2+ through voltage-gated Ca2+ channels [19]. This may explain why contraction to KCl in both aorta and vena cava is inhibited by KB-R7943, since membrane potential is an important regulator of NCX function. Membrane depolarization causes the NCX to favor the reverse-mode, while membrane hyperpolarization causes the NCX to favor the forward-mode [4]. The different effects of KB-R7943 on contraction in these tissues may also be because the agonists we tested increase Na+ influx in vena cava but not aorta, or that venous smooth muscle contraction relies more heavily on Na+ influx than aortic smooth muscle. Since the concentration gradient for Na+ is also an important regulator of NCX function, agonists that increase smooth muscle Na+ influx may be more likely to activate reverse-mode NCX. Also, KCl can increase Na+ influx in aorta, since the voltage-gated Na+ channel blocker TTX inhibited KCl-induced contraction in rat aortic rings [20]. Together, these data suggest that reverse-mode NCX is a significant source of Ca2+ influx in vena cava, but is minimally active – or activated only by certain agonists or depolarizing conditions – in aorta.
KB-R7943 caused a rightward shift in the CaCl2 concentration-response curve in the presence of ET-1 (100 nM) in both aorta and vena cava. However, at a CaCl2 concentration equivalent to that of our ‘normal’ PSS (1.6 mM), there was a ~25% reduction in contraction to Ca2+ in vena cava but no reduction in aorta. These data support a greater role for NCX-mediated Ca2+ influx during contraction in vena cava as opposed to aorta.
The differences between aorta and vena cava could also be due to receptor-mediated activation of protein kinase C (PKC), which enhances NCX function by phosphorylating the central cytosolic domain of the NCX [3]. However, it is currently unknown if only certain PKC isoforms interact with the NCX, if these isoforms are expressed in RA and RVC, and if activation of different PKC isoforms could be responsible for our experimental outcomes. While ET-1 can activate PKC and up-regulate expression of several different PKC isoforms in smooth muscle [21], it is unknown if these isoforms could specifically target the NCX when activated by ET-1. ET-1 can enhance NCX function in renal epithelial cells by activating PKC, but this same effect is not yet reported to occur in vascular smooth muscle [22]. Future studies are necessary to determine what PKC isoforms are expressed in aorta and vena cava, if these isoforms can phosphorylate the NCX, and if the differences in NCX function between aorta and vena cava are due to PKC-dependent modification of the NCX as opposed to differences in Na+ handling.
4.3. Secondary effects of KB-R7943
An increasing number of off-target effects of KB-R7943 have been discovered, leading to doubts about the selectivity of KB-R7943 for the NCX. At concentrations similar to those shown to block reverse-mode NCX, KB-R7943 has been described to inhibit L-type Ca2+ channels, TRPC channels, and ryanodine receptors [23–26]. Nevertheless, KB-R7943 continues to be used extensively to study the contribution of NCX to a variety of cellular functions, due in part to the lack of other potent, commercially available NCX inhibitors. Our intention was to validate our findings using other NCX inhibitors, but this proved extremely difficult. We attempted to obtain SEA-0400, another NCX inhibitor reported to have increased potency and selectivity as compared to KB-R7943, but it is not available for researchers at this time. The only other available NCX inhibitor, SN-6, was found to be insoluble in PSS for the duration of incubation required for our experimental paradigm, and thus useless as another NCX inhibitor for comparison to the experiments using KB-R7943. While we recognize the power of confirming our findings with the use of another NCX inhibitor, the insolubility and lack of availability of such compounds made us unable to do so. Instead, we conducted several experiments to differentiate the specific and non-specific effects of KB-R7943 in aorta and vena cava because of the potential for off-target effects.
4.4. Limitations
This study is not without limitations. As another means of activating reverse-mode NCX, we attempted to increase intracellular Na+ by inhibiting the Na+/K+ ATPase with ouabain. Ouabain did not contract aorta or vena cava (data not shown) at concentrations used to contract smaller vessels with myogenic tone [27]. This is similar to other reports in the literature that show rat aorta contracts very little to ouabain unless K+ is removed [28]. At extremely high concentrations (>100 μM), contraction to ouabain was minimal (20–40 mg in vena cava) as compared to ET-1 (400–600 mg). These responses, however, were very inconsistent between tissues. Thus, we were unable use ouabain to corroborate our findings that suggested KB-R7943 was inhibiting contraction in an NCX-dependent manner in vena cava.
While we did not assess the depletion of intracellular Ca2+ stores in our Ca2+ influx experiments (fig. 8), it is likely that sarcoplasmic reticular stores were depleted by incubation in Ca2+-free PSS. Since Ca2+ influx via the reverse-mode NCX is activated after depletion of calcium stores, the rightward shift caused by KB-R7943 in aorta may be because NCX-mediated Ca2+ influx is important for replenishment of sarcoplasmic Ca2+ stores. Further experiments will be required to investigate KB-R7943-dependent inhibition of store-operated Ca2+ influx directly.
We did not investigate differences in NCX isoform expression between aorta and vena cava. It is unknown if our findings are due to differential expression of NCX-1, -2 and -3 in aorta and vena cava, even though KB-R7943 is believed to inhibit all three NCX isoforms at the concentration used in this study [29,30]. NCX-1 is often reported to be the predominant isoform expressed in most vascular smooth muscle, although other reports debate if NCX-1 is the most important to vascular function [31,32]. We also did not investigate if the NCX-1 protein expressed in aorta and vena cava was an NCX-1 splice variant. Of the 12 NCX splice variants currently identified, NCX1.3 is described as “smooth-muscle specific” [33], although pulmonary arterial smooth muscle cells can express a variety of variants within the same cell [34]. To our knowledge, the NCX-1 antibody we used in these experiments does not differentiate between the splice variants of NCX-1, nor have such antibodies been developed. Any differences in the pharmacology of KB-R7943 on these NCX splice variants have not been established, but cannot be ruled out as another possible explanation for the differences seen between aorta and vena cava.
We did not directly examine the effects of KB-R7943 on TRP channel function. There is an increasing amount of evidence that NCX and TRP channels are either functionally or physically linked [35]. Na+ entry through TRP channels is a driving force for Ca2+ entry caused by the reverse-mode NCX [36]. This association makes distinguishing between inhibition of TRP channels by KB-R7943 and inhibition of the NCX extremely difficult. Thus, we cannot be certain that our results are due solely to the actions of KB-R7943 on NCX and not at TRP channels, since the function of the NCX and the opening of TRP channels may be closely intertwined.
Because these experiments were performed in endothelium-intact tissues, we cannot determine the role of endothelial NCX versus smooth muscle cell NCX during contraction in aorta and vena cava. Vena cava contain only a single layer of smooth muscle cells, making reliable denudation of the endothelium extremely difficult to accomplish without destroying vessel function. Thus, we elected to use endothelium-intact tissues in these experiments. Even though the endothelium was present in these tissues, it is unlikely that inhibition of endothelial NCX would attenuate contraction in aorta and vena cava. Activation of reverse-mode NCX in endothelial cells causes nitric oxide release and endothelium dependent-dependent vasorelaxation [37–39]. Thus, inhibition of endothelial NCX would likely potentiate, and not inhibit, contraction. However, Inhibition of nitric oxide synthase and cyclooxygenase actually potentiated the inhibitory effects of KB-R7943 on ET-1-induced contraction in aorta and vena cava, suggesting that a portion of the effects of KB-R7943 on ET-1-induced contraction is endothelium-dependent. Further investigation will be required to precisely distinguish between the contributions of endothelial NCX versus smooth muscle NCX to the contractile function of these tissues.
5. Conclusions
Our findings suggest that the NCX, while often considered only as a means of Ca2+ extrusion, has a prominent role in Ca2+ influx and contraction in the vena cava. While activation of reverse-mode NCX alone is incapable of contracting aorta, NCX activation increases intracellular Ca2+ and contracts vena cava. Reverse-mode NCX function in vena cava is also suspected during KCl- and ET-1-induced contraction, since the reverse-mode NCX inhibitor KB-R7943 markedly attenuated contraction to these agonists in vena cava. We propose that the effects of KB-R7943 on vena cava and aorta contraction are due to reverse-mode NCX inhibition and not because of one possible secondary action of KB-R7943, inhibition of voltage-gated Ca2+ channels. These studies suggest that there is an important difference between arteries and veins in terms of regulation of Ca2+ influx during contraction, and that this difference represents potential therapeutic targets for specifically targeting venous smooth muscle to treat vascular diseases such as hypertension.
Acknowledgments
Supported by NIH P01HL70687.
Abbreviations
- BKCa channels
large-conductance Ca2+-activated potassium channels
- ET-1
endothelin-1
- KB-R7943
2-(2-(4-(4-nitrobenzyloxy)phenyl)ethyl)-isothiourea methanesulfonate
- NCX
Na+/Ca2+ exchanger
- PKC
protein kinase C
- RA
rat thoracic aorta
- RVC
rat vena cava
- SD
Sprague-Dawley
- TEA
tetraethylammonium
- TRPC channels
transient receptor potential (canonical) channels
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Philipson KD, Nicoll DA, Ottolia M, Quednau BD, Reuter H, John S, Qiu Z. The Na+/Ca2+ exchange molecule: an overview. Ann N Y Acad Sci. 2002;976:1–10. doi: 10.1111/j.1749-6632.2002.tb04708.x. [DOI] [PubMed] [Google Scholar]
- 2.Annunziato L, Pignataro G, Di Renzo GF. Pharmacology of brain Na+/Ca2+ exchanger: from molecular biology to therapeutic perspectives. Pharmacol Rev. 2004;56:633–54. doi: 10.1124/pr.56.4.5. [DOI] [PubMed] [Google Scholar]
- 3.Iwamoto T, Pan Y, Wakabayashi S, Imagawa T, Yamanaka HI, Shigekawa M. Phosphorylation-dependent regulation of cardiac Na+/Ca2+ exchanger via protein kinase C. J Biol Chem. 1996;271:13609–15. doi: 10.1074/jbc.271.23.13609. [DOI] [PubMed] [Google Scholar]
- 4.Blaustein MP, Lederer WJ. Sodium/calcium exchange: its physiological implications. Physiol Rev. 1999;79:763–854. doi: 10.1152/physrev.1999.79.3.763. [DOI] [PubMed] [Google Scholar]
- 5.Blaustein MP. Sodium ions, calcium ions, blood pressure regulation, and hypertension: a reassessment and a hypothesis. Am J Physiol. 1977;232:C165–73. doi: 10.1152/ajpcell.1977.232.5.C165. [DOI] [PubMed] [Google Scholar]
- 6.Iwamoto T, Kita S, Zhang J, Blaustein MP, Arai Y, Yoshida S, Wakimoto K, Komuro I, Katsuragi T. Salt-sensitive hypertension is triggered by Ca2+ entry via Na+/Ca2+ exchanger type-1 in vascular smooth muscle. Nat Med. 2004;10:1193–9. doi: 10.1038/nm1118. [DOI] [PubMed] [Google Scholar]
- 7.Zhang J, Ren C, Chen L, Navedo MF, Antos LK, Kinsey SP, Iwamoto T, Philipson KD, Kotlikoff MI, Santana LF, Wier WG, Matteson DR, Blaustein MP. Knockout of Na+/Ca2+ exchanger in smooth muscle attenuates vasoconstriction and L-type Ca2+ channel current and lowers blood pressure. Am J Physiol Heart Circ Physiol. 2010;298:H1472–83. doi: 10.1152/ajpheart.00964.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Davis KA, Samson SE, Hammel KE, Kiss L, Fulop F, Grover AK. Functional linkage of Na+-Ca2+-exchanger to sarco/endoplasmic reticulum Ca2+ pump in coronary artery: comparison of smooth muscle and endothelial cells. J Cell Mol Med. 2009;13:1775–83. doi: 10.1111/j.1582-4934.2008.00480.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fink GD, Arthur C. Corcoran Memorial Lecture. Sympathetic activity, vascular capacitance, and long-term regulation of arterial pressure. Hypertension. 2009;53:307–12. doi: 10.1161/HYPERTENSIONAHA.108.119990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fameli N, van Breemen C, Kuo K-H. A quantitative model for linking Na+/Ca2+ exchanger to SERCA during refilling of the sarcoplasmic reticulum to sustain [Ca2+] oscillations in vascular smooth muscle. Cell Calcium. 2007;42:565–75. doi: 10.1016/j.ceca.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 11.Poburko D, Liao C-H, Lemos VS, Lin E, Maruyama Y, Cole WC, van Breemen C. Transient receptor potential channel 6-mediated, localized cytosolic [Na+] transients drive Na+/Ca2+ exchanger-mediated Ca2+ entry in purinergically stimulated aorta smooth muscle cells. Circ Res. 2007;101:1030–8. doi: 10.1161/CIRCRESAHA.107.155531. [DOI] [PubMed] [Google Scholar]
- 12.Watts SW, Fink GD, Northcott CA, Galligan JJ. Endothelin-1-induced venous contraction is maintained in DOCA-salt hypertension; studies with receptor agonists. Br J Pharmacol. 2002;137:69–79. doi: 10.1038/sj.bjp.0704831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thakali K, Fink GD, Watts SW. Arteries and veins desensitize differently to endothelin. J Cardiovasc Pharmacol. 2004;43:387–93. doi: 10.1097/00005344-200403000-00009. [DOI] [PubMed] [Google Scholar]
- 14.Tykocki NR, Gariepy CE, Watts SW. Endothelin ET(B) receptors in arteries and veins: multiple actions in the vein. J Pharmacol Exp Ther. 2009;329:875–81. doi: 10.1124/jpet.108.145953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thakali K, Demel SL, Fink GD, Watts SW. Endothelin-1-induced contraction in veins is independent of hydrogen peroxide. Am J Physiol Heart Circ Physiol. 2005;289:H1115–22. doi: 10.1152/ajpheart.00086.2005. [DOI] [PubMed] [Google Scholar]
- 16.Li L, Watts SW, Banes AK, Galligan JJ, Fink GD, Chen AF. NADPH oxidase-derived superoxide augments endothelin-1-induced venoconstriction in mineralocorticoid hypertension. Hypertension. 2003;42:316–21. doi: 10.1161/01.HYP.0000084853.47326.F2. [DOI] [PubMed] [Google Scholar]
- 17.Watts SW, Webb RC. Mechanism of ergonovine-induced contraction in the mesenteric artery from deoxycorticosterone acetate-salt hypertensive rat. J Pharmacol Exp Ther. 1994;269:617–25. [PubMed] [Google Scholar]
- 18.Ashida T, Blaustein MP. Regulation of cell calcium and contractility in mammalian arterial smooth muscle: the role of sodium-calcium exchange. J Physiol (Lond) 1987;392:617–35. doi: 10.1113/jphysiol.1987.sp016800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bozler E. Role of calcium in initiation of activity of smooth muscle. Am J Physiol. 1969;216:671–4. doi: 10.1152/ajplegacy.1969.216.3.671. [DOI] [PubMed] [Google Scholar]
- 20.Fort A, Cordaillat M, Thollon C, Salazar G, Mechaly I, Villeneuve N, Vilaine J-P, Richard S, Virsolvy A. New insights in the contribution of voltage-gated Na(v) channels to rat aorta contraction. PLoS ONE. 2009;4:e7360. doi: 10.1371/journal.pone.0007360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wolf SC, Brodbeck C, Sauter G, Risler T, Brehm BR. Endothelin-1 regulates protein kinase C isoforms differently in smooth muscle cells and in cardiomyocytes. J Cardiovasc Pharmacol. 2004;44 (Suppl 1):S301–3. doi: 10.1097/01.fjc.0000166278.78162.e5. [DOI] [PubMed] [Google Scholar]
- 22.Kita S, Katsuragi T, Iwamoto T. Endothelin-1 enhances the activity of Na+/Ca2+ exchanger type 1 in renal epithelial cells. J Cardiovasc Pharmacol. 2004;44 (Suppl 1):S239–43. doi: 10.1097/01.fjc.0000166262.19852.b5. [DOI] [PubMed] [Google Scholar]
- 23.Kraft R. The Na+/Ca2+ exchange inhibitor KB-R7943 potently blocks TRPC channels. Biochem Biophys Res Commun. 2007;361:230–6. doi: 10.1016/j.bbrc.2007.07.019. [DOI] [PubMed] [Google Scholar]
- 24.Liang GH, Kim JA, Seol GH, Choi S, Suh SH. The Na+/Ca2+ exchanger inhibitor KB-R7943 activates large-conductance Ca2+-activated K+ channels in endothelial and vascular smooth muscle cells. Eur J Pharmacol. 2008;582:35–41. doi: 10.1016/j.ejphar.2007.12.021. [DOI] [PubMed] [Google Scholar]
- 25.Cheng H, Smith GL, Hancox JC, Orchard CH. Inhibition of spontaneous activity of rabbit atrioventricular node cells by KB-R7943 and inhibitors of sarcoplasmic reticulum Ca2+ ATPase. Cell Calcium. 2011;49:56–65. doi: 10.1016/j.ceca.2010.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Barrientos G, Bose DD, Feng W, Padilla I, Pessah IN. The Na+/Ca2+ exchange inhibitor 2-(2-(4-(4-nitrobenzyloxy)phenyl)ethyl)isothiourea methanesulfonate (KB-R7943) also blocks ryanodine receptors type 1 (RyR1) and type 2 (RyR2) channels. Mol Pharmacol. 2009;76:560–8. doi: 10.1124/mol.109.057265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Linde CI, Karashima E, Raina H, Zulian A, Wier WG, Hamlyn JM, Ferrari P, Blaustein MP, Golovina VA. Increased arterial smooth muscle Ca2+ signaling, vasoconstriction, and myogenic reactivity in Milan hypertensive rats. Am J Physiol Heart Circ Physiol. 2012;302:H611–20. doi: 10.1152/ajpheart.00950.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gallo LC, Davel APC, Xavier FE, Rossoni LV. Time-dependent increases in ouabain-sensitive Na+, K+-ATPase activity in aortas from diabetic rats: The role of prostanoids and protein kinase C. Life Sci. 2010;87:302–8. doi: 10.1016/j.lfs.2010.07.005. [DOI] [PubMed] [Google Scholar]
- 29.Amran MS, Homma N, Hashimoto K. Pharmacology of KB-R7943: a Na+-Ca2+ exchange inhibitor. Cardiovascular Drug Reviews. 2003;21:255–76. doi: 10.1111/j.1527-3466.2003.tb00121.x. [DOI] [PubMed] [Google Scholar]
- 30.Iwamoto T. Forefront of Na+/Ca2+ exchanger studies: molecular pharmacology of Na+/Ca2+ exchange inhibitors. J Pharmacol Sci. 2004;96:27–32. doi: 10.1254/jphs.fmj04002x6. [DOI] [PubMed] [Google Scholar]
- 31.Lytton J. Na+/Ca2+ exchangers: three mammalian gene families control Ca2+ transport. Biochem J. 2007;406:365–82. doi: 10.1042/BJ20070619. [DOI] [PubMed] [Google Scholar]
- 32.Levitsky DO. Three types of muscles express three sodium-calcium exchanger isoforms. Ann N Y Acad Sci. 2007;1099:221–5. doi: 10.1196/annals.1387.063. [DOI] [PubMed] [Google Scholar]
- 33.Hurtado C, Prociuk M, Maddaford TG, Dibrov E, Mesaeli N, Hryshko LV, Pierce GN. Cells expressing unique Na+/Ca2+ exchange (NCX1) splice variants exhibit different susceptibilities to Ca2+ overload. Am J Physiol Heart Circ Physiol. 2006;290:H2155–62. doi: 10.1152/ajpheart.00958.2005. [DOI] [PubMed] [Google Scholar]
- 34.Zheng Y-M, Wang Y-X. Sodium-calcium exchanger in pulmonary artery smooth muscle cells. Ann N Y Acad Sci. 2007;1099:427–35. doi: 10.1196/annals.1387.017. [DOI] [PubMed] [Google Scholar]
- 35.Rosker C, Graziani A, Lukas M, Eder P, Zhu MX, Romanin C, Groschner K. Ca2+ signaling by TRPC3 involves Na+ entry and local coupling to the Na+/Ca2+ exchanger. J Biol Chem. 2004;279:13696–704. doi: 10.1074/jbc.M308108200. [DOI] [PubMed] [Google Scholar]
- 36.Lemos VS, Poburko D, Liao C-H, Cole WC, van Breemen C. Na+ entry via TRPC6 causes Ca2+ entry via NCX reversal in ATP stimulated smooth muscle cells. Biochem Biophys Res Commun. 2007;352:130–4. doi: 10.1016/j.bbrc.2006.10.160. [DOI] [PubMed] [Google Scholar]
- 37.Teubl M, Groschner K, Kohlwein SD, Mayer B, Schmidt K. Na(+)/Ca(2+) exchange facilitates Ca(2+)-dependent activation of endothelial nitric-oxide synthase. J Biol Chem. 1999;274:29529–35. doi: 10.1074/jbc.274.41.29529. [DOI] [PubMed] [Google Scholar]
- 38.Fameli N, Kuo K-H, van Breemen C. A model for the generation of localized transient [Na+] elevations in vascular smooth muscle. Biochem Biophys Res Commun. 2009;389:461–5. doi: 10.1016/j.bbrc.2009.08.166. [DOI] [PubMed] [Google Scholar]
- 39.Schneider J-C, Kebir El D, Chéreau C, Mercier J-C, Dall’Ava-Santucci J, Dinh-Xuan AT. Involvement of Na(+)/Ca(2+) exchanger in endothelial NO production and endothelium-dependent relaxation. Am J Physiol Heart Circ Physiol. 2002;283:H837–44. doi: 10.1152/ajpheart.00789.2001. [DOI] [PubMed] [Google Scholar]