

Abstract
Rhamnolipids have multiple potential applications as “green” surfactants for industry, remediation, and medicine. As a result, they have been intensively investigated to add to our understanding of their biosynthesis and improve yields. Several studies have noted that the addition of a fatty acid cosubstrate increases rhamnolipid yields, but a metabolic explanation has not been offered, partly because biosynthesis studies to date have used sugar or sugar derivatives as the carbon source. The objective of this study was to investigate the role of fatty acid cosubstrates in improving rhamnolipid biosynthesis. A combination of stable isotope tracing and gene expression assays was used to identify lipid precursors and potential lipid metabolic pathways used in rhamnolipid synthesis when fatty acid cosubstrates are present. To this end, we compared the rhamnolipids produced and their yields using either glucose alone or glucose and octadecanoic acid-d35 as cosubstrates. Using a combination of sugar and fatty acids, the rhamnolipid yield was significantly higher (i.e., doubled) than when glucose was used alone. Two patterns of deuterium incorporation (either 1 or 15 deuterium atoms) in a single Rha-C10 lipid chain were observed for octadecanoic acid-d35 treatment, indicating that in the presence of a fatty acid cosubstrate, both de novo fatty acid synthesis and β-oxidation are used to provide lipid precursors for rhamnolipids. Gene expression assays showed a 200- to 600-fold increase in the expression of rhlA and rhlB rhamnolipid biosynthesis genes and a more modest increase of 3- to 4-fold of the fadA β-oxidation pathway gene when octadecanoic acid was present. Taken together, these results suggest that the simultaneous use of de novo fatty acid synthesis and β-oxidation pathways allows for higher production of lipid precursors, resulting in increased rhamnolipid yields.
INTRODUCTION
Rhamnolipids are surface-active secondary metabolites secreted by Pseudomonas aeruginosa and related species in the stationary phase. They are the most intensively studied type of biosurfactant and have some documented advantages over synthetic surfactants in terms of biodegradability, toxicity, structural diversity, surfactancy, and stability in extremes of temperature, pH, and salt concentration (e.g., 1, 19). Rhamnolipids contain one (monorhamnolipid) or two (dirhamnolipid) rhamnose sugars linked to a β-d-(β-d-hydroxyalkanoyloxy)alkanoic acid (HAA) (42). The exact physiological functions of rhamnolipids remain unclear, but studies have shown that they serve as a virulence factor for Pseudomonas infection (43) and also affect biofilm formation (2, 5) and the swarming motility of bacteria (3, 13). Additionally, these versatile molecules have antimicrobial activities (9) and enhance the aqueous solubilities and biodegradation of slightly soluble organic compounds (20) and complex heavy metal contaminants (20).
Rhamnolipids are produced as a mixture of congeners with conserved hydrophilic head groups and variable hydrophobic tail groups (6). Rhamnolipid production is transcriptionally regulated by quorum sensing (26, 27). The details of the pathways and genes involved in rhamnolipid biosynthesis have been largely studied using radioactive tracer-labeled carbon sources (11, 12) and P. aeruginosa rhamnolipid null mutants (25, 29). These studies have shown that rhamnolipids are synthesized by diverting precursors for the rhamnosyl and lipid moieties from central metabolic pathways to the rhamnolipid biosynthesis pathway (Fig. 1). The donor of the rhamnosyl moiety is dTDP-l-rhamnose, produced from d-glucose by a series of reactions catalyzed by the AlgC and Rml enzymes (20). It has been suggested that the lipid moiety precursor is diverted from de novo fatty acid synthesis since the stereochemistry of the β-hydroxy fatty acid in the lipid moiety matches that of the intermediates in de novo fatty acid synthesis but not intermediates in the catabolic fatty acid β-oxidation pathway (42). RhlA has been identified as the enzyme that diverts the (R)-β-hydroxyacyl-acyl carrier protein (ACP) from de novo fatty acid synthesis to the rhamnolipid biosynthesis pathway by catalyzing the formation of HAA (42). RhlB (25) and RhlC (29) are two rhamnosyltransferases which are responsible for the consecutive additions of two rhamnosyl groups to HAA to form monorhamnolipid first and then dirhamnolipid.
Fig 1.

Biosynthesis of rhamnolipids, highlighting the proposed contributions of de novo fatty acid biosynthesis and β-oxidation to the fatty acids incorporated into rhamnolipids. (A) Normally, in de novo fatty acid biosynthesis, the initial four-carbon β-ketoacyl-ACP is formed from the condensation of acetyl-CoA and malonyl-ACP. This reaction is catalyzed by FabH. Thereafter, the fatty acid is elongated in a cyclic fashion, each cycle adding two carbons [R(n+2), R(n+4), R(n+6), etc.], by the condensation of acyl-ACP (shown as Rn acyl-ACP) with malonyl-ACP [intermediates shown as R(n+2)]. We propose that when a fatty acid is added as a cosubstrate, the β-ketoacyl-ACP can also be formed from a bypass route linking the β-oxidation pathway with the de novo fatty acid pathway, likely catalyzed by a FabH-like enzyme. Specifically, the acyl-CoA β-oxidation intermediate, in particular the C8-acyl-CoA, condenses with malonyl-ACP to form the C10-β-ketoacyl-ACP, which is then recruited by RhlA to form HAA. The dashed arrow indicates the proposed bypass route for recruiting fatty acids from β-oxidation for rhamnolipid biosynthesis. (B) Structures of HAA, monorhamnolipid, and dirhamnolipid.
The rhamnolipid biosynthesis pathway has been elucidated using carbohydrates or their derivatives, e.g., glucose (4, 29), glycerol (10, 11), or mannitol (7), as the growth substrate. The catabolism of these carbon sources does not involve β-oxidation. However, P. aeruginosa is well known for its ability to metabolize a range of fatty acids (C4 to C18) (37). As an opportunistic pathogen in cystic fibrosis patients, P. aeruginosa has also been reported to induce the degradation of lung surfactant lipids to fatty acids (C14 to C18) to help sustain high cell density in vivo (35). A high cell density allows this bacterium to induce quorum sensing, which in turn results in the production of virulence factors. A recent study showed that virulence factor expression, including that of rhamnolipids, is affected by fatty acid β-oxidation enzymes. For example, a P. aeruginosa PAO1 mutant that inactivates the fadD2 gene showed a significantly lower than normal production of rhamnolipid (17). Additionally, rhamnolipid yields can be increased by the addition of oils or oil waste products, which are metabolized through β-oxidation, as a carbon source (23). A recent study showed that rhamnolipids synthesized from fatty acids with an odd number of carbons consisted of both odd- and even-number β-hydroxy fatty acid units, while rhamnolipids synthesized from fatty acids with an even number of carbons consisted of only even-number β-hydroxy fatty acid units (15). Taken together, these studies suggest the potential for a metabolic link between β-oxidation and rhamnolipid biosynthesis.
The goal of this work was to investigate rhamnolipid biosynthesis under conditions that stimulate the β-oxidation pathway during growth. Specifically, we compared the rhamnolipid yields, structural information, and expressions of selected genes with three treatments: 1.25% glucose alone (glucose) and 1% glucose in combination with either 0.25% octadecanoic acid (C18) or 0.25% deuterated octadecanoic acid-d35 (C18-d35). The glucose treatment was used to confirm that fatty acid amendment increased rhamnolipid yields and to provide a baseline control of the major rhamnolipid congeners produced. The C18-d35 treatment was used to trace the pathways used in octadecanoic acid metabolism and incorporation into the rhamnolipid. The C18 treatment served as a control for the C18-d35 treatment to confirm that there were no effects of deuteration on rhamnolipid yields, congener distributions, or gene expressions. The deuterium distributions in the rhamnolipids produced were analyzed by mass spectrometry. To complement this analysis, the expression levels of key genes involved in β-oxidation (fadD1, fadD2, fadA), de novo fatty acid synthesis (fabH1, fabG), and rhamnolipid biosynthesis (rhlA, rhlB) were monitored during bacterial growth using quantitative PCR (q-PCR).
MATERIALS AND METHODS
Bacterial strain and culture conditions.
Pseudomonas aeruginosa strain ATCC 9027, which produces only monorhamnolipids (18, 41), was purchased from the American Type Culture Collection (Rockville, MD) and stored as a glycerol freezer stock at −80°C. A peptone, yeast extract, glucose (PTYG) plate (Difco) was prepared from the freezer stock, and a single colony was inoculated into a preculture composed of NH4H2PO4 (0.3%), K2HPO4 (0.2%), glucose (0.2%), FeSO4 · 7H2O (0.5 mg Fe per liter), and MgSO4 · 7H2O (0.1%). The preculture was incubated at 37°C for 24 h with constant shaking at 200 rpm, and then 200 μl was inoculated into 20 ml growth culture containing phosphate-buffered mineral salts medium (MSM) composed of solutions A and B. Solution A contained 2.5 g NaNO3, 0.4 g MgSO4 · 7H2O, 1.0 g NaCl, 1.0 g KCl, 0.05 g CaCl2 · 2H2O, and 5 ml H3PO4 (85.0%) per liter. Solution B contained 0.5 g FeSO4 · 7H2O, 1.5 g ZnSO4 · 7H2O, 1.5 g MnSO4 · H2O, 0.3 g H3BO3, 0.15 g CoCl2 · 6H2O, 0.15 g CuSO4 · 5H2O, and 0.10 g Na2MoO4 · 2H2O per liter. One milliliter of solution B was added to 1 liter of solution A. The final pH of the MSM was adjusted to 7.0 with KOH pellets. The MSM was supplemented with either 1.25% glucose alone, 1% glucose plus 0.25% octadecanoic acid, or 1% glucose plus 0.25% octadecanoic acid-d35. The octadecanoic acid was purchased from Sigma-Aldrich Chemical Company, Inc. (Milwaukee, WI), and the octadecanoic acid-d35 was purchased from Cambridge Isotope Laboratories, Inc. (Andover, MA). The growth cultures were incubated at 37°C for 96 h with constant shaking at 200 rpm. Samples (100 μl each) were taken at regular intervals and plated on tryptic soy agar (TSA) plates (Difco) for enumeration. All treatments were repeated in triplicate, and each experiment was performed a minimum of two times.
Rhamnolipid yields.
Rhamnolipids were harvested and purified after 96 h as described previously (18). Briefly, cells were separated from the supernatant by centrifugation at 10,000 rpm for 10 min, subjected to acid precipitation, extracted with a 9:1 ratio of chloroform to methanol, and concentrated by rotoevaporation. The samples were further purified by column chromatography using a silica gel (60-Å pore size) packed glass column and a 6:6:6:1:1 (vol/vol/vol/vol/vol) mixture of hexane, dichloromethane, ethyl acetate, chloroform, and methanol with 0.1% acetic acid. The fractions were tested for the presence of rhamnolipids with anthrone reagent. The fractions containing rhamnolipids were combined, rotoevaporated, transferred to a preweighed 1.5-ml plastic microcentrifuge tube, and dried by air. The mass of each sample was recorded.
Electrospray ionization-mass spectrometry.
Electrospray ionization-mass spectrometry (ESI-MS) was performed using a Thermo Finnigan LCQ high-performance liquid chromatography (HPLC)-MS system equipped with an ion trap. The sample solvent, a 1:1 ratio of acetonitrile to methanol, was also used as the carrier solvent; the samples were introduced via direct infusion. Typical flow rates ranged from 400 to 500 μl h−1 with a capillary temperature of 200°C and under standard ESI tuning conditions. All experiments were performed in the negative-ion mode. Duplicate experiments were run on all rhamnolipid samples; relative abundances for all ions were reproducible to 3 to 5%.
Fourier transform ion cyclotron resonance mass spectrometry.
Fourier transform ion cyclotron resonance mass spectrometry (FT-ICR) was performed for high-resolution and accurate mass measurements on a Bruker Apex Qh 9.4T FT-ICR instrument. The samples were introduced using a conventional Bruker ESI source under standard conditions for negative ions. Tandem mass spectrometry (MS/MS) was performed using quadrupole collision-induced dissociation (QCID) with collision energies of 12 eV and He as the bath gas. Duplicate experiments were run on all rhamnolipid samples; relative abundances for all ions were reproducible to 3 to 5%.
Bacterial RNA extraction and cDNA synthesis.
Transcriptional expressions of the selected target genes (fadD1, fadD2, fadA, fabH1, fabG, rhlA, and rhlB) were evaluated by the quantification of mRNA transcripts as described previously (22). Briefly, 500-μl samples were taken from each treatment at 8, 20, 24, 30, and 48 h, mixed with 1 ml of RNAprotect bacterial reagent (Qiagen, Valencia, CA), and stored at −20°C. RNA was extracted using the RNeasy minikit (Qiagen, Valencia, CA) from extraction volumes that were standardized at a cell number of 1 × 109 CFU. Genomic DNA contaminants were removed by DNase treatment, and the integrity of the RNA was confirmed by gel electrophoresis. RNA concentrations were measured using a TBS 380 fluorometer (Turner Biosystems, Sunnyvale, CA) following staining with RiboGreen (Invitrogen, Carlsbad, CA). The RNA was reverse transcribed into cDNA using the iScript cDNA synthesis kit (Bio-Rad Laboratories, Hercules, CA) in a total volume of 20 μl containing 4 μl of 5× iScript reaction mix buffer, 1 μl of iScript reverse transcriptase, and 68 ng of DNase-treated total RNA. Negative reactions were performed for each sample using the reaction mixture without reverse transcriptase. The conditions for the reverse transcription reactions were 25°C for 5 min, 42°C for 30 min, and 85°C for 5 min. All samples taken at the same time point were processed together to avoid bias (22).
Quantification of transcriptional gene expression by qPCR.
The transcript levels of the target genes were quantified as the relative expression ratios of the target genes with respect to two housekeeping genes, proC and rpoD (28). The primers used in the quantitative reverse transcriptase (qRT) PCR were taken from the literature or designed using Primer3 (http://frodo.wi.mit.edu/primer3) (32) and are listed in Table 1. Duplicate qRT-PCRs were performed for target and reference genes from the same time point using a CFX96 quantitative PCR detection system (Bio-Rad) using the SsoFast EvaGreen supermix kit as recommended by the manufacturer. The reaction mixture volume was 10 μl and contained 5 μl of SsoFast EvaGreen supermix (Bio-Rad), 0.4 μM each primer, 2 μl of cDNA template, and 2.2 μl nuclease-free water. The amplification conditions were 1 min at 95°C, followed by 40 cycles at 95°C for 6 s, 60°C for 6 s, and 72°C for 6 s, followed by melt curve analysis for the presence of a unique PCR product. The reaction efficiencies were calculated for each amplicon using the equation E = 10(−1/slope) from the slope of each calibration curve (28) that was generated using 5-fold serial dilutions of cDNA (Table 1). To check for residual contaminating genomic DNA, negative controls were analyzed in the same way using cDNA templates that were synthesized from RT-negative reactions.
Table 1.
Primer sequences used in qRT-PCR analysis
| Gene | Locus | Primer | Sequence (5′— 3′) | Amplicon length (bp) | PCR efficiencya | Source |
|---|---|---|---|---|---|---|
| proC | PA0393 | proC-F | CAG GCC GGG CAG TTG CTG TC | 180 | 1.96 | 33 |
| proC-R | GGT CAG GCG CGA GGC TGT CT | |||||
| rpoD | PA0576 | rpoD-F | GGG CGA AGA AGG AAA TGG TC | 178 | 1.99 | 33 |
| rpoD-R | CAG GTG GCG TAG GTG GAG AA | |||||
| fadD1 | PA3299 | fadD1-F | CTT CCG CAA GCT GGA CTT CT | 181 | 2.01 | This study |
| fadD1-R | GGT GCC GAC CTG GAT GTT | |||||
| fadD2 | PA3300 | fadD2-F | AGG CGC AGG AGG TGA TGA T | 193 | 2.01 | This study |
| fadD2-R | GCG ACG AAC AGG GTG TTG A | |||||
| fadA | PA3013 | fadA-F | AGG CAA GTT CAA GGA CGA GA | 210 | 2.04 | This study |
| fadA-R | AGG GTC TCG ACG GTG GTT | |||||
| fabH1 | PA0999 | fabH1-F | AGG TGC TGT CCA AGC GCA T | 211 | 2.06 | This study |
| fabH1-R | ACA TTC TCG TCG AGG AAG GTC G | |||||
| fabG | PA2967 | fabG-F | AGA TCG CCG AAA CCC TCA | 195 | 1.98 | This study |
| fabG-R | CAT CGA ACC ACT CGT CGT CT | |||||
| rhlA | PA3479 | rhlA-F | ATC ACC AAG GAC GAC GAG GT | 183 | 2.00 | This study |
| rhlA-R | GTC GAG CAT CGC CTG GTT | |||||
| rhlB | PA3478 | rhlB-F | CAC CGC GTG AGC CTC TGC ACC ATC CC | 241 | 2.06 | 22 |
| rhlB-R | CCC AGA GCG AGC CGA CCA CCA CGA TGT |
PCR efficiency (E) = 10[−1/slope].
The relative expression ratio (RER) of each target gene (T) was calculated from control (glucose) and treatment (C18 or C18-d35) CT values of the target and two reference (R1 and R2) genes at each growth stage using the Nordgård model (equation 1) (24). An effect on gene expression was considered significant when the corresponding ratios were >2 or <0.5.
| (1) |
where E is the qRT-PCR efficiency, CT is the cycle threshold, R1 is proC, R2 is rpoD, and ΔCT is the difference in the CT values between the control (glucose) and the octadecanoic acid (C18 or C18-d35) treatments for the gene transcripts.
Statistical analysis.
Statistical analyses were performed using the JMP Statistical Discovery v8.0 software (SAS Institute, Inc., Cary, NC). Treatment effects of the carbon sources on the rhamnolipid yields were evaluated by one-way analysis of variance (ANOVA) using the Tukey-Kramer honestly significant difference (HSD) test (α = 0.05). Significant differences between the means for the RER of each target gene of C18 and C18-d35 treatments were evaluated separately at each time point using a two-tailed t test (P ≤ 0.05).
RESULTS
Bacterial growth and rhamnolipid yields.
The growth rates of strain ATCC 9027 in the three treatments, glucose, C18, and C18-d35, were similar (Fig. 2), suggesting that fatty acid treatments had little effect on bacterial growth. Rhamnolipids were harvested after 96 h, and the yields were compared by one-way ANOVA using the Tukey-Kramer HSD test. The results revealed that the yields of rhamnolipid were doubled in treatments containing either octadecanoic acid or octadecanoic acid-d35 in comparison to the yield of rhamnolipid treated with glucose alone (Table 2). No significant differences were found in rhamnolipid yields between the two octadecanoic acid treatments.
Fig 2.
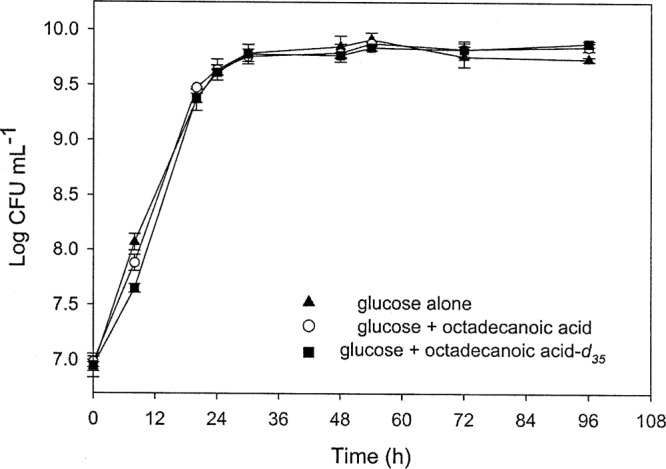
Growth of strain ATCC 9027 in MSM supplemented with 1.25% glucose alone, 1% glucose plus 0.25% octadecanoic acid, or 1% glucose plus 0.25% octadecanoic acid-d35.
Table 2.
The effects of octadecanoic acid and octadecanoic acid-d35 on rhamnolipid yields at 96 h
| Substrate | Rhamnolipid yielda (g/liter) | Molar carbon content yieldb (g/mol substrate carbon) |
|---|---|---|
| 1.25% glucose alone | 1.0 ± 0.07 a | 2.4 |
| 1% glucose plus 0.25% octadecanoic acid | 2.1 ± 0.41 b | 4.3 |
| 1% glucose plus 0.25% octadecanoic acid-d35 | 2.6 ± 0.26 b | 5.5 |
Values reported are the means of triplicate flasks ± the SD of the mean. Significant treatment differences, indicated by different numbers of letters, were determined by one-way ANOVA using the Tukey-Kramer HSD test to compare means for experiments with different carbon sources (α = 0.05).
Molar carbon content yield is the (mass of rhamnolipid produced)/(moles of substrate carbon added).
Rhamnolipid characterization by mass spectrometry.
The distributions of the major monorhamnolipid species harvested from strain ATCC 9027 grown in each of the three treatments can be ascertained from ESI-LCQ mass spectrometry. These data are shown in Fig. 3a to c and tabulated in Table 3. It is noted that the mass spectral data shown here reflect both contributions from deuterium additions and contributions from natural-abundance 13C atoms in the chains, although the relative abundance values reported in Table 3 have been corrected for the latter.
Fig 3.

Negative-ion ESI-LCQ-MS of the monorhamnolipid mixture harvested from P. aeruginosa strain ATCC 9027 grown in 1.25% glucose (a), 1% glucose plus 0.25% octadecanoic acid (b), and 1% glucose plus 0.25% octadecanoic acid-d35 (c).
Table 3.
Ion assignments, obtained from ESI-LCQ-MS, of monorhamnolipid species produced by strain ATCC 9027 grown in one of three carbon sources
| Carbon source | Major species observed | Observed ion assignment (m/z) | Relative abundancea | Fragment ions observed from MS/MS (m/z) |
|---|---|---|---|---|
| 1.25% glucose | [(Rha-C10-C8)—H]−, [(Rha-C8-C10)—H]− | 475 | 7 | 333, 305, 169, 163, 141 |
| [(Rha-C10-C10)—H]− | 503 | 100 | 333, 169, 163 | |
| [(Rha-C10-C12:1)—H]−, [(Rha-C12:1-C10)—H]− | 529 | 10 | ||
| [(Rha-C10-C12)—H]−, [(Rha-C12-C10)—H]− | 531 | 16 | ||
| 1% glucose plus 0.25% octadecanoic acid | [(Rha-C10-C8)—H]−, [(Rha-C8-C10)—H]− | 475 | 4 | |
| [(Rha-C10-C10)—H]− | 503 | 100 | ||
| [(Rha-C10-C12:1)—H]−, [(Rha-C12:1-C10)—H]− | 529 | 9 | ||
| [(Rha-C10-C12)—H]−, [(Rha-C12-C10)—H]− | 531 | 16 | ||
| 1% glucose plus 0.25% octadecanoic acid-d35 | [(Rha-C10-C8)—H]−, [(Rha-C8-C10)—H]− | 475 | 9 | |
| {[(Rha-C10-C8)-d1]—H}−, {[(Rha-C8-C10)-d1]—H}− | 476 | 4 | 305, 170, 169, 163, 142, 141 | |
| {[(Rha-C10-C8)-d15]—H}−, {[(Rha-C8-C10)-d15]—H}− | 490 | 2 | 305, 184, 163, 141 | |
| [(Rha-C10-C10)—H]− | 503 | 100 | ||
| {[(Rha-C10-C10)-d1]—H}− | 504 | 59 | 340, 334, 333, 170, 169, 163 | |
| {[(Rha-C10-C10)-d2]—H}− | 505 | 36 | ||
| {[(Rha-C10-C10)-d15]—H}− | 518 | 53 | 518, 354, 348, 333, 184, 169, 163 | |
| {[(Rha-C10-C10)-d16]—H}− | 519 | 42 | 519, 355, 348, 334, 184, 170, 169, 163 | |
| {[(Rha-C10-C10)-d17]—H}− | 520 | 20 | ||
| [(Rha-C10-C12:1)—H]−, [(Rha-C12:1-C10)—H]− | 529 | 9 | ||
| [(Rha-C10-C12)—H]−, [(Rha-C12-C10)—H]− | 531 | 22 | ||
| {[(Rha-C10-C12)-d1]—H}−, {[(Rha-C12-C10)-d1]—H}− | 532 | 15 | ||
| {[(Rha-C10-C10)-d30]—H}− | 533 | 26 | 369, 348, 184, 163 | |
| {[(Rha-C10-C10)-d31]—H}− | 534 | 23 | ||
| {[(Rha-C10-C12:1)-d15]—H}−, {[(Rha-C12:1-C10)-d15]—H}− | 544 | 3 | ||
| {[(Rha-C10-C12)-d15]—H}−, {[(Rha-C12-C10)-d15]—H}− | 546 | 9 | ||
| {[(Rha-C10-C12)-d16]—H}−, {[(Rha-C12-C10)-d16] —H}− | 547 | 7 | ||
| {[(Rha-C10-C12)-d17]—H}−, {[(Rha-C12-C10)-d17]—H}− | 548 | 5 | ||
| {[(Rha-C10-C12)-d30]—H}−, {[(Rha-C12-C10)-d30]—H}− | 561 | 3 | ||
| {[(Rha-C10-C12)-d31]—H}−, {[(Rha-C12-C10)-d31]—H}− | 562 | 6 |
Relative abundances have been corrected for the expected contributions of naturally occurring 13C species in the chains.
Monorhamnolipids that were harvested from strain ATCC 9027 grown in either the glucose or C18 treatments exhibited identical spectra, each with four major monorhamnolipid species: [(Rha-C10-C8)—H]−/[(Rha-C8-C10)—H]−, [(Rha-C10-C10)—H]−, [(Rha-C10-C12:1)—H]−/[(Rha-C12:1-C10)—H]−, and [(Rha-C10-C12)—H]−/[(Rha-C12-C10)—H]− at m/z 475, 503, 529, and 531, respectively. For the C18-d35 treatment, the same four major congener ions were observed. Additionally, however, the {[(Rha-C10-C10)-d1, -d2, -d15, -d16, -d17, -d30, -d31]—H}− isotopologues were observed at m/z 504, 505, 518, 519, 520, 533, and 534, respectively. Similarly, the {[(Rha-C10-C12:1)/(Rha-C12:1-C10)-d1, -d2, -d15, -d16, -d17, -d30, -d31]—H}− and {[(Rha-C10-C12)/(Rha-C12-C10)-d1, -d2, -d15, -d16, -d17, -d30, -d31]—H}− isotopologues were observed at their corresponding m/z values. In contrast, only the {[(Rha-C10-C8)/(Rha-C8-C10)-d1, -d2, -d15]—H}− isotopologues were observed at m/z 476, 477, and 490, respectively. The presence of a mixture of nondeuterated and deuterated monorhamnolipids harvested from the C18-d35 treatment suggests that both glucose and octadecanoic acid-d35 were involved in the rhamnolipid biosynthesis pathway.
The selected parent ions [M—H]− were analyzed further to identify the positions of the incorporated deuteriums. Negative-ion Fourier transform mass spectrometry (FTMS)-QCID spectra for [(Rha-C10-C8)—H]−/[(Rha-C8-C10)—H]−, {[(Rha-C10-C8)-d1]—H]}−/{[(Rha-C8-C10)-d1]—H]}−, and {[(Rha-C10-C8)-d15]—H}−/{[(Rha-C8-C10)-d15]—H}− at m/z 475, 476, and 490, respectively, are shown in Fig. 4a to c. The selection and fragmentation of m/z 475 (Fig. 4a) resulted in five daughter ions at m/z 333, 305, 169, 163, and 141; the ions at m/z 333 and 305 corresponded to [(Rha-C10)—H]− and [(Rha-C8)—H]− resulting from the loss of the terminal C8 and C10 chains, respectively. The [Rha—H]−, [C8—H]−, and [C10—H]− chain fragment ions were observed at m/z 163, 141, and 169, respectively. The relative intensities of the peaks at m/z 305 and 333 suggest that the Rha-C8-C10 congener is about twice as abundant as the Rha-C10-C8 congener.
Fig 4.

Negative-ion FTMS QCID of [(Rha-C10-C8)—H]− at m/z 475 (a), {[(Rha-C10-C8)-d1—H}− at m/z 476 (b), and {[(Rha-C10-C8)-d15]—H}− at m/z 490 (c) from a monorhamnolipid mixture harvested from strain ATCC 9027 grown in 1% glucose plus 0.25% octadecanoic acid-d35. Asterisks denote ions corresponding to chemical background noise.
The fragmentation of {[(Rha-C10-C8)-d1]—H]}−/{[(Rha-C8-C10)-d1]—H]}− at m/z 476 (Fig. 4b) resulted in six ions at m/z 305, 170, 169, 163, 142, and 141. Although of low signal-to-noise ratios, additional peaks at 334 and 306 were also observed. Ions at m/z 169 and 170 corresponded to the [C10—H]− and [(C10-d1)—H]− species, respectively. Similarly, m/z 141 and 142 corresponded to the [C8—H]− and [(C8-d1)—H]− species, respectively. The low-intensity peaks at 305 and 306 are attributed to the [(Rha-C8)—H]− and {[(Rha-C8)-d1]—H}− ions, while at 334, that is assigned to the corresponding {[(Rha-C10)-d1]—H}− species. The location of the single site of deuteration within the C8 and C10 chains could not be definitively determined; however, this single deuterium was incorporated equally into both chains based on the identical intensity ratios of the m/z 142 to m/z 141 ions for the [(C8-d1)—H]− to [C8—H]− and the m/z 170 to m/z 169 ions for the [(C10-d1)—H]− to [C10—H]− species, respectively.
The fragmentation of {[(Rha-C10-C8)-d15]—H}−/{[(Rha-C8-C10)-d15]—H}− at m/z 490 (Fig. 4c) resulted in five ions at 348, 305, 184, 163, and 141, although the m/z 348 peak was almost buried in the noise, because the (Rha-C10-C8)-d15 congener is expected to be only about half as abundant as the (Rha-C8-C10)-d15 congener, as noted above. Also particularly noteworthy about this spectrum was the absence of a peak at m/z 169 that would be assigned to the [C10—H]− ion. The only C10 chain peak observed was at m/z 184, corresponding to [(C10-d15)—H]−. Thus, unlike the {[(Rha-C10-C8)-d1]—H]}−/{[(Rha-C8-C10)-d1]—H]}− isotopologues, in which the single deuterium is incorporated equally into both chains, the 15 deuterium atoms are incorporated solely into the C10 chain. This assertion is substantiated further by the absence of any ions associated with C8 chains that contain deuterium atoms. The only C8-containing ions observed were the [C8—H]− at m/z 141 and the [(Rha-C8)—H]− at m/z 305.
As shown in Fig. 5a, the fragmentation of nondeuterated [(Rha-C10-C10)—H]− at m/z 503 resulted in four ions at m/z 163, 169, 333, and 339 corresponding to [Rha—H]−, [C10—H]−, [(Rha-C10)—H]−, and [(C10-C10)—H]− fragments, the latter ion resulting from the loss of a neutral rhamnose from the parent ion. The fragmentation of the deuterated analogue {[(Rha-C10-C10)-d1]—H}− at m/z 504 (Fig. 5b) indicates that the single deuterium is distributed equally between the two C10 chains as for {[(Rha-C10-C8)-d1]—H]}−, as shown by the equal ion intensity of the [(Rha-C10)—H]− and {[(Rha-C10)-d1]—H}− ions at m/z 333 and 334, respectively. In addition, peaks at 340, 170, and 169 indicate the presence of the {[(C10-C10)-d1]—H}−, [(C10-d1)—H]−, and [C10—H]− ions, respectively. One important point to note, however, is the unequal intensities of the m/z 169 and 170 ions; these peaks would be expected to be of equal intensities for equal distributions of the single deuterium between the two C10 chains. The fact that they were not suggests some contribution from the m/z 503 species to this spectrum, which is consistent with mass selection to only a precision of ∼0.5 to 1 mass units in our system.
Fig 5.

Negative-ion FTMS QCID of [(Rha-C10-C10)—H]− at m/z 503 (a), {[(Rha-C10-C10)-d1]—H}− at m/z 504 (b), {[(Rha-C10-C10)-d15]—H}− at m/z 518 (c), {[(Rha-C10-C10)-d16]—H}− at m/z 519 (d), and {[(Rha-C10-C10)-d30]—H]}− at m/z 533 (e) from a monorhamnolipid mixture harvested from strain ATCC 9027 grown in 1% glucose plus 0.25% octadecanoic acid-d35.
The fragmentation of {[(Rha-C10-C10)-d15]—H}− at m/z 518 (Fig. 5c) resulted in several ions, the most notable being the m/z 184 species, which is 15 mass units higher than the m/z 169 species. This species is due to the [(C10-d15)—H]− ion; along with the observation of the [C10—H]− at m/z 169, its presence clearly indicates the incorporation of the 15 deuterium atoms into a single C10 chain. These assignments are further supported by observation of the {[(C10-C10)-d15]—H}− at m/z 354, the {[(Rha-C10)-d15]—H}− at m/z 348, and the [(Rha-C10)—H]− at m/z 333.
The fragmentation of {[(Rha-C10-C10)-d16]—H}− at m/z 519 (Fig. 5d) showed the absence of an ion at m/z 185, which would be expected if [(C10-d16)—H]− ions were present, and the presence of an m/z 170 peak was due to the [(C10-d1)—H]− ion. These observations indicate that the 16th deuterium is not incorporated into the C10-d15 chain but, rather, is incorporated into the second C10 chain. Peaks at m/z 355, 348, and 334 due to the {[(C10-C10)-d16]—H}−, {[(Rha-C10)-d15]—H}−, and {[(Rha-C10)-d1]—H}− ions, respectively, further support this conclusion. Also of significant abundance in this spectrum was a peak at m/z 169 attributed to the [C10—H]− ion; this ion is clearly unexpected for the {[(Rha-C10-C10)-d16]—H}− parent ion and suggests some contribution from the m/z 518 species to this spectrum, similar to what was noted above for the m/z 504 species. Further evidence of a contribution from the m/z 518 species to this spectrum was the presence of a small peak at m/z 333 due to the [(Rha-C10)—H]− ion.
The fragmentation of the {[(Rha-C10-C10)-d30]—H}− ion at m/z 533 (Fig. 5e) showed the presence of m/z 348 and 369 ions corresponding to the {[(Rha-C10)-d15]—H}− and {[(C10-C10)-d30]—H}− species. The presence of these ions, along with the absence of an m/z 169 ion, supports the hypothesis that 15 deuterium atoms are incorporated into each of the two C10 chains for the {[(Rha-C10-C10)-d30]—H}− isotopologue.
Finally, the fragmentation behavior of the {[(Rha-C10-C10)-d17]—H}− was similar to that of the {[(Rha-C10-C10)-d16]—H}− ion, in which 15 deuteriums were incorporated into one C10 chain and the additional two deuteriums were incorporated into the second C10 chain (data not shown).
Gene expression studies.
The expressions of selected key genes involved in β-oxidation (fadA, fadD1, fadD2), de novo fatty acid synthesis (fabH1, fabG), and rhamnolipid biosynthesis (rhlA, rhlB) were monitored using quantitative reverse transcriptase PCR (qRT-PCR) to (i) investigate how octadecanoic acid influences rhamnolipid production and (ii) to look for evidence of a possible “bypass route” from β-oxidation to de novo fatty acid synthesis, which is suggested by the deuterium tracer study described above. For each gene examined, the expression patterns for each time point were compared by calculating the RER values of the target genes in the control (glucose alone) and octadecanoic acid (C18 or C18-d35) treatment samples normalized with respect to the two reference genes, proC and rpoD (equation 1).
Three β-oxidation genes were studied: fadD1, fadD2, and fadA. For fadD1, the RER values were close to or slightly less than 1.0 in all growth phases for the C18 treatment, indicating the absence of an octadecanoic acid-induced effect on this gene. The results were similar for the C18-d35 treatment except for an apparent isotopic effect in the log phase (8 h), at which time the RER of fadD1 was clearly upregulated in comparison to the nondeuterated treatment (Fig. 6A). Both the C18 and the C18-d35 treatments showed similar results for fadD2: a gradual downregulation over the course of the study resulting in a >2-fold reduction in the RER at 48 h (Fig. 6B). In contrast, the fadA gene was slightly upregulated in the early phases of growth, and expression increased to a maximum at 24 h. Specifically, fadA showed increased RER values in the log (8 h, 1.4-fold), late log (20 h, 1.6- to 2.0-fold), and early stationary (24 h, 2.5- to 3.8-fold) growth phases, followed by a return to RER values of ∼1 in the stationary (30 h) and late stationary (48 h) phases of growth (Fig. 6C). There were no significant differences (P > 0.05) in fadA expression levels between the C18 and C18-d35 treatments.
Fig 6.

Relative expression ratios of the target genes fadD1 (A), fadD2 (B), fadA (C), fabH1 (D), fabG (E), rhlA (F), and rhlB (G) at 8, 20, 24, 30, and 48 h. The control was glucose alone, and the two treatments were glucose plus octadecanoic acid (black bars) and glucose plus octadecanoic acid-d35 (gray bars). The dashed line shows an expression ratio of 1, which indicates that the effect of octadecanoic acid or octadecanoic acid-d35 on the target gene was equal to the effect on the reference genes.
There was no effect of octadecanoic acid on the expression of the de novo fatty acid synthesis gene fabH1 (Fig. 6D). The fabG gene encodes an NADPH-dependent 3-ketoacyl reductase, which is the enzyme that catalyzes the reduction of β-ketoacyl-ACP to β-hydroxyacyl-ACP, which, in turn, is the substrate for RhlA and the precursor for HAA synthesis (9, 32). The results show that the presence of octadecanoic acid did not change the fabG RER values at 8 or 20 h in the log or early stationary phase and that there was an approximate 2-fold reduction in the RER values after the stationary phase was reached, including at 24, 30, and 48 h (Fig. 6E).
For the rhamnolipid biosynthesis genes, the RER values were close to 1.0 for rhlA and rhlB throughout the log (8 h), late log (20 h), and early stationary (24 h) growth phases for both the C18 and C18-d35 treatments, indicating the absence of an octadecanoic acid-induced effect on rhlA and rhlB expression during these growth stages (Fig. 6F). However, in the stationary (30 h) and midstationary (48 h) phases, there were 2- to 3-fold and 330- to 600-fold increases, respectively, in the RER values for rhlA in the octadecanoic acid treatments (Fig. 6F). For rhlB, there was no change at 30 h but a 225- to 290-fold increase at 48 h (Fig. 6G). There were no significant differences (P > 0.05) in the RER values of either rhlA or rhlB between the C18 or C18-d35 treatments, confirming the absence of a deuterium-induced effect on gene expression.
DISCUSSION
Both de novo fatty acid synthesis and β-oxidation contribute to deuterium tracing.
Information concerning rhamnolipid biosynthesis has been derived primarily from studies using carbohydrate carbon sources which require the de novo synthesis of lipid precursors for the molecule. However, recent studies have suggested a metabolic link between fatty acid degradation and rhamnolipid biosynthesis (15, 17), and other studies have shown increased rhamnolipid yields in the presence of oils, which contain various fatty acids (23, 30, 38). Similarly, the results of this study show that octadecanoic acid as a cosubstrate increased rhamnolipid yield, and based on these results, we describe a possible physiological basis for this increase. Specifically, mass spectroscopy results suggest that when a fatty acid cosubstrate is present, de novo fatty acid synthesis and β-oxidation pathways both contribute lipid precursors for rhamnolipid biosynthesis. This conclusion derives from the observation that the deuterium distribution in a single rhamnolipid carbon chain follows one of two patterns. The first pattern is that a small number of deuterium atoms (1 or 2) are incorporated into a single carbon chain, e.g., rhamnolipid-d1 or -d2 congeners. The second pattern observed is 15 deuterium atoms incorporated into a single carbon chain.
To explore whether de novo fatty acid synthesis alone could explain these two patterns, an analysis of the potential C10 isotopologues that could be generated by de novo fatty acid synthesis was performed, assuming that the C18-d35 substrate is first subjected to β-oxidation, and the resulting acetyl-coenzyme A (CoA) and malonyl-CoA moieties then undergo de novo fatty acid synthesis. This analysis showed that isotopologues ranging from C10-d0 to C10-d8 are all possible (see Table S1 in the supplemental material). This analysis is consistent with the results shown by mass spectrometry, which identified rhamnolipid isotopologues ranging from m/z 504 to m/z 508. However, these results suggest that if only the de novo fatty acid synthesis pathway is used, the maximum possible number of deuterium atoms that could be incorporated into one single carbon chain is eight (see Table S1). Thus, the C10 isotopologue with 15 deuteriums could not come from de novo fatty acid synthesis.
We therefore suggest that the second pattern of deuterium incorporation, 15 deuterium atoms in a single carbon chain, is explained by involvement of the β-oxidation pathway. In this case, we propose that the d15 unit, C8-d15-acyl CoA, which results from β-oxidation, is recruited into the de novo fatty acid synthesis pathway by a yet-unidentified β-ketoacyl-ACP synthase, for example, a FabH-like enzyme (Fig. 1A, proposed bypass route). This would allow the C8-d15-acyl CoA to be drained directly from the β-oxidation pathway into de novo fatty acid synthesis, followed by the addition of malonyl-ACP to produce C10-d15 β-ketoacyl-ACP. This in turn would be converted to C10-d15 β-hydroxyacyl-ACP, the substrate recruited by the RhlA enzyme to make HAA and then rhamnolipid (Fig. 1A). A C12-d15 β-hydroxyacyl-ACP could be synthesized in a similar manner by leaving the C10-d15 β-hydroxyacyl-ACP in the de novo fatty acid synthesis cycle for one more round. Thus, when examining the synthesized rhamnolipid congeners, it would be expected that both the Rha-C10-d15 and the Rha-C12-d15 lipid chains would be observed, but there would be no Rha-C8-d15 lipid chains.
This proposed pathway is supported by examination of the four major rhamnolipid congeners observed in this study: Rha-C10-C8, Rha-C10-C10, Rha-C10-C12:1, and Rha-C10-C12. In the presence of octadecanoic acid-d35, the proposed pathway would result in the formation of Rha-C10-C10-d15 (one chain labeled) or Rha-C10-C10-d30 (both chains labeled), Rha-C10-C12:1-d15 (one chain labeled) or Rha-C10-C12:1-d30 (both chains labeled), and Rha-C10-C12-d15 (one chain labeled) or Rha-C12-C12-d30 (both chains labeled). In contrast, for Rha-C8-C10, only the Rha-C8-C10-d15 would be observed because only the C10 chain can be d15 labeled. These results are consistent with our mass spectrometry results (Fig. 3 to 5). We speculate, based on these results, that the enzyme(s) involved in the “bypass route” has the highest affinity for the C8-acyl-CoA β-oxidation intermediate. Hori et al. (15) recently suggested a different bypass route from β-oxidation to de novo synthesis. In their study, fatty acids with C7 to C12 carbon chains were used as growth substrates, and it was proposed that the bypass works by directly converting the 3-ketoacyl-CoA β-oxidation intermediate to a 3-ketoacyl-ACP. The deuterium-tracing results from this study help to clarify, and suggest specifically, that it is the C8-acyl-CoA β-oxidation intermediate that is utilized. This is supported by recent evidence showing that P. aeruginosa can directly shunt C8-acyl-CoA into the fatty acid synthesis pathway (39).
Gene expression studies of fatty acid degradation genes.
Two of the three β-oxidation pathway genes tested (fadD1, fadD2, fadA) showed expression differences in the C18 treatments in comparison to treatment with glucose alone. This, combined with the deuterium tracer results, suggests that these genes serve two purposes when octadecanoic acid is present: first, to process fatty acid substrates for energy production and cell growth, and second, to supply fatty acid intermediates for rhamnolipid biosynthesis. These genes were chosen for analysis based on the hypothesis that the d15 unit is drained directly from the β-oxidation pathway as an acyl-CoA intermediate and because it is known that FadD and FadA are directly involved in the reactions producing acyl-CoA intermediates that could then be diverted to the de novo fatty acid synthesis pathway (8).
The fadD gene encodes an acyl-CoA-synthetase which activates fatty acid substrates for β-oxidation by the addition of coenzyme A (Fig. 1). The pathway and associated fad genes for fatty acid β-oxidation have been well characterized in Escherichia coli, but less is known about this system in P. aeruginosa. One difference is that P. aeruginosa has 6- to 8-fold more genes involved in fatty acid catabolism than does E. coli (36). Pseudomonas aeruginosa has two fadD genes, fadD1 and fadD2, and it has been reported that they can be cotranscribed on a single larger transcript or as smaller independent transcripts (17). While both genes can use fatty acid substrates ranging from C4 to C18:1, it has been reported that fadD1 is induced most strongly by C18:1 followed by C10:0, while fadD2 was induced very strongly by C8:0 and C10:0 fatty acids (17). The disruption of the fadD2 gene was also shown to decrease the production of virulence factors, including rhamnolipids, suggesting that it is necessary for normal rhamnolipid production (17). Our results did not show a significant upregulation of either the fadD gene in the early stages of growth and actually showed a pattern of downregulation of the fadD genes over the course of the study, particularly for fadD2. It is not yet clear why the fadD2 gene is downregulated in the C18 treatment but not in glucose alone in the later stages of growth.
The third β-oxidation pathway gene examined, fadA, encodes a 3-ketoacyl-CoA thiolase, which participates in β-oxidation by catalyzing the removal of acetyl-CoA from 3-ketoacyl-CoA; this results in a shortening of the parent acyl-CoA by two carbons (Fig. 1) (8). The addition of octadecanoic acid caused a strong increase in the expression of this gene at 20 and 24 h coinciding with the early stationary phase (Fig. 2) and the onset of rhamnolipid production. By 30 h, this effect had disappeared. The increased expression of fadA in the early stages of growth suggests that the addition of octadecanoic acid results in activation of the β-oxidation pathway to provide intermediates for both cell growth and eventual recruitment into the rhamnolipid biosynthesis pathway. Further examination of the role of the fadD gene and other β-oxidation genes is warranted.
Gene expression studies—de novo fatty acid synthesis.
Two genes, fabH1 and fabG, involved in de novo fatty acid synthesis were chosen to begin examining a possible bypass route between β-oxidation and de novo synthesis (Fig. 1). These genes were chosen to examine whether FabH is involved in catalyzing the condensation reaction of malonyl-ACP with the octanoyl-CoA-d15 intermediate of β-oxidation to form β-ketodecanoyl k-ACP-d15. This would be followed by a reduction reaction catalyzed by FabG to form β-hydroxydecanoyl-ACP-d15.
In E. coli, the FabH enzyme catalyzes the condensation of malonyl-ACP with acetyl-CoA to form β-ketoacyl-ACP, which initiates de novo fatty acid synthesis (Fig. 5) (31). However, for other bacteria, e.g., Mycobacterium tuberculosis, FabH catalyzes the condensation of longer fatty acyl-CoAs (e.g., octanoyl-CoA) with malonyl-ACP (42). We hypothesized that in P. aeruginosa, the fabH1 gene might be a candidate to use for examining the diversion of octanoyl-CoA-d15 from β-oxidation to de novo fatty acid synthesis. However, octadecanoic acid did not affect the expression of the fabH1 gene in this study. It should be noted that the FabH homologs in P. aeruginosa have been only tentatively identified; thus, other FabH homologs need to be investigated for this role in future studies. A recent paper suggested that FabY, a new class of β-ketoacyl ACP synthases, can catalyze the initial cycle of fatty acid synthesis in P. aeruginosa, indicating that other genes encoding a β-ketoacyl ACP synthase may also needed to be investigated for their roles in rhamnolipid biosynthesis (40).
There was a reduction in the expression of the fabG gene at 20 to 24 h. This suggests that the bypass route does not use FabG to catalyze the conversion of the β-ketodecanoyl-ACP-d15 to the β-hydroxydecanoyl-ACP-d15. However, a consequence of this downregulation of fabG would be to slow biosynthesis of long-chain fatty acids, resulting in the accumulation of short-chain fatty acids that could be used for the synthesis of other molecules (14). One such molecule is the quorum-sensing signal N-butanoyl–l-homoserine lactone (C4-HSL), which activates the expression of the rhlAB operon (16). As discussed below, there is a large upregulation of this operon in the stationary phase.
Though fabG was downregulated, a FabG-like enzyme activity is needed to convert β-ketoacyl-ACP to the β-hydroxyacyl-ACP substrate for RhlA (Fig. 1). We propose that there must be an alternative enzyme that has not yet been identified with NADPH-dependent β-ketoacyl-ACP reductase activity that participates in supplying the substrate for RhlA when fatty acids are present in the medium. As an example, one possibility is RhlG, which has been reported to be a FabG homologue (4), although several studies since this report have shown that RhlG is not necessary for rhamnolipid production (21). The expression of the rhlG gene was therefore tested, but octadecanoic acid addition reduced the expression of this gene after 20 h and through the stationary phase (data not shown). Thus, it is likely that there is another yet-unidentified enzyme with β-ketoacyl-ACP reductase activity that is used in the bypass route.
Gene expression studies of rhlA and rhlB.
Octadecanoic acid had a large effect on expressions of the rhlA and rhlB genes beginning at 30 h, suggesting that the enhanced yield of rhamnolipid in this treatment results, at least in part, from the upregulation of rhlA and rhlB. One possible mechanism by which rhlA and rhlB could be upregulated by octadecanoic acid is suggested by recent work that showed an interaction between long-chain fatty acids, specifically oleate (C18:1Δ9), and the P. aeruginosa PsrA autorepressor (16). This study showed that oleate binds to PsrA, resulting in decreased expression of the rpoS gene from the middle-log to early stationary phase. The rpoS gene encodes a stationary-phase sigma factor, RpoS, which has been shown to regulate quorum sensing, virulence, and several hundred stationary-phase genes (34). Of specific interest here is that the decreased expression of rpoS resulted in an increase in the production of the quorum-sensing signal N-butanoyl–l-homoserine lactone (C4-HSL) (16), which in turn binds to its transcriptional regulator, RhlR, and activates the expression of the rhlAB operon (20). Thus, the observed increase in the RER values of the rhlA and rhlB for the C18 and C18-d35 treatments may be partly due to a long-chain fatty acid-induced increase in production of the C4-HSL signal.
In summary, rhamnolipid yields were doubled when octadecanoic acid was added as a cosubstrate with glucose. The deuterium tracer results presented in the current study suggest that there is a “bypass route” through which an octanoyl-CoA intermediate of β-oxidation is incorporated into rhamnolipids without de novo fatty acid biosynthesis. The bypass route was induced when octadecanoic acid was added as a cosubstrate, and qRT-PCR results suggest that production of β-oxidation pathway enzymes, including FadD2 and FadA, is regulated to generate intermediates that are then recruited for rhamnolipid biosynthesis. This is the first demonstration that both de novo fatty acid synthesis and β-oxidation can provide precursors for rhamnolipid biosynthesis. An insight from the bypass route is that P. aeruginosa is able to choose the most efficient way to synthesize rhamnolipids depending on the substrates present in the growth environment. Thus, in the presence of octadecanoic acid, P. aeruginosa did not completely catabolize the fatty acid but rather partially catabolized it into intermediates of the correct chain length to use in rhamnolipid production.
Supplementary Material
ACKNOWLEDGMENT
This work was supported by grant CHE-0714245 from the National Science Foundation Collaborative Research in Chemistry program.
Footnotes
Published ahead of print 5 October 2012
Supplemental material for this article may be found at http://aem.asm.org/.
REFERENCES
- 1. Banat IM, Makkar RS, Cameotra SS. 2000. Potential commercial applications of microbial surfactants. Appl. Microbiol. Biotechnol. 53:495–508 [DOI] [PubMed] [Google Scholar]
- 2. Boles BR, Thoendel M, Singh PK. 2005. Rhamnolipids mediate detachment of Pseudomonas aeruginosa from biofilms. Mol. Microbiol. 57:1210–1223 [DOI] [PubMed] [Google Scholar]
- 3. Caiazza NC, Shanks RMQ, O'Toole GA. 2005. Rhamnolipids modulate swarming motility patterns of Pseudomonas aeruginosa. J. Bacteriol. 187:7351–7361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Campos-Garcia J, et al. 1998. The Pseudomonas aeruginosa rhlG gene encodes an NADPH-dependent beta-ketoacyl reductase which is specifically involved in rhamnolipid synthesis. J. Bacteriol. 180:4442–4451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. de Kievit TR. 2009. Quorum sensing in Pseudomonas aeruginosa biofilms. Environ. Microbiol. 11:279–288 [DOI] [PubMed] [Google Scholar]
- 6. Déziel E, et al. 1999. Liquid chromatography/mass spectrometry analysis of mixtures of rhamnolipids produced by Pseudomonas aeruginosa strain 57RP grown on mannitol or naphthalene. Biochim. Biophys. Acta 1440:244–252 [DOI] [PubMed] [Google Scholar]
- 7. Déziel E, Lépine F, Milot S, Villemur R. 2003. rhlA is required for the production of a novel biosurfactant promoting swarming motility in Pseudomonas aeruginosa: 3-(3-hydroxyalkanoyloxy)alkanoic acids (HAAs), the precursors of rhamnolipids. Microbiology 149:2005–2013 [DOI] [PubMed] [Google Scholar]
- 8. Fujita Y, Matsuoka H, Hirooka K. 2007. Regulation of fatty acid metabolism in bacteria. Mol. Microbiol. 66:829–839 [DOI] [PubMed] [Google Scholar]
- 9. Haba E, et al. 2003. Physicochemical characterization and antimicrobial properties of rhamnolipids produced by Pseudomonas aeruginosa 47T2 NCBIM 40044. Biotechnol. Bioeng. 81:316–322 [DOI] [PubMed] [Google Scholar]
- 10. Hauser G, Karnovsky ML. 1954. Studies on the production of glycolipide by Pseudomonas aeruginosa. J. Bacteriol. 68:645–654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hauser G, Karnovsky ML. 1957. Rhamnose and rhamnolipide biosynthesis by Pseudomonas aeruginosa. J. Biol. Chem. 224:91–105 [PubMed] [Google Scholar]
- 12. Hauser G, Karnovsky ML. 1958. Studies on the biosynthesis of l-rhamnose. J. Biol. Chem. 233:287–291 [PubMed] [Google Scholar]
- 13. Heurlier K, et al. 2004. Positive control of swarming, rhamnolipid synthesis, and lipase production by the posttranscriptional RsmA/RsmZ system in Pseudomonas aeruginosa PAO1. J. Bacteriol. 186:2936–2945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hoang TT, Sullivan SA, Cusick JK, Schweizer HP. 2002. β-Ketoacyl acyl carrier protein reductase (FabG) activity of the fatty acid biosynthetic pathway is a determining factor of 3-oxo-homoserine lactone acyl chain lengths. Microbiology 148:3849–3856 [DOI] [PubMed] [Google Scholar]
- 15. Hori K, Ichinohe R, Unno H, Marsudi S. 2011. Simultaneous syntheses of polyhydroxyalkanoates and rhamnolipids by Pseudomonas aeruginosa IFO3924 at various temperatures and from various fatty acids. Biochem. Eng. J. 53:196–202 [Google Scholar]
- 16. Kang Y, et al. 2009. The long-chain fatty acid sensor, PsrA, modulates the expression of rpoS and the type III secretion exsCEBA operon in Pseudomonas aeruginosa. Mol. Microbiol. 73:120–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kang Y, Zarzycki-Siek J, Walton CB, Norris MH, Hoang TT. 2010. Multiple FadD acyl-CoA synthetases contribute to differential fatty acid degradation and virulence in Pseudomonas aeruginosa. PLoS One 5:e13557 doi:10.1371/journal.pone.0013557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lebrón-Paler A, et al. 2006. Determination of the acid dissociation constant of the biosurfactant monorhamnolipid in aqueous solution by potentiometric and spectroscopic methods. Anal. Chem. 78:7649–7658 [DOI] [PubMed] [Google Scholar]
- 19. Lovaglio RB, dos Santos FJ, Jafelicci M, Contiero J. 2011. Rhamnolipid emulsifying activity and emulsion stability: pH rules. Colloids Surf. B Biointerfaces 85:301–305 [DOI] [PubMed] [Google Scholar]
- 20. Maier RM, Soberon-Chavez G. 2000. Pseudomonas aeruginosa rhamnolipids: biosynthesis and potential applications. Appl. Microbiol. Biotechnol. 54:625–633 [DOI] [PubMed] [Google Scholar]
- 21. Miller DJ, Zhang YM, Rock CO, White SW. 2006. Structure of RhlG, an essential β-ketoacyl reductase in the rhamnolipid biosynthetic pathway of Pseudomonas aeruginosa. J. Biol. Chem. 281:18025–18032 [DOI] [PubMed] [Google Scholar]
- 22. Neilson JW, et al. 2010. Cadmium effects on transcriptional expression of rhlB/rhlC genes and congener distribution of monorhamnolipid and dirhamnolipid in Pseudomonas aeruginosa IGB83. Appl. Microbiol. Biotechnol. 88:953–963 [DOI] [PubMed] [Google Scholar]
- 23. Nitschke M, Costa SGVAO, Contiero J. 2005. Rhamnolipid surfactants: an update on the general aspects of these remarkable biomolecules. Biotechnol. Prog. 21:1593–1600 [DOI] [PubMed] [Google Scholar]
- 24. Nordgard O, Kvaloy JT, Farmen RK, Heikkila R. 2006. Error propagation in relative real-time reverse transcription polymerase chain reaction quantification models: the balance between accuracy and precision. Anal. Biochem. 356:182–193 [DOI] [PubMed] [Google Scholar]
- 25. Ochsner UA, Fiechter A, Reiser J. 1994. Isolation, characterization, and expression in Escherichia coli of the Pseudomonas aeruginosa rhlAB genes encoding a rhamnosyltransferase involved in rhamnolipid biosurfactant synthesis. J. Biol. Chem. 269:19787–19795 [PubMed] [Google Scholar]
- 26. Ochsner UA, Reiser J. 1995. Autoinducer-mediated regulation of rhamnolipid biosurfactant synthesis in Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. U. S. A. 92:6424–6428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pearson JP, Pesci EC, Lglewshi BH. 1997. Roles of Pseudomonas aeruginosa las and rhl quorum-sensing systems in control of elastase and rhamnolipid biosynthesis genes. J. Bacteriol. 179:5756–5767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pfaffl MW. 2001. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29:e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rahim R, et al. 2001. Cloning and functional characterization of the Pseudomonas aeruginosa rhlC gene that encodes rhamnosyltransferase 2, an enzyme responsible for di-rhamnolipid biosynthesis. Mol. Microbiol. 40:708–718 [DOI] [PubMed] [Google Scholar]
- 30. Robert M, et al. 1989. Effect of the carbon source on biosurfactant production by Pseudomonas aeruginosa 44T1. Biotechnol. Lett. 11:871–874 [Google Scholar]
- 31. Rock CO, Jackowski S. 2002. Forty years of bacterial fatty acid synthesis. Biochem. Biophys. Res. Commun. 292:1155–1166 [DOI] [PubMed] [Google Scholar]
- 32. Rozen S, Skaletsky HJ. 2000. Primer3 on the WWW for general users and for biologist programmers, p 365–386 In Krawetz S, Misener S. (ed), Bioinformatics methods and protocols: methods in molecular biology. Humana Press, Totowa, NJ: [DOI] [PubMed] [Google Scholar]
- 33. Savli H, et al. 2003. Expression stability of six housekeeping genes: a proposal for resistance gene quantification studies of Pseudomonas aeruginosa by real-time quantitative RT-PCR. J. Med. Microbiol. 52:403–408 [DOI] [PubMed] [Google Scholar]
- 34. Schuster M, Hawkins AC, Harwood CS, Greenberg EP. 2004. The Pseudomonas aeruginosa RpoS regulon and its relationship to quorum sensing. Mol. Microbiol. 51:973–985 [DOI] [PubMed] [Google Scholar]
- 35. Son MS, Matthews WJ, Jr, Kang Y, Nguyen DT, Hoang TT. 2007. In vivo evidence of Pseudomonas aeruginosa nutrient acquisition and pathogenesis in the lungs of cystic fibrosis patients. Infect. Immun. 75:5313–5324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Son MS, Nguyen DT, Kang Y, Hoang TT. 2008. Engineering of FRT-lacZ fusion constructs: induction of the Pseudomonas aeruginosa fadAB1 operon by medium and long chain-length fatty acids. Plasmid 59:111–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Stanier RY, Palleroni NJ, Doudoroff M. 1966. The aerobic pseudomonads: a taxonomic study. J. Gen. Microbiol. 43:159–271 [DOI] [PubMed] [Google Scholar]
- 38. Trummler K, Effenberger F, Syldatk C. 2003. An integrated microbial/enzymatic process for production of rhamnolipids and l-(+)-rhamnose from rapeseed oil with Pseudomonas sp. DSM 2874. Eur. J. Lipid Sci. Technol. 105:563–571 [Google Scholar]
- 39. Yuan Y, Leeds JA, Meredith TC. 2012. Pseudomonas aeruginosa directly shunts β-oxidation degradation intermediates into de novo fatty acid biosynthesis. J. Bacteriol. 194:5185–5196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yuan Y, Sachdeva M, Leeds JA, Meredith TC. 2012. Fatty acid biosynthesis in Pseudomonas aeruginosa is initiated by the FabY class of β-ketoacyl acyl carrier protein synthases. J. Bacteriol. 194:5171–5184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhang Y, Miller RM. 1992. Enhanced octadecane dispersion and biodegradation by a Pseudomonas rhamnolipid surfactant (biosurfactant). Appl. Environ. Microbiol. 58:3276–3282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhu K, Rock CO. 2008. RhlA converts β-hydroxyacyl-acyl carrier protein intermediates in fatty acid synthesis to the β-hydroxydecanoyl-β-hydroxydecanoate component of rhamnolipids in Pseudomonas aeruginosa. J. Bacteriol. 190:3147–3154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zulianello L, et al. 2006. Rhamnolipids are virulence factors that promote early infiltration of primary human airway epithelia by Pseudomonas aeruginosa. Infect. Immun. 74:3134–3147 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.