

Abstract
Identifying molecular biomarkers has become one of the important tasks for scientists to assess the different phenotypic states of cells or organisms correlated to the genotypes of diseases from large-scale biological data. In this paper, we proposed a text-mining-based method to discover biomarkers from PubMed. First, we construct a database based on a dictionary, and then we used a finite state machine to identify the biomarkers. Our method of text mining provides a highly reliable approach to discover the biomarkers in the PubMed database.
1. Introduction
Identifying molecular biomarkers has become an essential task for bioinformatics scientists to assess the different phenotypic states of cells or organisms correlated to the genotypes of diseases from large-scale biological data [1]. The text mining technique has become a critical technique for designing future predictive and personalized medicine. At the same time, the PubMed database which comprises more than 21 million citations for biomedical literature offers an enriched source for us to explore the biomarkers across human disease and to mine the biomarkers related to diseases. Therefore, integrating automatic literature searches, and text mining is a fast emerging research area in epigenetics, DNA methylation, and more specifically biomarker discovery studies. For almost every cancer type, new publications that discover biomarker candidates are updated frequently, especially with advanced high-throughput methodologies. Efficient text mining tools and algorithm development are extremely needed.
Many text mining technologies that are proposed by different groups, such as machine learning technologies including support vector machine [2], decision tree [3], Bayes classifier [4], and random forest [5], are used for text mining. Also the natural language processing technique is used to determine the structures and linguistic components of sentences and then parses the sentences in a bag of words, together with a statistic approach to get the matched results from the text databases. OMIM database [6] is one of the important databases for biomarker-related disease research. The MeSH Browser [7] is used to map disease associations to MeSH IDs.
In this paper, we use a state machine to simulate the transforms of the biomarkers from individual entities to associated diseases and pathways as well as networks. Several abstracted templates are summarized from known expert experience and knowledge. The biomarkers are ranked based on the importance to the diseases and the citations of the literature from PubMed. Based on this template, every mined biomarker-related pathways, networks, and disease will be collected and matched with the templates.
2. Method
All the biomarkers mentioned in this paper are mined from the PubMed database. For each biomarker candidate, we use a finite state machine (FSM) [8] to identify biomarker, pathways, and associated diseases. Only the candidates which are accepted by FSM are viewed as biomarkers. The association between the biomarkers and the diseases can be output to refine the biomarkers.
As shown in Figure 1. The first step is to create a biomarker dictionary, the second step is construct a DBXML [9] database, and the third step is using the finite state machine to conform the disease-related biomarkers. We first create our DBXML database from the PubMed database. The Lucence technique is used to split the document into a bag of words and extract terms such as gene names, interaction relationships, pathways, and network names. In the mean time, based on our domain knowledge, we construct the dictionary for further analysis. Based on the Lucence parsed terms and dictionary, the DBXML database is created for biomarker extraction. To retrieve the keyword from the DBXML database, exact matching, fuzzy matching, and list matching methods are used to match the terms saved in DBXML. If the end state of the FMS is in an acceptance state, the keywords-related genes, proteins, or small molecules are marked as biomarkers.
Figure 1.
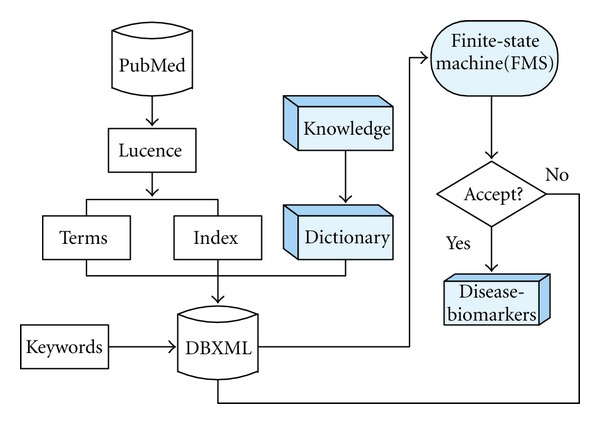
The flow chart of the biomarker discovery.
2.1. Construct a Database for Gene/Protein and Disease
We first construct a database which includes categories according to their names, diseases, interactions, pathways, and network information. Then, we collect a list of diseases, gene/protein and so on, and then put them into the dictionary. The structure of the dictionary is shown as Table 1.
Table 1.
The dictionary of the biomarkers.
| Gene | Protein | Pathway | Disease |
|---|---|---|---|
| P53 | P53 | Ras | Diabetes |
| APC | APC | Wnt | Breast cancer |
| MDM2 | Pten | Death receptor pathway | Liver cancer |
| Ras | HCC | Ether lipid metabolism | Huntington |
| Axin-1 | HPR | Thiamine metabolism | Liver cirrhosis |
| LCE2B |
Porphyrin and chlorophyll | Prostate cancer | |
| AXIN1 | Metabolism | Leukemia | |
| SLC22A1 |
We use a dynamic method to collect the full-text document, and then the Lucence is applied to split the word. For Lucence, we need to delete the old document and create new Lucence document index. The Lucence document contains three paths, content and the index of the document, the terms, and the modified date.
Each word is separated by a series of phrases, and we use the dictionary to parse the full-text and then divide them into several primary categories: molecule names, interaction keywords, and verbs. After we extract the keywords, we construct the segment of the xml document for those keywords. The protein name is an entity, and the interaction represented the relation of the entity which is used to extract the relationships between diseases, genes, mutations, and proteins. We give an example of an xml segment extracted from PubMed as follows:
-
<protein id=010>
-
<name>P53 </name>
-
<interact>MDM2</interact>
-
</protein>
If the words cannot match the dictionary, it will be ignored. Some keywords can be removed from the database as they are not suitable for our definition. Additional tags can also be added by the users. Table 1 shows the dictionary of the biomarkers.
Our database does not contain interaction pairs and pathways. We will dynamically parse online databases for the protein/gene names and build the interaction network.
2.2. Using the FSM to Identify Biomarkers
We used the finite state machine (FSM) to identify the biomarkers in our database. The FSM is a state machine which has a start node, accepting node, input entities, and relations. The roles contain the information of each entity such as genes, proteins, and small molecules.
In this paper, the FSM for identifying biomarkers is regarded as a template which serves to match corresponding biomarkers as shown in Figure 2. In addition, the template can be modified by users. Our methods include exact matching, fuzzy matching and list matching. For disease, we use the exact match method, for all molecules, we use fuzzy matching, and for interaction, we use list-member matching.
Figure 2.
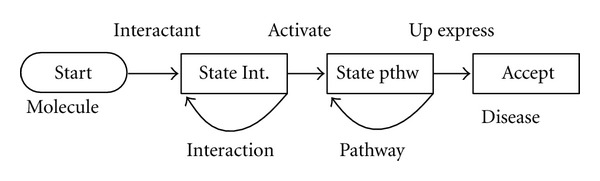
The identification of the biomarker using the finite state machine.
For the list member of interactions, the list members are defined as ILIST(P a)∶ = (P 1, P 2, P 3,…P n), where P a interacts with P 1, P 2, … P n, which dynamically parse online databases. We construct the protein-protein interaction network around P a in the FSM. We also obtain the pathway from the KEGG database.
The FSM includes <left-context FSM>, <entity FSM>, and 〈right-context-FSM〉. The roles of the entities are determined by the context of the left and right neighbors of the entities.
For example, for the entity P53 which is a protein, we determine the role of the entity as follows:
-
If < right-context>
-
= <(“express”“present”)(“in”“at pathway”)>
-
Then entity role = in the pathway
The output of the FSM is the track nodes between <Disease> ⇔ <Potential Biomarker> which include paper name and author name. The FSM is shown in Figure 2.
3. Experimental Results
Based on our framework, a query on liver cancer and the candidate biomarkers are report as Table 2.
Table 2.
The list of biomarker-disease associations mined from PubMed.
| EntrezID | Gene name | Symbol |
|---|---|---|
| 11914 | ALPHA 1,4-GALACTOSYLTRANSFERASE | A4GALT |
| 3558 | ACETOACETYL-COA SYNTHETASE | AACS |
| 5758 | ABHYDROLASE DOMAIN CONTAINING 1 | ABHD1 |
| 18925 | ACYL-COA THIOESTERASE 12 | ACOT12 |
| 18925 | ACYL-COA THIOESTERASE 12 | ACOT12 |
| 17809 | ACYL-COA THIOESTERASE 2 | ACOT2 |
| 17766 | ACYL-COA THIOESTERASE 4 | ACOT4 |
| 15426 | ACYL-COA SYNTHETASE BUBBLEGUM FAMILY MEMBER 1 | ACSBG1 |
| 11191 | ACYL-COA SYNTHETASE BUBBLEGUM FAMILY MEMBER 2 | ACSBG2 |
In a query process, we dynamically parse the identified genes/proteins and construct the interact network. We then use Cytoscape software [10] to display the interaction network shown as Figure 3.
Figure 3.
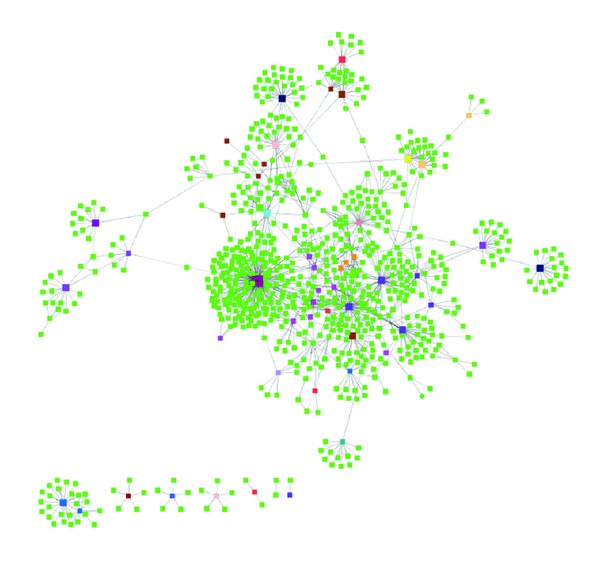
A diseases-related gene associated network. Green nodes are genes, and the nodes in other colors are diseases.
4. Conclusions
The proposed method is based on text mining technique from the PubMed database, combined with the full text search-engine technology (Lucence), a complex network of biological and signaling pathways. First, we construct a database based on a dictionary; second, we use a FSM to identify the biomarkers; finally, we output the disease-associated biomarkers. This research offers a comprehensive text mining to discover biomarkers.
Acknowledgments
This work was supported by NSF CAREER (CCF-0845888) (H. Li and C. Liu) and by the Center for Science of Information (CSoI), an NSF Science and Technology Center, under Grant Agreement CCF-0939370.
References
- 1.Chervona Y, Costa M. Histone modifications and cancer: biomarkers of prognosis? American Journal of Cancer Research. 2012;2(5):589–597. [PMC free article] [PubMed] [Google Scholar]
- 2.Qi Z, Tian Y, Shi Y. Laplacian twin support vector machine for semi-supervised classification. Neural Network. 2012;35:46–53. doi: 10.1016/j.neunet.2012.07.011. [DOI] [PubMed] [Google Scholar]
- 3.Taniguchi E, Kawaguchi T, Sakata M, Itou M, Oriishi T, Sata M. Lipid profile is associated with the incidence of cognitive dysfunction in viral cirrhotic patients: a data-mining analysis. doi: 10.1111/j.1872-034X.2012.01076.x. Hepatology Research. In press. [DOI] [PubMed] [Google Scholar]
- 4.Zhang H, Liu G, Chow TWS, Liu W. Textual and visual content-based anti-phishing: a Bayesian approach. IEEE Transactions on Neural Networks. 2012;22(10):1532–1546. doi: 10.1109/TNN.2011.2161999. [DOI] [PubMed] [Google Scholar]
- 5.Touw WG, Bayjanov JR, Overmars L, et al. Data mining in the Life Sciences with Random Forest: a walk in the park or lost in the jungle? doi: 10.1093/bib/bbs034. Briefings in Bioinformatics. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hamosh A, Scott AF, Amberger JS, Bocchini CA, McKusick VA. Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Research. 2005;33:D514–D517. doi: 10.1093/nar/gki033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Crespo Azcarate M, Mata Vazquez J, Mana Lopez M. Improving image retrieval effectiveness via query expansion using MeSH hierarchical structure. doi: 10.1136/amiajnl-2012-000943. Journal of the American Medical Informatics Association. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Garcia-Remesal M, Maojo V, Crespo J. A knowledge engineering approach to recognizing and extracting sequences of nucleic acids from scientific literature. Proceedings of the 32nd Annual International Conference of the IEEE Engineering in Medicine and Biology Society; 2010; pp. 1081–1084. [DOI] [PubMed] [Google Scholar]
- 9. Oracle: Oracle Berkeley DB XML, 2012.
- 10.Shannon P, Markiel A, Ozier O, et al. Cytoscape: a software Environment for integrated models of biomolecular interaction networks. Genome Research. 2003;13(11):2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]