

Abstract
Autophagy is a lysosomal degradation pathway that can degrade bulk cytoplasm and superfluous or damaged organelles, such as mitochondria, to maintain cellular homeostasis. It is now known that dysregulation of autophagy can cause pathogenesis of numerous human diseases. Here, we discuss the critical roles that autophagy plays in the pathogenesis of liver diseases such as nonalcoholic and alcoholic fatty liver, drug-induced liver injury, protein aggregate-related liver diseases, viral hepatitis, fibrosis, aging and liver cancer. In particular, we discuss the emerging therapeutic potential by pharmacological modulation of autophagy for these liver diseases.
Keywords: autophagy, mitophagy, liver diseases
Introduction
Autophagy is an intracellular degradation process by which lysosomes degrade proteins, cellular organelles and invading microbes. Three different types of autophagy have been identified in mammalian cells: macroautophagy (here after referred to as autophagy), chaperone-mediated autophagy (CMA), and microautophagy, and they differ in how the target cargos are delivered to lysosomes [1]. Autophagy involves the formation of a double-membrane structure termed an autophagosome, which fuses with a lysosome to form an autolysosome where the cellular contents sequestered in the autophagosome are degraded by lysosomal enzymes. Microautophagy results in the direct uptake of cytoplasm or organelles at the lysosomal surface by invagination and protrusion of the lysosomal membrane. While autophagy and microautophagy can degrade proteins and large structures in a selective and non-selective manner, CMA only selectively degrades a subset of proteins that have the specific motif KFERQ [1–4].
Most tissues have a basal level autophagy which contributes to cellular homeostasis by regulating the routine turnover of cytoplasmic components. However, autophagy can also be induced by various conditions and stresses such as starvation, protein aggregates, virus infection, and oxidative and endoplasmic reticulum (ER) stresses [5–7]. Increasing evidence now indicates that autophagy is involved in many pathophysiological conditions in various organisms including development, differentiation, tissue remodeling, tissue injury, aging and cancer [1,2,8].
Liver is one of the most dynamic organs in mammals and humans, and autophagy plays important roles in liver physiology and pathology [2,3,9]. In fact, autophagy was discovered from experiments conducted using the liver [10]. This review will focus on recent progress regarding the role of autophagy in liver pathophysiology and the emerging therapeutic approaches for liver diseases that target autophagy.
Targeting autophagy for non-alcoholic and alcoholic fatty liver disease
Nonalcoholic Fatty Liver Disease (NAFLD) is currently the most common chronic liver disease worldwide. In western countries, approximately 20~30% of adults have NAFLD, and its prevalence increases to approximately 80% among people who are obese or have diabetes [11–14]. NAFLD is defined as the accumulation of fat in the liver in the absence of secondary causes, such as alcohol consumption. NAFLD can increase the risk for development of fibrosis and cirrhosis and can eventually lead to development of hepatocellular carcinoma (HCC) [13]. NAFLD is also associated with insulin resistance and metabolic syndrome, which increase the risk for type II diabetes and cardiovascular disease. Currently, there is no effective treatment available for NAFLD. Therefore, development of effective therapies for NAFLD would have significant impact.
Although autophagy was generally thought to degrade intracellular proteins (non-selectively) or organelles (selectively), recent evidence indicates that autophagy may also selectively degrade lipid droplets, which is a process termed lipophagy [15]. Cellular lipids are generally stored as triglycerides (TG) in a lipid droplet and are surrounded by a phospholipid monolayer. Evidence that autophagy may regulate hepatic TG contents is based on the following observations: (i) pharmacological inhibition of autophagy by 3-methyladenine (3MA), which targets the mammalian PI3-kinase vps34, increases TG contents in hepatocytes either cultured in regular medium or treated with an unsaturated fatty acid (oleic acid) [15]; (ii) pharmacological inhibition of lysosomal degradation by chloroquine (CQ), which increases lysosomal pH, also elevates TG contents in hepatocytes that are treated with fatty acids [16]; (iii) genetic inhibition of autophagy by siRNA knockdown of Atg5 enhances TG content in fatty acid-treated hepatocytes [15]; and (iv) liver-specific Atg7 knockout mice have increased hepatic TG contents in either fed mice or in mice starved for 24 hours [15]. In addition to hepatocytes, inhibition of autophagy can also lead to the accumulation of lipid droplets in hepatic stellate cells resulting in decreased fibrogenesis ([17] see below).
Autophagy is negatively regulated by mTOR. Rapamycin, an mTOR inhibitor, induces autophagy in many cultured cells and in many tissues in vivo. In cultured hepatocytes, treatment with rapamycin increases co-localization of lipid droplets with LC3 positive autophagosomes or with LAMP1 positive autolysosomes and lysosomes, and it decreases oleic acid-induced increase of TG levels [15,16]. In addition to in vitro studies, rapamycin also attenuates high-fat diet (HFD)-induced fatty liver in a mouse model [18]. Mice treated with rapamycin have reduced body weight and lower serum leptin and insulin levels compared with HFD control mice [18]. However, autophagy was not assessed in these rapamycin-treated HFD fed mice. Nevertheless, these data suggest that pharmacological induction of autophagy by rapamycin may attenuate fatty liver by induction of lipophagy.
In addition to regulating autophagy and protein synthesis, increasing evidence indicates that mTOR is also essential for regulating lipid biosynthesis [19]. There are at least two mTOR complexes: mTOR complex1 (mTOR1) and mTOR complex 2 (mTOR2). mTOR1 is rapamycin-sensitive whereas mTOR2 is rapamycin-insensitive [20]. mTOR1, negatively regulates autophagy by interacting with the ULK1-Atg13-FIP200 complex in mammalian cells [21]. mTOR1 also regulates protein translation by phosphorylating 70 kDa ribosomal protein S6 kinase (p70S6K) and the eukaryotic translation initiation factor 4E binding protein (4EBP1). In contrast, mTOR2 is activated by growth factors and regulates the activation of AKT as well as cytoskeletal organization [20]. mTOR2 directly phosphorylates AKT to allow the maximal activation of AKT resulting in the inhibition of the transcription factor FoxO [22,23]. FoxO has been demonstrated to be able to regulate the expression of autophagy genes in mouse skeletal muscle. Inhibition of mTOR2 by siRNA knockdown of rictor increased FoxO3 nuclear localization and autophagy in skeletal muscle [23]. However, more studies are needed to determine whether mTOR2 would also be important in other tissues in addition to skeletal muscle.
Lipid biosynthesis is regulated by a family of transcription factors designated sterol regulatory element-binding proteins (SREBPs) [24]. There are three isoforms of SREBP in the liver that regulate gene expression for fatty acid and cholesterol biosynthesis: SREBP-1a, SREBP-1c, and SREBP-2. Insulin has been shown to activate AKT, which increases expression of lipogenic genes by inducing nuclear accumulation of SREBP-1 [25]. Akt activates mTOR by directly phosphorylating tuberous sclerosis complex1/2 (TSC1/2) and PRAS40. It is now known that mTOR positively regulates SREBP-1 by controlling the nuclei location of Lipin 1, a phosphatic acid phosphatase [26]. Pharmacological suppression of mTOR by Torin 1 causes dephosphorylation of Lipin 1, which promotes its nuclear localization [26]. The nuclear localized dephosphorylated Lipin 1 decreases SREBP-1-mediated lipogenesis by reducing the abundance of nuclear SREBP-1 protein [26]. Mice with liver-specific deletion of raptor, a key component of the mTOR1 complex, are similar to rapamycin-treated HFD mice because they are highly resistant to HFD-induced weight gain and hepatic steatosis [18]. Therefore, it is possible that suppression of mTOR may have two beneficial effects: suppression of SREBP-mediated lipogenesis and induction of lipophagy. Pharmacological targeting of mTOR may thus provide a new avenue for NAFLD.
In addition to NAFLD, alcoholic liver disease (ALD) is another major liver disease in the United States and worldwide. One of the early major pathological changes of ALD is steatosis, and some heavy alcohol drinkers can have further progression to fibrosis and cirrhosis [27]. Interestingly, not all alcohol drinkers develop ALD, and only approximately 30% of heavy drinkers develop fibrosis and cirrhosis [28,29]. These data suggest that either genetic factors affect the susceptibility to advanced ALD or liver cells activate some protective pathways against its detrimental effects. Although some evidence indicates that patatin-like phospholipase domain-containing protein 3 (PNPLA3) may be involved in ALD in Caucasian alcohol drinkers [30,31], data from animal experiments failed to support such a notion [32]. Future studies are needed to identify other genetic factors in ALD. In addition to genetic factors, recent data from our lab suggest that autophagy is activated to attenuate acute ethanol-induced steatosis and liver injury. Acute ethanol-induced autophagy selectively targets damaged mitochondria and lipid droplets, but it does not seem to target general protein degradation because long-lived protein degradation is not changed in ethanol-treated primary hepatocytes [28,33]. More importantly, induction of autophagy by rapamycin completely suppresses acute alcohol-induced steatosis (Figure 1). As discussed above, in addition to induction of autophagy, rapamycin may also suppress lipogenesis by inhibiting mTOR 1. However, it seems that induction of autophagy would play a more important role in alcoholic steatosis because it has been reported that rapamycin does not affect lipogenesis gene expression due to its less potent inhibition on mTOR compared to Torin 1 [26]. Although the role of autophagy is relatively clear in acute alcohol-induced liver injury, it is less clear how the autophagy process is modulated in the chronic alcohol context. It is suggested that long time alcohol consumption may lead to autophagy suppression by affecting intracellular traffic and lysosomal functions [34]. Nevertheless, mTOR inhibitors could be very promising preventive or therapeutic drugs for both NAFLD and ALD because they can induce autophagy and may also suppress lipogenesis.
Figure 1. Induction of autophagy by rapamcyin inhibits acute ethanol-induced steatosis.
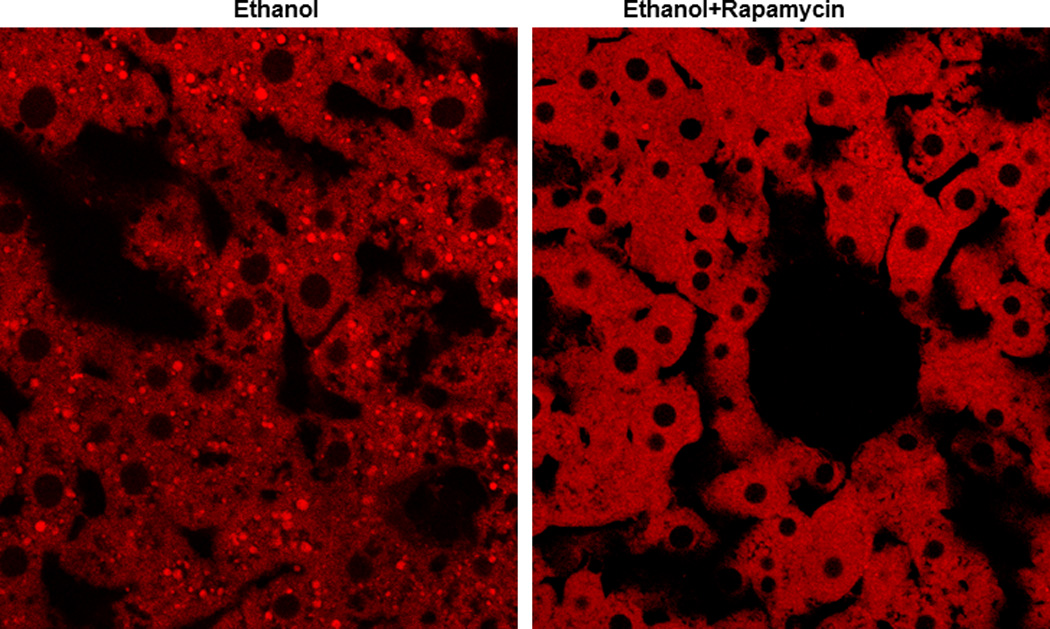
C57BL/6J mice were treated with ethanol (4.5 g/kg) or ethanol plus rapamycin (2 mg/kg) for 16 hours. Cryosection of livers were prepared and stained with Bodipy 581/591-C11 (50 nM) followed by confocal microscopy.
Although the above evidence strongly supports a role for autophagy in the regulation of lipid homeostasis in hepatocytes, autophagy may have additional roles in regulating adipocyte differentiation and in determining the balance between white and brown fat. Two independent groups have reported that knockout of either Atg5 or Atg7 suppresses adipocyte differentiation [35,36]. Similar effects are observed when using pharmacological inhibitors for autophagy or lysosomal functions. Decreased white adipose mass and enhanced insulin sensitivity are observed in the adipocyte-specific Atg7 knockout mouse. White adipocytes in Atg7 knockout mice have increased features of brown adipocytes because they are smaller and have more mitochondria with increased rates of fatty acid beta-oxidation. Consequently, adipocyte-specific Atg7 knockout mice have a lean body mass and are resistant to HFD-induced obesity [35]. Supporting the findings in animal models, autophagy is up-regulated due to decreased mTOR signaling in adipose tissue of obese people with or without diabetes [37,38]. Therefore, suppression of autophagy in different tissues may lead to different outcomes. While it would be beneficial to induce autophagy for treatment of existing fatty liver by degrading lipid droplets, suppression of autophagy in adipocyte tissue may also be beneficial to decrease adipocyte differentiation for protection against obesity and improvement of insulin sensitivity. How to differentially target and modulate autophagy activity in different tissues is thus a very important topic for future studies.
Targeting autophagy for drug-induced liver injury
Liver is the major organ to metabolize and detoxify drugs. Thus, the liver is often the target damaged by drugs. Drug-induced liver injury has been a major problem leading to a halt in drug development and withdrawal of approved drugs from the market. More than 50% of acute liver failure cases in the US are due to drug-induced hepatotoxicity. Acetaminophen (APAP) is a widely used drug and is an antipyretic and analgesic. An overdose of APAP can cause severe liver injury in animals and man although it is generally safe at therapeutic levels [39]. The molecular mechanisms and signaling pathways that regulate APAP-induced liver injury have been well studied [40]. APAP-induced hepatotoxicity is mainly mediated by N-acetyl-p-benzoquinone imine (NAPQI), a highly reactive metabolite that is generated from the metabolism of APAP by the cytochrome P450 system (such as CYP2E1). NAPQI depletes hepatic glutathione (GSH), a major molecule regulating cellular redox homeostasis. NAPQI also reacts with many cellular proteins, including mitochondrial proteins, to form protein adducts that trigger mitochondrial damage and subsequent necrosis. We recently found that APAP induces autophagy to remove damaged mitochondria to mitigate APAP-induced necrosis [41]. Treatment with rapamycin inhibits APAP-induced necrosis in primary cultured hepatocytes and in mouse livers (Figure 2). Interestingly, rapamycin does not affect APAP metabolism, suggesting its protective role is downstream of APAP metabolism and is likely mediated by the induction of mitophagy. More importantly, APAP-induced necrosis and liver injury are suppressed by post-treatment with rapamycin (2 hours after APAP administration) [41]. These observations may have potential therapeutic applications since most APAP-overdose patients in the emergency room have already surpassed the metabolism phase. Although the exact mechanisms by which mitophagy protects against cell death are not clear, it is thought that mitophagy may mitigate mitochondria-derived reactive oxygen species (ROS) formation and the release of pro-cell death factors from mitochondria. Indeed, we found that APAP-induced ROS are suppressed by rapamycin but exacerbated by CQ treatment [41].
Figure 2. Induction of autophagy by rapamcyin inhibits APAP-induced liver injury.
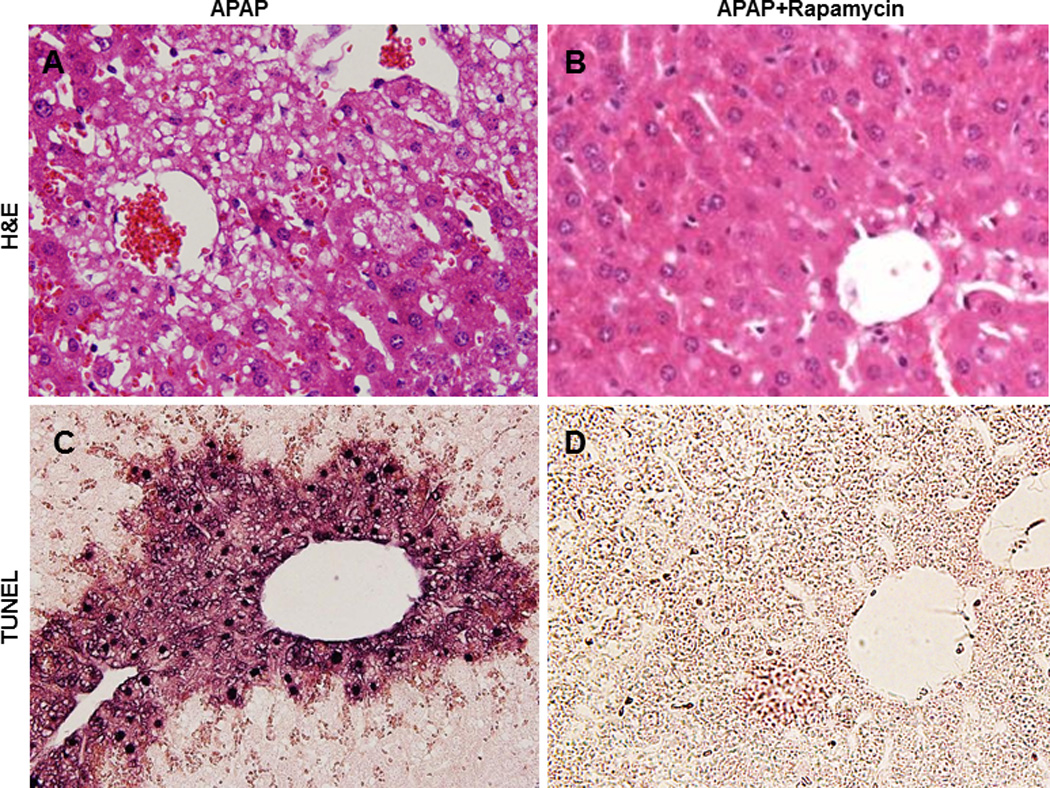
C57BL/6J mice were treated with APAP (500 mg/kg) or APAP plus rapamycin (2 mg/kg) for 6 hours. Tissue sections were stained with hematoxylin and eosine (H&E, A–B). Panel A shows typical central lobule necrotic cells with vacuolization, cell swelling and nuclear disintegration in APAP-treated mice, which are all eliminated by rapamycin (B). (C) TUNEL staining shows condensed positive nuclear DNA, and the diffuse staining in the area of necrosis indicates the release of nuclear DNA into the cytoplasm and extracellular in APAP-treated mice, whereas (D) all of these changes are inhibited by rapamycin treatment.
Since mitochondrion is a common target for various drug-induced hepatotoxicities, modulation of autophagy to remove the damaged mitochondria would be a promising approach to attenuate drug-induced liver injury. In addition to APAP, it is reported that Efavirenz (EFV), one of the most widely used non-nucleoside reverse transcriptase inhibitors for HIV, also induces hepatotoxicity via mitochondrial damage. Interestingly, EFV also triggers mitophagy as a cell survival mechanism, and pharmacological suppression of autophagy enhanced EFV-induced cell death [42].
Although autophagosome enveloped mitochondria was observed by Dr. De Duve in the 1960s, the molecular mechanisms of mitophagy have only recently been revealed. Research works from yeast have identified several molecules that are important for mitophagy including Uth1p, an outer mitochondrial membrane protein and member of the so-called “SUN” family, and Au1p, a member of the family of protein phosphatase homologs that localizes to the mitochondrial intermembrane space [43,44]. More recently, Atg32 was found to be a mitochondrial receptor for mitophagy in yeast [45,46]. Interestingly, Uth1p, Aup1p and Atg32 are necessary for mitophagy but not for nonspecific autophagy, indicating the selective nature of mitophagy. No Mammalian homologues of Atg32, Uth1p and Aup1p have been found. In general, it seems that multiple molecules and signaling pathways could be involved in mitophagy in mammalian cells. Among these pathways, Nix, BH-3 only Bcl-2 family proteins, and mitochondrial permeability transition have been suggested to play roles in mitophagy [47–49]. More recently, FUNDC1, an outer mitochondria membrane protein which has a typical LC3-binding motif YXXL, was found to play a critical role in hypoxia-induced mitophagy [50]. In addition, the role of the PTEN-induced mitochondrial protein kinase 1 (Pink1)-Parkin signaling pathway in mitophagy has recently been extensively studied. Pink1 and Parkin are two genes that are often mutated in recessive Parkinson’s disease (PD). Pink1 is a serine/threoine kinase with a mitochondrial targeting sequence, whereas Parkin is an E3 ubiquitin ligase. Recent evidence indicates that Pink1 is stabilized on the outer mitochondrial membrane where it recruits Parkin to impaired mitochondria when mitochondrial membrane potential is dissipated [51–54]. Parkin then promotes the ubiquitination of a subset of outer mitochondrial membrane proteins including VDAC, Tom20, mitofusin 1 (Mfn1) and mitofusin 2 (Mfn2) [55–57]. Parkin also recruits p62/SQSTM1 (Sequestosome 1), an autophagy receptor molecule, to mitochondria likely through the direct binding of p62 with ubiquitinated mitochondrial proteins because of its direct interaction with LC3 [54]. However, it has been found that p62 is not essential for mitophagy in mammalian cells [58]. One possibility that Pink1-Parkin promotes mitophagy could be due to Parkin-mediated Mfn1 and Mfn2 degradation. Mfn1 and Mfn2 are GTPase proteins that are essential for mitochondria fusion. Their degradation prevents fusion of mitochondria and thus promotes mitophagy. More intriguingly, it seems that different portions of mitochondrial proteins may be degraded via different mechanisms depending on the location of the protein on mitochondria. Whereas outer mitochondrial membrane and intermembrane space proteins are degraded by the proteasome, mitochondrial matrix proteins seem to be degraded by autophagy [56,57]. Further works are needed to elucidate how the ubiquitin proteasome system and autophagy are coordinated to selectively degrade mitochondria. Nevertheless, it appears that targeting autophagy to remove damaged mitochondria could be a promising approach for mitochondria-mediated hepatoxicity induced by various drugs.
Targeting autophagy for protein aggregate-mediated liver injury
In response to starvation, autophagy is activated to degrade proteins in order to provide the necessary nutrients for cells to survive. Although this process is in general a nonselective process similar to the selective removal of damaged/or excess organelles, autophagy may also selectively remove intracellular protein aggregates. Although cells can also utilize the ubiquitin-proteasome system in addition to autophagy for protein degradation, the ubiquitin-proteasome system can only degrade soluble proteins. In contrast, autophagy degrades all forms of misfolded proteins and can have a degree of specificity via receptor molecules, such as p62 and NBR1 (neighbor of BRCA1 gene 1). p62 and NBR1 directly use their C-terminal ubiquitin binding domains (UBA) to bind with poly- or mono-ubiquitin, and they also bind directly with the autophagy protein LC3 with their LIR (LC3-interacting region). Thus, p62 and NBR1 act as autophagy receptors for ubiquitinated proteins and link them to autophagy degradation [59–61]. If not timely removed, misfolded proteins can be toxic to cells by inducing ER stress to trigger cell death. Several protein aggregate-related liver diseases exist in humans, such as alpha-1-antitrypsin (AT) deficiency, hypofibrinogenemia and alcoholic Mallory body formation. AT protein is synthesized in hepatocytes and secreted into the blood where it acts as an inhibitor for neutrophil proteases. A mutation in the AT gene results in the misfolding of AT protein and causes its retention in the ER as an aggregate form. Although a portion of AT aggregates may be degraded by the ubiquitin-proteasome system, recent evidence indicates that autophagy plays a critical role in removal of AT aggregates [62]. Autophagosomes with enveloped AT aggregates and increased autophagosome numbers in the liver cells of AT deficient patients have been observed. Moreover, when mutant AT proteins are expressed in Atg5-knockout cells, the degradation of mutant AT proteins is decreased compared with wild type cells. More importantly, carbamazepine (CBZ), a widely used anti-seizure drug with low toxicity, significantly decreases AT protein aggregates by inducing autophagy in mouse livers [63]. As a result, CBZ significantly reduces liver injury and fibrosis in AT mutant mice. Interestingly, in addition to increased autophagy, CBZ also increases proteasome-mediated protein degradation. Although CBZ has no effects on insoluble mutant AT protein aggregate degradation in Atg5-knockout MEFs, CBZ increases the degradation of soluble mutant AT protein aggregates in these Atg5-knockout cells likely through increased proteasomal degradation. Intriguingly, rapamcyin has no effect on mutant AT protein aggregate degradation, suggesting CBZ may induce autophagy through an mTOR-independent pathway. As discussed above, efficient mitophagy seems to require both the proteasome and autophagy. Therefore, it will be interesting to examine whether CBZ is an ideal drug to induce mitophagy in future studies. Several other mTOR-independent autophagy inducers have been identified from small molecule screening using either clearance of autophagy substrates, such as mutant huntingtin, or by using the GFP-LC3 puncta assay [64–66]. A group of chemicals including lithium, sodium valproate and CBZ induce autophagy for the clearance of mutant huntingtin by reducing intracellular inositol levels in an mTOR-independent manner [65]. In addition, L-type Ca2+ channel antagonists such as verapamil, loperamide, amiodarone, nimodipine, and nitrendipine, can also enhance mTOR independent autophagy by modulating intracellular Ca2+ levels [64]. However, it remains to be tested whether these drugs also attenuate AT deficiency-induced liver injury.
Similar to the pathological changes of AT deficiency, hypofibrinogenemia is another liver ER storage disease due to a mutant form of fibrinogen. Fibrinogen is synthesized in hepatocytes and secreted into blood to regulate blood coagulation. A mutant form results from the single substitution of Arg375 to Trp in the γ-chain and causes the formation of misfolded fibrinogen protein aggregates, which are named Aguadilla γD [67,68]. Aguadilla γD aggregates accumulate in the ER, and a subset of individuals with this mutant develop liver fibrosis [67,68]. Using yeast strains that are either deficient of proteasome components (pre1-1 and pre2-2) or an autophagy gene (atg14), it was found that both the proteasome and autophagy are responsible for clearance of Aguadilla Γd [67]. However, it remains unknown whether pharmacological modulation of autophagy, such as by using CBZ, would also attenuate hypofibrinogenemia and its related liver pathogenesis.
Chronic alcohol consumption also leads to the formation of inclusion bodies known as “Mallory Bodies” in hepatocytes, which is often accompanied with alcoholic hepatitis and cirrhosis [69]. Mallory Bodies are ubiquitin positive protein aggregates which also contain cytokeratin 8 and 18. They share similar features with other inclusion bodies found in neuronal degenerative diseases, such as Lewy bodies in Alzheimer's disease and Huntingtin inclusions in Huntington's disease [70]. Although the mechanisms for the induction of Mallory Bodies are not completely known, it is suggested that inhibition of proteasome activity may play a role [71]. Indeed, proteasome inhibitors can induce Mallory body-like structures in cultured cells and in mouse liver. Interestingly, induction of autophagy by the mTOR inhibitor rapamycin significantly suppresses Mallory body formation both in vitro and in vivo [71,72]. These findings suggest that induction of autophagy may help to attenuate Mallory body formation [71,72].
It should also be noted that among individuals that carry the gene mutant of either AT or fibrinogen, only a subset of the individuals develop liver disease. Therefore, other genetic factors, such as a second gene mutation, or other pathological complications, such as viral infection that affects either proteasome or autophagy function, may determine the development of these diseases. It is possible that different individuals may have varied proteasome or basal autophagy functions. Nevertheless, targeting autophagy may be a beneficial therapeutic approach for these protein aggregate-induced liver diseases.
Targeting autophagy for viral hepatitis
Autophagy and/or autophagy genes might have both anti-viral and pro-viral functions against various viruses. Autophagy directly recognizes viral components and promotes their degradation in lysosomes, a process termed “xenophagy” [73–76]. Autophagy can also influence the innate and adaptive immune system in response to viral infections. Some viruses have adapted and use mechanisms that either inhibit autophagy or allow their escape from autophagy recognition to survive [76]. In contrast, other viruses, including the hepatitis C and B viruses (HCV and HBV), use components of the autophagy machinery to promote their own replication [76–80]. It is estimated that more than 130 million people are infected with HCV worldwide, and HCV is a major cause of chronic liver disease [81]. HCV is an enveloped single-strand RNA virus with a 9.6kb genome that encodes a large polyprotein and is processed by cellular and viral proteases to produce mature core and non-structured proteins [82]. While the structured core proteins are the major components of the viral particle, the non-structured proteins are required for HCV genome replication because they organize the replication complexes associated with a multi-membrane structure called the membranous web [83]. Moreover, HCV infection is also associated with hepatic steatosis due to decreased mitochondrial β-oxidation and lipoprotein secretion as well as enhanced lipogenesis [84,85]. Lipid droplets may also play a role in HCV replication because the HCV core protein and HCV replication complex are often closely associated with lipid droplets [85]. Furthermore, inhibition of lipid biosynthesis suppresses HCV replication, supporting a role for lipid droplets in HCV replication [86].
Increasing evidence indicates that HCV infection is associated with the autophagic process although controversy exists regarding whether HCV infection induces incomplete, complete or selective autophagy. Accumulation of early autophagosomes, but not late autolysosomes, are observed in HCV viral RNA-transfected Huh7 cells, and this induction seems dependent on HCV virus-induced ER stress and unfolded protein response (UPR) [78]. In patients with chronic HCV infection, an electron microscopy analytic study found an increased number of early autophagic vesicles but did not find late autolysosomes [87]. Furthermore, long-lived protein degradation was not changed, which suggests that accumulated autophagosomes are either due to a defect in fusion with lysosomes or alterations of lysosomes due to HCV infection [78]. Another study using the same HCV-infected Huh7 cells found increased UPR-mediated autophagy by HCV, and this autophagic process was completed by assessing autophagic flux and using EM analysis to determine the number of autophagosomes and autolysosomes present [88]. The exact explanation for these discrepancies is not clear, but it could be due to limitations of in vitro cell culture conditions used in these studies. Nevertheless, it is generally agreed that autophagy may favor HCV replication. Genetic suppression of autophagy by using siRNAs against essential autophagy genes such as Atg7, LC3, Beclin-1, Atg5 and Atg12 suppresses HCV replication [78,80,88]. Since UPR is required for HCV-induced autophagy, it was found that genetic silencing of UPR components also leads to inhibition of HCV replication and infectious virus production [88]. It is proposed that HCV replication may utilize autophagosomal membranes as the membranous web for organizing the HCV replication complex. Thus, knockdown of essential autophagy genes may lead to a decreased number of autophagosomes and, in turn, result in less HCV replication. Moreover, like other viruses, HCV also has the capacity to evade innate immune defenses by using its serine protease NS3/4A-dependent proteolysis of the mitochondria-associated antiviral signaling protein (MAVS). Interestingly, suppression of autophagy by either knockdown of Atg5 or CHOP (a UPR gene) enhances HCV-triggered innate immunity activation. Induction of autophagy by rapamycin inhibits, whereas suppression of autophagy by CQ enhances, HCV-mediated innate immunity activation. Furthermore, blocking the fusion of autophagosomes with lysosomes by knockdown of lysosome-associated membrane protein 2 (LAMP2) and Rab7, two genes that are essential for the maturation step of autolysosomes, also suppresses the HCV-induced innate immune response. Consequently, knockdown of LAMP2 and Rab7 also suppresses HCV replication in HCV-infected Huh7 cells [88]. Thus, it seems that a complete autophagic process is indispensable for productive HCV replication and for suppression of the HCV-mediated innate immune response.
In addition to in vitro studies using Huh7 cells, Desai et al. generated a transgenic mouse with hepatocyte-specific expression of NS3/4A proteins to study the role of the viral protease on interferon (IFN) and autophagy in HCV-infected mouse liver [89]. Interestingly, no significant immunosuppression or delayed viral clearance was found in transgenic mice expressing NS3/4A despite increased NS3/4A-mediated cleavage of MAVS. Mice treated with IFN-α or IFN-β had differential effects on the autophagic process. Through cellular fractionation studies, it was found that IFN-β promotes autophagic flux and autolysosome-mediated NS3/4A protein degradation. In contrast, IFN-α steered autophagosomal compartments away from lysosomes. Thus, it is suggested that a balance between IFN-α and IFN-β is crucial for an efficient anti-HCV host response.
Although HCV-mediated autophagy does not induce long-lived protein degradation, a recently study demonstrated that selective autophagy for lipids is induced in HCV replicon and HCV-infected cells in vitro [90]. Using a double-tagged green and red fluorescent LC3 to determine autophagic flux, this study showed that HCV-induced autophagosomes are capable of maturing, and that the LC3-positive vesicles contained cholesterol. It is speculated that inhibition of autophagy may alter the flux of cholesterol from lysosomes to the endoplasmic reticulum and, in turn, affect the biogenesis of lipid droplets and impair HCV virion assembly. Collectively, it seems that autophagy is required for HCV replication, delivery of incoming viral RNA to the translational apparatus and suppression of the innate anti-viral immune response. Pharmacological suppression of autophagy may help to restrict HCV replication, but caution needs to be taken for this approach because suppression of autophagy may lead to excessive lipid accumulation.
In contrast to HCV, HBV is a small partially double-stranded DNA virus which is able to integrate into the cellular chromosomal DNA. It is estimated that 350 million people are chronically infected with HBV, and 25% of the HBV-infected individuals will develop HCC [91]. Similar to HCV, HBV also induces ER stress-mediated autophagy without affecting long-lived protein degradation in hepatoma cells that are transfected with HBV plasmids [79,92]. Expression of HBV protein induces Beclin-1 up-regulation, and HBV protein also binds to phosphatidylinositol 3 Kinase class III (PI3K) to enhance its activity and, in turn, promote autophagy [79,93]. Moreover, mutation analysis shows that HBV small surface protein (SHBs) is also required for HBV-induced autophagy [92]. Pharmacological inhibition of PI3K by 3MA or siRNA knockdown of Vps34 (the mammalian PI3K) suppresses autophagy, which leads to the suppression of HBV DNA replication. This suggests that HBV can use autophagy to enhance its DNA replication. Furthermore, it has been found that autophagy machinery is critical for envelope and release of HBV. Pharmacological induction of autophagy by rapamycin promotes HBV envelopment and production, whereas 3MA inhibits HBV envelopment and release [92]. Unlike HCV, HBV DNA replication does not involve the membranous web or require association with lipid droplets. It seems that the role of autophagy in enhancing HBV viral replication may be mainly to regulate HBV envelopment and release. Based on the above discussion, it appears that autophagy promotes the replication of both HCV and HBV even though the two viruses have different structures. It is possible that pharmacological suppression of autophagy may inhibit hepatitis viral replication.
Targeting autophagy for liver fibrosis
Liver fibrosis is the final outcome of chronic liver diseases, which is the result of a reversible wound-healing response toward repeated liver injury [94,95]. Liver fibrosis is characterized by the accumulation of extracellular matrix (ECM), which often leads to the disruption of liver parenchyma by forming scar tissue. Fibrosis can progress to cirrhosis, chronic liver failure, portal hypertension, and HCC. The pace of this progression ranges from 20 to 40 years, which is influenced by several environmental and certain genetic factors. Every year, approximately 30,000 people die of cirrhosis in the US, which is the most common non-neoplastic cause of mortality [96]. Using cell culture and animal models, multiple mechanisms have been suggested to be critical for the pathogenesis of liver fibrosis and include: oxidative stress, increased fibrogenic cytokines (i.e., transforming growth factor-β (TGF-β)), hepatocyte apoptosis and chronic inflammation [95]. It is generally believed that the activated myofibroblast is responsible for fibrosis in all chronic liver diseases., Overwhelming evidence indicates that quiescent hepatic stellate cells (HSC) are activated to myofibroblasts to produce the scar tissue found in fibrotic liver diseases including viral hepatitis, alcohol and non-alcoholic liver diseases as well as biliary disease However, it has also been suggested that bone marrow-derived fibrocytes or circulating mesenchymal cells may also be the source for myofibroblasts. In addition, cells (hepatocytes, cholangiocytes and endothelial cells) may undergo a transition to mesenchymal cells to become activated myofibroblasts [94,95,97]. The majority of retinoids in the body are stored in the quiescent HSC, and retinoids are lost from HSC when they are activated. Activated HSC express new receptors such as the platelet- derived growth factor (PDGF) receptor and TGF-β receptor as well as new proteins such as α-smooth muscle actin [95]. Activated HSC also produce collagen and ECM to replace parenchymal tissue by scarring. Multiple mechanisms have been suggested to be important for the activation of HSC, such as activation of immune cells including cytokines and ECM components following liver injury [94].
Another well-known feature of HSC activation is the loss of cytoplasmic lipid droplets, which are mainly composed of lipids in the form of fatty esters such as triglycerides. Since autophagy can breakdown lipid droplets via lipophagy, it is hypothesized that autophagy may play a role in regulating the loss of lipid droplets during HSC activation. Indeed, two different groups recently independently reported that induction of autophagy in HSCs promotes HSC activation and proliferation [17,98]. Treatment with Bafilomycin A1, a vacuole ATPase inhibitor, suppresses autophagy and results in the inhibition of proliferation and activation of both cultured mouse and human HSC. Other autophagy inhibitors, such as 3MA and CQ, also inhibit HSC activation in vitro [17,98]. Administration of CCl4 or thioacetamide (TAA) has been well-known to induce liver fibrosis in mouse livers. Interestingly, autophagy is increased in either CCl4 or TAA-treated HSC in vivo. To further elucidate the role of HSC autophagy in liver fibrosis in vivo, Dr. Friedman’s group generated HSC-specific Atg7 knockout mice using the glial fribrillary acidic protein promoter (GFAP-cre) [17]. They found that liver fibrosis is attenuated in either CCl4 or TAA-treated HSC-specific Atg7 knockout mice compared with wild type mice. However, there was no difference in liver injury between HSC-specific Atg7 knockout mice and wild type mice, suggesting the suppression of liver fibrosis was not secondary to the reduced liver injury. Since HSC activation is an energy consuming process, the authors hypothesized that lipophagy in HSC cells may provide a key energy source of free fatty acids from the breakdown of lipid droplets to fuel HSC activation. This notion is further supported by the observation that addition of oleic acid to HSC significantly rescued the decrease of fibrogenesis induced by a block in autophagy. Theoretically, it is therefore possible that selective inhibition of autophagy in liver fibrogenic cells might be used to treat patients with liver fibrosis. However, since the fibrogenic cells only account for a small portion of the cells in the liver, it is not clear how the drug would specifically target fibrogenic cells without affecting other cell types. Moreover, conflicting data also exist that support an anti-fibrosis role of autophagy. Increased collagen deposition and fibrosis is observed in Beclin 1 heterozygous deletion mice, suggesting autophagy may suppress fibrosis in the kidney [99]. Further analysis from primary cultured mouse mesangial cells (MMC) reveals that pharmacological inhibition of autophagy (by Bafilomycin A1, (BAF)) or genetic knockdown of Beclin 1 (by siRNA) leads to increased protein levels of collagen in TGF-β-treated cells. Interestingly, collagen is found to be co-localized with LC3 positive vesicles and in LAMP1 positive lysosomes, suggesting that collagen might be degraded via the autophagy pathway. As discussed above, induction of autophagy by CBZ also attenuates liver fibrosis in the mouse mutant AT model [63]. However, it should be noted that mutant AT-induced liver fibrosis might be triggered by the chronic inflammatory response induced by the mutant AT protein aggregates whereas CCl4-induced liver fibrosis is mainly mediated by the post-liver injury. Moreover, autophagy is activated by CBZ in hepatocytes, but it is not clear whether autophagy is also increased in HSC cells in CBZ–treated mice. Therefore, autophagy might suppress liver fibrosis by preventing excess collagen accumulation or promote liver fibrosis by enhancing HSC activation. These conflicting data could be due to different models used. Therefore, further studies are needed to clarify the role of autophagy in the process of liver fibrosis before a therapeutic approach targeting autophagy can be used to treat patients with liver fibrosis.
Targeting autophagy for aged liver
While deficient or impaired autophagy can contribute to aging, aged tissues also have declined autophagy activity. The aging-related degeneration process is improved by conditions that activate autophagy, such as caloric restriction. The life span in multiple organisms including yeast, C elegans, mice and primates is significantly extended by stimulation of autophagy [100–102]. The expression levels of many autophagy genes or related proteins (such as Atg7, Atg5, Atg4B and Lamp2) are decreased in the brains and livers of aging humans and mice [103–105]. Zhang and Cuervo found that there is increased liver injury in aged mice (22 month old) compared to young mice (6 month old), and that the increased injury is likely due to increased apoptosis as demonstrated by increased capase-3 activity, TUNEL positive cells and serum alanine aminotransferase (ALT) activity. Part of the impaired liver function could be due to declined Lamp-2A levels in aged livers, which results in decreased chaperone-mediated autophagy. Restoration of Lamp-2A levels in the mouse liver using an inducible-transgenic mouse model significantly decreases apoptosis and improves the impaired detoxification rate of xenobiotics in the aged liver [105]. Moreover, aged mice are more susceptible to ischemia/reperfusion-induced liver injury. Overexpression of either Atg4B or Beclin 1 significantly inhibits ischemia/reperfusion-induced liver injury by increased autophagy activity and blockage of the onset of mitochondrial permeability transition [104].
In addition to caloric restriction, resveratrol has shown beneficial effects against the aging process in many experimental systems [106]. The anti-aging effects of both caloric restriction and resveratrol might be due to the activation of Sirtuin 1 (SIRT1) [106]. SIRT1 is a NAD+-dependent deacetylase that acts both in the nucleus and in the cytoplasm. SIRT1 deacetlyates a subset of autophagy proteins such as Atg5, Atg7 and LC3, but the exact role of the acetylation modification of these autophagy proteins on the regulation of the autophagic process is not clear [107]. SIRT1 can also regulate several transcription factors, such as p53, NF-κB, HSF1, FOXO1, -3, -4, and PGC1α, which are all involved in life span regulation and simultaneously influence autophagy [108]. Long term: future work is needed to demonstrate whether liver functions would be improved in long time resveratrol fed animals or by other pharmacological means that increase autophagy activity.
Targeting Autophagy for liver cancer
HCC is one of the major cancers and a major health problem worldwide. HCC accounts for more than 600,000 deaths per year [109]. HCC is very common in southeast Asia and Africa due to their high HBV infection rate. However, the incidence of HCC has increased in the US and western Europe over the past 25 years. Viral infection, such as HBV (mainly in Asia and Africa) and HCV (mainly in western countries and Japan), as well as alcohol abuse are responsible for the majority of HCC, but the exact molecular pathogenesis is not yet well understood [110,111]. HCC is a highly malignant and fatal neoplasia. Treatments such as surgical resection, ablation and transplantation can significantly improve survival in patients diagnosed at an early HCC stage. However, no effective treatments are available for patients with advanced or intermediate stage HCC [112,113].
As an essential regulator for the homeostasis of cellular nutrients, energy and organelles, it is not surprising that autophagy plays an important role in tumorigenesis and cancer therapy. It has been well-documented that autophagy is a tumor suppression mechanism. Mechanistically it is suggested that autophagy works as a sentinel to remove damaged proteins and organelles, especially damaged and senescent mitochondria, which are the major cellular sources of ROS. Autophagy can also protect against cellular metabolic stress and genome-instability and thus prevent tumorigenesis. One of the earliest and best evidence that autophagy acts as a tumor suppressor is from the observations made from Beclin 1 knockout mice [114,115]. Beclin 1 is the yeast homologue of Atg6, which forms a complex with Vps34, Vps15 and Atg14. This complex promotes autophagic membrane nucleation by producing phosphatidylinositol 3-phosphate (PI-3-P). While homozygous Beclin 1 knockout mice are embryonic lethal, Beclin 1 heterozygous mice survive and develop spontaneous tumors in multiple tissues including liver [115]. Subsequently, it is further found that Beclin 1 is frequently monoallelically deleted in many human cancers such as breast, prostate and ovarian cancers [114,116]. Moreover, loss of other autophagy regulatory genes, such as Bif-1 and Atg4C, increases tumorigenesis in mice [117,118], and loss of heterozygosity of UVRAG, a Beclin 1 interacting protein, is also frequently observed in colon cancers in human [117,119]. To further support the concept that autophagy is a tumor suppressor, many other known tumor suppressor genes, such as LKT, AMPK, and phosphatase and tensin homologue (PTEN), are positive regulators of autophagy [120–123], whereas many oncogenes, including phosphatidylinosital 3-kinase, Akt and anti-apoptotic Bcl-2 family proteins, suppress autophagy [124]. More recently, it was demonstrated that mice lacking either Atg5 or Atg7 in the liver also develop spontaneous benign tumors. More importantly, it is found that increased p62 protein due to the lack of autophagy in these mouse livers plays a critical role in liver tumor development [125,126]. p62 competes with kelch-like ECH-associated protein 1 (KEAP1) for its binding with nuclear factor (erythroid-derived 2)-like 2 (NFE2L2/NRF2) resulting in the release and persistent activation of NRF2 in the autophagy deficient mouse liver [127]. In addition to the regulation of NRF2, recent evidence suggests that p62 may have multiple functions, such as the regulation of NF-kB [128], apoptosis [129], Wnt signaling [130] and mTOR [131]. Interestingly, deletion of p62 in Atg7-deficient mouse livers significantly decreases the incidence of liver tumors [126]. By analyzing human HCC samples from more than 100 patients, it was found that the protein level of p62 and the activation of NRF2 are closely associated with HCC development [125]. Although it has long been known that NRF2 plays a critical role in regulating induction of cellular detoxification enzymes against various cellular stresses, somatic mutations in NRF2 and KEAP1 are often found in gall bladder, lung, and head and neck tumors [132]. These mutations all lead to the persistent activation of NRF2 and increased expression of cellular antioxidants, detoxification enzymes and multi-drug efflux pumps [132,133]. ROS have long been thought to promote tumorigenesis by inducing DNA damage, activating inflammatory pathways and stabilizing the hypoxia inducing factor. However, ROS can also be harmful to cancer cells by inducing cell death. Therefore, it is not surprising that cancer cells also use mechanisms to avoid death by adaptive up-regulation of the KEAP1-NRF2 pathway. Indeed, a recent study found that NRF2 promotes oncogene-driven tumorigenesis by reducing ROS production in a mouse model. Genetic deletion of NRF2 impairs K-Ras (G12D)-induced pancreas cancer in vivo [134]. It remains unknown whether deletion of NRF2 would also rescue liver tumors in Atg5 or Atg7-deficient mouse livers. Accumulation of p62 has been found to be associated with poor prognosis in patients with lung carcinoma, but not squamous cell carcinoma, suggesting some evolutionary differences between different types of cancers [135]. Nevertheless, understanding the cellular functions of p62-KEAP1-NRF2 pathways may provide a new therapeutic basis for the development of treatments for HCC.
While normal cells can use autophagy to inhibit the process of tumorigenesis, tumor cells can also use autophagy as a cell survival mechanism against cellular stress or apoptosis induced by many traditional chemotherapeutic drugs. This is particularly crucial because many current cancer therapy agents activate autophagy. As discussed above, mTOR plays a central role in the regulation of cellular growth, proliferation, and survival by phosphorylating downstream p70S6K and 4-EBP1 to activate protein synthesis. Mutations of negative mTOR regulators, such as TSC1-TSC2, LKB1 and PTEN, are often found in many cancers. Moreover, many cancer cells have aberrant activation of AKT and PI3K, which leads to the activation of mTOR. mTOR has an important role in regulating cancer cell proliferation, therefore, targeting mTOR inhibition has been used for anti-cancer treatment. However, use of mTOR inhibitors, such as rapamycin, for cancer treatment in clinical trials has generated very limited success. One possible explanation for these disappointing clinical trials is that rapamcyin activates autophagy as a cell survival mechanism, which may counteract its inhibitory effects on tumor cell growth. Future studies are needed to test the efficacy of mTOR inhibitors on tumor therapy in combination with autophagy inhibitors.
While no effective therapeutic treatment is available for advanced stage HCC patients, one new drug named sorafenib has recently proven to have some survival benefits [112]. Sorafenib is a multi-kinase inhibitor that targets the Ras/Raf/MEK/ERK signaling and vascular endothelial growth factor receptors (VEGFRs) and platelet-derived growth factor receptor to inhibit tumor proliferation and angiogenesis as well as to induce apoptosis in a variety of tumor models [136,137]. We recently found that in addition to inducing apoptosis in cultured HCC cells, sorafenib also induces autophagy through ER stress independent of the MEK1/2-ERK1/2 pathway. Inhibition of autophagy by CQ markedly increased sorafenib-induced tumor suppression in vitro and in vivo [138]. We also found that oxaliplatin, a new platinum-based chemotherapy drug, induces autophagy in various HCC cell lines. Combination of oxaliplatin with the autophagy inhibitor CQ further increases oxaliplatin-induced ROS production and, in turn, results in more pronounced tumor suppression in both cultured HCC cells and in HCC xenografts [139]. Similarly, inhibition of autophagy has also been shown to sensitize histone deacetylase inhibitor-induced cell death in HCC cells [140]. Moreover, most tumor cells live in a hypoxic environment in solid tumors including HCC. Autophagy is activated under hypoxic conditions in HCC cells, and thus makes HCC cells more resistant to chemotherapy [141]. Therefore, combination of autophagy inhibitors and other molecular targeted therapies may be a promising strategy to increase the efficiency of chemotherapy or to overcome the drug-resistance that occurs in many treatments due to the induction of autophagy in HCC treatment.
Concluding Remarks
Our knowledge of the molecular mechanisms regulating autophagy and its impact on human diseases has substantially improved due to the rapid research progress on autophagy. As outlined in this review, autophagy plays significant roles in multiple aspects related to liver pathophysiology and liver diseases: removal of misfolded proteins and balance of nutrients and energy, sentinel for organelle turn over, maintenance of lipid homeostasis, and participation in hepatitis virus infection and replication . Defects or suppression of autophagy can lead to hepatocyte cell death, steatohepatitis and HCC. As summarized in this review, several potential chemicals have been used as a pharmacological approach to modulate autophagy in hepatocytes cultured in vitro and in mouse liver (Table 1). Moreover, many high-throughput screenings for small molecules that modulate autophagy have been conducted and are undergoing, and these will definitely generate more productive ways to modulate autophagy to benefit patients with liver diseases.
Table 1.
Agents Targeting Autophagy for Liver Diseases
| Agent | Autophagy | Disease | Pathophysiologic Effect | References |
|---|---|---|---|---|
| 3MA | Suppresses | Steatosis/NAFLD | Increases TG content in hepatocytes | (Singh et al., 2009a) |
| HBV | Suppresses viral DNA replication, envelopment and release | (Li et al., 2011) | ||
| Liver Fibrosis | Suppresses HSC activation | (Hernandez-Gea et al., 2012; Thoen et al., 2011) | ||
| CQ | Suppresses | Steatosis/NAFLD | Increases TG content in hepatocytes | (Mei et al., 2011) |
| APAP-induced hepatotoxicity | Increases APAP-induced ROS formation | (Ni et al., 2012) | ||
| HCV | Increases HCV-mediated innate immunity activation | (Ke et al., 2011) | ||
| Liver Fibrosis | Suppresses HSC activation | (Hernandez-Gea et al., 2012; Thoen et al., 2011) | ||
| HCC | Increases oxaliplatin and sorafenib-induced tumor suppression | (Ding et al., 2011, Shi et al., 2011) | ||
| Rapamycin | Induces | Steatosis/ALD | Suppresses acute alcohol-induced steatosis | (Ding et al., 2010) |
| Steatosis/NAFLD | Decreases TG content in hepatocytes | (Mei et al., 2011; Singh et al., 2009a) | ||
| APAP-induced hepatotoxicity | Suppresses APAP-induced necrosis, liver injury and ROS production | (Ni et al., 2012) | ||
| Protein aggregate-induced disease | Reduces formation of Mallory Bodies | (Harada et al., 2008a; Harada et al., 2008b) | ||
| HCV/HBV | Suppresses HCV-mediated innate immunity activation, promotes HBV envelopment and production | (Ke et al., 2011; Li et al., 2011) | ||
| Torin 1 | Induces | Steatosis/ALD | Decreases SREBP1-mediated lipogenesis | (Peterson et al., 2011) |
| CBZ | Induces | AT-protein aggregation | Decreases AT-protein aggregates and associated liver fibrosis in a mTOR-independent manner | (Hidvegi et al., 2010) |
| Huntington's Disease | Clear mutant Huntington proteins | (Sarkar et al., 2007) | ||
| Lithium | Induces | Huntington's Disease | Clear mutant Huntington proteins in a mTOR independent manner | (Sarkar et al., 2007) |
| Valproate | Induces | Huntington's Disease | Clear mutant Huntington proteins in a mTOR independent manner | (Sarkar et al., 2007) |
| CCL4 | Induces | Liver Fibrosis | Increases fibrosis | (Hernandez-Gea et al., 2012) |
| TAA | Induces | Liver Fibrosis | Increases fibrosis | (Hernandez-Gea et al., 2012) |
| Resveratrol | Induces | Liver Aging | Slows aging process | (Rubinsztein et al., 2011) |
| BAF | Suppresses | Liver Fibrosis | Suppresses proliferation and activation of HSC | (Hernandez-Gea et al., 2012; Thoen et al., 2011) |
Acknowledgement
The research work in W.X Ding’s lab was supported in part by the NIAAA funds R01 AA020518-01, R21 AA017421, National Center for Research Resources (5P20RR021940-07), and P20 RR016475 from the IDeA Networks of Biomedical Research Excellence (INBRE) program of the National Center for Research Resources. J. Williams was supported by the “Training Program in Environmental Toxicology” [grant 5 T32 ES007079] from the National Institute of Environmental Health Sciences. The authors would like to thank Margitta Lebofsky for her excellent assistance for the TUNEL assay.
List of Abbreviations
- APAP
Acetaminophen
- ALD
Alcoholic liver disease
- ALT
Alanine aminotransferase
- AT
Alpha-1-antitrypsin
- BAF
Bafilomycin A 1
- CBZ
Carbamazepine
- CQ
Chloroquine
- CMA
Chaperone-mediated autophagy
- 4EBP1
4E translational initiation factor 4E binding protein-1
- EFV
Efavirenz
- ECM
Extracellular matrix
- ER
Endoplasmic reticulum
- GSH
Glutathione
- HCV
Hepatitis C Virus
- HBV
Hepatitis B Virus
- HCC
Hepatocellular carcinoma
- HFD
High-fat diet
- HSC
Hepatic stellate cell
- IFN
Interferon
- KEAP1
Kelch-like ECH-associated protein 1
- LIR
LC3-interacting region
- LAMP2
Lysosome-associated membrane protein 2
- MAVS
Mitochondria-associated antiviral signaling protein
- Mfn1
Mitofusin1
- Mfn2
Mitofusin2
- 3MA
3-methyladenine
- mTOR
Mammalian target of rapamycin
- MTOR1
mTOR Complex 1
- MTOR2
mTOR Complex 2
- NAPQI
N-acetyl-p-benzoquinone imine
- NFE2L2/NRF2
Nuclear factor erythroid-derived 2-like 2
- NBR1
Neighbor of BRCA gene 1
- NAFLD
Nonalcoholic fatty liver disease
- p70SGK
70-kDa ribosomal protein S6 kinase-1
- PI3K
Phosphatidylinositol 3 kinase class III
- PI-3-P
phosphatidylinositol 3-phosphate
- PNPLA3
Patatin-like phospholipase domain-containing protein 3
- Pink1
PTEN-induced mitochondrial protein kinase 1
- PD
Parkinson’s Disease
- PDGF
Platelet-derived growth factor
- PTEN
phosphatase and tensin homologue
- ROS
Reactive oxygen species
- SQSTM1
Sequestosome 1
- SREBPs
Sterol regulatory element-binding protein
- SHBs
HBV small surface protein
- SIRT1
Sirtuin 1
- TSC1/2
Tuberous sclerosis complex 1 / 2
- TAA
Thioacetaminide
- TG
Triglyceride
- TGF-β
Transforming Growth Factor β
- UBA
Ubiquitin binding domain
- UPR
Unfolded protein response
- VEGFR
Vascular endothelial growth factor receptors
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–1075. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ding WX. Role of autophagy in liver physiology and pathophysiology. World J Biol Chem. 2010;1:3–12. doi: 10.4331/wjbc.v1.i1.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yin XM, Ding WX, Gao W. Autophagy in the liver. Hepatology. 2008;47:1773–1785. doi: 10.1002/hep.22146. [DOI] [PubMed] [Google Scholar]
- 4.Arias E, Cuervo AM. Chaperone-mediated autophagy in protein quality control. Curr Opin Cell Biol. 2011;23:184–189. doi: 10.1016/j.ceb.2010.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ding WX, Ni HM, Gao W, Yoshimori T, Stolz DB, Ron D, Yin XM. Linking of autophagy to ubiquitin-proteasome system is important for the regulation of endoplasmic reticulum stress and cell viability. Am J Pathol. 2007;171:513–524. doi: 10.2353/ajpath.2007.070188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sumpter R, Jr, Levine B. Selective autophagy and viruses. Autophagy. 2011;7:260–265. doi: 10.4161/auto.7.3.14281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Singh R, Cuervo AM. Autophagy in the cellular energetic balance. Cell Metab. 2011;13:495–504. doi: 10.1016/j.cmet.2011.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shintani T, Klionsky DJ. Autophagy in health and disease: a double-edged sword. Science. 2004;306:990–995. doi: 10.1126/science.1099993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rautou PE, Mansouri A, Lebrec D, Durand F, Valla D, Moreau R. Autophagy in liver diseases. J Hepatol. 2010;53:1123–1134. doi: 10.1016/j.jhep.2010.07.006. [DOI] [PubMed] [Google Scholar]
- 10.Deter RL, Baudhuin P, De Duve C. Participation of lysosomes in cellular autophagy induced in rat liver by glucagon. J Cell Biol. 1967;35:C11–C16. doi: 10.1083/jcb.35.2.c11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.de Alwis NM, Day CP. Non-alcoholic fatty liver disease: the mist gradually clears. J Hepatol. 2008;48(Suppl 1):S104–S112. doi: 10.1016/j.jhep.2008.01.009. [DOI] [PubMed] [Google Scholar]
- 12.Marchesini G, Moscatiello S, Di Domizio S, Forlani G. Obesity-associated liver disease. J Clin Endocrinol Metab. 2008;93:S74–S80. doi: 10.1210/jc.2008-1399. [DOI] [PubMed] [Google Scholar]
- 13.Malhi H, Gores GJ. Molecular mechanisms of lipotoxicity in nonalcoholic fatty liver disease. Semin Liver Dis. 2008;28:360–369. doi: 10.1055/s-0028-1091980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Neuschwander-Tetri BA. Hepatic lipotoxicity and the pathogenesis of nonalcoholic steatohepatitis: the central role of nontriglyceride fatty acid metabolites. Hepatology. 2010;52:774–788. doi: 10.1002/hep.23719. [DOI] [PubMed] [Google Scholar]
- 15.Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, Tanaka K, Cuervo AM, Czaja MJ. Autophagy regulates lipid metabolism. Nature. 2009;458:1131–1135. doi: 10.1038/nature07976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mei S, Ni HM, Manley S, Bockus A, Kassel KM, Luyendyk JP, Copple BL, Ding WX. Differential roles of unsaturated and saturated fatty acids on autophagy and apoptosis in hepatocytes. J Pharmacol Exp Ther. 2011;339:487–498. doi: 10.1124/jpet.111.184341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hernandez-Gea V, Ghiassi-Nejad Z, Rozenfeld R, Gordon R, Fiel MI, Yue Z, Czaja MJ, Friedman SL. Autophagy Releases Lipid That Promotes Fibrogenesis by Activated Hepatic Stellate Cells in Mice and in Human Tissues. Gastroenterology. 2012 doi: 10.1053/j.gastro.2011.12.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chang GR, Chiu YS, Wu YY, Chen WY, Liao JW, Chao TH, Mao FC. Rapamycin protects against high fat diet-induced obesity in C57BL/6J mice. J Pharmacol Sci. 2009;109:496–503. doi: 10.1254/jphs.08215fp. [DOI] [PubMed] [Google Scholar]
- 19.Laplante M, Sabatini DM. An emerging role of mTOR in lipid biosynthesis. Curr Biol. 2009;19:R1046–R1052. doi: 10.1016/j.cub.2009.09.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011;12:21–35. doi: 10.1038/nrm3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mizushima N. The role of the Atg1/ULK1 complex in autophagy regulation. Curr Opin Cell Biol. 2010;22:132–139. doi: 10.1016/j.ceb.2009.12.004. [DOI] [PubMed] [Google Scholar]
- 22.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 23.Mammucari C, Milan G, Romanello V, Masiero E, Rudolf R, Del Piccolo P, Burden SJ, Di Lisi R, Sandri C, Zhao J, Goldberg AL, Schiaffino S, Sandri M. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab. 2007;6:458–471. doi: 10.1016/j.cmet.2007.11.001. [DOI] [PubMed] [Google Scholar]
- 24.Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest. 2002;109:1125–1131. doi: 10.1172/JCI15593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Porstmann T, Griffiths B, Chung YL, Delpuech O, Griffiths JR, Downward J, Schulze A. PKB/Akt induces transcription of enzymes involved in cholesterol and fatty acid biosynthesis via activation of SREBP. Oncogene. 2005;24:6465–6481. doi: 10.1038/sj.onc.1208802. [DOI] [PubMed] [Google Scholar]
- 26.Peterson TR, Sengupta SS, Harris TE, Carmack AE, Kang SA, Balderas E, Guertin DA, Madden KL, Carpenter AE, Finck BN, Sabatini DM. mTOR complex 1 regulates lipin 1 localization to control the SREBP pathway. Cell. 2011;146:408–420. doi: 10.1016/j.cell.2011.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zakhari S, Li TK. Determinants of alcohol use and abuse: Impact of quantity and frequency patterns on liver disease. Hepatology. 2007;46:2032–2039. doi: 10.1002/hep.22010. [DOI] [PubMed] [Google Scholar]
- 28.Ding WX, Manley S, Ni HM. The emerging role of autophagy in alcoholic liver disease. Exp Biol Med (Maywood) 2011;236:546–556. doi: 10.1258/ebm.2011.010360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gao B, Bataller R. Alcoholic liver disease: pathogenesis and new therapeutic targets. Gastroenterology. 2011;141:1572–1585. doi: 10.1053/j.gastro.2011.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tian C, Stokowski RP, Kershenobich D, Ballinger DG, Hinds DA. Variant in PNPLA3 is associated with alcoholic liver disease. Nat Genet. 2010;42:21–23. doi: 10.1038/ng.488. [DOI] [PubMed] [Google Scholar]
- 31.Stickel F, Buch S, Lau K, Meyer zu Schwabedissen H, Berg T, Ridinger M, Rietschel M, Schafmayer C, Braun F, Hinrichsen H, Gunther R, Arlt A, Seeger M, Muller S, Seitz HK, Soyka M, Lerch M, Lammert F, Sarrazin C, Kubitz R, Haussinger D, Hellerbrand C, Broring D, Schreiber S, Kiefer F, Spanagel R, Mann K, Datz C, Krawczak M, Wodarz N, Volzke H, Hampe J. Genetic variation in the PNPLA3 gene is associated with alcoholic liver injury in caucasians. Hepatology. 2011;53:86–95. doi: 10.1002/hep.24017. [DOI] [PubMed] [Google Scholar]
- 32.Chen W, Chang B, Li L, Chan L. Patatin-like phospholipase domain-containing 3/adiponutrin deficiency in mice is not associated with fatty liver disease. Hepatology. 2010;52:1134–1142. doi: 10.1002/hep.23812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ding WX, Li M, Chen X, Ni HM, Lin CW, Gao W, Lu B, Stolz DB, Clemens DL, Yin XM. Autophagy reduces acute ethanol-induced hepatotoxicity and steatosis in mice. Gastroenterology. 2010;139:1740–1752. doi: 10.1053/j.gastro.2010.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Donohue TM., Jr Autophagy and ethanol-induced liver injury. World J Gastroenterol. 2009;15:1178–1185. doi: 10.3748/wjg.15.1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang Y, Goldman S, Baerga R, Zhao Y, Komatsu M, Jin S. Adipose-specific deletion of autophagy-related gene 7 (atg7) in mice reveals a role in adipogenesis. Proc Natl Acad Sci U S A. 2009;106:19860–19865. doi: 10.1073/pnas.0906048106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Singh R, Xiang Y, Wang Y, Baikati K, Cuervo AM, Luu YK, Tang Y, Pessin JE, Schwartz GJ, Czaja MJ. Autophagy regulates adipose mass and differentiation in mice. J Clin Invest. 2009;119:3329–3339. doi: 10.1172/JCI39228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kovsan J, Bluher M, Tarnovscki T, Kloting N, Kirshtein B, Madar L, Shai I, Golan R, Harman-Boehm I, Schon MR, Greenberg AS, Elazar Z, Bashan N, Rudich A. Altered autophagy in human adipose tissues in obesity. J Clin Endocrinol Metab. 2011;96:E268–E277. doi: 10.1210/jc.2010-1681. [DOI] [PubMed] [Google Scholar]
- 38.Ost A, Svensson K, Ruishalme I, Brannmark C, Franck N, Krook H, Sandstrom P, Kjolhede P, Stralfors P. Attenuated mTOR signaling and enhanced autophagy in adipocytes from obese patients with type 2 diabetes. Mol Med. 2010;16:235–246. doi: 10.2119/molmed.2010.00023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Larson AM, Polson J, Fontana RJ, Davern TJ, Lalani E, Hynan LS, Reisch JS, Schiodt FV, Ostapowicz G, Shakil AO, Lee WM. Acetaminophen-induced acute liver failure: results of a United States multicenter, prospective study. Hepatology. 2005;42:1364–1372. doi: 10.1002/hep.20948. [DOI] [PubMed] [Google Scholar]
- 40.Jaeschke H, Bajt ML. Intracellular signaling mechanisms of acetaminophen-induced liver cell death. Toxicol Sci. 2006;89:31–41. doi: 10.1093/toxsci/kfi336. [DOI] [PubMed] [Google Scholar]
- 41.Ni HM, Bockus A, Boggess N, Jaeschke H, Ding WX. Activation of autophagy protects against acetaminophen-induced hepatotoxicity. Hepatology. 2012;55:222–232. doi: 10.1002/hep.24690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Apostolova N, Gomez-Sucerquia LJ, Gortat A, Blas-Garcia A, Esplugues JV. Compromising mitochondrial function with the antiretroviral drug efavirenz induces cell survival-promoting autophagy. Hepatology. 2011;54:1009–1019. doi: 10.1002/hep.24459. [DOI] [PubMed] [Google Scholar]
- 43.Camougrand N, Kissova I, Velours G, Manon S. Uth1p: a yeast mitochondrial protein at the crossroads of stress, degradation and cell death. FEMS Yeast Res. 2004;5:133–140. doi: 10.1016/j.femsyr.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 44.Tal R, Winter G, Ecker N, Klionsky DJ, Abeliovich H. Aup1p, a yeast mitochondrial protein phosphatase homolog, is required for efficient stationary phase mitophagy and cell survival. J Biol Chem. 2007;282:5617–5624. doi: 10.1074/jbc.M605940200. [DOI] [PubMed] [Google Scholar]
- 45.Okamoto K, Kondo-Okamoto N, Ohsumi Y. Mitochondria-anchored receptor Atg32 mediates degradation of mitochondria via selective autophagy. Dev Cell. 2009;17:87–97. doi: 10.1016/j.devcel.2009.06.013. [DOI] [PubMed] [Google Scholar]
- 46.Kanki T, Wang K, Cao Y, Baba M, Klionsky DJ. Atg32 is a mitochondrial protein that confers selectivity during mitophagy. Dev Cell. 2009;17:98–109. doi: 10.1016/j.devcel.2009.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lemasters JJ, Qian T, He L, Kim JS, Elmore SP, Cascio WE, Brenner DA. Role of mitochondrial inner membrane permeabilization in necrotic cell death, apoptosis, and autophagy. Antioxid Redox Signal. 2002;4:769–781. doi: 10.1089/152308602760598918. [DOI] [PubMed] [Google Scholar]
- 48.Sandoval H, Thiagarajan P, Dasgupta SK, Schumacher A, Prchal JT, Chen M, Wang J. Essential role for Nix in autophagic maturation of erythroid cells. Nature. 2008;454:232–235. doi: 10.1038/nature07006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schweers RL, Zhang J, Randall MS, Loyd MR, Li W, Dorsey FC, Kundu M, Opferman JT, Cleveland JL, Miller JL, Ney PA. NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc Natl Acad Sci U S A. 2007;104:19500–19505. doi: 10.1073/pnas.0708818104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu L, Feng D, Chen G, Chen M, Zheng Q, Song P, Ma Q, Zhu C, Wang R, Qi W, Huang L, Xue P, Li B, Wang X, Jin H, Wang J, Yang F, Liu P, Zhu Y, Sui S, Chen Q. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat Cell Biol. 2012;14:177–185. doi: 10.1038/ncb2422. [DOI] [PubMed] [Google Scholar]
- 51.Jin SM, Lazarou M, Wang C, Kane LA, Narendra DP, Youle RJ. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J Cell Biol. 2010;191:933–942. doi: 10.1083/jcb.201008084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lazarou M, Jin SM, Kane LA, Youle RJ. Role of PINK1 Binding to the TOM Complex and Alternate Intracellular Membranes in Recruitment and Activation of the E3 Ligase Parkin. Dev Cell. 2012 doi: 10.1016/j.devcel.2011.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183:795–803. doi: 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ding WX, Ni HM, Li M, Liao Y, Chen X, Stolz DB, Dorn GW, 2nd, Yin XM. Nix is critical to two distinct phases of mitophagy, reactive oxygen species-mediated autophagy induction and Parkin-ubiquitin-p62-mediated mitochondrial priming. J Biol Chem. 2010;285:27879–27890. doi: 10.1074/jbc.M110.119537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gegg ME, Cooper JM, Chau KY, Rojo M, Schapira AH, Taanman JW. Mitofusin 1 and mitofusin 2 are ubiquitinated in a PINK1/parkin-dependent manner upon induction of mitophagy. Hum Mol Genet. 2010;19:4861–4870. doi: 10.1093/hmg/ddq419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yoshii SR, Kishi C, Ishihara N, Mizushima N. Parkin mediates proteasome-dependent protein degradation and rupture of the outer mitochondrial membrane. J Biol Chem. 2011;286:19630–19640. doi: 10.1074/jbc.M110.209338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chan NC, Salazar AM, Pham AH, Sweredoski MJ, Kolawa NJ, Graham RL, Hess S, Chan DC. Broad activation of the ubiquitin-proteasome system by Parkin is critical for mitophagy. Hum Mol Genet. 2011;20:1726–1737. doi: 10.1093/hmg/ddr048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Narendra D, Kane LA, Hauser DN, Fearnley IM, Youle RJ. p62/SQSTM1 is required for Parkin-induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy. 2010;6:1090–1106. doi: 10.4161/auto.6.8.13426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bjorkoy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, Stenmark H, Johansen T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol. 2005;171:603–614. doi: 10.1083/jcb.200507002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Overvatn A, Bjorkoy G, Johansen T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282:24131–24145. doi: 10.1074/jbc.M702824200. [DOI] [PubMed] [Google Scholar]
- 61.Ichimura Y, Kumanomidou T, Sou YS, Mizushima T, Ezaki J, Ueno T, Kominami E, Yamane T, Tanaka K, Komatsu M. Structural basis for sorting mechanism of p62 in selective autophagy. J Biol Chem. 2008;283:22847–22857. doi: 10.1074/jbc.M802182200. [DOI] [PubMed] [Google Scholar]
- 62.Perlmutter DH. Alpha-1-antitrypsin deficiency: importance of proteasomal and autophagic degradative pathways in disposal of liver disease-associated protein aggregates. Annu Rev Med. 2011;62:333–345. doi: 10.1146/annurev-med-042409-151920. [DOI] [PubMed] [Google Scholar]
- 63.Hidvegi T, Ewing M, Hale P, Dippold C, Beckett C, Kemp C, Maurice N, Mukherjee A, Goldbach C, Watkins S, Michalopoulos G, Perlmutter DH. An autophagy-enhancing drug promotes degradation of mutant alpha1-antitrypsin Z and reduces hepatic fibrosis. Science. 2010;329:229–232. doi: 10.1126/science.1190354. [DOI] [PubMed] [Google Scholar]
- 64.Williams A, Sarkar S, Cuddon P, Ttofi EK, Saiki S, Siddiqi FH, Jahreiss L, Fleming A, Pask D, Goldsmith P, O'Kane CJ, Floto RA, Rubinsztein DC. Novel targets for Huntington's disease in an mTOR-independent autophagy pathway. Nat Chem Biol. 2008;4:295–305. doi: 10.1038/nchembio.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sarkar S, Perlstein EO, Imarisio S, Pineau S, Cordenier A, Maglathlin RL, Webster JA, Lewis TA, O'Kane CJ, Schreiber SL, Rubinsztein DC. Small molecules enhance autophagy and reduce toxicity in Huntington's disease models. Nat Chem Biol. 2007;3:331–338. doi: 10.1038/nchembio883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhang L, Yu J, Pan H, Hu P, Hao Y, Cai W, Zhu H, Yu AD, Xie X, Ma D, Yuan J. Small molecule regulators of autophagy identified by an image-based high-throughput screen. Proc Natl Acad Sci U S A. 2007;104:19023–19028. doi: 10.1073/pnas.0709695104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kruse KB, Dear A, Kaltenbrun ER, Crum BE, George PM, Brennan SO, McCracken AA. Mutant fibrinogen cleared from the endoplasmic reticulum via endoplasmic reticulum-associated protein degradation and autophagy: an explanation for liver disease. Am J Pathol. 2006;168:1299–1308. doi: 10.2353/ajpath.2006.051097. quiz 1404-1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Brennan SO, Maghzal G, Shneider BL, Gordon R, Magid MS, George PM. Novel fibrinogen gamma375 Arg-->Trp mutation (fibrinogen aguadilla) causes hepatic endoplasmic reticulum storage and hypofibrinogenemia. Hepatology. 2002;36:652–658. doi: 10.1053/jhep.2002.35063. [DOI] [PubMed] [Google Scholar]
- 69.Jensen K, Gluud C. The Mallory body: theories on development and pathological significance (Part 2 of a literature survey) Hepatology. 1994;20:1330–1342. [PubMed] [Google Scholar]
- 70.Riley NE, Li J, Worrall S, Rothnagel JA, Swagell C, van Leeuwen FW, French SW. The Mallory body as an aggresome: in vitro studies. Exp Mol Pathol. 2002;72:17–23. doi: 10.1006/exmp.2001.2413. [DOI] [PubMed] [Google Scholar]
- 71.Harada M, Strnad P, Toivola DM, Omary MB. Autophagy modulates keratin-containing inclusion formation and apoptosis in cell culture in a context-dependent fashion. Exp Cell Res. 2008;314:1753–1764. doi: 10.1016/j.yexcr.2008.01.035. [DOI] [PubMed] [Google Scholar]
- 72.Harada M, Hanada S, Toivola DM, Ghori N, Omary MB. Autophagy activation by rapamycin eliminates mouse Mallory-Denk bodies and blocks their proteasome inhibitor-mediated formation. Hepatology. 2008;47:2026–2035. doi: 10.1002/hep.22294. [DOI] [PubMed] [Google Scholar]
- 73.Alexander DE, Leib DA. Xenophagy in herpes simplex virus replication and pathogenesis. Autophagy. 2008;4:101–103. doi: 10.4161/auto.5222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Talloczy Z, Virgin HWt, Levine B. PKR-dependent autophagic degradation of herpes simplex virus type 1. Autophagy. 2006;2:24–29. doi: 10.4161/auto.2176. [DOI] [PubMed] [Google Scholar]
- 75.Nakagawa I, Amano A, Mizushima N, Yamamoto A, Yamaguchi H, Kamimoto T, Nara A, Funao J, Nakata M, Tsuda K, Hamada S, Yoshimori T. Autophagy defends cells against invading group A Streptococcus. Science. 2004;306:1037–1040. doi: 10.1126/science.1103966. [DOI] [PubMed] [Google Scholar]
- 76.Kudchodkar SB, Levine B. Viruses and autophagy. Rev Med Virol. 2009;19:359–378. doi: 10.1002/rmv.630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Czaja MJ. Functions of autophagy in hepatic and pancreatic physiology and disease. Gastroenterology. 2011;140:1895–1908. doi: 10.1053/j.gastro.2011.04.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sir D, Chen WL, Choi J, Wakita T, Yen TS, Ou JH. Induction of incomplete autophagic response by hepatitis C virus via the unfolded protein response. Hepatology. 2008;48:1054–1061. doi: 10.1002/hep.22464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sir D, Tian Y, Chen WL, Ann DK, Yen TS, Ou JH. The early autophagic pathway is activated by hepatitis B virus and required for viral DNA replication. Proc Natl Acad Sci U S A. 2010;107:4383–4388. doi: 10.1073/pnas.0911373107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Dreux M, Gastaminza P, Wieland SF, Chisari FV. The autophagy machinery is required to initiate hepatitis C virus replication. Proc Natl Acad Sci U S A. 2009;106:14046–14051. doi: 10.1073/pnas.0907344106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Alter MJ. Epidemiology of hepatitis C virus infection. World J Gastroenterol. 2007;13:2436–2441. doi: 10.3748/wjg.v13.i17.2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chisari FV. Unscrambling hepatitis C virus-host interactions. Nature. 2005;436:930–932. doi: 10.1038/nature04076. [DOI] [PubMed] [Google Scholar]
- 83.Gosert R, Egger D, Lohmann V, Bartenschlager R, Blum HE, Bienz K, Moradpour D. Identification of the hepatitis C virus RNA replication complex in Huh-7 cells harboring subgenomic replicons. J Virol. 2003;77:5487–5492. doi: 10.1128/JVI.77.9.5487-5492.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Piver E, Roingeard P, Pages JC. The cell biology of hepatitis C virus (HCV) lipid addiction: molecular mechanisms and its potential importance in the clinic. Int J Biochem Cell Biol. 2010;42:869–879. doi: 10.1016/j.biocel.2010.01.005. [DOI] [PubMed] [Google Scholar]
- 85.Syed GH, Amako Y, Siddiqui A. Hepatitis C virus hijacks host lipid metabolism. Trends Endocrinol Metab. 2010;21:33–40. doi: 10.1016/j.tem.2009.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gastaminza P, Cheng G, Wieland S, Zhong J, Liao W, Chisari FV. Cellular determinants of hepatitis C virus assembly, maturation, degradation, and secretion. J Virol. 2008;82:2120–2129. doi: 10.1128/JVI.02053-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Rautou PE, Cazals-Hatem D, Feldmann G, Mansouri A, Grodet A, Barge S, Martinot-Peignoux M, Duces A, Bieche I, Lebrec D, Bedossa P, Paradis V, Marcellin P, Valla D, Asselah T, Moreau R. Changes in autophagic response in patients with chronic hepatitis C virus infection. Am J Pathol. 2011;178:2708–2715. doi: 10.1016/j.ajpath.2011.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ke PY, Chen SS. Activation of the unfolded protein response and autophagy after hepatitis C virus infection suppresses innate antiviral immunity in vitro. J Clin Invest. 2011;121:37–56. doi: 10.1172/JCI41474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Desai MM, Gong B, Chan T, Davey RA, Soong L, Kolokoltsov AA, Sun J. Differential, type I interferon-mediated autophagic trafficking of hepatitis C virus proteins in mouse liver. Gastroenterology. 2011;141:674–685. 685, e671–e676. doi: 10.1053/j.gastro.2011.04.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Vescovo T, Romagnoli A, Perdomo AB, Corazzari M, Ciccosanti F, Alonzi T, Nardacci R, Ippolito G, Tripodi M, Garcia-Monzon C, Lo Iacono O, Piacentini M, Fimia GM. Autophagy Protects Cells From HCV-Induced Defects in Lipid Metabolism. Gastroenterology. 2012;142:644–653. e643. doi: 10.1053/j.gastro.2011.11.033. [DOI] [PubMed] [Google Scholar]
- 91.Custer B, Sullivan SD, Hazlet TK, Iloeje U, Veenstra DL, Kowdley KV. Global epidemiology of hepatitis B virus. J Clin Gastroenterol. 2004;38:S158–S168. doi: 10.1097/00004836-200411003-00008. [DOI] [PubMed] [Google Scholar]
- 92.Li J, Liu Y, Wang Z, Liu K, Wang Y, Liu J, Ding H, Yuan Z. Subversion of cellular autophagy machinery by hepatitis B virus for viral envelopment. J Virol. 2011;85:6319–6333. doi: 10.1128/JVI.02627-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Tang H, Da L, Mao Y, Li Y, Li D, Xu Z, Li F, Wang Y, Tiollais P, Li T, Zhao M. Hepatitis B virus X protein sensitizes cells to starvation-induced autophagy via up-regulation of beclin 1 expression. Hepatology. 2009;49:60–71. doi: 10.1002/hep.22581. [DOI] [PubMed] [Google Scholar]
- 94.Hernandez-Gea V, Friedman SL. Pathogenesis of liver fibrosis. Annu Rev Pathol. 2011;6:425–456. doi: 10.1146/annurev-pathol-011110-130246. [DOI] [PubMed] [Google Scholar]
- 95.Brenner DA. Molecular pathogenesis of liver fibrosis. Trans Am Clin Climatol Assoc. 2009;120:361–368. [PMC free article] [PubMed] [Google Scholar]
- 96.El-Serag HB, Rudolph KL. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology. 2007;132:2557–2576. doi: 10.1053/j.gastro.2007.04.061. [DOI] [PubMed] [Google Scholar]
- 97.Dranoff JA, Wells RG. Portal fibroblasts: Underappreciated mediators of biliary fibrosis. Hepatology. 2010;51:1438–1444. doi: 10.1002/hep.23405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Thoen LF, Guimaraes EL, Dolle L, Mannaerts I, Najimi M, Sokal E, van Grunsven LA. A role for autophagy during hepatic stellate cell activation. J Hepatol. 2011;55:1353–1360. doi: 10.1016/j.jhep.2011.07.010. [DOI] [PubMed] [Google Scholar]
- 99.Kim SI, Na HJ, Ding Y, Wang Z, Lee SJ, Choi ME. Autophagy promotes intracellular degradation of type I collagen induced by Transforming Growth Factor (TGF)-beta1. J Biol Chem. 2012 doi: 10.1074/jbc.M111.308460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Alberti A, Michelet X, Djeddi A, Legouis R. The autophagosomal protein LGG-2 acts synergistically with LGG-1 in dauer formation and longevity in C. elegans. Autophagy. 2010;6 doi: 10.4161/auto.6.5.12252. [DOI] [PubMed] [Google Scholar]
- 101.Kissova I, Deffieu M, Manon S, Camougrand N. Uth1p is involved in the autophagic degradation of mitochondria. J Biol Chem. 2004;279:39068–39074. doi: 10.1074/jbc.M406960200. [DOI] [PubMed] [Google Scholar]
- 102.Madeo F, Tavernarakis N, Kroemer G. Can autophagy promote longevity? Nat Cell Biol. 2010;12:842–846. doi: 10.1038/ncb0910-842. [DOI] [PubMed] [Google Scholar]
- 103.Lipinski MM, Zheng B, Lu T, Yan Z, Py BF, Ng A, Xavier RJ, Li C, Yankner BA, Scherzer CR, Yuan J. Genome-wide analysis reveals mechanisms modulating autophagy in normal brain aging and in Alzheimer's disease. Proc Natl Acad Sci U S A. 2010;107:14164–14169. doi: 10.1073/pnas.1009485107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wang JH, Ahn IS, Fischer TD, Byeon JI, Dunn WA, Jr, Behrns KE, Leeuwenburgh C, Kim JS. Autophagy suppresses age-dependent ischemia and reperfusion injury in livers of mice. Gastroenterology. 2011;141:2188–2199. e2186. doi: 10.1053/j.gastro.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zhang C, Cuervo AM. Restoration of chaperone-mediated autophagy in aging liver improves cellular maintenance and hepatic function. Nat Med. 2008;14:959–965. doi: 10.1038/nm.1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Rubinsztein DC, Marino G, Kroemer G. Autophagy and aging. Cell. 2011;146:682–695. doi: 10.1016/j.cell.2011.07.030. [DOI] [PubMed] [Google Scholar]
- 107.Lee IH, Cao L, Mostoslavsky R, Lombard DB, Liu J, Bruns NE, Tsokos M, Alt FW, Finkel T. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc Natl Acad Sci U S A. 2008;105:3374–3379. doi: 10.1073/pnas.0712145105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Saunders LR, Verdin E. Cell biology. Stress response and aging. Science. 2009;323:1021–1022. doi: 10.1126/science.1170007. [DOI] [PubMed] [Google Scholar]
- 109.Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA Cancer J Clin. 2005;55:74–108. doi: 10.3322/canjclin.55.2.74. [DOI] [PubMed] [Google Scholar]
- 110.Thorgeirsson SS, Lee JS, Grisham JW. Molecular prognostication of liver cancer: end of the beginning. J Hepatol. 2006;44:798–805. doi: 10.1016/j.jhep.2006.01.008. [DOI] [PubMed] [Google Scholar]
- 111.de Lope CR, Tremosini S, Forner A, Reig M, Bruix J. Management of HCC. J Hepatol. 2012;56(Suppl):S75–S87. doi: 10.1016/S0168-8278(12)60009-9. [DOI] [PubMed] [Google Scholar]
- 112.Forner A, Llovet JM, Bruix J. Hepatocellular carcinoma. Lancet. 2012 doi: 10.1016/S0140-6736(11)61347-0. [DOI] [PubMed] [Google Scholar]
- 113.Bruix J, Sherman M. Management of hepatocellular carcinoma: an update. Hepatology. 2011;53:1020–1022. doi: 10.1002/hep.24199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Aita VM, Liang XH, Murty VV, Pincus DL, Yu W, Cayanis E, Kalachikov S, Gilliam TC, Levine B. Cloning and genomic organization of beclin 1, a candidate tumor suppressor gene on chromosome 17q21. Genomics. 1999;59:59–65. doi: 10.1006/geno.1999.5851. [DOI] [PubMed] [Google Scholar]
- 115.Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, Troxel A, Rosen J, Eskelinen EL, Mizushima N, Ohsumi Y, Cattoretti G, Levine B. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest. 2003;112:1809–1820. doi: 10.1172/JCI20039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, Levine B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 1999;402:672–676. doi: 10.1038/45257. [DOI] [PubMed] [Google Scholar]
- 117.Marino G, Salvador-Montoliu N, Fueyo A, Knecht E, Mizushima N, Lopez-Otin C. Tissue-specific autophagy alterations and increased tumorigenesis in mice deficient in Atg4C/autophagin-3. J Biol Chem. 2007;282:18573–18583. doi: 10.1074/jbc.M701194200. [DOI] [PubMed] [Google Scholar]
- 118.Takahashi Y, Coppola D, Matsushita N, Cualing HD, Sun M, Sato Y, Liang C, Jung JU, Cheng JQ, Mule JJ, Pledger WJ, Wang HG. Bif-1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nat Cell Biol. 2007;9:1142–1151. doi: 10.1038/ncb1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Liang C, Feng P, Ku B, Dotan I, Canaani D, Oh BH, Jung JU. Autophagic and tumour suppressor activity of a novel Beclin1-binding protein UVRAG. Nat Cell Biol. 2006;8:688–699. doi: 10.1038/ncb1426. [DOI] [PubMed] [Google Scholar]
- 120.Hezel AF, Bardeesy N. LKB1; linking cell structure and tumor suppression. Oncogene. 2008;27:6908–6919. doi: 10.1038/onc.2008.342. [DOI] [PubMed] [Google Scholar]
- 121.Liang J, Shao SH, Xu ZX, Hennessy B, Ding Z, Larrea M, Kondo S, Dumont DJ, Gutterman JU, Walker CL, Slingerland JM, Mills GB. The energy sensing LKB1-AMPK pathway regulates p27(kip1) phosphorylation mediating the decision to enter autophagy or apoptosis. Nat Cell Biol. 2007;9:218–224. doi: 10.1038/ncb1537. [DOI] [PubMed] [Google Scholar]
- 122.Cully M, You H, Levine AJ, Mak TW. Beyond PTEN mutations: the PI3K pathway as an integrator of multiple inputs during tumorigenesis. Nat Rev Cancer. 2006;6:184–192. doi: 10.1038/nrc1819. [DOI] [PubMed] [Google Scholar]
- 123.Degtyarev M, De Maziere A, Orr C, Lin J, Lee BB, Tien JY, Prior WW, van Dijk S, Wu H, Gray DC, Davis DP, Stern HM, Murray LJ, Hoeflich KP, Klumperman J, Friedman LS, Lin K. Akt inhibition promotes autophagy and sensitizes PTEN-null tumors to lysosomotropic agents. J Cell Biol. 2008;183:101–116. doi: 10.1083/jcb.200801099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Maiuri MC, Tasdemir E, Criollo A, Morselli E, Vicencio JM, Carnuccio R, Kroemer G. Control of autophagy by oncogenes and tumor suppressor genes. Cell Death and Differentiation. 2009;16:87–93. doi: 10.1038/cdd.2008.131. [DOI] [PubMed] [Google Scholar]
- 125.Inami Y, Waguri S, Sakamoto A, Kouno T, Nakada K, Hino O, Watanabe S, Ando J, Iwadate M, Yamamoto M, Lee MS, Tanaka K, Komatsu M. Persistent activation of Nrf2 through p62 in hepatocellular carcinoma cells. J Cell Biol. 2011;193:275–284. doi: 10.1083/jcb.201102031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Takamura A, Komatsu M, Hara T, Sakamoto A, Kishi C, Waguri S, Eishi Y, Hino O, Tanaka K, Mizushima N. Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 2011;25:795–800. doi: 10.1101/gad.2016211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Komatsu M, Kurokawa H, Waguri S, Taguchi K, Kobayashi A, Ichimura Y, Sou YS, Ueno I, Sakamoto A, Tong KI, Kim M, Nishito Y, Iemura S, Natsume T, Ueno T, Kominami E, Motohashi H, Tanaka K, Yamamoto M. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol. 2010;12:213–223. doi: 10.1038/ncb2021. [DOI] [PubMed] [Google Scholar]
- 128.Moscat J, Diaz-Meco MT. p62 at the crossroads of autophagy, apoptosis, and cancer. Cell. 2009;137:1001–1004. doi: 10.1016/j.cell.2009.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Jin Z, Li Y, Pitti R, Lawrence D, Pham VC, Lill JR, Ashkenazi A. Cullin3-based polyubiquitination and p62-dependent aggregation of caspase-8 mediate extrinsic apoptosis signaling. Cell. 2009;137:721–735. doi: 10.1016/j.cell.2009.03.015. [DOI] [PubMed] [Google Scholar]
- 130.Gao C, Cao W, Bao L, Zuo W, Xie G, Cai T, Fu W, Zhang J, Wu W, Zhang X, Chen YG. Autophagy negatively regulates Wnt signalling by promoting Dishevelled degradation. Nat Cell Biol. 2010;12:781–790. doi: 10.1038/ncb2082. [DOI] [PubMed] [Google Scholar]
- 131.Duran A, Amanchy R, Linares JF, Joshi J, Abu-Baker S, Porollo A, Hansen M, Moscat J, Diaz-Meco MT. p62 is a key regulator of nutrient sensing in the mTORC1 pathway. Mol Cell. 2011;44:134–146. doi: 10.1016/j.molcel.2011.06.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Hayes JD, McMahon M. NRF2 and KEAP1 mutations: permanent activation of an adaptive response in cancer. Trends Biochem Sci. 2009;34:176–188. doi: 10.1016/j.tibs.2008.12.008. [DOI] [PubMed] [Google Scholar]
- 133.Shibata T, Kokubu A, Gotoh M, Ojima H, Ohta T, Yamamoto M, Hirohashi S. Genetic alteration of Keap1 confers constitutive Nrf2 activation and resistance to chemotherapy in gallbladder cancer. Gastroenterology. 2008;135:1358–1368. 1368, e1351–e1354. doi: 10.1053/j.gastro.2008.06.082. [DOI] [PubMed] [Google Scholar]
- 134.DeNicola GM, Karreth FA, Humpton TJ, Gopinathan A, Wei C, Frese K, Mangal D, Yu KH, Yeo CJ, Calhoun ES, Scrimieri F, Winter JM, Hruban RH, Iacobuzio-Donahue C, Kern SE, Blair IA, Tuveson DA. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature. 2011;475:106–109. doi: 10.1038/nature10189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Inoue D, Suzuki T, Mitsuishi Y, Miki Y, Suzuki S, Sugawara S, Watanabe M, Sakurada A, Endo C, Uruno A, Sasano H, Nakagawa T, Satoh K, Tanaka N, Kubo H, Motohashi H, Yamamoto M. Accumulation of p62/SQSTM1 is associated with poor prognosis in patients with lung adenocarcinoma. Cancer Sci. 2012 doi: 10.1111/j.1349-7006.2012.02216.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Chang YS, Adnane J, Trail PA, Levy J, Henderson A, Xue D, Bortolon E, Ichetovkin M, Chen C, McNabola A, Wilkie D, Carter CA, Taylor IC, Lynch M, Wilhelm S. Sorafenib (BAY 43-9006) inhibits tumor growth and vascularization and induces tumor apoptosis and hypoxia in RCC xenograft models. Cancer Chemother Pharmacol. 2007;59:561–574. doi: 10.1007/s00280-006-0393-4. [DOI] [PubMed] [Google Scholar]
- 137.Wilhelm SM, Carter C, Tang L, Wilkie D, McNabola A, Rong H, Chen C, Zhang X, Vincent P, McHugh M, Cao Y, Shujath J, Gawlak S, Eveleigh D, Rowley B, Liu L, Adnane L, Lynch M, Auclair D, Taylor I, Gedrich R, Voznesensky A, Riedl B, Post LE, Bollag G, Trail PA. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004;64:7099–7109. doi: 10.1158/0008-5472.CAN-04-1443. [DOI] [PubMed] [Google Scholar]
- 138.Shi YH, Ding ZB, Zhou J, Hui B, Shi GM, Ke AW, Wang XY, Dai Z, Peng YF, Gu CY, Qiu SJ, Fan J. Targeting autophagy enhances sorafenib lethality for hepatocellular carcinoma via ER stress-related apoptosis. Autophagy. 2011;7:1159–1172. doi: 10.4161/auto.7.10.16818. [DOI] [PubMed] [Google Scholar]
- 139.Ding ZB, Hui B, Shi YH, Zhou J, Peng YF, Gu CY, Yang H, Shi GM, Ke AW, Wang XY, Song K, Dai Z, Shen YH, Fan J. Autophagy activation in hepatocellular carcinoma contributes to the tolerance of oxaliplatin via reactive oxygen species modulation. Clin Cancer Res. 2011;17:6229–6238. doi: 10.1158/1078-0432.CCR-11-0816. [DOI] [PubMed] [Google Scholar]
- 140.Liu YL, Yang PM, Shun CT, Wu MS, Weng JR, Chen CC. Autophagy potentiates the anti-cancer effects of the histone deacetylase inhibitors in hepatocellular carcinoma. Autophagy. 2010;6:1057–1065. doi: 10.4161/auto.6.8.13365. [DOI] [PubMed] [Google Scholar]
- 141.Song J, Qu Z, Guo X, Zhao Q, Zhao X, Gao L, Sun K, Shen F, Wu M, Wei L. Hypoxia-induced autophagy contributes to the chemoresistance of hepatocellular carcinoma cells. Autophagy. 2009;5:1131–1144. doi: 10.4161/auto.5.8.9996. [DOI] [PubMed] [Google Scholar]