

Abstract
Five preventative HIV vaccine efficacy trials have been conducted over the last 12 years, all of which evaluated vaccine efficacy (VE) to prevent HIV infection for a single vaccine regimen versus placebo. Now that one of these trials has supported partial VE of a prime-boost vaccine regimen, there is interest in conducting efficacy trials that simultaneously evaluate multiple prime-boost vaccine regimens against a shared placebo group in the same geographic region, for accelerating the pace of vaccine development. This article proposes such a design, which has main objectives (1) to evaluate VE of each regimen versus placebo against HIV exposures occurring near the time of the immunizations; (2) to evaluate durability of VE for each vaccine regimen showing reliable evidence for positive VE; (3) to expeditiously evaluate the immune correlates of protection if any vaccine regimen shows reliable evidence for positive VE; and (4) to compare VE among the vaccine regimens. The design uses sequential monitoring for the events of vaccine harm, non-efficacy, and high efficacy, selected to weed out poor vaccines as rapidly as possible while guarding against prematurely weeding out a vaccine that does not confer efficacy until most of the immunizations are received. The evaluation of the design shows that testing multiple vaccine regimens is important for providing a well-powered assessment of the correlation of vaccine-induced immune responses with HIV infection, and is critically important for providing a reasonably powered assessment of the value of identified correlates as surrogate endpoints for HIV infection.
Keywords: HIV vaccine efficacy clinical trial, immune correlate of protection, one-way crossover design, surrogate endpoint for HIV infection, two-phase sampling
Introduction
Background on Past HIV Vaccine Efficacy Trials, with Emphasis on the Sequential Monitoring Plans
Five randomized, double-blinded, placebo-controlled preventative HIV vaccine efficacy trials have been conducted, all with HIV infection as a primary endpoint, four of which yielded results on the vaccine efficacy (VE) to reduce the rate of HIV infection [VE = (1 – HR)×100%, where HR is the hazard ratio (vaccine/placebo) of HIV infection diagnosis]. The Vax004, Vax003, and Step trials indicated that VE was zero or very low at best (Flynn et al., 2005; Pitisuttithum et al., 2006; Buchbinder et al., 2008), whereas the RV144 trial provided modest evidence for positive VE (estimated VE = 31%, 95% confidence interval (CI) 1% to 51%, 2-sided p-value = 0.04) (Rerks-Ngarm et al., 2009). RV144 evaluated a prime-boost vaccine regimen, and several products are becoming available that may be combined into novel prime-boost regimens, generating enthusiasm for a follow-up efficacy trial (or trials) that will evaluate multiple such regimens. Here we propose a Phase 2b design for a follow-up trial configured to accelerate the pace of answering key scientific questions and hence to shorten the time until the eventual licensure of an efficacious HIV vaccine. The main features of the proposed design are to evaluate multiple vaccine regimens versus a shared placebo group, adaptive two-stage evaluation of vaccine efficacy against infections occurring proximal or distal to the immunization series, tailored sequential monitoring for optimizing efficiency of vaccine efficacy evaluation, augmented design features to improve the assessment of immune correlates of protection, and head-to-head comparisons of vaccine efficacy among vaccine regimens.
The previous efficacy trials used group sequential designs, wherein an independent Data Safety Monitoring Board (DSMB) periodically reviewed interim results on estimation and inference for VE (Table 1). Vax004 and Vax003 had essentially the same Phase 3 design, whereas Step and Phambili (Gray et al., 2009) (Phambili did not yield a result on VE) had essentially the same Phase 2b design. All four trials evaluated VE at a single interim analysis; Vax004 and Vax003 used O'Brien-Fleming monitoring (O'Brien and Fleming, 1979) to recommend early stopping based on strong evidence for reasonably high efficacy (test H0: VE ≤ 30% vs. H1: VE > 30%), whereas Step and Phambili used a customized monitoring procedure to recommend early stopping based on strong evidence for positive efficacy on either the infection endpoint (test H0: VE ≤ 0% vs. H1: VE > 0%) or on the set-point viral load co-primary endpoint. At the sole interim analysis Step was also monitored for low efficacy at best (we refer to this as “non-efficacy monitoring”). In particular, conditional power monitoring was used to recommend early stopping if there was less than a 20% chance to reject the composite null hypothesis of both VE ≤ 0% and no vaccine effect on mean viral load, if in future follow-up the true VE would be 60% and the true viral load effect would be a 1 log10 lower mean viral load in the infected vaccine group compared to the infected placebo group. By only allocating a small part of the overall type I error rate to the interim analysis, this monitoring procedure, similar to the O'Brien-Fleming approach, only recommended stopping based on strong interim evidence. Phambili planned similar non-efficacy monitoring, but the trial was un-blinded before the planned interim analysis (the un-blinding was precipitated by evidence from the Step trial that the vaccine may cause an increased risk of HIV acquisition, Buchbinder et al., 2008).
Table 1.
Approaches to Group Sequential Monitoring of HIV Vaccine Efficacy in Past Efficacy Trials.
| Efficacy Trial | Monitoring Type | Number and Timing of Interim Analyses | Null and Alternative Hypotheses | Alpha level | Boundary Type |
|---|---|---|---|---|---|
|
Vax004 Phase 3 1998-2003 |
Efficacy | 1; when 50% infections expected | H0: VE ≤ 30% vs. H1: VE > 30% |
0.025 | O'Brien-Fleming |
|
Vax003 Phase 3 1999-2003 |
Efficacy | 1; when 50% infections expected | H0: VE ≤ 30% vs. H1: VE > 30% |
0.025 | O'Brien-Fleming |
|
Step/HVTN 502 Phase 2b 2004-2007 |
Non-efficacy | 1; 30 PP infections1 | H0: VE ≤ 0% vs. H1: VE > 60% |
0.05 | Conditional Power < 20% |
| Efficacy | 1; 30 PP infections | H0: VE ≤ 0% vs. H1: VE > 0% |
0.05 | Custom | |
|
Phambili/HVTN 503 Phase 2b 2005-2007 |
Non-efficacy | 1; 60 PP infections | H0: VE ≤ 50% vs. H1: VE > 50% |
N/A | Conditional Power < 20% |
| Efficacy | 1; 60 PP infections | H0: VE ≤ 0% vs. H1: VE > 0% |
0.05 | Custom | |
|
RV144 Phase 2b 2004-2009 |
Harm | Monthly | H0: VE ≥ 0% vs. H1: VE < 0% |
0.05 | Pocock-type2 |
| Non-efficacy | 8; every 6 to 12 months | H0: VE ≤ 0% vs. H1: VE > 50% |
N/A | Conditional Power < 10% | |
| Efficacy | 1; 2/3 of follow-up information | H0: VE ≤ 30% vs. H1: VE > 30% |
0.025 | O'Brien-Fleming |
Per-protocol (PP) infections are those diagnosed after the Week 12 visit in volunteers HIV negative at baseline and who received the first two doses of either vaccine or placebo, excluding those who were either diagnosed with HIV infection before or at the Week 12 visit or who violated the protocol on the basis of pre-defined criteria (Buchbinder et al., 2008). The interim analysis was triggered by the 30th PP infection in the primary analysis group of subjects with Adenovirus-5 neutralization titers ≤ 200.
Continuous stopping boundary of Betensky (1998).
RV144 used O'Brien-Fleming monitoring for reasonably high efficacy (test H0: VE ≤ 30% vs. H1: VE > 30%) at one interim analysis, and also used conditional-power monitoring for non-efficacy at eight interim analyses (every 6-12 months). At each interim analysis the conditional power to reject H0: VE ≤ 0% was calculated under five assumptions about the true VE for the future period of follow-up: (1) VE = 0%, (2) VE = 50%, (3) the current estimate of VE, (4) the current lower 95% confidence limit for VE, and (5) the current upper 95% confidence limit for VE. Stopping was recommended if the conditional power under both assumptions (2) and (3) was less than 10%.
A common feature of the two VaxGen trials and to a lesser extent the Step and Phambili trials is that they either used no monitoring for non-efficacy or conservative monitoring, hence implicitly betting (from a utility perspective) on a reasonable chance for moderate efficacy (Gilbert, 2010), a gamble given the lack of clear scientific rationale (Burton, 2004). In contrast, the proposed design, closer to RV144, uses more aggressive monitoring for non-efficacy, which, had it been applied to the previous three trials that concluded lack of efficacy, would have delivered the conclusion sooner, without incurring an unacceptable risk of prematurely abandoning a promising vaccine candidate such as that identified in RV144. This is illustrated below (see section, “Application of the Proposed Design to Past HIV Vaccine Efficacy Trials”).
Summary of Objectives of the Proposed Design
The previous efficacy trials all evaluated a single vaccine regimen versus placebo. Now that more vaccine regimens are on the near-term horizon for potential efficacy testing, the proposed design evaluates multiple such regimens simultaneously in the same geographic region, sharing a placebo group, with purpose to accelerate the pace of answering key scientific questions about multiple candidate vaccine regimens and hence to accelerate the pace of vaccine development. The primary objective of the design is to expeditiously evaluate VE against HIV infection diagnosed within 18 months of randomization [a parameter we refer to as VE(0-18)] for each vaccine regimen versus placebo, using a sequential monitoring approach fitting to scientific, ethical, and operational considerations. The primary objective focuses on evaluating protection against HIV exposures proximal to the immunization series because the level of protection is plausibly greatest while the vaccine-induced immune responses are at their peak levels, and many immunological parameters wane after the last immunization. The interval 18 months is selected anticipating that the tested vaccine regimens will have HIV envelope protein immunizations at Months 3, 6, and 12. Reasons for counting all infections after randomization rather than only counting infections after a time-point by which full immunity is expected to accrue include: (1) to assure a fair comparison of vaccine regimens that may have different temporal immunity dynamics; and (2) to obviate the need to select a potentially arbitrary starting time. If issues (1) and (2) are not problematic for the particular vaccine regimens under study, then it would be reasonable to assess VE(6-18) (say) for the primary analysis, albeit as for the analysis of VE(0-18) an intention-to-treat approach is used. Further discussion on this issue is provided in the section, “Intention-to-Treat and Per-Protocol Analysis of VE.”
The secondary objectives of the design include: (1) to evaluate durability of vaccine efficacy for each regimen showing reliable evidence for positive VE(0-18); (2) to expeditiously and rigorously evaluate immune correlates of protection if any of the vaccine regimens show reliable evidence for positive VE(0-18); and (3) to compare vaccine efficacy among the vaccine regimens. For secondary objective 1, the durability of vaccine efficacy is evaluated via estimation and inference about the curve VE(t) = (1 – HR(t))×100%, where HR(t) is the hazard ratio (vaccine/placebo) of HIV infection diagnosis at time t, ranging from 0 to 36 months post-randomization. For secondary objective 2, immune correlates are evaluated if at least one vaccine regimen shows reliable evidence for positive VE(0-18), with all vaccine regimens included in the assessment, and all available follow-up information included. For secondary objective 3, VE(0-18) is compared among the vaccine regimens, and, if multiple regimens show evidence for positive VE(0-18), durability of VE(t) is compared among the positively efficacious regimens for t ranging between 18 and 36 months.
Secondary objective 1 is important because any vaccine showing positive efficacy proximal to the immunization series merits assessment for the durability of the efficacy, since durability largely influences a vaccine's public health utility (Anderson, Swinton, and Garnett, 1995; Anderson and Garnett, 1996; Abu-Raddad et al., 2007), and, due to data from past HIV vaccine trials showing that many measured vaccine-induced immune responses tend to wane over time, waning efficacy is a ubiquitous concern. Moreover, RV144 motivates this objective, as there was a non-significant trend suggesting that efficacy waned after the first year (Rerks-Ngarm et al., 2009). Secondary objective 2 is important because as soon as there is reliable evidence that a vaccine confers some protective efficacy, it becomes a scientific priority to develop immunological biomarkers that predict the level of VE (one of the “Grand Challenges in Global Health” of the Foundation of the NIH and the Gates Foundation). Such VE-predictive biomarkers would be used as primary endpoints in subsequent Phase I/II trials of refined vaccine candidates, providing a rational basis for iterative improvement of vaccine regimens. There is perception that the one trial showing positive efficacy (RV144) is taking a long time to deliver answers about immune correlates, motivating building planned processes into the proposed design to deliver these answers sooner. Secondary objective 3 is important because head-to-head concurrent comparisons of VE within the same trial provides the most rigorous data evidence for decisions about whether and which vaccine regimens to advance to a Phase 3 licensure trial. Furthermore, concurrent assessment of multiple vaccine regimens is expected to shorten the time to a Phase 3 trial compared to separate single-vaccine regimen trials. Additional objectives assess HIV vaccine effects on post-infection endpoints such as viral load; however it is beyond the scope of this article to address approaches for these objectives.
The remainder of this article describes the proposed design and reports on its operating characteristics, with main sections: Description of proposed Phase 2b study design; Sequential monitoring of VE(0-18); Accrual and trial duration for the proposed design implemented in South Africa; Application of the proposed design to past HIV vaccine efficacy trials; Statistical power for assessing an immune correlate of HIV infection; Statistical power for detecting a valuable specific surrogate of protection; Comparing vaccine efficacy among the vaccine regimens; Additional issues; Summary of the proposed design; Other issues of interest that merit further research.
Description of Proposed Phase 2b Study Design
Set-Up of Design
HIV uninfected volunteers are randomized in equal allocation to a placebo regimen and to between 1 and 3 vaccine regimens, and are followed for up to 36 months for diagnosis of the primary endpoint of HIV infection. While our main interest is in the 2- and 3-vaccine arm trials, we include the 1-vaccine arm trial for comparison. Volunteers receive immunizations at Month 0, 1, 3, 6, and 12 and receive HIV tests monthly starting at Month 0. (A rationale for monthly testing is described below in the section, “Why Monthly HIV Diagnostic Tests?”, and has precedent in PrEP trials, e.g., Grant et al., 2010.) We assume that T-cell based prime vaccinations are delivered at the Month 0 and 1 visits (and possibly later visits), and antibody-based envelope protein boosts are delivered at the Month 3, 6, and 12 visits. The trial is event-driven, with the requisite number of HIV infection events in the first 18 months (pooled over a vaccine regimen and placebo) selected such that vaccine regimens with VE(0-18) at least 40% will be identified with high power. Specifically, for each vaccine regimen the design is defined by the characteristic that it has 90% power to reject H0: VE(0-18) ≤ 0% if VE(0-18) = 40%, using a 1-sided alpha = 0.025-level log-rank test.
At the end of each vaccine regimen's evaluation, the estimated VE(0-18), 95% CI, and 2-sided p-value, all adjusted for the interim monitoring, will be reported. The reported 95% CI for VE(0-18) is guaranteed to exclude one of the points VE(0-18) = 0% or VE(0-18) = 46%. Thus, the trial will provide reliable evidence either that VE(0-18) is above 0% or below 46%. For a vaccine regimen that just barely meets the efficacy criterion, the trial will report an estimated VE(0-18) of 30% (Rao-Blackwell adjusted unbiased estimate), 95% CI of 0% to 46%, and 2-sided p-value of 0.05. Each vaccine regimen showing statistically significant positive VE(0-18) will be evaluated for efficacy durability by way of never reaching the non-efficacy boundary described below in the sequential monitoring section. Therefore, for each vaccine regimen the design may be viewed as a two-stage design, wherein vaccine efficacy over 18 months is evaluated in stage 1, and, if and only if positive efficacy is demonstrated, then vaccine efficacy over the extended period of 36 months is evaluated in stage 2. The premise of the two-stage design is that vaccine efficacy is expected to be at least as high proximal to the immunization series as distal. Moreover, the design may be viewed as multiple concurrent two-stage designs, each of which evaluates a vaccine regimen versus placebo, with resource savings accrued via a shared placebo group.
The above approach uses the same type I error rate for each vaccine regimen versus placebo regardless of the number of vaccine arms. Consequently, the risk of any type I errors increases with the number of arms. An alternative design would control the overall type I error rate at 0.025 by using a 1-sided 0.025/M-level test, where M is the number of vaccine arms. This design would require substantially more participants, however, and may be overly stringent, given the trial is not a Phase 3 licensure trial, but rather is a Phase 2b “discovery trial” (Self, 2006; Gilbert et al., 2010) with goals to discover and characterize partially efficacious vaccines and the immune correlates of protection, as well as to provide preliminary comparative assessments of vaccine efficacy.
More Rigorous Evaluation of Immune Correlates via Crossover of Placebo Recipients
An ultimate goal for HIV vaccine research is development of a measurable characteristic of the vaccine-induced immune response that reliably predicts VE (Plotkin, 2008), a so-called “surrogate of protection (SoP)” or a surrogate endpoint for HIV infection (Qin et al., 2007). In the first tier (least rigorous) of immune correlates assessment, the goal is to discover biomarkers that predict the subsequent rate of HIV infection in the vaccine group(s), named a correlate of risk (CoR). However, a discovered CoR may have no value to predict VE because it may merely correlate with an intrinsic factor such as innate immunity or host genetics that determines whether individuals are more or less naturally resistant to infection (Follmann, 2006; Qin et al., 2007). Recognizing this limitation of the first-tier correlates assessment, statistical approaches have been developed to assess a more rigorous kind of correlate, a second-tier correlate named a SoP, defined as a CoR that reliably predicts VE, otherwise known as a partially valid surrogate endpoint for HIV infection (Follmann, 2006; Gilbert and Hudgens, 2008; Gilbert, Qin, and Self, 2008; Qin et al., 2008; Wolfson and Gilbert, 2010). Assessment of a second-tier correlate requires predicting the ‘counterfactual’ values of the vaccine-induced immunological biomarker for a subset of placebo recipients. As proposed by Follmann (2006), these predictions may be derived based either on (1) Modeling the relationship between baseline subject characteristics and the biomarker (baseline immunogenicity predictor approach, BIP), and/or on (2) Crossing over a subset of uninfected placebo recipients to the vaccine group and directly measuring their vaccine-induced biomarkers (crossover placebo vaccination approach, CRPV). For a given biomarker the second-tier methods yield an estimate of the “VE curve,” VE(s), which describes how VE changes with the level of the vaccine-induced biomarker. A biomarker valuable for guiding refinement of a vaccine regimen showing some efficacy in the trial will have VE(s) varying widely across levels of s, for example VE(s) will be near 0 for s near 0 (e.g., “negative” immune response) and VE(s) will be large (e.g., 70-90%) for a large immune response s.
We believe both the BIP and CRPV approaches merit use in the proposed efficacy trial design. In particular, if at least one vaccine regimen demonstrates positive VE(0-18), then we propose to cross-over random samples of uninfected placebo subjects to each vaccine regimen that is advanced to Stage 2. While various time-points of cross-over could be considered, the default approach [originally proposed by Follmann (2006)] is appealing, wherein cross-over occurs at the last study visit (the Month 36 visit in our prototype design). The crossed-over subjects are immunized on the same schedule as when they entered the trial, which is necessary for credibility of the ‘time-constancy’ assumption, which states that for crossed-over placebo subjects, the measured immune response is the same as it would have been had it been measured approximately three years earlier on the same schedule relative to the first vaccination.
An alternative approach would cross-over subjects at various times starting at the Month 18 visit. The advantage of this approach is that availability of immune response data at multiple cross-over points would facilitate diagnostic tests of the time-constancy assumption mentioned above (Follmann, 2006). However, the disadvantage is that no post-crossover information from these subjects would be used for the analysis of VE(t) for t > 18 months. That is, in analyses of VE(t) for t > 18 months, the crossed over subjects would be counted in the placebo group only and would be censored at the time of crossover. While this crossover would have no effect on the evaluation of VE(0-18), it would attenuate the statistical power for evaluating VE(t) for t > 18 months. More research is needed to determine the optimal fraction of placebo recipients to crossover, balancing the needs of assessing an immunological surrogate endpoint with the needs of assessing durability of vaccine efficacy. The default approach that waits until the Month 36 visit to cross-over placebo subjects is appealing given the importance of maximizing power for assessing waning vaccine efficacy. It is also appealing for simplifying the study, avoiding the complexity of multiple random cross-over times.
Sequential Monitoring of VE(0-18)
Sequential Monitoring for Non-Efficacy
For each vaccine regimen, the proposed design monitors for non-efficacy at several analyses at evenly spaced numbers of infections diagnosed within 18 months pooled over the vaccine group and the placebo group. We require the number of infections n1 triggering the first interim analysis to be at least 37% of the maximum information, to ensure that a decision to complete a vaccine's evaluation has a minimum level of data support (Freidlin, Korn, and Gray, 2010). In particular, following the suggestion of Freidlin, Korn, and Gray (2010), 37% of the maximum infections was chosen as the first point because, if the estimated VE(0-18) is less than or equal to zero, then the unadjusted/nominal 95% confidence interval for VE(0-18) will exclude the design alternative VE(0-18) = 40% for which the design has 90% power to detect. Because the proposed design requires a maximum of 176 infections within 18 months, this rule equates to the earliest non-efficacy interim analysis taking place at the 65th infection. This approach is an informal way to ensure that, if the reported point estimate indicates non-efficacy, then there will be enough precision about the inference to reliably rule out the design alternative of 40% vaccine efficacy. Completing a vaccine regimen's evaluation prior to this point would be problematic because, given the wide confidence interval, some interpreters of the published result may not be convinced that low efficacy at best was reliably established. This could raise thorny questions about whether additional efficacy trials would be needed, counter to an objective of the design to provide sufficiently definitive evidence about low efficacy such that another efficacy trial would not be needed. Note that with the proposed design the reported monitoring-adjusted 95% confidence interval for VE(0-18) for a weeded-out vaccine regimen is guaranteed to lie below 46%.
To ensure that vaccines with weak efficacy during the ramp-up period of immunity (while the immunizations are being received) but substantial efficacy later are not prematurely weeded out (i.e., the reported 95% confidence interval for VE(0-18) does not lie above 0) based on inter-current infections, we define n1 as the maximum of 65 and the first infection diagnosis event within 18 months such that at least 20% occurred after the ramp-up period (i.e., post-Month 6 visit). Below we show that with this approach the design has less than 20% risk of incorrectly weeding out a vaccine with VE(0-18) = 40% and halved VE during the pre-defined ramp-up period of 0-6 months (see the entry Avg VE(0-18) = 40% in Table 2 Scenario B, where the estimated probability of weed-out is 0.008 + 0.179 = 0.187). If VE(0-18) = 40%, the infection count in the first 18 months when 20% occur post 6 months has median 70, inter-quartile range 58−82, and 10th−90th percentiles 49−92. If VE(0-18) = 0%, the infection count when 20% occur post 6 months has median 79, inter-quartile range 68−92, and 10th−90th percentiles 58−103.
Table 2.
Probabilities (×100%) that the Trial Will Report Each of the Results Efficacy, Potential Harm, Non-Efficacy, and High Efficacy: Scenario (A) [Time-Constant VE(0-18)]*; Scenario (B) [Halved VE in First 6 Months]*.
| Scenario A [Time-Constant VE(0-18)] | ||||||||
|---|---|---|---|---|---|---|---|---|
| Avg VE (0-18) | Avg RR (1-18) | Eff | Harm | Harm Time | Non-Eff | Non-Eff Time | High-Eff | High-Eff Time |
| - | 3.0 | 0.0 | 100.0 | 6.8 (4.9-9.2) | 0.0 | 14.1 (14.1-14.1) | 0.0 | - (---) |
| - | 2.5 | 0.0 | 99.3 | 7.6 (5.5-10.5) | 0.7 | 12.8 (11.8-13.9) | 0.0 | - (---) |
| - | 2.0 | 0.0 | 88.9 | 9.2 (6.2-12.3) | 11.1 | 13.1 (12.3-14.2) | 0.0 | - (---) |
| - | 1.5 | 0.0 | 42.9 | 10.1 (6.4-13.0) | 57.1 | 13.4 (12.5-14.8) | 0.0 | - (---) |
| 0% | 1.0 | 2.7 | 4.2 | 8.6 (6.1-12.4) | 93.0 | 14.6 (13.1-17.8) | 0.0 | - (---) |
| 20% | 0.8 | 30.5 | 1.2 | 7.4 (5.9-10.5) | 68.3 | 16.7 (13.7-22.0) | 0.0 | - (---) |
| 30% | 0.7 | 63.0 | 0.6 | 7.0 (5.8-10.2) | 36.4 | 18.1 (14.2-23.4) | 0.0 | 17.0 (17.0-17.0) |
| 40% | 0.6 | 89.5 | 0.2 | 6.7 (5.8-9.2) | 9.9 | 19.5 (14.5-24.8) | 0.4 | 20.0 (15.5-21.3) |
| 50% | 0.5 | 94.8 | 0.1 | 6.8 (5.8-9.1) | 1.0 | 18.1 (14.2-24.8) | 4.1 | 21.0 (16.8-29.7) |
| 60% | 0.4 | 68.1 | 0.0 | 6.9 (5.9-8.9) | 0.0 | 20.0 (15.8-21.3) | 31.9 | 22.7 (17.4-29.8) |
| 70% | 0.3 | 14.5 | 0.0 | - (---) | 0.0 | - (---) | 85.5 | 22.4 (17.1-29.6) |
| 80% | 0.2 | 0.2 | 0.0 | - (---) | 0.0 | - (---) | 99.8 | 18.8 (13.4-23.8) |
| Scenario B [Halved VE in First 6 Months] | ||||||||
|---|---|---|---|---|---|---|---|---|
| Avg VE (0-18) | Avg RR (1-18) | Eff | Harm | Harm Time | Non-Eff | Non-Eff Time | High-Eff | High-Eff Time |
| - | 3.0 | 0.0 | 96.0 | 8.6 (6.1-11.2) | 4.0 | 12.3 (11.1-12.9) | 0.0 | - (---) |
| - | 2.5 | 0.0 | 84.9 | 9.5 (6.3-12.1) | 15.1 | 12.5 (11.7-13.3) | 0.0 | - (---) |
| - | 2.0 | 0.0 | 57.5 | 10.1 (6.5-12.6) | 42.5 | 12.7 (12.1-13.7) | 0.0 | - (---) |
| - | 1.5 | 0.0 | 22.5 | 10.2 (6.4-12.9) | 77.5 | 13.2 (12.4-14.4) | 0.0 | - (---) |
| 0% | 1.0 | 2.7 | 4.2 | 8.6 (6.1-12.4) | 93.0 | 15.8 (13.6-21.2) | 0.0 | - (---) |
| 20% | 0.8 | 25.9 | 1.9 | 7.7 (6.0-10.9) | 72.3 | 15.8 (13.6-21.2) | 0.0 | - (---) |
| 30% | 0.7 | 54.3 | 1.2 | 7.3 (5.9-10.8) | 44.4 | 16.1 (13.8-22.0) | 0.0 | - (---) |
| 40% | 0.6 | 81.3 | 0.8 | 7.0 (5.9-9.8) | 17.9 | 15.9 (13.9-21.5) | 0.1 | 21.3 (19.3-24.4) |
| 50% | 0.5 | 92.5 | 0.6 | 6.8 (5.8-8.9) | 4.6 | 15.6 (13.8-18.4) | 2.3 | 28.5 (20.2-29.9) |
| 60% | 0.4 | 72.4 | 0.3 | 6.6 (5.4-8.7) | 0.8 | 15.3 (14.1-16.7) | 26.5 | 29.1 (21.2-29.2) |
| 70% | 0.3 | 16.2 | 0.2 | 6.4 (5.1-7.4) | 0.1 | 15.3 (14.1-16.3) | 83.5 | 25.2 (21.4-29.8) |
| 80% | 0.2 | 0.2 | 0.2 | 6.6 (5.0-7.9) | - | - (---) | 99.7 | 24.1 (18.2-29.3) |
Efficacy (Eff in the third column) is the result that VE(0-18) > 0% with reported 95% confidence interval lying above 0%. Potential Harm (Harm) is the result that the potential harm boundary is reached. Non-efficacy (Non-Eff) is the result that the reported 95% confidence interval for VE(0-18) does not lie above 0%; this occurs if the non-efficacy boundary is reached at an interim analysis or the final analysis for assessing VE(0-18). High efficacy (High-Eff) is the result that the reported 95% confidence interval for VE(0-18) lies above 50%. The Times for the various events show the 50th (10th-90th) percentiles of the number of months until the event is reached.
An alternative approach would determine n1 based on a minimal percentage of person-time at-risk occurring after the ramp-up period. This approach is motivated by two potential down-sides of the infections-based approach: n1 has relatively high variance, because it depends on the unknown HIV incidence in each study arm; and n1 depends on the relative level of VE(0-18) during and after the ramp-up period, such that the timing of n1 could indirectly leak information on vaccine efficacy to individuals outside of the DSMB. However, the infections-based approach has the advantage of defining the milestone based on the information scale for a survival analysis, whereas the person-time at-risk approach could start the analysis based on a small number of infections. Therefore we select the infections-based approach, and in limited simulations we found that the two approaches had very similar false-weed-out rates concordant within 1%. Another potential approach would monitor for non-efficacy at evenly spaced numbers of total infections, and use a weighted log-rank statistic that down-weights infections occurring during the ramp-up period. While this approach could be configured to give satisfactory operating characteristics, it is not clear that this weighting scheme would be desirable for assessing positive efficacy, such that different test statistics may be warranted for testing the two alternative directions. In contrast, the selected approach allows a symmetric monitoring design with the un-weighted log-rank test used for testing in both directions (Emerson and Fleming, 1989).
Once n1 is determined for a vaccine regimen, the timing of the subsequent analyses for evaluating non-efficacy are defined to satisfy all of the criteria: (1) achieve 90% power to detect VE(0-18) = 40%; (2) use as many analyses as possible up to nine; and (3) evenly space the interim analyses at intervals of at least five infections. Based on these criteria all 9 analyses are scheduled if and only if n1 ≤ 127. In the case that VE(0-18) = 40%, there is a > 99.9% chance that all 9 analyses will be scheduled.
Several stopping boundaries were considered, and we select the “P = 0.6 stopping boundary” (Emerson and Fleming, 1989), which is slightly less aggressive than the Pocock (1977) boundary for early stopping, chosen to balance the objectives of rapidly weeding out non-efficacious vaccines and protecting against the false weed-out error mentioned above. The operating characteristics of the non-efficacy monitoring plan are described below (in the section, “Accrual and Trial Duration for the Proposed Design Implemented in South Africa”). Based on expectations for accrual, HIV incidence, and dropout for the proposed design implemented in South Africa (described below) for a vaccine regimen with VE(0-18) = 40%, the median value of n1 is 75, in which case there are 9 analyses with the last one occurring at nmax = 176 infections. For n1 = 75, Figure 1 shows the non-efficacy stopping boundary on the scale of the nominal estimated hazard ratio over 18 months [HR(0-18)]; the boundary is reached as soon as an interim estimate of HR(0-18) goes below the boundary.
Figure 1.
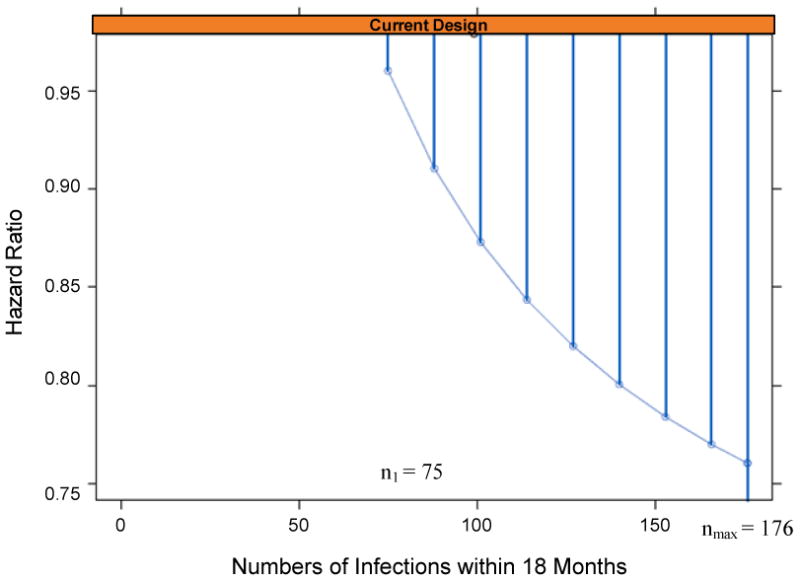
P = 0.6 non-efficacy boundary comparing a vaccine regimen versus placebo, for the scenario where the first interim analysis occurs at n1=75 infections diagnosed within 18 months. If the final analysis at nmax infections is reached before the boundary is crossed, then the reported 95% confidence interval for VE(0-18) will be above 0%.
The Lan-DeMets (Lan and DeMets, 1983) implementation of the stopping boundary is used so as to allow flexibility in the timing and number of analyses. For validity this approach requires that the future analysis times are selected to be independent of the current estimate of VE(0-18) (Betensky, 1998). Given that the interim analyses are fairly frequent and it is not pressing to detect a non-efficacy signal a few months earlier, this assumption is acceptable.
Should Sequential Monitoring for any Vaccine Efficacy be Performed?
A goal of the trial design is to facilitate expeditious assessment of immune correlates for all vaccines showing some efficacy. One technique for helping achieve this is sequential monitoring for positive efficacy (test H0: VE ≤ 0% vs. H1: VE > 0%), and to initiate the immune correlates assessment (i.e., commence measuring the pre-specified candidate immune correlates from infected vaccine-group subjects and from frequency matched uninfected vaccine-group subjects) when the efficacy signal is reached. However, a potential problem with this approach is that, in order to initiate the immune correlates analysis, many individuals would need to know that the positive efficacy signal is achieved (e.g., lab personnel and the managers of specimen processing and shipments), and it may be difficult to ensure that dissemination of this knowledge would not damage study conduct (Ellenberg, Fleming, and DeMets, 2002).
Given this potential problem, we expect that a simpler approach may be more effective, wherein for each vaccine regimen the immune correlates assessment is automatically initiated 9-12 months before all of the information is available for evaluating VE(0-18) (i.e., when the last enrolled participant has 6-9 months of follow-up). The immunologic work is only initiated for vaccine regimens that did not earlier reach the non-efficacy boundary, for which some positive efficacy is likely. Vaccines not hitting the non-efficacy boundary will have estimated VE(0-18) at least 20-25% (as demonstrated in Figure 1: for example if the non-efficacy boundary is not reached at 151 infections than the estimated hazard ratio must be less than 0.78, i.e., VE(0-18) must exceed 22%), supporting at least low-level efficacy that would make a correlates analysis worthwhile. This approach would straightforwardly maintain confidentiality, as no one but the independent statistician(s) and DSMB would know whether reliable evidence for positive efficacy had been achieved. Moreover, the known date for a go/no-go decision would help study personnel prepare for the correlates analyses, and this approach may provide results sooner than the interim monitoring-based approach, because the analysis may begin before an efficacy signal would be detected.
Sequential Monitoring for High Efficacy
While it is unlikely that the prime–boost HIV vaccine regimens under preparation for efficacy testing will confer high levels of protective efficacy, for scientific and ethical reasons it may be prudent to monitor for this event, which, if detected, would lead to un-blinding of participants and reporting of the result (see section “Timing of Reporting of Results and of Un-blinding” for additional discussion on un-blinding). We define “high enough efficacy to warrant un-blinding” as reliable evidence that VE > 50%, operationalized by a log-rank test rejecting H0: VE ≤ 50% vs. H1: VE > 50% at 1-sided 0.025-alpha level. The proposed design tests H0 at three interim analyses, at evenly spaced numbers of arm-pooled infections diagnosed between 0 and 18 months with final number fixed at the median nmax if VE(0-18) = 50% (176 in the prototype design). An O'Brien-Fleming stopping boundary is used so as to require strong early evidence for VE(0-18) > 50% (shown in Figure 2). As for the non-efficacy monitoring, the Lan-DeMets (1983) implementation is used so as to allow flexibility in the timing and number of analyses. Unlike the non-efficacy monitoring, if the VE(0-18) estimate is near the boundary then the DSMB may request an additional interim analysis, in which case the Lan-DeMets implementation could be swapped with Betensky's (1998) continuous stopping boundary to ensure valid type I error control. Figures 3 and 4 show the power curve for detecting VE(0-18) > 50% and the cumulative probabilities of reaching the high efficacy boundary by the four analysis times. For vaccines with VE(0-18) in the range 0-50%, this monitoring has negligible impact on the operating characteristics of the design.
Figure 2.

O'Brien-Fleming high-efficacy boundary comparing a vaccine regimen versus placebo, for the scenario where the first non-efficacy interim analysis occurs at n1=75 infections diagnosed within 18 months such that nmax = 176 infections (and the first high-efficacy interim analysis starts at 44 infections).
Figure 3.

Statistical power for rejecting H0: VE(0-18) ≤ 50% in favor of H1: VE(0-18) > 50% as a function of the true VE(0-18) (1-sided 0.025-level test). Here VE(0-18) = [1 – hazard ratio over the first 18 months] × 100%.
Figure 4.
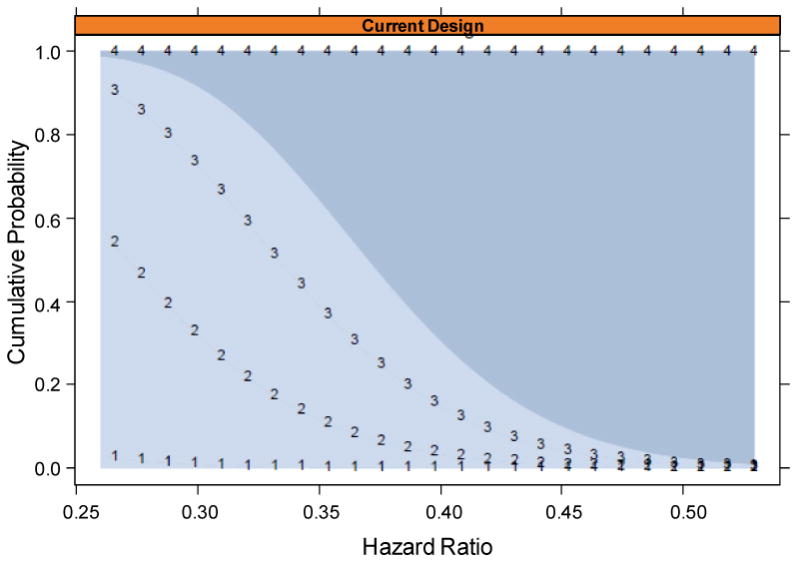
Cumulative probability of reaching the high-efficacy boundary by the first, second, third, and final analyses (lines demarked by 1, 2, 3, 4, respectively), as a function of the true HR(0-18), for the scenario where the first non-efficacy interim analysis occurs at n1=75 infections diagnosed within 18 months. For example, if the true HR(0-18) = 0.3 [i.e., VE(0-18) = 70%], then there is a 30% chance to reach the boundary by the second analysis and a 75% chance to reach the boundary by the third analysis.
Sequential Monitoring for Potential Vaccine Harm
Given the potential vaccine-enhancement of HIV infection risk observed in the Step trial (Buchbinder et al., 2008), it is prudent to closely monitor for VE(0-18) < 0%. To provide maximally close monitoring for each vaccine regimen, the proposed design performs interim analyses after every HIV infection event diagnosed between 0 and 18 months ranging from the 7th to the n1th (pooled over a vaccine regimen and placebo). Similar monitoring was performed by Heyse et al. (2008) in a rotavirus vaccine trial and is being used in an ongoing HIV Vaccine Trials Network (HVTN) trial. Such “continuous” monitoring is performed by an un-blinded statistician (independent from the protocol statisticians) who observes whether, after each confirmed HIV infection event, the stopping boundary is reached. The monitoring applies exact one-sided binomial tests of H0: p ≤ 0.5 versus H1: p > 0.5, where p is the probability that an infected subject was assigned to the vaccine group. Each test is performed at the same pre-specified nominal/unadjusted alpha-level, chosen based on simulations such that, for each vaccine regimen, the overall type I error rate by the 99th arm-pooled infection (i.e., the probability that the potential-harm boundary is reached when the vaccine is actually safe, p = 0.5) equals 0.05. The number 99 is selected because, under the null [VE(0-18) = 0%], there is a 90% chance that the non-efficacy monitoring would commence by the 99th infection in the first 18 months (n1 ≤ 99). If n1 is below 99, then the effect is that less than 0.05 overall type I error rate is spent; for example, with n1 = 75 the overall error rate is about 0.045. The impact on the potential harm monitoring is a slight loss of power to detect a harmful vaccine. If n1 exceeds 100, then the tests continue to be applied (using the same critical value), which slightly increases the overall type I error rate during the trial (estimated at 0.0532 for n1 = 120 and at 0.0558 for n1 = 140).
Figure 5 shows the potential-harm stopping boundary, and the upper rows of Table 2 describe the power of the monitoring plan to reach the boundary under different HRs > 1. For example, for a vaccine with time-constant HR = 1.5 (50% elevation in the infection hazard rate over the first 18 months) there is a 43% chance to stop before the n1th infection, and the median stopping time is 10.1 months (Table 2 Scenario A). In addition, if the vaccine doubles the risk of infection (HR = 2.0), there is a 89% chance to stop before the n1th infection, and the median stopping time is 9.2 months (Table 2 Scenario A).
Figure 5.

Potential-harm boundary comparing a vaccine regimen versus placebo. For each infection diagnosed within 18 months of randomization from the 7th to the 99th (x) the boundary is reached if at least y of the infections were assigned to the vaccine arm. The red and blue lines illustrate the analyses at the 30th and 60th infection, with stopping boundary ≥ 22 and ≥ 40 of the infections in the vaccine arm, respectively.
The potential-harm boundary is only defined out to the n1th infection because the non-efficacy boundary serves the function to stop harmful vaccines at all later infection counts, in fact much more aggressively than would an extended harm-boundary [e.g., a vaccine with estimated VE(0-18) < -2% at the first non-efficacy interim analysis is guaranteed to reach the stopping boundary]. An alternative approach to monitoring for potential vaccine-harm would use a repeated generalized likelihood ratio test (Siegmund, 1985, Chapter 4; Wald, 1947) applied at the same analysis times, with potential advantages that the procedure is approximately asymptotically efficient and the critical value is obtained analytically. The boundaries (based on the binomial proportion p) are almost identical to the exact binomial-test-based boundaries (not shown).
The potential-harm monitoring is not intended to reliably establish harm [i.e., VE(0-18) < 0%], as a vaccine regimen could meet the boundary and the reported 95% confidence interval for VE(0-18) would include 0% (although the 90% confidence interval, if constructed correspondent to the testing procedure, would exclude 0%). Rather, the objective is to apply extra caution and prudence for a prevention trial that enrolls healthy volunteers. More discussion may be needed to determine whether this degree of caution is warranted, given that an error to reach the potential-harm boundary for a truly safe vaccine [with VE(0-18) ≥ 0%] may cause undue damage to the HIV vaccine field.
Operational Considerations for the Timing of Interim Analyses
On the surface, the timing of interim analyses is complex, because it is separately determined for each vaccine regimen based on the rate of infection event, and differs across the monitoring types. However, for the purpose of continuous potential-harm monitoring in the current HVTN trial (HVTN 505), the HVTN developed an effective procedure for rapid adjudication of HIV infection events and for automatic generation of monitoring reports after each confirmed infection event. The existence of this system makes straightforward the accommodation of multiple monitoring schedules. In particular, after each adjudicated infection the un-blinded statistician creates the routine reports and notes whether any interim analyses are due, and, if so, whether any boundaries are reached. Reaching a boundary prompts the statistician to immediately notify the DSMB, which may request a more complete analysis that includes secondary endpoints, collated into a report for the next DSMB meeting. Based on this report the DSMB will make recommendations about continuing or stopping each vaccine regimen. Given the complexity of the pros and cons of continuing or stopping each vaccine regimen, the DSMB might be asked to report to a predetermined Oversight Group as well as the Team given the complexity and implications that may be beyond the DSMB's immediate purview (Ellenberg, Fleming, and DeMets, 2002; Emerson, 2006; Fleming, 2006; Emerson and Fleming, 2010). The Oversight Group includes critical stake-holders, such as representatives of the sponsor, the vaccine manufacturer, and the research group conducting the efficacy trial.
Given that an effective system for accurately and rapidly identifying HIV infection endpoints is in place, it would also be feasible to use continuous monitoring for all of the monitored events, although more work would be needed to delineate the pros and cons.
Monitoring for Operational Futility
Achieving the primary objectives in a timely manner requires sufficiently high rates of accrual, HIV incidence, and ascertainment of the primary endpoint of HIV infection. Therefore, the design monitors these three types of data, and at each DSMB meeting presents an analysis of the projected time until the final analysis, with a prediction interval to assess uncertainty in the projection. Because the projection method is based only on blinded data (pooling over study groups), and the guidelines for what outcomes constitute operational futility are pre-specified and pre-vetted with various stake-holders including the sponsor, vaccine-manufacturers, DSMB, and experts in the field, the operational futility monitoring poses minimal risk to study integrity and is widely used in clinical research. Developing a statistical approach to projecting operational futility was an important aspect of designing the current small Phase 2 HIV vaccine efficacy trial (HVTN 505). While we consider it beyond the scope of this manuscript to describe details of potential operational futility monitoring plans, it is important to note that such monitoring would be employed.
Accrual and Trial Duration for the Proposed Design Implemented in South Africa
Because the proposed design is event-driven, the required number of subjects to enroll and the anticipated trial duration are estimated based on anticipated rates of accrual, HIV incidence in the placebo group and dropout. We illustrate these calculations for South Africa, where based on HVTN experience we assume: uniform accrual over a 12 month period, with halved accrual in the first 3 months; 4% annual HIV incidence in the placebo group; and 5% annual dropout. Ten thousand trials were simulated, assuming the HIV incidence and dropout rates have Poisson distributions, and assuming each vaccine regimen has VE(0-18) = 50% with either (A) constant VE throughout 0−18 months or (B) constant VE throughout 0−6 months at VE(0-6) = 30% and constant VE throughout 6−18 months at VE(6-18) = 60%, both scenarios for which early stopping is unlikely and hence a relatively large sample size N is needed, which should be planned for. In particular, N = 2150 is chosen as the number enrolled (per arm) such that for each vaccine regimen, under either Scenario A or B, there is at least an 85−90% chance that at least nmax = 176 infections will be diagnosed within 18 months (combined across the vaccine and placebo arms). In particular, with N = 2150 per group, there is probability 0.025, 0.10, 0.25, 0.50, and 0.75 of 165, 173, 181, 189, and 198 infections diagnosed within 18 months, respectively, and this result is the same for Scenarios A and B. For N = 2000 per group these numbers decrease by about 12 while for a sample size of N = 2250 these numbers increase by about 10.
Based on the 10,000 simulated trials under Scenario A using the sample sizes and accrual rates shown in Table 3, Figure 6a-c shows distributions of the trial duration under different values for true VE(0-18), for trials with 1, 2, or 3 vaccine regimens. Worthless vaccines [with VE(0-18) = 0%] are weeded out (i.e., reach the non-efficacy boundary) within 17 months with 50% probability, and within 20 months with 99% probability (Figure 6a). If a vaccine regimen has VE(0-18) ≥ 40%, then there is at least 82% probability that the regimen will be fully evaluated to the maximum duration of 48 months (Figure 6a). For a trial with 2 or 3 vaccine regimens each with VE(0-18) ≥ 40%, there is at least 93% probability that the trial will reach the full 48 months (Figure 6b,c). Furthermore, if a vaccine regimen has low efficacy in the range 20-30%, then both events of weed-out and continuation to the end are fairly likely. For example, if all vaccine regimens have VE(0-18) = 30%, then a trial with 1, 2, and 3 vaccine regimens will reach the full 48 months with probability approximately 55%, 67%, and 80% (black dashed lines in Figures 6a-c).
Table 3.
Projected Accrual Rate and Number Enrolled for the Proposed Design.
| Number of Study Arms (with One Placebo Arm) | Accrual Per Week During 52 Week Accrual period1 | Number Accrued Per Study Arm2 | Total Accrued (N) | |
|---|---|---|---|---|
| Initial 13 Weeks | Subsequent 39 Weeks | |||
| 2 | 47 | 95 | 2150 | 4300 |
| 3 | 71 | 142 | 2150 | 6450 |
| 4 | 95 | 189 | 2150 | 8600 |
These accrual rates lead to full accrual at 12 months since the first subject is enrolled, such that the maximum trial duration is 48 months.
Equal allocation of subjects to the study arms.
Figure 6.

Distributions of the total trial duration for trials with (a) 2, (b) 3, and (c) 4 study groups, all with one placebo group. (a) 1 vaccine regimen versus placebo. (b) 2 vaccine regimens versus placebo. (c) 3 vaccine regimens versus placebo. For trials with multiple vaccine groups, a trial completes once all of the vaccine groups reach the end of evaluation. The calculations for this figure assume the true VE(0-18) is constant over time.
Table 2 shows corresponding information on the probabilities that each individual vaccine regimen reaches each type of stopping boundary, and, if so, how long it takes. Our goal is to have high probability of weeding out vaccines with 0-15% efficacy and low probability of weeding out vaccines with at least 40-50% efficacy. Under either Scenario A and B there is a very low risk that the trial would report a 50% efficacious vaccine as non-efficacious, whereas for a 40% efficacious vaccine this risk is about 10% if VE(0-18) is constant and about 19% if VE(0-18) is halved in the first 6 months (Table 2).
For the design with two vaccine arms, the first with constant VE(0-18) = 20% and constant VE(18-36) = 10% and the second with constant VE(0-18) = 50% and constant VE(18-36) = 25%, Figure 7 shows the distributions of the number of HIV infections diagnosed during the time-intervals 0-36 months, 0-18 months, 0-6 months, 6-18 months, and 18-36 months. The distributions have many outliers due to every type of monitoring bound being reached with at least small positive probability.
Figure 7.

Distributions (box-plots) of the number of infections diagnosed in different time-intervals 0-36 months, 0-18 months, 0-6 months, 6-18 months, and 18-36 months, for the placebo arm and two vaccine arms. The first vaccine has constant VE(0-18) = 20% and constant VE(18-36) = 10% and the second vaccine has constant VE(0-18) = 50% and constant VE(18-36) = 25%.
Application of the Proposed Design to Past HIV Vaccine Efficacy Trials
We applied the proposed 2-arm version (one vaccine versus placebo) of the design to the Vax004, Vax003, Step, and RV144 data-sets. The needed results for determining whether and when any boundaries are crossed are the number of infections triggering the first interim analysis for non-efficacy (which turns out to be 65 for each trial), the infection split after each infection in 0−18 months from the 7th to the 64th (for potential harm monitoring), the estimated HRs over 0−18 months at each of the interim analyses for non-efficacy starting at the 65th infection, and the estimated HRs over 0−18 months at each of the interim analyses for high efficacy. Because Vax003, Step, and RV144 evenly randomized subjects to vaccine or placebo, the proposed boundaries could be directly applied [for Step we analyzed all subjects instead of focusing on the primary analysis cohort− the subgroup with low neutralization levels (≤200) to Adenovirus 5]. However, Vax004 used a 2:1 vaccine: placebo allocation, precluding their direct application. To allow direct application to Vax004, we created 10,000 1:1 allocation data-sets by increasing the placebo group by 33% and decreasing the vaccine group by 33%, the former achieved by random sampling the placebo group data with replacement and the latter achieved by random sampling the vaccine group data without replacement. All of the needed statistics for checking boundary-crossings were then computed for each of the 10,000 data-sets. A single data-set for analysis was then constructed by using for each statistic the median of the 10,000 statistics; for example, for non-efficacy monitoring, at each interim analysis we use the median of the 10,000 HR(0−18) estimates as the HR(0−18) estimate. This procedure approximately represents the real Vax004 trial because it preserves the expected vaccine efficacy at all time-points and preserves the total statistical information in the data (expected total number of infections).
For each trial, we evaluated infections diagnosed during the first 18 months to determine the time of the first non-efficacy interim analysis and hence n1 and nmax. Hazard ratio estimates were computed (with the proportional hazards model) at each scheduled interim analysis, and were compared to the non-efficacy boundary. In addition, 1-sided Fisher's exact test p-values were compared to the potential-harm boundary after each infection diagnosed within 18 months starting at the seventh, and hazard ratio estimates were compared to the high-efficacy boundary at the scheduled high-efficacy interim analyses. For each trial, SeqTrial software was used to make final inferences about VE(0-18) accounting for all of the monitoring, using the median unbiased estimator of the HR(0-18) with analysis time ordering. None of the trials would have reached the potential-harm boundary or the high-efficacy boundary, though Step came close (Figure 8c).
Figure 8.
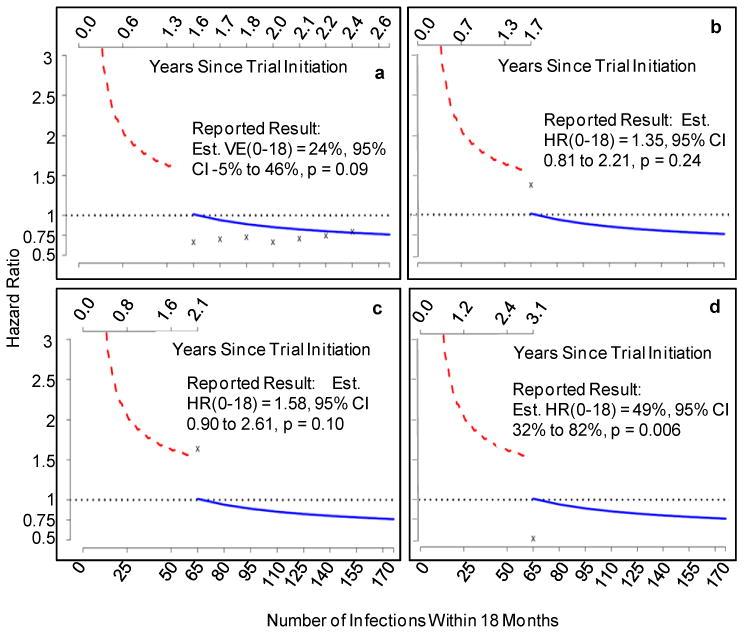
Proposed potential harm, non-efficacy and high efficacy sequential monitoring boundaries applied to the past HIV vaccine efficacy trials. red line = potential-harm boundary; blue line = non-efficacy boundary; x's indicate estimated hazard ratios over the first 18 months for (a) Vax004, (b) Vax003, (c) Step, (d) RV144.
The results are presented in Figure 8 and Table 4. For all four trials, the first interim analysis occurs at n1 = 65 infections (the earliest allowed) such that the final analysis is scheduled at nmax = 176 infections, with nine analyses, the first eight evenly spaced at intervals of 15 infections. Vax004, Vax003, and Step reach the non-efficacy boundary at the seventh, first, and first interim analysis, respectively, and a conclusion of low efficacy at best would have been determined about 24, 33, and 9 months sooner than the actual designs that were used. Therefore use of the proposed non-efficacy monitoring approach would have accelerated the delivery of the non-efficacy results to the field, especially for the VaxGen trials. Furthermore, the proposed non-efficacy monitoring would have resulted in completion of the trials before hundreds of subjects would reach the Month 6 visit, hence sparing them from receiving the Month 6 immunization. In particular, for Step 645 of the 1,836 randomized men (35%) would have been spared the recombinant adenovirus vector vaccination at 6 months (Table 4).
Table 4.
Application of the Proposed Sequential Design to the Past HIV Vaccine Efficacy Trials.
| Total Randomized and HIV Negative at Baseline | Total Reached Month 6 Visit | Number Infections Diagnosed in First 18 Months | ||||||
|---|---|---|---|---|---|---|---|---|
| Trial | Actual Trial | At Boundary Proposed Design | Actual Trial | At Boundary Proposed Design | Time First Analysis Proposed Design | Total Actual Trial | At First Analysis Proposed Design | At Boundary Proposed Design |
| Vax004 | 5,403 | 5,403 (100%) |
5,403 (100%) |
5,107 (94%) |
1 yr 7mo |
171 | 65 | 155 (69V: 86P) |
| Vax003 | 2,527 | 2,527 (100%) |
2,527 (100%) |
2019 (80%) |
1 yr 8 mo |
104 | 65 | 65 (37V: 28P) |
| Step | 1,836 | 1,771 (96%) |
1,836 (100%) |
1191 (65%) |
2 yrs 1 mo |
67 | 65 | 65 (40V: 25P) |
| RV144 | 16,395 | 16,395 (100%) |
16,395 (100%) |
16,165 (99%) |
3 yrs 1 mo |
67 | 65 | Not Crossed |
| Trial Duration | Trial Result | ||||
|---|---|---|---|---|---|
| Trial | Analysis of Boundary Crossing | Actual Trial | Proposed Design | Actual Trial Est. VE(0-18), 95% CI, 2-sided p-value | Proposed Design Est. VE(0-18), 95% CI, 2-sided p-value |
| Vax004 | 7th | 4 yrs 6 mos |
2 yrs 5 mos |
10%, -20% to 33%, p=0.48 |
24%, -5% to 46%, p=0.09 |
| Vax003 | 1st | 4 yrs 6 mos |
1 yr 8 mo |
1.03, 0.67 to 1.4, p=0.87 |
1.35*, 0.81 to 2.21, p=0.24 |
| Step | 1st | 2 yrs 10 mos |
2 yrs 1 mo |
1.47, 0.95 to 2.28, p=0.08 |
1.58*, 0.90 to 2.61, p=0.10 |
| RV144 | Not Crossed |
5 yrs 0 mos |
5 yrs 0 mos |
44%, 8% to 66%, p=0.02 |
49%, 32% to 82%, p=0.006 |
Reported as Est. HR(0-18) and 95% CI for HR(0-18)
In contrast to the other three trials, RV144 does not reach the non-efficacy boundary, thus indicating some positive efficacy on VE(0-18), such that the trial would have continued to stage 2, assessing vaccine efficacy over the full 36 months. As such, the monitoring plan used for RV144 would have led to similar results as the actual trial design, which is appropriate. In addition, note that of the four previous efficacy trials, Vax004 was approximately the same size as the proposed design, with 171 infections diagnosed within 18 months (compared to our target of 176 infections), whereas the other trials accrued too-few infections within 18 months to meet the infection requirements of the proposed design. This underscores the importance of conducting the proposed design in a high-incidence region.
This exercise also hints at possible low-level vaccine efficacy of the Vax004 vaccine regimen during the first 18 months of follow-up, with estimated VE(0-18) = 24% and p = 0.09. However, the data-set was a pseudo data-set. For the actual Vax004 data-set, Table 5 shows point and confidence interval estimates of VE(0-3), VE(0-6), VE(0-9), VE(0-12), VE(0-15), and VE(0-18), together with p-values. While the point estimates suggest 25%−30% vaccine efficacy during the first 12 months, the results are not statistically significant, and the estimated VE(0-18) is 10% with 95% CI -20% to 33%, p = 0.48. Figure 9 shows a complementary analysis, where vaccine efficacy based on the instantaneous hazard ratio at time t, VE(t), was estimated for all t between 0 and 36 months. Specifically, the vaccine and placebo group hazard functions of infection at time t since entry were separately estimated by nonparametric kernel smoothing (with Epanechnikov kernels) for all t between 0 and 36 months, and then VE(t) was estimated by one minus the ratio of hazard function estimates (vaccine/placebo) at time t. Pointwise and simultaneous 95% confidence intervals were constructed by the method of Gilbert et al. (2002), using the bias-adjustment procedure as described. The bandwidths were chosen to minimize the mean integrated squared error as described in Gilbert et al. (2002). This analysis differs from the analyses of VE(0-3), VE(0-6), VE(0-9), VE(0-12), VE(0-15), and VE(0-18), which evaluated time-averaged hazard-ratios rather than hazard-ratios at particular times.
Table 5.
Analysis of VE by Time Interval in the Vax004 Trial.
| VE Parameter | Estimated VE* | 95% Confidence Interval | p-value |
|---|---|---|---|
| VE(0-3) | -21% | -244% to 57% | 0.72 |
| VE(0-6) | 31% | -14% to 58% | 0.14 |
| VE(0-9) | 30% | -3% to 52% | 0.07 |
| VE(0-12) | 23% | -8% to 46% | 0.13 |
| VE(0-15) | 17% | -13% to 39% | 0.24 |
| VE(0-18) | 10% | -20% to 33% | 0.48 |
Based on a proportional hazards model for infections diagnosed within the specified time-interval.
Figure 9.
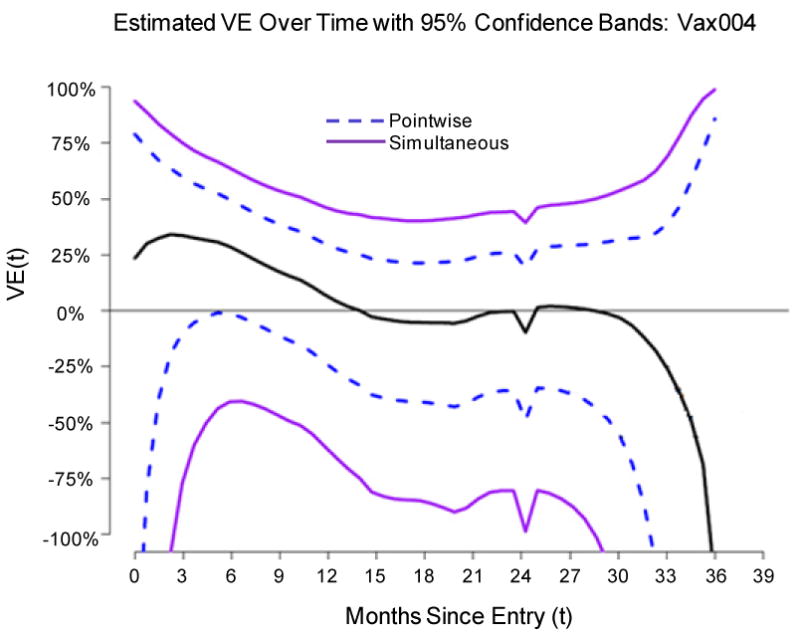
(Nonparametric kernel smoothing estimate of VE(t) for the Vax004 trial data, with 95% pointwise and simultaneous confidence intervals).
Statistical Power for Assessing an Immune Correlate of HIV Infection
Two main types of correlate analyses are conducted among vaccinated subjects, the first of which evaluates immunological measurements at a key fixed time-point (e.g., the Month 6.5 visit, approximate peak immunity) as predictors of HIV infection over a subsequent period of time (e.g., over the next 18 months), and the second of which evaluates time-dependent immune responses as predictors of infection during the next short interval of time extending to the next HIV test. The analyses are complementary, as the former aims to discover correlates that can be measured at a single time-point as close as possible to baseline and hence hold potential as practical surrogate endpoints; the latter addresses the relationship of the immune response near the time of exposure with the acute risk of infection. Given that vaccine-induced HIV antibodies tend to rapidly wane over time, the analyses could easily yield different answers. The Cox proportional hazards model provides an approach to assessing both types of correlates.
We computed power to assess a normally distributed quantitative HIV-specific immunological measurement taken 2 weeks after the Month 6 visit (referred to as the Month 6.5 visit) as a predictor of the subsequent rate of HIV infection. This assessment is performed only for the vaccine groups, as the immune responses will be negative/zeros for (almost) all placebo recipients. We assume the immunological measurement has no, low, medium, or high noise, (defined as 100%, 90%, 67%, or 50%, respectively, of the inter-subject variance in the measurement being protection-relevant), where the protection-irrelevant variance may stem from a variety of sources including technical assay measurement error and variability in the time between the last immunization and the sample-draw (this time is centered around 14 days with several days of variation). We show power results for the scenario where the hazard rate of HIV infection in all of the vaccine arms pooled follows a proportional hazards model and decreases by the fraction RR per 2 standard deviation increase in the protection-relevant variability of the immunological measurement, where RR is varied from 0.3 to 1.0. For simplicity, the identical proportional hazards model is assumed for each vaccine arm.
For each of the 10,000 simulated trials discussed above for 2-, 3-, and 4-arm trials and constant VE(0-18)=50% for each vaccine arm, we counted as cases vaccine recipients diagnosed with HIV infection between 6.5 and 24 months or between 6.5 and 36 months, and assumed the immune response was measured for 95% of these subjects. Addressing these two time periods evaluates correlates of infection for exposures proximal to the immunization series, and for exposures over the complete follow-up period, respectively. For the proximal time period it would be more consistent with the primary and secondary objectives to assess correlates over 6.5 to 18 months, and our decision to focus on 6.5 to 24 months is due to the greater number of infection events, which largely improves power to detect the same effect size. However, waning of vaccine-induced immunity from 18 to 24 months may imply a smaller plausible effect size for the 6.5 to 24 month analysis.
All vaccine arms were pooled into a single group for analysis, which allows detection of a correlate with a mechanism that is common across the vaccine regimens. To create a control group of uninfected vaccine recipients, we selected a random sample of vaccine recipients that tested HIV negative at the Month 6.5 visit and completed follow-up with an HIV negative test at the terminal Month 36 visit. This sample was chosen to provide a 5:1 ratio of uninfected to infected vaccine recipients in 6.5−24 or 6.5-36 months, which provides approximately 83% efficiency compared to an approach that would measure the immune response from all controls. For each data-set, a 1-sided Wald test (alpha = 0.025) in a proportional hazards model was used to test whether the hazard rate decreases with measured immune response level. To account for the two-phase/case-cohort sampling of immune responses, the Borgan et al. (2000) estimator II was used. Power was computed as the fraction of simulation runs with 1-sided p-value bounded by 0.025. Table 6 shows the number of vaccine recipients from which we expect to have the measured immune response available.
Table 6.
Number of Vaccine Recipients with Immune Response Measured at Month 6.5 Visit and Hence Used in the Evaluation of an Immunological Correlate of Risk, for Vaccine Regimens with Time-Constant VE of 50%*.
| Sample Size for Analysis Counting Infections Diagnosed Between 6.5 and 24 Months | |||
|---|---|---|---|
| Number of Vaccine Arms | Expected Number Infections Diagnosed 6.5−24 Months with Immunological Data | Number Uninfected Vaccinee Controls (5:1 Ratio) | Total Number of Immunological Measurements |
| 1 | 53 | 265 | 318 |
| 2 | 106 | 530 | 636 |
| 3 | 159 | 795 | 954 |
| Sample Size for Analysis Counting Infections Diagnosed Between 6.5 and 36 Months | |||
|---|---|---|---|
| Number of Vaccine Arms | Expected Number Infections Diagnosed 6.5−36 Months with Immunological Data | Number Uninfected Vaccinee Controls (5:1 Ratio) | Total Number of Immunological Measurements |
| 1 | 87 | 435 | 522 |
| 2 | 174 | 870 | 1044 |
| 3 | 261 | 1305 | 1566 |
Figures 10a-f show power curves for the 24 scenarios defined by the number of vaccine arms, assay noise levels, and time-period 6.5−24 or 6.5−36 months for diagnosing infections. Benchmarks for realistically-detectable effect sizes (RRs) are indicated on the plots, based on estimates observed in Vax004 for which there was an estimated 0.45 RR per 2 SD increase in the log10 50% MN neutralization titer (Gilbert et al., 2005) and an estimated 0.61 RR per 2 SD increase in the percent viral inhibition as measured by an antibody-dependent cell-mediated viral inhibition (ADCVI) assay (Forthal et al., 2007). The four plotted benchmarks are the estimated RRs per 2 SD protection-relevant variability (x-axis scale) that result under each of the four scenarios that the assay had noise-level equal to one of our supposed levels. The results show that assay-noise attenuates power, and that all of the designs have adequate power to detect a correlate with strength of the MN neutralization titer in Vax004, whereas the 3- and 4-arm designs but not the 2-arm design have adequate power to detect an ADCVI-like correlate. Power increases with the number of vaccine regimens.
Figure 10.
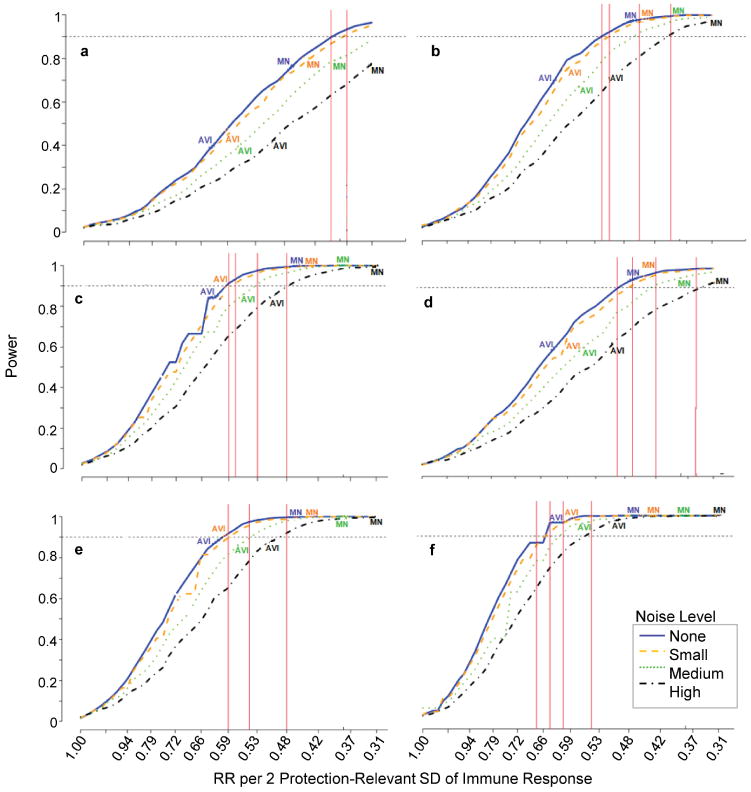
Power curves for the probability of detecting an immunological correlate of risk, assuming that all tested vaccines have true VE(0-18) = 50% and at least one achieves positive efficacy. A Noise Level of None, Small, Medium, and Large assumes the protection-irrelevant assay-variability is 0%, 50%, 67%, or 100% as large as the protection-relevant assay-variability, respectively. Dashed line indicates 90% Power. (a, b, c) include HIV infections diagnosed between 6.5 and 24 months post-randomization, for (a) 1, (b) 2, and (c) 3 vaccine regimens. (d, e, f) include HIV infections diagnosed between 6.5 and 36 months post-randomization, for (d) 1, (e) 2, and (f) 3 vaccine regimens.
Statistical Power for Detecting a Valuable Specific Surrogate of Protection
As described above, for immunological measurements discovered to be CoRs it is of interest to evaluate their value as specific SoPs. A CoR with surrogate value will have VE(s) varying in s; therefore, we evaluate the power of the proposed trial design to reject the null hypothesis of a useless surrogate [H0: VE(s) = VE] versus the alternative hypothesis of a biomarker with some surrogate value [H1: VE(s) varies in s]. We base the calculations on the parametric method for estimating VE(s) initially developed by Follmann (2006) and later extended by Gilbert and Hudgens (2008) to accommodate 2-phase sampling and assay censoring limits.
Power is calculated using 1,000 trials simulated the same as above using the no measurement error scenario, with additional data generated for allowing the BIP, CRPV, and BIP+CRPV designs. Similar to the above, we assess power for infections diagnosed in the periods 6.5−24 months and 6.5−36 months, pooling infections across all the vaccine regimens, and assuming each vaccine has time-constant VE= 50% through 36 months. The additional generated data are as follows: (1) a BIP W is simulated in all trial participants who reach the month 6.5 visit HIV negative, such that W and S have a bivariate normal distribution each with mean 2 and variance 1 and correlation 0.8; (2) for placebo recipients HIV negative at the terminal visit at 36 months, 10 times more than the number of placebo recipients infected over the first 36 months are crossed over to the vaccine arm and have S measured; (3) the time between month 6.5 and infection diagnosis in the placebo arm follows an exponential distribution with annual incidence of 4%; and (4) the time between the month 6.5 visit and infection diagnosis in the vaccine arm conditional on S and W follows an exponential distribution with hazard rate beta10 + beta11 S, with beta10 chosen such that VE = 50% at all follow-up times and beta11 chosen such that S is inversely correlated with the infection hazard in the vaccine group and either: (i) VE(s) = VE for all s; (ii) VE(0) = 25% and VE(4) = 75%; or (iii) VE(0)=0% and VE(4)=90%. These scenarios reflect biomarkers with no surrogate value, moderate surrogate value, and high surrogate value, respectively, and the corresponding true curves are illustrated in Figure 11. Note that this set-up assumes availability of subject characteristics highly predictive of S (linear correlation 0.8, which is plausible based on the correlation of 0.85 observed between hepatitis A vaccine titers and hepatitis B vaccine titers (Czeschinski et al., 2000) and power would be less if such characteristics were not available. For simplicity, for each scenario (i)−(iii), the same true coefficients beta10 and beta11 are assumed for each vaccine arm. It would also be of interest to evaluate scenarios where the VE(s) curve differed among the vaccine regimens.
Figure 11.

Three true VE(s) curves under which power is calculated for rejecting H0: VE(s) = VE in favor of H1: VE(s) varies in s [solid horizontal line = null scenario VE(s) = VE; dotted line = moderate surrogate value scenario; dashed line = high surrogate value scenario].
Table 7 shows the power estimates for these curves. The simulations confirm that the tests for all three designs have nominal size of 0.05. For a trial with one vaccine regimen, power is moderate to detect even high surrogate value; for the BIP + CRPV design power is 58% and 71% for follow-up through 24 and 36 months. This shows that greater statistical information is needed to assess a surrogate endpoint than to assess a correlate of risk, a point well known in the surrogate endpoint assessment literature. Increasing the number of vaccine arms substantially increases power, for example for the BIP + CRPV design there is power of 77% and 84% to detect high surrogate value for 2-vaccine and 3-vaccine arm trials over 24 months of follow-up. This illustrates that an important function of studying multiple vaccine regimens in the same trial is to improve the resolution of the degree to which a correlate of risk has value as a surrogate endpoint. This advantage is accrued only if the immunological predictor of VE is common among the multiple vaccine regimens, which is most likely to occur if the vaccine regimens have the same (or very similar) mechanism of protection. Given difficulty in assuring a common mechanism, it is prudent to carry out the surrogate endpoint analysis separately for each vaccine regimen, although power is limited as shown here. The efficacy trial may evaluate the same protein boost within each tested vaccine regimen, which would support plausibility of a common mechanism.
Table 7.
Power for Testing that an Immunological Biomarker has Some Surrogate Endpoint Value: H0: VE(s) = VE versus H1: VE(s) varies in s [Sub-Sampling]*.
| Analysis | Counts | Infections | Analysis | Counts | Infections | |
|---|---|---|---|---|---|---|
| Diagnosed Through 24 Months | Diagnosed Through 36 Months | |||||
| True VE(s) | 1Vac Arm |
2Vac Arms |
3Vac Arms |
1Vac Arm |
2Vac Arms |
3Vac Arms |
| BIP + CRPV Design | ||||||
|
VE(s) = 0.50 (Null) |
0.03 | 0.04 | 0.05 | 0.04 | 0.04 | 0.05 |
|
VE(s) = mod increase |
0.27 | 0.35 | 0.41 | 0.43 | 0.58 | 0.68 |
|
VE(s) = large increase |
0.58 | 0.77 | 0.84 | 0.71 | 0.86 | 0.94 |
| BIP Design | ||||||
|
VE(s) = 0.50 (Null) |
0.04 | 0.05 | 0.05 | 0.04 | 0.05 | 0.05 |
|
VE(s) = mod increase |
0.34 | 0.44 | 0.48 | 0.57 | 0.69 | 0.71 |
|
VE(s) = large increase |
0.71 | 0.84 | 0.89 | 0.86 | 0.94 | 0.96 |
| CRPV Design | ||||||
|
VE(s) = 0.50 (Null) |
0.05 | 0.04 | 0.05 | 0.03 | 0.06 | 0.04 |
|
VE(s) = mod increase |
0.1 | 0.11 | 0.13 | 0.15 | 0.17 | 0.2 |
|
VE(s) = large increase |
0.15 | 0.17 | 0.20 | 0.22 | 0.3 | 0.34 |
For the BIP + CRPV and BIP designs the BIP has linear correlation 0.8 with the immunological biomarker S. For the BIP + CRPV and CRPV designs, among placebo recipients uninfected at closeout, 10 times more than the number of placebo recipients infected over the first 36 months are crossed over to the vaccine arm and have S measured.
The power calculations also show that the designs with BIP provide much greater power than the CRPV design. This is expected because an excellent BIP was assumed, such that for the BIP and BIP + CRPV designs vaccine recipients outside the phase-2 sample and placebo recipients have considerable information about S; whereas in contrast for the CRPV design vaccine recipients outside the phase-2 sample have no information about S and infected placebo recipients have no information about S. Furthermore, for the CRPV design uninfected placebo subjects outside of the phase-2 sample have no information about S, and when the calculations were repeated using complete sampling of S for uninfected placebo recipients, power for the CRPV design improved considerably (results in Table 8). For example, for 3 vaccine arms and 24 month follow-up power to detect an excellent surrogate increases from 20% to 33%.
Table 8.
Power for Testing that an Immunological Biomarker has Some Surrogate Endpoint Value: H0: VE(s) = VE versus H1: VE(s) varies in s [Complete Sampling]*.
| Analysis | Counts | Infections | Analysis | Counts | Infections | |
|---|---|---|---|---|---|---|
| Diagnosed Through 24 Month | Diagnosed Through 36 Months | |||||
| True VE(s) | 1Vac Arm |
2Vac Arms |
3Vac Arms |
1Vac Arm |
2Vac Arms |
3Vac Arms |
| BIP + CRPV Design | ||||||
|
VE(s) = 0.50 (Null) |
0.04 | 0.05 | 0.04 | 0.05 | 0.05 | 0.04 |
|
VE(s) = mod. increase |
0.35 | 0.45 | 0.53 | 0.58 | 0.73 | 0.79 |
|
VE(s) = large increase |
0.81 | 0.91 | 0.95 | 0.9 | 0.97 | 0.99 |
| BIP Design | ||||||
|
VE(s) = 0.50 (Null) |
0.05 | 0.05 | 0.06 | 0.05 | 0.05 | 0.04 |
|
VE(s) = mod. increase |
0.41 | 0.49 | 0.52 | 0.61 | 0.71 | 0.76 |
|
VE(s) = large increase |
0.83 | 0.91 | 0.94 | 0.91 | 0.97 | 0.99 |
| CRPV Design | ||||||
|
VE(s) = 0.50 (Null) |
0.05 | 0.04 | 0.06 | 0.05 | 0.05 | 0.05 |
|
VE(s) = mod. increase |
0.13 | 0.14 | 0.14 | 0.18 | 0.24 | 0.25 |
|
VE(s) = large increase |
0.22 | 0.28 | 0.33 | 0.36 | 0.44 | 0.47 |
For the BIP + CRPV and BIP designs the BIP has linear correlation 0.8 with the immunological biomarker S. For the BIP + CRPV and CRPV designs, all placebo recipients uninfected at closeout are vaccinated and have S measured.
In addition, the power calculations in Table 7 show that the BIP design provides slightly higher power than the BIP + CRPV design. This result is counter-intuitive given that CRPV provides additional information under the assumption (which was made) that uninfected placebo recipients with immune response measured after crossover equals the immune response 6.5 months after randomization. Part of the explanation comes from the fact that an excellent BIP was used, such that it is not surprising that no improvement is conferred. In fact, Follmann's (2006) simulation study for the case of complete-sampling showed no efficiency improvement moving from BIP to BIP+CRPV when the linear correlation between the BIP and S exceeds 0.8. Moreover, for the case of complete-sampling the simulations were repeated for a modestly predictive BIP with linear correlation 0.25, and for a single vaccine power was 48% for BIP+CRPV design and 33% for the BIP design. This demonstrates that CRPV indeed augments power when only a modestly predictive BIP is available. Another part of the explanation comes from the fact that CRPV was only administered for a phase-2 sub-sample of uninfected placebo recipients; when complete CRPV sampling was used the power between the designs equalized, and sometimes power for the BIP + CRPV design exceeded that for the BIP design (Table 8).
We think that a full explanation is achieved by noting that the parametric method we used for the BIP + CRPV design uses different sets of samples to accomplish the two main estimation steps, i.e., the estimation of the conditional distribution of S given W, and the maximization of the estimated likelihood. Specifically, only samples with S and W measured in the vaccine group contribute to the former step, whereas samples with W measured in both the vaccine and placebo groups contribute to the latter step. This conjecture is partly supported by the fact that the BIP + CRPV design performs slightly better than the BIP design when we enter the information about the true conditional distribution of S into the parametric method. Moreover, in ongoing unpublished work, we are developing a nonparametric method based on a discretized W for estimating VE, which allows the information from crossed-over placebo subjects to contribute to the estimation of the conditional distribution of S. With inclusion of this extra information, we are finding that the BIP + CRPV design always provides greater efficiency than the BIP design.
Whereas the BIP and BIP + CRPV approaches require some modeling assumptions linking the risk of disease under each treatment assignment to S and other covariates, the CRPV approach can advantageously be implemented without making such assumptions. Indeed, Follmann (2006) developed nonparametric tests for any surrogate value based on the CRPV design. While appealing, we expect the BIP and BIP + CRPV designs to be most useful in practice, because the availability of a good BIP largely improves statistical power compared to the CRPV only design.
Comparing Vaccine Efficacy Among the Vaccine Regimens
Power to Compare VE(0-18) Among the Vaccine Regimens
Power for testing equality of VE(0-18) between two vaccine arms was evaluated in two ways, each of which uses all available blinded follow-up information through 18 months. The first way uses a standard log-rank test wherein the null hypothesis is rejected if the 2-sided p-value is less than 0.05. The second way is more stringent, wherein the null hypothesis is rejected if both the 2-sided p-value is less than 0.05 and the vaccine regimen showing superiority has VE(0-18) > 0% [based on the reported 95% confidence for VE(0-18) interval lying above 0%]. The two approaches give similar power, with slightly smaller power for the latter method if one of the vaccines has zero efficacy (Table 9).
Table 9.
Power for Comparing VE(0-18) Between Two Vaccine Regimens1.
| Vaccine 1 | Vaccine 2 | |||||
|---|---|---|---|---|---|---|
| VE(0-18) | Expected Number Infections 0-18 Months | VE(0-18) | Expected Number Infections 0-18 Months | Hazard Ratio (0-18) Vaccine 2 vs. 1 | Power of Log-Rank Test | Power of Log-Rank Test2 |
| 0% | 88 | 30% | 72 | 0.70 | 0.51 | 0.45 |
| 0% | 88 | 40% | 66 | 0.60 | 0.80 | 0.78 |
| 0% | 88 | 50% | 59 | 0.50 | 0.95 | 0.94 |
| 0% | 88 | 60% | 50 | 0.40 | 0.99 | 0.99 |
| 15% | 81 | 40% | 66 | 0.71 | 0.56 | 0.56 |
| 15% | 81 | 50% | 59 | 0.59 | 0.87 | 0.87 |
| 15% | 81 | 60% | 50 | 0.47 | 0.98 | 0.98 |
| 15% | 81 | 70% | 41 | 0.35 | >0.995 | >0.995 |
| 30% | 72 | 50% | 59 | 0.71 | 0.51 | 0.51 |
| 30% | 72 | 60% | 50 | 0.57 | 0.87 | 0.87 |
| 30% | 72 | 70% | 41 | 0.43 | 0.99 | 0.99 |
| 30% | 72 | 80% | 29 | 0.29 | >0.995 | >0.995 |
| 45% | 62 | 60% | 50 | 0.73 | 0.39 | 0.39 |
| 45% | 62 | 70% | 41 | 0.55 | 0.85 | 0.85 |
| 45% | 62 | 80% | 29 | 0.36 | >0.995 | >0.995 |
2-sided 0.05 level log-rank test, using all available blinded follow-up information between 0 and 18 months. In particular, if at least one vaccine regimen achieves positive efficacy [i.e., the reported 95% confidence interval for VE(0-18) lies above 0%] and no vaccine regimens reach the potential-harm boundary, then all vaccine regimens have the full 18 months of follow-up. Similarly, if no vaccine regimens achieve positive efficacy but none reach the potential-harm boundary, and none reach the non-efficacy boundary until the final analysis, then all vaccine regimens have the full 18 months of follow-up. If no vaccine regimens achieve positive efficacy and at least one hits the potential-harm or non-efficacy boundary before the final analysis, then all vaccine regimens have whatever follow-up information through 18 months is available up to the time the last regimen is weeded out.
The same test except that rejection of the null hypothesis requires both that the log-rank test reject and that the superior vaccine regimen achieves positive efficacy VE(0-18) > 0%.
The proposed design has high power to distinguish vaccines with 30% versus 60% VE(0-18) (power = 87%) and moderate power to distinguish vaccines with 30% versus 50% VE(0-18) (power = 51%) (Table 9).
Probability of Correctly Selecting the Vaccine Regimen with Highest VE(0-18)
In contrast to the above power results, under the objective to select-and-advance a high-performing vaccine regimen to a subsequent efficacy trial (perhaps Phase 3), without requiring reliable evidence for superiority of the advanced vaccine regimen, the design is adequately large for moderate differences in VE(0-18). In particular, suppose selection is based on the estimate of VE(0-18); for 3-arm and 4-arm designs, Table 10 shows probabilities that the truly best vaccine will be correctly selected under different scenarios for true VE(0-18) values. The design has high probability to select the best vaccine, especially if a tolerance limit of 10% VE is allowed for what constitutes a meaningful difference.
Table 10.
Probabilities of Correctly Selecting the Vaccine Regimen with Highest True VE(0-18).
| 2 Vaccine Regimens | |||
|---|---|---|---|
| VE(0−18) % (Vx1, Vx2) | Prob at least 1 vaccine achieves positive efficacy1 | Prob select best vaccine2 | Prob select best vaccine within 10% tolerance3 |
| (0, 40) | 0.81 | 0.81 | 0.81 |
| (20, 40) | 0.83 | 0.79 | 0.79 |
| (30, 40) | 0.87 | 0.71 | 0.87 |
| (30, 50) | 0.96 | 0.91 | 0.91 |
| (40, 50) | 0.98 | 0.80 | 0.98 |
| (40, 60) | 0.99 | 0.80 | 0.80 |
| (45, 60) | >0.995 | 0.74 | 0.74 |
| (50, 60) | >0.995 | 0.64 | >0.995 |
| (50, 65) | >0.995 | 0.51 | 0.51 |
| 3 Vaccine Regimens | |||
| VE(0-18) % (Vx1, Vx2, Vx3) | |||
| (0, 0, 40) | 0.81 | 0.81 | 0.81 |
| (0, 20, 40) | 0.83 | 0.80 | 0.80 |
| (0, 30, 40) | 0.88 | 0.71 | 0.87 |
| (20, 20, 40) | 0.85 | 0.79 | 0.79 |
| (29, 30, 40) | 0.89 | 0.71 | 0.86 |
| (0, 30, 60) | 0.99 | 0.73 | 0.73 |
| (0, 45, 60) | >0.995 | 0.68 | 0.68 |
| (30, 30, 60) | 0.99 | 0.72 | 0.72 |
| (40, 50, 60) | >0.995 | 0.60 | 0.77 |
A vaccine achieves some positive efficacy if the potential-harm boundary and non-efficacy boundary are never reached, such that the reported 95% confidence interval for VE(0-18) lies above 0%.
This column shows the probability that the vaccine regimen with the highest estimated VE(0-18) both achieves positive efficacy and has the highest true VE(0-18).
This column shows the probability that the vaccine regimen with the highest estimated VE(0-18) both achieves positive efficacy and has true VE(0-18) no more than 10 percentage points lower (on the additive scale) than the vaccine regimen with the highest true VE(0-18); e.g., if vaccines 1, 2, and 3 have true VE(0-18) of 20%, 30%, 40%, then selecting either vaccine 2 or 3 (but not vaccine 1) meets the criterion.
Additional Issues
Why Monthly HIV Diagnostic Tests?
The rationale for frequent HIV testing is to improve the assessment of immune correlates. The monthly schedule of HIV testing will allow catching 50-80% of the infected subjects in the acute-phase (antibody-negative phase) of infection, before HIV has undergone significant evolution, albeit some T cell escape may occur in the early weeks post-HIV acquisition (Goonetilleke et al., 2009). This allows analysis of the originating HIV sequences in the majority of infected subjects, thereby allowing a ‘sieve analysis’ to be conducted, which is a method of identifying how the vaccine efficacy on HIV acquisition depends on the genetics of the transmitting/founder HIV sequences relative to the insert HIV sequences represented in the tested vaccine (Gilbert, McKeague, and Sun, 2008); in particular to identify HIV amino acid sites and sets of sites in antibody epitopes or T cell epitopes that have an elevated rate of mismatch to the insert sequences in vaccine versus placebo recipients.
Sieve analysis is intrinsically tied with the evaluation of immune correlates of protection, as two sides of the same coin. Specifically, on the one hand, if VE > 0% and a sieve effect (i.e., elevated rate of amino acid mismatches to the insert sequence for vaccine versus placebo sequences) is detected, then the implication, given the fact the trial is randomized and double-blinded, is that vaccine-induced immune responses to certain HIV epitopes must have caused the protection. Therefore the detected sieve effect leads to follow-up explorations to identify measurable immune responses that capture (at least partially) these protective responses and thereby have some validity as surrogate endpoints for HIV infection. For example, identification of a sieve effect in 7 particular HIV antibody epitopes generates the hypothesis that the sum of neutralization levels to these 7 targets matched to the vaccine insert sequence would have high surrogate value.
On the other hand, sieve analysis is very useful for validating the degree to which an immunological measurement is a valid surrogate endpoint. To illustrate, suppose VE > 0% and the candidate surrogate, S, is a summary measure of the magnitude and breadth of neutralizing antibody titers to a panel of pseudo-viruses constructed from acute-phase HIV isolates from infected placebo recipients. If S has surrogate value to predict VE, it must be the case that protein differences to the vaccine-insert are larger in infected vaccine than placebo recipients; this logically follows because genetic mutations in antibody epitopes are known to effect neutralization levels. Therefore, sieve analysis is a tool for corroborating the surrogate value of S as a SoP. However, this sieve analysis would not be possible with infrequent HIV diagnostic testing such as the semi-annual schedule used by the previous efficacy trials, given that too-few infected subjects would be caught in the acute-phase to afford an assessment of the vaccine effect on transmitted sequences.
In addition, the sieve analysis may be directly incorporated into the surrogates assessment described above, by estimating the VE(s) curve with the endpoint definition restricted to HIV infection with a strain within a certain threshold of genetic distance to the vaccine-insert. This analysis would be repeated for a range of thresholds. Greater variation in the VE(s) curve for thresholds closer to the insert-sequence would support the value of the immune biomarker as a surrogate endpoint.
Intention-to-Treat and Per-Protocol Analysis of VE
Vaccine efficacy trials commonly assess VE in the intention-to-treat (ITT) cohort, which is all randomized subjects, as well as in the modified intention-to-treat (MITT) cohort, which is the subset of the ITT cohort that are later discovered to not have been HIV infected at baseline. Because blinded procedures are used for ascertaining baseline infection status, the MITT analysis has the same validity from randomization as the ITT analysis, such that the MITT analysis is generally preferred, given that it assesses vaccine efficacy in HIV uninfected persons. In addition, given the ubiquitous concern that a vaccine may not confer protection until all or at least some of the immunizations are received, most vaccine efficacy trials also assess vaccine efficacy in the sub-cohort that receives all of the immunizations and are disease-free after the immunization series; this sub-cohort may be referred to as the per-protocol (PP) cohort (Horne, Lachenbruch, and Goldenthal, 2001). All of the past HIV vaccine efficacy trials assessed VE in both the MITT and PP cohorts, with the MITT assessment the primary analysis in each case (Gilbert et al., 2010).
As stated above, the MITT analysis is primary because the comparator groups are guaranteed to have balanced prognostic factors on average due to randomization and double-blinding, such that the analysis assesses the causal effect of assignment to vaccine. In contrast, the standard analytic approach to assessing PP VE applies the same method as used for the MITT analysis, which compares HIV infection incidence between the subgroups of vaccine and placebo recipients that are observed to qualify for the PP sub-cohort. However, these comparator sub-cohorts are subsets of randomized subjects, such that the analysis is susceptible to possible post-randomization selection bias (Rosenbaum, 1984; Robins and Greenland, 1992; Frangakis and Rubin, 2002), hence making the results difficult to meaningfully interpret. To improve upon this standard analysis of VE in the PP cohort, an analytic method that adjusts for measured factors that simultaneously predict HIV infection and PP sub-cohort membership (such factors cause the selection bias) should be applied (e.g., Lu and Tsiatis, 2008; Tsiatis et al., 2008; Zhang, Tsiatis, and Davidian, 2008; Moore and van der Laan, 2009; Zhang and Gilbert, 2010), which in addition to correcting for bias can improve statistical power by leveraging prognostic factors. Moreover, because some simultaneously predictive factors may be unmeasured, the sensitivity of results to such factors should also be investigated, following the paradigm described in Scharfstein, Rotnitzky, and Robins (1999). Therefore, in our proposed design we assess VE in the MITT cohort for the primary analysis and conduct a causal sensitivity analysis of PP VE for the secondary analysis, wherein the answer is reported as a range of point estimates and a corresponding union of 95% confidence intervals (a so-called “sensitivity interval”), which account for a spectrum of potential levels of post-randomization selection bias (Shepherd, Gilbert, and Lumley, 2007).
Timing of Reporting of Results and of Un-blinding
With respect to reporting the results, the proposed design has two stages: for stage 1, results are reported on VE(0-18); and for stage 2 [which occurs if and only if at least one vaccine regimen achieves positive efficacy for VE(0-18)], results are reported on the durability of VE between 18 and 36 months. For stage 2 the issues are simple: all vaccine arms advanced to stage 2 plus the placebo arm continue blinded follow-up until the last enrolled subject has 36 months of follow-up, at which time the final analysis is conducted and the results reported.
The issues are more complicated for stage 1, with the approach to un-blinding dependent upon which boundaries are reached. As soon as a vaccine arm reaches a conclusion [either by reaching the potential-harm boundary, the non-efficacy boundary, the high efficacy boundary, or completing the evaluation of VE(0-18) without reaching a boundary], the result is reported. This conveys the result to the field as expeditiously as possible. If a vaccine arm completed its evaluation by reaching the potential-harm boundary, then the arm would be immediately un-blinded, given the ethical warrant to inform participants of the potential harm caused by exposure to the vaccine. The other study arms would continue blinded. If a vaccine arm reaches the high efficacy boundary, then the placebo group is immediately un-blinded and offered this vaccine. If it is the single vaccine arm design, then the sole vaccine group is also un-blinded. However if it is the multiple vaccine arm design, and at least two vaccine arms are still being evaluated, then the blind is maintained for all of the vaccine arms, which allows continuing accrual of data for comparing vaccine efficacy head-to-head among the vaccine regimens. Furthermore, if a vaccine arm reaches the high efficacy boundary, it may be worth continuing the vaccine's evaluation out to 36 months. While a rigorous assessment of durability of VE will likely be impossible (given that the contemporaneous comparator placebo group is being offered the vaccine), the additional follow-up may nonetheless provide useful data about the vaccine, which would be difficult to collect in follow-on studies. Further thought is needed on this issue, and on whether it is also warranted to offer subjects assigned to the other vaccine arms the highly efficacious vaccine.
Next we consider the scenario wherein a vaccine arm completes its evaluation by reaching the non-efficacy boundary. In this case, blinded follow-up under the original HIV diagnostic testing schedule would continue either until all other vaccine arms are weeded out, or, in the case that at least one vaccine arm achieves positive efficacy, until all enrolled subjects have 18 months of follow-up. This continued blinded follow-up would contribute information to the analyses of safety, VE(0-18) (including comparisons with other vaccine regimens), and immune correlates of protection. If, alternatively, the arm were un-blinded then the post-un-blinding data would be excluded from the main analyses of vaccine efficacy and of immunological surrogate endpoints, given that the un-blinding may lead to imbalances in HIV prognostic factors between the vaccine and placebo groups (and between vaccine arms), which could not be confidently corrected for statistically due to the inability to accurately measure HIV risk behavior and exposure. Given the scientific benefit accrued from maintaining the blind and the absence of evidence of harm caused to participants, it seems ethical to maintain blinding for subjects assigned a vaccine regimen shown to have low efficacy at best.
For operational reasons, ideally all study arms would be un-blinded at the same time, as un-blinding one study arm could compromise follow-up for the participants assigned to the other arms. As discussed above, by dividing the trial into two stages the design does not achieve this, as vaccine arms reaching a stopping boundary will be un-blinded once the evaluation of VE(0-18) is completed, whereas vaccine arms not reaching a stopping boundary will be un-blinded once stage 2 is completed (expected at least 18 months later). While one approach would keep vaccine arms reaching the non-efficacy boundary blinded all the way through stage 2, this seems like a poor use of resources, given that non-efficacy over 18 months is expected to predict non-efficacy from 18-36 months, such that it is prudent to complete the evaluation of non-efficacious vaccines at 18 months. Thus, our approach makes the un-blinding as simultaneous as ethically warranted within each stage. As discussed above, for stage 2 a completely simultaneous un-blinding is achieved, whereas for stage 1, if no vaccine arms reach the potential-harm boundary then a completely simultaneous un-blinding is achieved. The informed consent process would describe the events that would trigger un-blinding, and the approach to un-blinding would be vetted with local Institutional Review Boards and the DSMB.
In summary, the whole study is un-blinded at the first event of: (1) the last of the vaccine regimens is weeded out, either by reaching the potential-harm boundary or the non-efficacy boundary; (2) the last of the vaccine regimens reaches the high efficacy boundary; (3) the last enrolled subject reaches 36 months of follow-up, for the case that neither event (1) nor (2) occurs. For event (1), the trial has maximum duration of 18 months beyond the last enrolled subject, and minimum duration the time at which either the last weeded-out vaccine regimen reaches the potential-harm boundary or accrues n1 infections diagnosed within 18 months.
What Does Completing a Vaccine Regimen for Non-Efficacy Entail?
As described above, upon reaching the non-efficacy boundary, the primary result on VE(0-18) would be reported, thus providing data as expeditiously as possible. Figure 6 (a) shows that, by the time a vaccine regimen reaches the non-efficacy boundary, accrual is very likely to be complete, in which case weeding out a regimen would not spare enrollees, all of whom would have received at least one immunization. On the other hand a substantial fraction of enrollees will likely have not yet completed the immunization series, such that ceasing vaccinations upon reaching a non-efficacy boundary would spare immunizations. For example, at the median stopping time of a vaccine with VE(0-18) = 0%, approximately 3000 of the 4300 enrollees (pooled over a vaccine arm and placebo) would have completed the immunization series through Month 6 and approximately 1800 through Month 12. Moreover, regardless of the number of immunizations spared, it still may be warranted to cease immunizations at the time of reaching the non-efficacy boundary, as the primary question about VE(0-18) would have been answered. Furthermore, if accrual lags behind the planned accrual, then this approach may spare a great deal of immunizations and substantially decrease the total enrollment. Lastly, if a vaccine regimen reaches the potential-harm boundary then a large number of enrollments and immunizations would likely be spared. Therefore, the proposed design ceases immunizations and accrual to vaccine arms if and when they reach a non-efficacy boundary.
Equal Versus Unequal Allocation to the Vaccine and Placebo Groups
The design equally allocates subjects to each study group, which is inefficient for the two- and three-vaccine arm trials, for which the efficient design would randomize more subjects to the placebo arm. The rationale for equal allocation is to increase the information for the second and third secondary objectives to evaluate immunological correlates of infection rate in the vaccine groups and to compare vaccine efficacy among the vaccine regimens. Equal allocation results in efficiency loss for the primary objective in exchange for efficiency gain for key secondary objectives. This reflects the premise of the design that development of immune correlates of protection and head-to-head comparisons of vaccine efficacy are priorities for HIV vaccine research. More research is needed to thoroughly define the trade-offs of the equal-versus-unequal allocation approaches.
Accommodation of Pre-Exposure Prophylaxis (PrEP) and for Other HIV Prevention Interventions
Recently an efficacy trial in men who have sex with men in the Americas (mostly South America) demonstrated that daily oral PrEP use [fixed-dose combination tenofovir disoproxil fumarate (TDF) and emtricitabine (FTC)] provided an estimated 44% reduction in the incidence of HIV infection compared to placebo (Grant et al., 2010). Moreover, the incidence rate appeared especially low in men with detectable PrEP drug levels, suggesting that the PrEP efficacy is higher for adherent subjects. Because the PrEP drugs TDF and FTC are approved and some vaccine trial participants may take PrEP, it is relevant to consider how the design accommodates PrEP use. Moreover, several other efficacy trials of PrEP are ongoing, such that it is prudent to plan for how the trial design will respond to future results that will become available before or during the trial.
The baseline approach to accommodating PrEP does not alter the primary analysis, as it is intention-to-treat and compares HIV incidence among the vaccine and placebo groups while disregarding PrEP use. The event-driven design set-up is also unaltered, such that with or without PrEP the same numbers of HIV infections trigger the interim and final analyses. However, once the required numbers of events are fixed, PrEP use impacts the anticipated sample size needed to achieve the required number of infections in a timely manner via impact on the background HIV incidence. For example, if 10% PrEP use occurs, and we assume that PrEP users have a 50% reduction in incidence, then the sample size would need to be increased by approximately 5% (0.05 = 0.10×0.50) in order to deliver results within the same time-frame as the baseline scenario (no PrEP use). Alternatively, if all participants are offered PrEP and 80% accept it, then the sample size would need to be increased by approximately 40% (0.40 = 0.80×0.50).
Given the difficulty to predict the degree of PrEP use, the trial would monitor PrEP use through self-report questionnaires and PrEP drug level measurement. The enrollment target could be adjusted based on this monitoring; such an adaptation would pose minimal risk to study integrity because it is based on blinded data and a deterministic plan could be pre-specified for what data lead to what kinds of trial expansions. There is also uncertainty in the degree of PrEP efficacy, and this is addressed through the operational futility monitoring; the level of PrEP efficacy will affect the background HIV incidence, and the lower it is the more likely the operational futility guidelines will be met. The operational futility monitoring is primarily based on rates of accrual, HIV infection, and dropout during the study, regardless of the amount of PrEP use or PrEP efficacy.
It is relevant to evaluate whether PrEP is expected to enhance or diminish vaccine efficacy for trial design set-up, as this would impact the maximum plausible effect size VE, and hence could result in powering the trial for a different effect size. Currently the data on potential interaction of vaccines and PrEP are too scant to warrant altering effect size assumptions.
A second approach to accommodating PrEP use would offer a voluntary second randomization to PrEP or to PrEP placebo. This would form three analysis strata: subjects assigned PrEP, subjects assigned PrEP placebo, and subjects who declined the second randomization. The primary analyses would be intention-to-treat similar to the above, the difference being they would be stratified. For each regimen HIV incidence would be compared between vaccine and placebo within each of the three strata separately, and then aggregated into one overall estimate of VE; for example, assuming the same VE within each strata and using strata-specific baseline hazards in the Cox proportional hazards model. This analysis is valid because randomization and double-blinding guarantee balance in HIV prognostic factors within each stratum. While an interaction of PrEP and vaccine would complicate the interpretation, the assessment of the common VE still has useful interpretation as vaccine efficacy averaging over the three strata.
This primary analysis does not explicitly account for data on PrEP use or PrEP adherence, because of complications in achieving valid inferences adjusted for post-randomization intermediate variables that are subject to measurement error. However, secondary analyses using causal inference method would evaluate vaccine efficacy while subjects are actually using and not using PrEP. Additional secondary analyses would compare efficacy among each of the individual arms (Vaccine + PrEP, Placebo + PrEP Placebo, Vaccine + PrEP Placebo, Placebo + PrEP Placebo). A third approach would power the trial to compare efficacy among these individual arms, implicating a larger trial would be needed. These considerations for accommodating PrEP use are also relevant for use of other HIV prevention approaches. Accommodating microbicides may be particularly relevant given the recent report of a partially efficacious microbicide (point estimate of 39% reduction in HIV incidence compared to placebo) in the CAPRISA 004 Phase 2b efficacy trial of tenofovir gel conducted in South Africa (Karim et al., 2010).
Summary of the Proposed Design
The proposed design has the following main features:
-
Multiple vaccine regimens versus a shared placebo group
The design evaluates vaccine efficacy of multiple vaccine regimens using a shared placebo group in the same geographic region, and is akin to multiple Phase 2b two-arm vaccine versus placebo trials conducted simultaneously. The simultaneous evaluation of multiple vaccine regimens accelerates learning about vaccine efficacy compared to sequential two-arm trials.
The simultaneous evaluation of multiple vaccine regimens improves the assessment of immune correlates of protection compared to a two-arm design, both by increasing the amount of statistical information and by facilitating greater variability in vaccine-induced immune responses.
The design is well-powered to detect large differences but not moderate differences in VE(0-18) among the vaccine regimens. The design provides high probability of correctly selecting the vaccine regimen with the highest VE(0-18) within a 10% tolerance limit.
These advantages still attain if the vaccine regimens are initiated at different calendar times, as long as a concurrent placebo group is always used (see discussion in the section below, “Other Issues of Interest That Merit Further Research.”) However, precision for comparing VE among vaccine regimens may be reduced given the need to adjust for potential secular effects using the placebo group HIV incidence data.
-
Two-stage evaluation of vaccine efficacy
Vaccine efficacy of each vaccine regimen is evaluated in two stages, first over 18 months, and, if there is reliable evidence for VE(0-18) > 0%, over an additional 18 months. This design is efficient because assessment of durability of vaccine efficacy becomes a priority if, and only if, there is evidence for vaccine efficacy for infections occurring relatively soon after the immunization series.
-
Improved assessment of immune correlates of protection
Whereas even the design with one vaccine regimen has reasonable power for detecting correlates of HIV infection rate, testing multiple vaccine regimens is necessary for providing reasonably high power for assessing the surrogate value of identified correlates. Using identified correlates as immunogenicity study endpoints for follow-up, Phase I/II HIV vaccine trials may provide an unreliable basis for comparing and selecting vaccine regimens by their future protective efficacy. In contrast, vetting identified correlates by their estimated surrogate value provides a more rigorous basis for selecting follow-up study endpoints that are more likely to reliably predict protective efficacy in future efficacy trials.
Availability of baseline subject characteristics predictive of an immunological biomarker is crucial for enabling well-powered assessment of the biomarker as a surrogate endpoint.
The design provides for early initiation of the immune correlates assessment for promising vaccine regimens while maintaining confidentiality of the results and double-blind format.
The design uses frequent HIV testing, with advantages to improve the immune correlates assessment and the sieve analysis, as well as to allow assessment of the vaccine effect on acute-phase viral load. In addition, as PrEP use may increase over time, frequent HIV testing may also help in preventing drug resistance through PrEP use.
-
Sequential monitoring for efficient evaluation of vaccine efficacy
The design sequentially monitors each vaccine regimen for potential harm to elevate the rate of HIV infection, for non-efficacy and high efficacy. The design also conducts operational futility monitoring, to stop the trial if accrual, HIV incidence, or study quality metrics are inadequate.
The potential-harm monitoring plan is maximally vigilant by assessing a vaccine-associated elevation in HIV acquisition risk after each HIV infection.
The non-efficacy monitoring plan is designed to be as aggressive as possible while explicitly guarding against prematurely weeding out efficacious vaccine regimens with low efficacy during the initial immunizations. In particular, the monitoring plan is configured to bound the risk of weeding out a vaccine regimen with VE(0-18) = 40% and halved efficacy during the first six months at 20%.
For each tested vaccine regimen, the design will yield a result about vaccine efficacy over the first 18 months [VE(0-18)] that reliably distinguishes between greater than 0% efficacy versus less than 46% efficacy (i.e., the reported 95% confidence interval adjusting for the monitoring will exclude 0% or 46%). Therefore, weeded-out vaccine regimens will have reliable evidence that the vaccine efficacy is bounded by 46%. These statements are not absolute in that there is a tiny probability (< 1%) that a vaccine regimen would reach the potential-harm boundary very early (with the observed infection rate much higher in the vaccine than placebo arm) and the reported 95% confidence interval would cover both 0% and 46%.
Other Issues of Interest That Merit Further Research
Research is needed to identify the most appropriate methods for distinguishing waning VE from lack of waning VE in the presence of heterogeneous HIV exposure. For an identified method, power calculations are needed. Related research is needed for estimation of VE over time.
Power calculations are needed for comparing durability of vaccine efficacy [e.g., VE(18-36)] among vaccine regimens.
Power calculations are needed for assessing the vaccine effects on acute-phase viral load and on viral load at the HIV infection diagnosis visit.
Additional research is needed to ensure that the method for inference and estimation of VE(0-18) and of related vaccine efficacy parameters appropriately account for all of the sequential monitoring that is conducted.
This article assumed that all of the vaccine regimens were started simultaneously. While this would be ideal, practical considerations may necessitate staggered start times of one or more vaccine regimens. This event would require an extension of the total accrual period for the shared placebo group, to ensure concurrent randomized controls for all assessments of VE(t), and to provide a valid basis for comparing VE(t) among the vaccine arms. Research is needed to determine the impact of staggered vaccine start times on the ability to achieve the primary and secondary study objectives.
The statistical method for the primary and secondary analyses of VE(0-18) and of VE(t) may be made more efficient by leveraging baseline subject characteristics predictive of HIV infection, as discussed above in the section, “Intention-to-Treat and Per-Protocol Analysis of VE.” While valid published methods are available for accomplishing this task, research is needed to compare the methods to identify one with optimal characteristics for the context of the proposed trial design.
Acknowledgments
The authors thank the participants of the workshop, who provided insightful input into the proposed study design and into avenues of additional research. The NIAID-sponsored workshop, “Alternative Study Design for Early Efficacy Evaluation of HIV Prophylactic Vaccines,” was held in Bethesda on January 11, 2011. The authors also thank the workshop organizing committee, especially Elizabeth Adams and Mike Proschan. This research was funded by NIH grant 2 R37 AI054165-08 and by NIH NIAID 5 U01 AI068635. SMH was supported by NIH grant AI069470.
Author Notes: Peter B. Gilbert, Vaccine and Infectious Disease Division, Fred Hutchinson Cancer Research Center, and University of Washington, Seattle, WA. Douglas Grove, Vaccine Infectious Disease Division, Fred Hutchinson Cancer Research Center, Seattle, WA. Erin Gabriel, University of Washington. Ying Huang, Vaccine Infectious Disease Division, Fred Hutchinson Cancer Research Center, Seattle, WA. Glenda Gray, Perinatal HIV Research Unit, University of the Witwatersrand, Johannesburg, South Africa. Scott M. Hammer, Division of Infectious Diseases, Columbia University Medical Center, New York, NY. Susan P. Buchbinder, San Francisco Department of Public Health, University of California San Francisco, San Francisco, CA. James Kublin, Vaccine Infectious Disease Division, Fred Hutchinson Cancer Research Center, Seattle, WA. Lawrence Corey, Fred Hutchinson Cancer Research Center and University of Washington, Seattle, WA. Steven G. Self, Vaccine Infectious Disease Division, Fred Hutchinson Cancer Research Center and University of Washington, Seattle, WA.
Contributor Information
Peter B. Gilbert, Fred Hutchinson Cancer Research Center and University of Washington
Douglas Grove, Fred Hutchinson Cancer Research Center.
Erin Gabriel, University of Washington.
Ying Huang, Fred Hutchinson Cancer Research Center.
Glenda Gray, University of the Witwatersrand.
Scott M. Hammer, Columbia University Medical Center
Susan P. Buchbinder, San Francisco Department of Public Health and University of California, San Francisco
James Kublin, Fred Hutchinson Cancer Research Center.
Lawrence Corey, Fred Hutchinson Cancer Research Center and University of Washington.
Steven G. Self, Fred Hutchinson Cancer Research Center and University of Washington
References
- Anderson RM, Garnett GP. Low-efficacy HIV vaccines: potential for community-based intervention programmes. Lancet. 1996;348:1010–1013. doi: 10.1016/s0140-6736(96)07100-0. [DOI] [PubMed] [Google Scholar]
- Anderson RM, Swinton J, Garnett GP. Potential impact of low efficacy HIV-1 vaccines in populations with high rates of infection. Proc Biol Sci. 1995;261:147–151. doi: 10.1098/rspb.1995.0129. [DOI] [PubMed] [Google Scholar]
- Abu-Raddad L, Boily MC, Self SG, Longini IM., Jr Analytic insights into the population level impact of imperfect prophylactic HIV vaccines. J Acq Imm Def Synd Hum Retrov. 2007;45:454–467. doi: 10.1097/QAI.0b013e3180959a94. [DOI] [PubMed] [Google Scholar]
- Betensky RA. Construction of a continuous stopping boundary from an alpha spending function. Biometrics. 1998;54:1061–1071. [PubMed] [Google Scholar]
- Borgan O, Langholz B, Samuelsen SO, Goldstein L, Pogoda J. Exposure stratified case-cohort designs. Lifetime Data Analysis. 2000;6:39–58. doi: 10.1023/a:1009661900674. [DOI] [PubMed] [Google Scholar]
- Buchbinder SP, Mehrotra DV, Duerr A, et al. Efficacy assessment of a cell-mediated immunity HIV-1 vaccine (the Step Study): A double-blind, randomised, placebo-controlled, test-of-concept trial. Lancet. 2008;372:1881–1893. doi: 10.1016/S0140-6736(08)61591-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton DR, Desrosiers RC, Doms RW, et al. A sound rationale needed for phase III HIV-1 vaccine trials. Science. 2004;303:316. doi: 10.1126/science.1094620. [DOI] [PubMed] [Google Scholar]
- Czeschinski PA, Binding N, Witting U. Hepatitis A and hepatitis B vaccinations: immunogenicity of combined vaccine and of simultaneously or separately applied single vaccines. Vaccine. 2000;18:1074–1080. doi: 10.1016/s0264-410x(99)00354-0. [DOI] [PubMed] [Google Scholar]
- Ellenberg S, Fleming T, DeMets D. Data Monitoring Committees in Clinical Trials: A Practical Perspective. John Wiley & Sons, Ltd.; West Sussex, England: 2002. [Google Scholar]
- Emerson SS. Issues in the use of adaptive clinical trial designs. Statistics in Medicine. 2006;25:3270–3296. doi: 10.1002/sim.2626. [DOI] [PubMed] [Google Scholar]
- Emerson S, Fleming TR. Adaptive methods: Telling ‘the rest of the story’. Journal of Biopharmaceutical Statistics. 2010;20:1150–1165. doi: 10.1080/10543406.2010.514457. [DOI] [PubMed] [Google Scholar]
- Emerson SS, Fleming TR. Symmetric group sequential test designs. Biometrics. 1989;45:905–923. [PubMed] [Google Scholar]
- Freidlin B, Korn EL, Gray R. A general inefficacy interim monitoring rule for randomized trials. Clinical Trials. 2010;7:197–208. doi: 10.1177/1740774510369019. [DOI] [PubMed] [Google Scholar]
- Fleming TR. Standard versus adaptive monitoring procedures: a commentary. Statistics in Medicine. 2006;25:3305–3312. doi: 10.1002/sim.2641. [DOI] [PubMed] [Google Scholar]
- Flynn NM, Forthal DN, Harro CD, Judson FN, Mayer KH, Para MF the rgp120 HIV Vaccine Study Group. Placebo-controlled trial of a recombinant glycoprotein 120 vaccine to prevent HIV infection. J Infect Dis. 2005;191:654–665. doi: 10.1086/428404. [DOI] [PubMed] [Google Scholar]
- Follmann D. Augmented designs to assess immune response in vaccine trials. Biometrics. 2006;62:1161–1169. doi: 10.1111/j.1541-0420.2006.00569.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forthal D, Gilbert P, Landucci G, Phan T. Recombinant gp120 vaccine-induced antibodies inhibit clinical strains of HIV-1 in the presence of Fc receptor-bearing effector cells and correlate inversely with HIV infection rate. Journal of Immunology. 2007;178:6596–6603. doi: 10.4049/jimmunol.178.10.6596. [DOI] [PubMed] [Google Scholar]
- Frangakis CE, Rubin DB. Principal stratification in causal inference. Biometrics. 2002;58:22–29. doi: 10.1111/j.0006-341x.2002.00021.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert PB. Some design issues in Phase 2b prevention trials for testing efficacy of products or concepts. Statistics in Medicine. 2010;29:1061–1071. doi: 10.1002/sim.3676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert PB, Hudgens MG. Evaluating candidate principal surrogate endpoints. Biometrics. 2008;64:1146–1154. doi: 10.1111/j.1541-0420.2008.01014.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert PB, McKeague I, Sun Y. The two-sample problem for failure rates depending on a continuous mark: an application to vaccine efficacy. Biostatistics. 2008;9:263–276. doi: 10.1093/biostatistics/kxm028. [DOI] [PubMed] [Google Scholar]
- Gilbert PB, Peterson M, Follmann D, Francis D, Gurwith M, Heyward W, Hudgens M, Jobes D, Popovic V, Self S, Sinangil F, Burke D, Berman P. Immunologic responses to rgp120 vaccine correlate with the incidence of HIV-1 infection in a Phase 3 preventive HIV-1 vaccine trial. Journal of Infectious Diseases. 2005;191:666–677. doi: 10.1086/428405. [DOI] [PubMed] [Google Scholar]
- Gilbert PB, Qin L, Self SG. Evaluating a surrogate endpoint at three levels, with application to vaccine development. Stat Med. 2008;27:4758–4778. doi: 10.1002/sim.3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert P, Wei LJ, Kosorok MR, Clemens JD. Simultaneous inference on the contrast of two hazard functions with censored observations. Biometrics. 2002;58:773–780. doi: 10.1111/j.0006-341x.2002.00773.x. [DOI] [PubMed] [Google Scholar]
- Goonetilleke N, Liu MKP, Salazar-Gonzalez JF, et al. The first T cell response to transmitted/founder virus contributes to the control of acute viremia in HIV-1 infection. Journal of Experimental Medicine. 2009;206:1253–1272. doi: 10.1084/jem.20090365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray GE, Bekker L, Churchyard GJ, et al. Did unblinding affect HIV risk behaviour and risk perception in the HVTN503/Phambili study? Retrovirology. 2009;6(Suppl 3):P209. [Google Scholar]
- Heyse JF, Kuter BJ, Dallas MJ, Heaton P for the REST Study Team. Evaluating the safety of a rotavirus vaccine: the REST of the story. Clinical Trials. 2008;5:131–139. doi: 10.1177/1740774508090507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horne AD, Lachenbruch PA, Goldenthal KL. Intent-to-treat analysis and preventive vaccine efficacy. Vaccine. 2001;19:319–326. doi: 10.1016/s0264-410x(00)00152-3. [DOI] [PubMed] [Google Scholar]
- Lan KKG, DeMets DL. Discrete sequential boundaries for clinical trials. Biometrika. 1983;70:659–663. [Google Scholar]
- 694.Lu X, Tsiatis AA. Improving the efficiency of the log-rank test using auxiliary covariates. Biometrika. 2008;95:679. [Google Scholar]
- Karim QA, Karim SS, Frohlich JA, Grobler AC, Baxter C, Mansoor LE, Kharsany ABM, Sibeko S, Misana KP, Omar Z, Gengiah TN, Maarschalk S, Arulappan N, Mlotshwa M, Morris L, Taylor D on behalf of the CAPRISA 004 Trial Group. Effectiveness and safety of tenofovir gel, an antiretroviral microbicide, for the prevention of HIV infection in women. Science. 2010;3:1168–1174. doi: 10.1126/science.1193748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore KL, van der Laan MJ. Covariate adjustment in randomized trials with binary outcomes: Targeted maximum likelihood estimation. Stat Med. 2009;28:39–64. doi: 10.1002/sim.3445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien PC, Fleming TR. A multiple testing procedure for clinical trials. Biometrics. 1979;35:549–556. [PubMed] [Google Scholar]
- Pitisuttithum P, Gilbert P, Gurwith M, et al. Randomized, double-blind, placebo-controlled efficacy trial of a bivalent recombinant glycoprotein 120 HIV-1 vaccine among injection drug users in Bangkok, Thailand. J Infect Dis. 2006;194:1661–1671. doi: 10.1086/508748. [DOI] [PubMed] [Google Scholar]
- Plotkin SA. Vaccines: Correlates of vaccine-induced immunity. Clin Infect Dis. 2008;47:401–409. doi: 10.1086/589862. [DOI] [PubMed] [Google Scholar]
- Pocock SJ. Group sequential methods in the design and analysis of clinical trials. Biometrika. 1977;64:191–199. [Google Scholar]
- Qin L, Gilbert P, Corey L, McElrath J, Self S. A framework for assessing immunological correlates of protection in vaccine trials. J Infect Dis. 2007;196:1304–1312. doi: 10.1086/522428. [DOI] [PubMed] [Google Scholar]
- Qin L, Gilbert P, Follmann D, Li D. Assessing surrogate endpoints in vaccine trials with case-cohort sampling and the Cox model. Annals of Applied Statistics. 2008;2:386–407. doi: 10.1214/07-AOAS132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rerks-Ngarm S, Pitisuttithum P, Nitayaphan S, et al. Vaccination with ALVAC and AIDSVAX to prevent HIV-1 infection in Thailand. N Engl J Med. 2009;361:2209–2220. doi: 10.1056/NEJMoa0908492. [DOI] [PubMed] [Google Scholar]
- Robins JM, Greenland S. Identifiability and exchangeability of direct and indirect effects. Epidemiology. 1992;3:143–155. doi: 10.1097/00001648-199203000-00013. [DOI] [PubMed] [Google Scholar]
- Rosenbaum PR. The consequences of adjustment for a concomitant variable that has been affected by the treatment. J Roy Stat Soc Ser A. 1984;147:656–666. [Google Scholar]
- Self SG. Issues in the design of HIV vaccine efficacy trials. In: Patricia Kahn, Ian Gust, Wayne Koff., editors. Accelerating AIDS Vaccine Development: Challenges and Opportunities. Horizon Scientific Press; Norfolk: United Kingdom: 2006. [Google Scholar]
- Siegmund D. Sequential Analysis: Tests and Confidence Intervals. Springer-Verlag; New York: 1985. [Google Scholar]
- Scharfstein DO, Rotnitzky A, Robins JM. Adjusting for nonignorable dropout using semiparametric nonresponse models. Journal of the American Statistical Association. 1999;94:1096–1146. [Google Scholar]
- Shepherd B, Gilbert P, Lumley T. Sensitivity analyses comparing time-to-event outcomes only existing in a subset selected post-randomization, conditional on covariates, with application to HIV vaccine trials. Journal of the American Statistical Association. 2007;102:573–582. doi: 10.1198/016214507000000130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsiatis AA, Davidian M, Zhang M, Lu X. Covariate adjustment for two-sample treatment comparisons in randomized clinical trials: a principled yet flexible approach. Statistics in Medicine. 2008;27:4658–4677. doi: 10.1002/sim.3113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wald A. Sequential Analysis. Wiley; New York: 1947. [Google Scholar]
- Wolfson J, Gilbert P. Statistical identifiability and the surrogate endpoint problem, with application to vaccine trials. Biometrics. 2010;66:1153–1161. doi: 10.1111/j.1541-0420.2009.01380.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Gilbert P. Increasing the Efficiency of Prevention Trials by Incorporating Baseline Covariates. Statistical Communications in Infectious Diseases. 2010;2 doi: 10.2202/1948-4690.1002. Available at: http://www.bepress.com/scid/vol2/iss1/art1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Tsiatis AA, Davidian M. Improving efficiency of inferences in randomized clinical trials using auxiliary covariates. Biometrics. 2008;64:707–715. doi: 10.1111/j.1541-0420.2007.00976.x. [DOI] [PMC free article] [PubMed] [Google Scholar]