

Abstract
In this study, we investigated the metabolism of ethylene glycol in the Pseudomonas putida strains KT2440 and JM37 by employing growth and bioconversion experiments, directed mutagenesis, and proteome analysis. We found that strain JM37 grew rapidly with ethylene glycol as a sole source of carbon and energy, while strain KT2440 did not grow within 2 days of incubation under the same conditions. However, bioconversion experiments revealed metabolism of ethylene glycol by both strains, with the temporal accumulation of glycolic acid and glyoxylic acid for strain KT2440. This accumulation was further increased by targeted mutagenesis. The key enzymes and specific differences between the two strains were identified by comparative proteomics. In P. putida JM37, tartronate semialdehyde synthase (Gcl), malate synthase (GlcB), and isocitrate lyase (AceA) were found to be induced in the presence of ethylene glycol or glyoxylic acid. Under the same conditions, strain KT2440 showed induction of AceA only. Despite this difference, the two strains were found to use similar periplasmic dehydrogenases for the initial oxidation step of ethylene glycol, namely, the two redundant pyrroloquinoline quinone (PQQ)-dependent enzymes PedE and PedH. From these results we constructed a new pathway for the metabolism of ethylene glycol in P. putida. Furthermore, we conclude that Pseudomonas putida might serve as a useful platform from which to establish a whole-cell biocatalyst for the production of glyoxylic acid from ethylene glycol.
INTRODUCTION
Ethylene glycol is an important starting material for many applications in the chemical industry and is readily available in large quantities at low cost. Besides its application in antifreeze solutions, it is used for the chemical synthesis of glyoxal, glycolic acid, and glyoxylic acid. Glyoxylic acid serves as an important building block for the synthesis of many molecules of industrial interest. It is used for the production of agrochemicals, cosmetic ingredients, polymers, and pharmaceuticals, as well as for flavors and fragrance products. Different routes for the chemical synthesis of glyoxylic acid have been developed (29, 34). Besides the formation of by-products, all these chemical approaches share the major drawback of low yields. Industrial biotechnology using biocatalytic-based approaches provides an interesting alternative to overcome these difficulties. Examples for the enzymatic production of glyoxylic acid are the conversion of glycolic acid by glycolic acid oxidase from spinach, selectively yielding as much as 98% glyoxylic acid in a whole-cell system. Further, a two-step chemoenzymatic route using base catalysis and glycolic acid oxidase for the synthesis of glyoxylic acid from glyoxal, as well as the synthesis of glycolic acid by using glycerol oxidase from Aspergillus japonicus, has been described (15, 21, 39).
An attractive alternative to these procedures is combined one-step whole-cell biotransformation using ethylene glycol as a readily available and economically priced substrate. Various organisms that can utilize ethylene glycol as a sole source of carbon and energy under oxic as well as anoxic conditions have been described (13, 14). Metabolic sequential oxidation of ethylene glycol into glyoxylic acid via the intermediate glycolic acid has been shown in a variety of different bacterial species (8, 11, 17, 23). These reactions can be catalyzed either by dehydrogenases, by oxidases, or by a combination of these enzymes (6, 21, 22). Among the alcohol dehydrogenases (ADHs), the pyrroloquinoline quinone (PQQ)-dependent enzymes are of specific interest, since some of these enzymes are known to be involved in the oxidation of various short-chain alcohols (2, 12). One of the best-studied of these enzymes is Pseudomonas aeruginosa ExaA (formerly QedH), which is a homologue of Pseudomonas putida U PedE and PedH (3). This periplasmic enzyme has been found to be essential for growth utilizing ethanol as a carbon source, since it catalyzes the initial oxidation of the substrate into acetaldehyde (18).
ExaA has recently been reported to accept a broad spectrum of substrates in addition to ethanol, including primary and secondary alcohols, as well as diols such as 1,3-propanediol (9). From these results, together with the fact that a broad substrate spectrum represents a common feature of alcohol dehydrogenases, one could speculate that ethylene glycol could serve as an alternative substrate for ExaA. Because the absence of an appropriate cofactor regeneration system limits the use of the purified enzyme, the use of a whole-cell system is essential for applications based on this pathway. In this regard, P. aeruginosa is problematic due to its pathogenic potential. In contrast, strains of Pseudomonas putida are generally considered to be model organisms for biotechnological approaches due to their metabolic versatility and low pathogenic potential. Sequence analysis showed that all P. putida strains sequenced so far harbor at least one homologue of the exaA gene. Therefore, these strains might serve as good platforms for the establishment of a fermentative route from ethylene glycol to glyoxylic acid.
To test this hypothesis, we investigated, characterized, and compared the metabolisms of ethylene glycol in the two P. putida strains KT2440 and JM37. This was conducted via growth and bioconversion experiments, as well as directed mutagenesis and proteome analysis.
MATERIALS AND METHODS
Strains, plasmids, and growth conditions.
A list of the strains and plasmids used in this study is provided in Table 1. For the maintenance of P. putida strains, cells were grown on Pseudomonas isolation agar (BD, Franklin Lakes, NJ) at 30°C and were subsequently stored at 4°C until further use. For liquid growth, LB (35) or a modified M12 medium was used. One liter of modified M12 medium contained 1 g (NH4)2SO4, 0.1 g NaCl, 0.2 g MgSO4 · 7H2O, 0.02 g CaCl2 · 2H2O, 1 ml of SL4 trace element solution (31), and 2 g KH2PO4; the medium was finally adjusted to a pH of 6.7. Additionally, 10 ml of a sterile vitamin solution containing 100 mg/liter biotin, 200 mg/liter folic acid, 20 mg/liter aminobenzoic acid, 20 mg/liter riboflavin, 40 mg/liter calcium pantothenate, 140 mg/liter nicotinic acid, 40 mg/liter pyridoxine hydrochloride, 200 mg/liter myo-inositol, and 40 mg/liter thiamine hydrochloride was added. Unless indicated otherwise, precultures were grown overnight in test tubes with 5 ml medium by using a rotary shaker (CH-4103; Infors AG, Switzerland) at 30°C and 180 rpm. For growth experiments, 20 ml of M12 minimal medium supplemented with 10 mM ethylene glycol, glycolic acid, or glyoxylic acid as a carbon and energy source was inoculated with precultures to an initial optical density at 600 nm (OD600) of 0.01, and growth was monitored by measuring the OD600.
Table 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Genotype or relevant characteristics | Reference or source |
|---|---|---|
| Strains | ||
| Escherichia coli JM109 | recA1 supE44 endA1 hsdR17 gyrA96 relA1 thi Δ(lac-proAB) [F′ traD36 proAB+ lacIq lacZΔM15] | 40 |
| P. putida | ||
| KT2440 | Wild type | ATCC 47054 |
| ΔUPP4 | Δupp | 20 |
| GN104 | Δupp ΔpedH | This study |
| GN116 | Δupp ΔpedE | This study |
| GN127 | Δupp ΔpedE ΔpedH | This study |
| GN133 | Δupp ΔprpB | This study |
| GN140 | Δupp ΔprpB Δgcl | This study |
| GN187 | Δupp ΔprpB Δgcl ΔglcB | This study |
| GN259 | Δupp ΔprpB Δgcl ΔglcB ΔaceA | This study |
| JM37 | Pseudomonas putida strain originally isolated from an m-xylene-enriched soil sample | 28 |
| Plasmids | ||
| pJOE6261.2 | pIC20HE backbone with a kanamycin resistance gene and a copy of upp from P. putida KT2440 | 20 |
| pNG91.2 | pJOE6261.2 with the up- and downstream regions of PP_2674 cloned into a BamHI site | This study |
| pNG98.4 | pJOE6261.2 with the up- and downstream regions of PP_2679 cloned into a BamHI site | This study |
| pNG122.1 | pJOE6261.2 with the up- and downstream regions of prpB cloned into a BamHI site | This study |
| pNG123.6 | pJOE6261.2 with the up- and downstream regions of gcl cloned into an NdeI site | This study |
| pNG141.8 | pJOE6261.2 with the up- and downstream regions of glcB cloned into a BamHI site | This study |
| pNG214.4 | pJOE6261.2 with the up- and downstream regions of aceA cloned into a BamHI site | This study |
Generation of mutants.
For the deletion of various chromosomal genes in P. putida KT2440, the previously described upp/5-fluorouracil (5-FU) counterselection system was used (20). Briefly, the up- and downstream regions, including the start and stop codons, of the target gene were cloned into pJOE6261.2. Transformants were first selected on LB agar plates supplemented with 50 μg/ml of kanamycin (Kan). One of the Kanr 5-FUs clones obtained was incubated in LB at 30°C for 24 h and was subsequently plated onto M9 minimal plates (35) containing 20 μg/ml 5-FU and 0.2% glucose. 5-FUr and Kans clones were checked by colony PCR, and a clone that tested positive for the deletion of the corresponding gene was selected for further experiments. Oligonucleotides for the different strain constructions are shown in Table S1 in the supplemental material.
Bioconversion experiments.
For bioconversion experiments, 1-ml portions of precultures were transferred to 1-liter Erlenmeyer flasks containing 250 ml LB medium, and cells were grown to stationary phase. The cultures were harvested by centrifugation at 4°C and 13,700 × g for 20 min (SLA-3000; Sorvall) and were then washed with 40 ml of M12 medium without a carbon source. Finally, the cells were resuspended in M12 medium containing either 50 mM ethylene glycol, 25 mM glycolic acid, or 25 mM glyoxylic acid, and the OD600 was adjusted to 7.5. In order to provide a stable pH of 6.7 during the entire experiment, sodium phosphate buffer was used at 0.1 M for experiments with ethylene glycol and at 0.2 M for experiments with glycolic acid or glyoxylic acid. Fifty milliliters of this cell suspension was transferred to 250-ml Erlenmeyer flasks and was incubated in a rotary shaker at 30°C and 150 rpm for 120 h. Samples (1.5 ml) were taken at regular time intervals, and cells were removed by centrifugation at 20,000 × g for 1 min (centrifuge 5417 C; Eppendorf) for subsequent high-performance liquid chromatographic (HPLC) analysis of the supernatant.
HPLC analysis.
For HPLC analysis, samples were mixed with the internal standard xylitol at a final concentration of 10 mM and were filtered through a 0.2-μm-pore-size filter. HPLC analysis was carried out on an Agilent system (1200 series) using an Aminex HPX-87H cation-exchange resin column (300 by 7.8 mm; Bio-Rad) at 60°C, with 5 mM H2SO4 as the mobile phase and a flow rate of 0.5 ml/min. The substrates and products were quantified using the corresponding standards and a refractive index detector (Agilent 1200 series, G1262A). The detector temperature was set to 35°C. For the identification of unknown products, the corresponding fractions were pooled and were further analyzed by gas chromatography-mass spectrometry (GC-MS).
GC-MS analysis.
GC-MS analysis was performed using a Shimadzu system (GC2010, GCMS-QP2010) equipped with an FS-Supreme-5 capillary column (30 m by 0.25 mm; film thickness, 0.25 μm; CS-Chromatographie, Germany). Prior to GC-MS analysis, 500-μl portions of the samples were mixed with 10 μl HCl (37%), supplemented with 50 μg NaCl, and extracted with 2× 500 μl ethyl acetate. Samples were first extracted with 500 μl ethyl acetate, and the organic phase was removed. The residual aqueous phase was extracted again with 500 μl ethyl acetate, and the organic phase was removed again. Finally, both organic phases were pooled, evaporated with a stream of nitrogen, and derivatized using 40 μl TMS agent (at 70°C for 30 min). Chromatography was performed using helium as the carrier gas at a linear velocity of 30 cm/s. The temperature program started with an initial temperature of 60°C for 3 min, followed by a ramp to 200°C at a rate of 8°C/min. The column temperature was maintained for 1 min, raised to 300°C at a rate of 50°C/min, and held for 5 min. The injector and detector temperatures were 250°C and 200°C, respectively. The volume injected was 1 μl (split ratio, 1:50). Components were characterized in full-scan mode and were identified by comparison of their mass fragmentation patterns with those of the corresponding reference substances.
Culture conditions and preparation of samples for proteomics experiments.
For proteomics experiments, precultures of P. putida strains KT2440, GN259, and JM37 were grown in 20 ml LB medium overnight. From these precultures, 100-μl portions were transferred to 100 ml of M12 medium in 500-ml Erlenmeyer flasks and were then incubated at 30°C and 180 rpm with 10 mM glucose as the sole source of carbon and energy. After 12 h of incubation, cultures either were treated with 80 mM ethylene glycol or 10.8 mM glyoxylic acid or were left untreated for control experiments. After an additional incubation for 5 h, cells were harvested by centrifugation at 3,200 × g and were washed in three steps with 20 ml, 10 ml, and 1.5 ml of 5 mM magnesium acetate in 10 mM Tris-HCl at pH 8. For 2-dimensional differential in-gel electrophoresis (2D-DIGE) analysis, cell pellets were resuspended in 1 ml DIGE lysis buffer {30 mM Tris-HCl [pH 8.5], 7 M urea, 2 M thiourea, 4% 3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate [CHAPS]} and were lysed by sonication on ice (five intervals with a duration of 30 s) (Sonifier 450; Branson). Lysed samples were centrifuged at 14,000 × g for 10 min at 4°C, followed by ultracentrifugation (Beckman Coulter, Germany) at 4°C and 100,000 × g for 1 h. The protein concentration of the supernatant was determined using the Bradford assay (7).
2D-DIGE analysis.
All 2D-DIGE experiments were performed with three biological replicates. In each replicate, a treated and an untreated sample were compared. Minimal labeling was performed with Cy dyes according to the manufacturer's instructions (GE Healthcare, Germany). In one experiment (see Fig. S3 in the supplemental material), G-dyes (NH DyeAgnostics GmbH, Germany) were used instead of Cy dyes (GE Healthcare, Germany). After minimal labeling, samples were loaded onto 24-cm Immobiline DryStrips (pH 4 to 7 or pH 3 to 11) by rehydration loading in a DryStrip reswelling tray (GE Healthcare, Germany) according to the manufacturer's instructions. Isoelectric focusing was performed on an IPGphor 3 system (GE Healthcare, Germany) using the following protocol: step 1, 150 V for 2 h; step 2, 300 V for 2 h; step 3, 300 to 1,000 V for 8 h; step 4, 1,000 to 8,000 V for 3 h; step 5, 8,000 V for 7 h. Afterwards, strips were equilibrated according to the manufacturer's protocol. The second dimension was run on a 12% sodium dodecyl sulfate (SDS) gel in an Ettan DALTsix electrophoresis unit (GE Healthcare, Germany). After electrophoresis, 2D-DIGE gels were scanned on a Typhoon Trio+ imaging system (GE Healthcare, Germany) with excitation and emission wavelengths corresponding to those of the respective dyes.
Protein expression analysis.
Gel images were analyzed using Progenesis SameSpots software (version 4.0; Nonlinear Dynamics, United Kingdom). For image alignment and spot detection, the default settings of the software were used. Cy3 and Cy5 images from each gel were normalized to the corresponding internal standard (Cy2). Fold changes between treated and untreated samples were calculated for all spots detected and statistically analyzed by a one-way analysis of variance (ANOVA). Spots showing a differential expression of at least 1.8-fold (upregulation) or 0.6-fold (downregulation) with a P value of <0.05 were considered significant and were selected for spot picking. Spots were picked by use of an Ettan TM spot picker (GE Healthcare, Germany). Spot picking was validated by subsequent silver staining of gels (10). Spots not precisely picked by the picking robot were excised manually. All excised protein spots were in-gel digested with trypsin according to the work of Shevchenko et al. (37) and were analyzed by mass spectrometry (for further details, see “Protein identification by mass spectrometry” in the supplemental material).
RESULTS
Growth and bioconversion experiments with Pseudomonas putida strains JM37 and KT2440.
In order to test the abilities of the P. putida strains JM37 and KT2440 to use ethylene glycol or its putative degradation intermediates glycolic acid and glyoxylic acid as sole sources of carbon and energy, growth experiments were performed. These experiments revealed that strain JM37 is able to grow with ethylene glycol, glycolic acid, and glyoxylic acid, finally yielding optical densities of 0.6, 0.54, and 0.41, respectively. Notably, the initial lag phase during growth with ethylene glycol was longer than that with the other substrates (Fig. 1). In contrast to JM37, strain KT2440 did not show growth with ethylene glycol, glycolic acid, or glyoxylic acid, even after incubation for 24 h (Fig. 1) or 48 h (data not shown).
Fig 1.
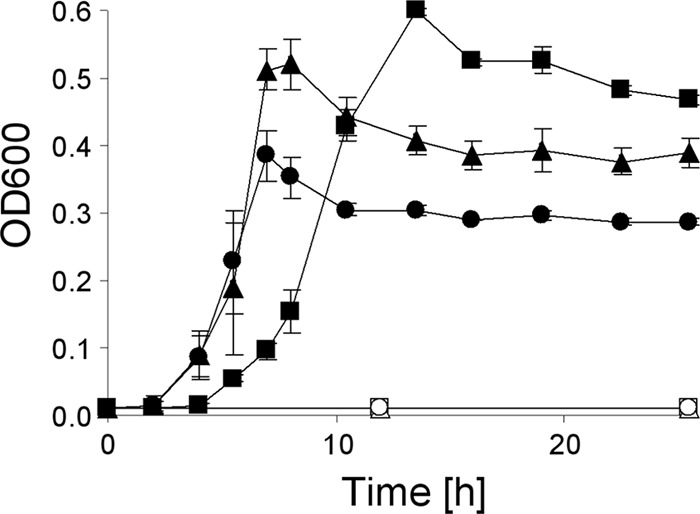
Growth of P. putida strains JM37 (filled symbols) and KT2440 (open symbols) in M12 minimal medium with 50 mM sodium phosphate buffer (pH 6.7) and either 10 mM ethylene glycol (squares), 10 mM glycolic acid (triangles), or 10 mM glyoxylic acid (circles). Cultures were incubated at 30°C with shaking at 180 rpm, and growth was measured as the OD600. Experiments were performed in triplicate. Error bars indicate standard deviations.
Experiments with resting cells of strain JM37 showed complete conversion of 50 mM ethylene glycol, 25 mM glycolic, acid or 25 mM glyoxylic acid within 24 h of incubation and a maximal conversion rate of 3.2 mM/h for ethylene glycol (Fig. 2A; see also Fig. S1A and B in the supplemental material). Increasing optical densities during these experiments indicated growth-permitting conditions (data not shown). For the identification of intermediates emerging from ethylene glycol metabolism, we analyzed the supernatants from bioconversion experiments by HPLC. In these experiments, no accumulation of putative intermediates could be observed.
Fig 2.

Bioconversion of 50 mM ethylene glycol in resting-cell suspension experiments (OD600, 7.5). LB-grown cells were washed and adjusted to an OD600 of 7.5 in M12 medium (100 mM sodium phosphate buffer, pH 6.7). The consumption of ethylene glycol (■) and the formation of the metabolites glycolic acid (□), glyoxylic acid (△), and oxalic acid (●) were analyzed by HPLC with refractive index detection. (A) P. putida strain JM37; (B) P. putida strain KT2440; (C) deletion mutant GN259 (Δupp ΔprpB Δgcl ΔglcB Δace), derived from strain KT2440; (D) deletion mutant GN127 (Δupp ΔpedE ΔpedH), derived from strain KT2440. Error bars represent standard deviations for three independent experiments.
Even though strain KT2440 did not grow with any of the substrates, resting-cell suspension experiments revealed complete conversion of ethylene glycol, glycolic acid, and glyoxylic acid (Fig. 2B; see also Fig. S1C and D in the supplemental material). However, compared to the results for strain JM37, strain KT2440 showed reduced conversion rates for all substrates and no increase in optical density during the time course of the experiment. As a consequence, the time for complete substrate conversion was significantly increased (>48 h), with 8.8 mM ethylene glycol still present in KT2440 supernatants after 24 h (Fig. 2B). At this time point, no substrate was left in supernatants of strain JM37 (Fig. 2A). Another difference from the results with strain JM37 was the detection of glycolic acid, glyoxylic acid, and oxalate in strain KT2440 supernatants during incubation with ethylene glycol. The maximal temporal accumulations of 3.6 mM glycolic acid and 12.6 mM glyoxylic acid were observed at 24 h after the start of the experiments by the addition of 50 mM ethylene glycol (Fig. 2B). In contrast, the concentration of oxalate was found to increase continuously, finally reaching 12.8 mM after 118 h of incubation.
When 25 mM glycolic acid was used, temporal accumulation of 2.5 mM glyoxylic acid was found after 24 h of incubation. Furthermore, 8.5 mM oxalic acid was detected as the final product after 118 h of incubation (see Fig. S1C in the supplemental material). When 25 mM glyoxylic acid was used, oxalic acid was found as the only product, with a final concentration of 11.3 mM after 118 h of incubation (see Fig. S1D). Interestingly, we found a carbon imbalance in all bioconversion experiments when substrate degradation and product formation were compared. Moreover, we found that the imbalance increased over the time course of the experiment, finally reaching 72%, 63%, and 51% of the initial carbon concentration with ethylene glycol, glycolic acid, and glyoxylic acid, respectively (Fig. 2B; see also Fig. S1C and D).
Bioconversion experiments with mutants of P. putida strain KT2440 defective in canonical degradation pathways for glyoxylic acid.
The carbon imbalance, together with the fact that no growth could be detected with any of the substrates, indicated the activity of an additional degradation pathway other than those leading to the formation of oxalic acid. For constant accumulation of the central metabolite, glyoxylic acid, the disruption of all potential degradation pathways is necessary. To achieve this, we generated the mutant strain GN259, which lacks the genes for glyoxylate carboligase (gcl), malate synthase (glcB), isocitrate lyase (aceA), and 2-methylisocitrate lyase (prpB). The additional deletion of prpB was chosen due to recent enzymatic evidence showing that Mycobacterium tuberculosis isocitrate lyase (ICL1) is able to function as a 2-methylisocitrate lyase, and thus, these two enzymes might functionally complement each other (19). Strain GN259 was then used in bioconversion experiments with ethylene glycol, glycolic acid, or glyoxylic acid as the substrate. These experiments revealed a significant reduction in the ethylene glycol conversion rate from that with strain KT2440 (Fig. 2B and C). Consequently, 16.2 mM ethylene glycol—twice the amount found in supernatants of strain KT2440—could be identified in supernatants of strain GN259 after 24 h of incubation. While strain KT2440 showed complete conversion of ethylene glycol, 1.8 mM ethylene glycol was still present in supernatants of strain GN259 after 118 h. Most importantly, strain GN259 showed constant accumulation of glycolic acid over the time course of the experiment. Whereas the concentration of glycolic acid increased constantly over time, reaching a final concentration of 14.1 mM after 118 h, the concentration of glyoxylic acid increased only over the first 24 h of the experiment and then remained at a constant level of about 10 mM. Oxalic acid accumulated constantly during the entire experiment, to a final concentration of 8.1 mM after 118 h. Finally, strain GN259 also showed an improved carbon balance during bioconversion experiments with ethylene glycol (83%) compared to that of strain KT2440.
Proteome analysis for identification of pathways involved in ethylene glycol metabolism.
In order to gain insight into the pathways involved in the metabolism of ethylene glycol and glyoxylic acid, quantitative proteomics experiments were carried out. We compared the proteomes of the untreated P. putida strains KT2440, JM37, and GN259 with their ethylene glycol- and glyoxylic acid-treated counterparts. Figures S2 to S9 in the supplemental material show 2D-DIGE images of the different strains under the different conditions. In the presence of ethylene glycol, 50 up- or downregulated proteins were identified for strain KT2440 and 21 for JM37. All quantitative proteomics results are given in the tables accompanying Fig. S2 to S9 in the supplemental material. For a subset of nine spots, quantitative data are given in Table 2.
Table 2.
Selected proteins regulated in response to ethylene glycol or glyoxylic acid treatment
| Protein spot no. | Protein | Fold change in expression of the following strain after the indicated treatmenta: |
|||||
|---|---|---|---|---|---|---|---|
| KT2440 |
GN259b |
JM37 |
|||||
| EG | GXA | EG | GXA | EG | GXA | ||
| 1 | Isocitrate lyase (AceA) | 2.8 | 2.8 | KO | KO | 1.9 | − |
| 2 | Glyoxylate carboligase (Gcl) | − | − | KO | KO | 9.6 | 16 |
| 3 | Malate synthase G (GlcB) | − | − | KO | KO | 3.7 | 17 |
| 4 | Quinoprotein alcohol dehydrogenase (PedH) | 21 | ND | ND | ND | ND | ND |
| 5 | Quinoprotein alcohol dehydrogenase (PedE) | 34 | − | 8.2 | − | 31 | 0.5 |
| 6 | Aldehyde dehydrogenase family protein (PedI) | 5.2 | − | − | − | 7.9 | − |
| 7 | Aldehyde dehydrogenase family protein (PP_0545) | − | − | 5.5 | − | 2 | − |
| 8 | Transcriptional regulator (PedR1) | 4.8 | − | − | − | 4.8 | − |
| 9 | PAS/PAC sensor hybrid histidine kinase (PedS1) | 7.7 | − | − | − | − | − |
As determined by 2D-DIGE. EG, ethylene glycol; GXA, glyoxylic acid. Each experiment comprised three biological replicates. Proteins were considered upregulated when they showed an increase of >1.8-fold or downregulated with a decrease to <0.6-fold. Regulation was assumed to be significant with a P value of ≤0.05. Data for EG treatment of KT2440 were taken from Fig. S2 and S3 in the supplemental material, except in the case of PedH, for which data were obtained from Fig. S4 in the supplemental material. Data for all other strains and conditions were taken from Fig. S5 to S9 in the supplemental material. If a protein was detected in more than one spot of a gel, the data for the spot with the highest fold change are shown in this table. Fold changes of all differentially regulated protein spots and the proteins identified therein are given in Fig. S2 to S9 in the supplemental material and in the corresponding tables. ND, not determined; −, the spot was not regulated in this experiment.
A KT2440 mutant (Δupp ΔprpB Δgcl ΔglcB Δace). KO, knockout (the gene was deleted in this mutant).
In ethylene glycol-treated cells of strain JM37, significant induction of enzymes involved in glyoxylic acid conversion, namely, isocitrate lyase AceA (1.9-fold), glyoxylate carboligase Gcl (9.6-fold), and malate synthase GlcB (3.7-fold), was observed. When cells were treated with glyoxylic acid, significant induction of Gcl (16-fold) and GlcB (17-fold) was detected. Conversely, in strain KT2440, isocitrate lyase AceA (2.8-fold), but not Gcl or GlcB, was found to be significantly upregulated with both substrates. Besides enzymes involved in the degradation of glyoxylic acid, the PQQ-dependent ADHs PP_2674 and PP_2679 were upregulated 34-fold and 21-fold, respectively, in strain KT2440 in the presence of ethylene glycol (Table 2). Under the same conditions, the protein homologous to PP_2674 was also found to be upregulated 31-fold in strain JM37. Sequence analysis revealed that PP_2674 and PP_2679 have 84% and 52% amino acid sequence identities, respectively, to the Pseudomonas aeruginosa ethanol dehydrogenase ExaA, as well as 98% and 99% sequence identities to the previously described Pseudomonas putida U enzymes PedE and PedH, respectively (Table 3) (3). Comparison of the genetic context of these genes in P. putida KT2440 further revealed high structural similarity to the phenylethanol degradation cluster (Ped) of P. putida U (Fig. 3).
Table 3.
Amino acid sequence comparison of the two quinoproteins PedE and PedH, the aldehyde dehydrogenase PedI, and the response regulator PedR1 of P. putida KT2440 with the homologues of P. putida U and P. aeruginosa PAO1
| Protein in P. putida KT2440 | % amino acid identity (name) of the homologue in: |
|
|---|---|---|
| P. putida U | P. aeruginosa PAO1 | |
| PedR1 (PP_2665) | 91 (PedR1) | 86 (ErbR) |
| PedE (PP_2674) | 98 (PedE) | 84 (ExaA) |
| PedH (PP_2679) | 99 (PedH) | 52 (ExaA) |
| PedI (PP_2680) | 97 (PedI) | 88 (ExaC) |
Fig 3.

Comparison of the gene cluster controlled by the ErbR regulator in P. aeruginosa PAO1 with the homologous clusters of P. putida U and KT2440. Genes represented by shaded arrows show significant similarities between the three strains. Homologues of the P. putida U proteins PedR1, PedS1, PedE, and PedI were found to be regulated under ethylene glycol treatment in P. putida KT2440. These genes represent homologues of the P. aeruginosa proteins ErbR, ErcS′, ExaA, and ExaC. Open reading frames indicated by dashed lines do not reflect the actual size of this region.
Due to this homology, we will use the Ped nomenclature of P. putida U for the corresponding genes PP_2664 to PP_2680 in P. putida KT2440 (Fig. 3).
Interestingly, PedH has a rather basic isoelectric point (pI) of 8.75, while PedE has a pI of 6.52. Thus, PedH eludes detection on the 2D-DIGE gels used routinely in our experiments, which cover the pH range of 4 to 7. It was detected only in a single ethylene glycol experiment carried out with strain KT2440 in which the protein extracts were separated with a pH gradient of 3 to 11 (see Fig. S4 in the supplemental material). In addition to the PQQ-dependent ADHs PedE and PedH, two aldehyde dehydrogenases (ALDHs), PedI and PP_0545, were also found to be upregulated in response to ethylene glycol. While only one of these ALDHs was found to be upregulated in strains KT2440 (PedI) and GN259 (PP_0545), both enzymes were induced in JM37 (Table 2). In contrast, PedE and the two ALDHs PedI and PP_0545 were not found to be induced after treatment with glyoxylic acid in any strain tested (Table 2; see also Fig. S5, S7, and S9 in the supplemental material).
In P. aeruginosa, the expression of the homologous ethanol degradation pathway is controlled by a complex regulatory system including the transcriptional regulator ErbR (30). In our experiments, we found that the expression of PedE, PedH, and PedI was coregulated with that of the ErbR homologue PedR1 (4.8-fold) in JM37 and KT2440 after ethylene glycol treatment (Table 2). The PAS/PAC sensor kinase PedS1 could be detected only in KT2440. No regulation of PedR1 could be detected in the mutant GN259. Surprisingly, this mutant still showed upregulation of PedE, though to a much lower degree than that for the wild-type strain (Table 2).
Bioconversion experiments with mutants of P. putida strain KT2440 harboring deletions of the quinoprotein alcohol dehydrogenases PedE and PedH.
As described above, our proteome analysis showed induction of the two quinoprotein ADHs PedE and PedH in cells treated with ethylene glycol. In order to test the importance of PedE and PedH in the metabolism of ethylene glycol, we constructed mutant strains in which either pedE (strain GN116), pedH (strain GN104), or both genes (strain GN127) were deleted. These deletion mutants were then subjected to bioconversion experiments in resting-cell suspensions. In these experiments, we found that single deletions had no effect; the mutants showed conversion rates similar to that of strain KT2440 (data not shown). In contrast, the conversion of ethylene glycol was significantly impaired in the double deletion mutant GN127; 35% of the initial substrate concentration could be found in supernatants even after incubation for 118 h (Fig. 2D). While oxalate accumulated over time in supernatants of the double deletion mutant to a final concentration of 11.7 mM, no traces of glycolic acid or glyoxylic acid could be detected. When glycolic acid or glyoxylic acid was used as a substrate, no differences between strain GN127 and strain KT2440 could be observed (see Fig. S1C, D, E, and F in the supplemental material).
DISCUSSION
The ability to grow with ethylene glycol as a sole source of carbon and energy has been demonstrated for various kinds of microorganisms, some of which initially oxidize it to glyoxylic acid via glycolic acid (11, 13). In this work, we investigated the potential of the Pseudomonas putida strains KT2440 and JM37 for the microbial oxidation of ethylene glycol into the more valuable product glyoxylic acid. In contrast to strain KT2440, strain JM37 showed rapid growth with ethylene glycol, glycolic acid, or glyoxylic acid as the sole source of carbon and energy. The observation of a longer initial lag phase for strain JM37 during growth on ethylene glycol suggests the induction of additional enzymes, such as dehydrogenases and oxidases, which are reported to be important for the first steps of ethylene glycol metabolism via glyoxylic acid (6, 8). Despite the differences in growth, resting-cell suspension experiments revealed complete conversion of ethylene glycol, glycolic acid, and glyoxylic acid for P. putida KT2440 and JM37, albeit with substantial differences in the time needed for complete conversion. While strain JM37 accumulated no potential metabolic intermediates during the conversion of ethylene glycol, glycolic acid and glyoxylic acid were found in supernatants of strain KT2440. This clearly indicates the roles of glycolic acid and glyoxylic acid as intermediates in the metabolism of ethylene glycol in strain KT2440. Previous studies had demonstrated glycolaldehyde to be a product of ethylene glycol oxidation (6, 22). Surprisingly, this first expected intermediate in a three-step oxidation of ethylene glycol to glyoxylic acid was not found to accumulate in our experiments. A possible explanation for this observation could be that the formation of glycolic acid from ethylene glycol is actually catalyzed in two consecutive steps by the same enzyme. Alternatively, free glycolaldehyde might not be detected due to the formation of Schiff base adducts with amino groups of nearby proteins (1). In both cases, the expected glycolaldehyde concentrations would be very low and thus could be below the detection limit of our system.
Even though further experiments are needed to prove this hypothesis, we propose that strain KT2440 actually metabolizes ethylene glycol via glycolaldehyde to glycolic acid and glyoxylic acid (Fig. 4). Subsequently, some of the glyoxylic acid is oxidized to oxalic acid, whereas about three-quarters of the substrate could not be found in any of the products identified.
Fig 4.
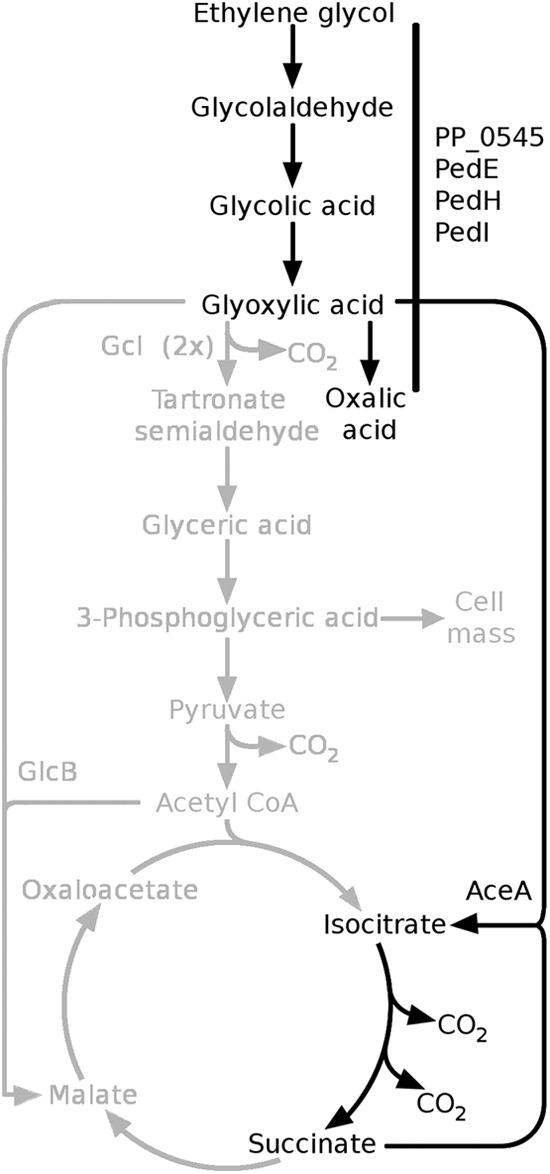
Postulated pathway for the metabolism of ethylene glycol in Pseudomonas putida strains KT2440 and JM37. The enzymes and/or metabolites identified in response to ethylene glycol in KT2440 are depicted in black. Additional pathways identified in strain JM37 are shown in gray. Detailed descriptions of the pathways are given in the text.
From our differential proteomics approach, increased expression levels of tartronate semialdehyde synthase (Gcl), malate synthase (GlcB), and isocitrate lyase (AceA) were found in strain JM37 after treatment with ethylene glycol or glyoxylic acid. In these experiments, isocitrate lyase showed the smallest fold change after treatment with ethylene glycol and was not significantly upregulated after treatment with glyoxylic acid. This suggests that the “glycerate pathway” and the “malate pathway” play key roles in glyoxylic acid conversion in JM37. It has been reported that the pathway initiated by Gcl enables growth on glyoxylic acid, while the pathways using GlcB and AceA can lead only to the production of energy (Fig. 4) (4, 24, 26). In a recent study, Li and coworkers tried to prevent the metabolism of glyoxylic acid in strain JM37 by disrupting all the degradation pathways mentioned above. However, no single mutations of aceA, glcB, or glc, and none of the corresponding double mutations, had any effect on glyoxylic acid degradation (28). The authors suggested that an additional degradation pathway might exist, since a number of such alternative pathways have been described for different microorganisms (5, 25, 38). Despite the possibility of such an alternative in JM37, our proteome analysis showed no induction of enzymes that could be related to one of these pathways. Since we cannot rule out the possibility that alternative metabolic pathways could be constitutively expressed, this issue needs further investigations, which are under way in our laboratory.
Interestingly, in strain KT2440, AceA, but not Gcl (PP_4297) or GlcB (PP_0356), was upregulated after treatment with ethylene glycol or glyoxylic acid. On the one hand, the absence of Gcl induction might explain the lack of growth with ethylene glycol, glycolic acid, or glyoxylic acid found for strain KT2440. On the other hand, the induction of AceA suggests that this enzyme is active under the conditions used. This enzyme catalyzes the first step of the glyoxylate shunt for the generation of energy but does not support cell mass generation. Together with the absence of detectable growth, the activity of AceA could therefore be the explanation for the carbon imbalance observed in all bioconversion experiments. This is further supported by the observation that conversions with the deletion mutant GN259 resulted in an improved carbon balance compared to that in strain KT2440. In addition, strain GN259 accumulated the intermediates glycolic acid and glyoxylic acid over the entire time course of the experiment. This underlines the importance of at least one of these pathways for ethylene glycol metabolism via the central intermediate glyoxylic acid in strain KT2440. At present, we cannot rule out the possibility that more than one pathway might be active or become active during incubation. However, our data clearly support the hypothesis that the degradation of ethylene glycol leads to the formation of oxalic acid and the production of energy from glyoxylic acid via the glyoxylate shunt.
In addition to the upregulation of proteins involved in the degradation of glyoxylic acid, our proteome analysis of ethylene glycol-induced cells of KT2440 and JM37 revealed the upregulation of the two PQQ-dependent ADHs PedE and PedH, as well as that of the NADH-dependent ALDHs PedI and PP_0545. As indicated by the name, the quinoprotein alcohol dehydrogenases PedE and PedH harbor the cofactor pyrroloquinoline quinone (PQQ) as a prosthetic group. These periplasmic enzymes have been shown to play a crucial role in the degradation of primary alcohols (32, 36) and have been reported to be induced by their corresponding substrates. The best-studied example of a PQQ-dependent ADH is ExaA, an ethanol dehydrogenase from P. aeruginosa (33). In this organism, ExaA has been found to catalyze the initial oxidation of ethanol to acetaldehyde, thus enabling growth on ethanol (36). In P. aeruginosa, the transcriptional regulator ErbR (formerly known as AgmR) controls the expression of ExaB (cytochrome c), ExaC (aldehyde dehydrogenase), and the operon for PQQ biosynthesis (pqqABCDEH) directly, whereas ExaA expression is regulated indirectly via the sensor kinase/response regulator pair EraS/EraR (formerly ExaD/ExaE) (16). Upstream of ErbR, the two histidine kinases ErcS and ErcS′, as well as the response regulator ErdR, have been identified as additional regulatory elements (30).
Our results indicate that the oxidation of ethylene glycol in KT2440 could be controlled by such an ErbR-type regulon. This hypothesis is supported by several lines of evidence: (i) induction of ErbR and ErcS′ homologues (PedR1, PedS1), (ii) induction of several enzyme homologues known to be regulated by ErbR in P. aeruginosa (e.g., PedE, PedI), and (iii) similarities in the corresponding genetic contexts of P. putida KT2440, P. aeruginosa, and P. putida U (Fig. 3). In addition, we found strongly reduced PedE expression and no induction of PedI in the P. putida aceA deletion strain GN259 after treatment with ethylene glycol and no induction compared to strain KT2440. This observation is in agreement with the findings of a recent study demonstrating that the deletion of aceA in P. aeruginosa ATCC 17933 results in downregulation of all genes necessary for ethanol oxidation, including ExaA (27). Deletion of the ErbR homologue PedR1 and subsequent characterization of the PedE and PedH induction pattern in the presence of ethylene glycol could finally prove this hypothesis; experiments for this purpose are currently in progress. Despite similarities between the ErbR-like regulon in P. aeruginosa and the putative homologue in different P. putida strains, some major differences exist. One such difference is the presence of two quinoproteins in P. putida strains (e.g., PedE and PedH), compared to only one in P. aeruginosa. Arias and coworkers have shown that PedE and PedH from strain P. putida U are involved in the conversion of phenylethanol and alcohols with a chain length of C6 to C9 (3). However, only a double mutant strain defective in both PQQ-dependent enzymes was unable to grow with the substrates mentioned above. Interestingly, growth with alcohols with chain lengths shorter than C6 was not altered for this double mutant, indicating the specificity and redundancy of this system. In analogy to this, our bioconversion experiments revealed that either PedE or PedH from P. putida strain KT2440 can catalyze the conversion of ethylene glycol in this organism. This can be concluded from the observation that only in the double mutant strain GN127 was the conversion of ethylene glycol significantly affected, whereas the single mutants exhibited rates similar to that of the parental strain. The fact that the conversion rates of glycolic acid and glyoxylic acid were not altered in strain GN127 highlights the importance of PedE and PedH for the first steps of ethylene glycol metabolism. The residual ethylene glycol conversion rate observed in strain GN127 is most likely due to the activity of unspecific dehydrogenases, partly compensating for the activities of the deleted PQQ-dependent ADHs. The absence of glycolic acid and glyoxylic acid accumulation in this mutant can be explained by the rapid oxidation of emerging small amounts of intermediates by subsequent enzymes of the oxidation chain, most likely the two ALDHs PedI and PP_0545.
In summary, we were able to reconstruct novel pathways for ethylene glycol metabolism in the Pseudomonas putida strains KT2440 and JM37 (Fig. 4). In KT2440, this pathway is initiated by the PQQ-dependent ADHs PedE and PedH and the ALDHs PedI and PP_0545 producing glyoxylic acid via the intermediates glycolaldehyde and glycolic acid. Subsequently, glyoxylic acid either is further oxidized to the “dead-end” product oxalic acid or is metabolized via AceA, leading to the generation of energy. In strain JM37, the activity of two additional pathways, namely, Gcl and GlcB, leads to rapid metabolism of ethylene glycol without accumulation of the corresponding intermediates or oxalic acid. The elimination of key enzymes for glyoxylic acid metabolism in strain KT2440 resulted in an improved carbon balance, most likely due to the deletion of aceA and the accumulation of glyoxylic acid over the entire experimental time course. From these results, we propose that P. putida KT2440 is a promising candidate for further metabolic engineering approaches to the establishment of a biocatalytic route for the production of glyoxylic acid from ethylene glycol.
Supplementary Material
ACKNOWLEDGMENTS
We gratefully acknowledge generous financial support by the German Federal Ministry of Education and Research (BMBF) in the frame of the project “Systems Biology in Pseudomonas for Industrial Biocatalysis.”
We thank Herbert Platsch (BASF AG, Ludwigshafen, Germany) for the exchange of preliminary studies and Yvonne Beck for valuable support in the generation of growth curves.
Footnotes
Published ahead of print 28 September 2012
Supplemental material for this article may be found at http://aem.asm.org/.
REFERENCES
- 1. Acharya AS, Manning JM. 1983. Reaction of glycolaldehyde with proteins: latent crosslinking potential of alpha-hydroxyaldehydes. Proc. Natl. Acad. Sci. U. S. A. 80:3590–3594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Adachi O, Ano Y, Toyama H, Matsushita K. 2007. Biooxidation with PQQ- and FAD-dependent dehydrogenases, p 1–35 In Schmid RD, Urlacher VB. (ed), Modern biooxidation: enzymes, reactions, and applications. Wiley-VCH Verlag GmbH & Co KGaA, Weinheim, Germany [Google Scholar]
- 3. Arias S, Olivera ER, Arcos M, Naharro G, Luengo JM. 2008. Genetic analyses and molecular characterization of the pathways involved in the conversion of 2-phenylethylamine and 2-phenylethanol into phenylacetic acid in Pseudomonas putida U. Environ. Microbiol. 10:413–432 [DOI] [PubMed] [Google Scholar]
- 4. Bailey E, Hullin RP. 1966. The metabolism of glyoxylate by cell-free extracts of Pseudomonas sp. Biochem. J. 101:755–763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Blackmore MA, Quayle JR. 1970. Microbial growth on oxalate by a route not involving glyoxylate carboligase. Biochem. J. 118:53–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Boronat A, Caballero E, Aguilar J. 1983. Experimental evolution of a metabolic pathway for ethylene glycol utilization by Escherichia coli. J. Bacteriol. 153:134–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bradford MM. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72:248–254 [DOI] [PubMed] [Google Scholar]
- 8. Caskey WH, Taber WA. 1981. Oxidation of ethylene glycol by a salt-requiring bacterium. Appl. Environ. Microbiol. 42:180–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chattopadhyay A, Förster-Fromme K, Jendrossek D. 2010. PQQ-dependent alcohol dehydrogenase (QEDH) of Pseudomonas aeruginosa is involved in catabolism of acyclic terpenes. J. Basic Microbiol. 50:119–124 [DOI] [PubMed] [Google Scholar]
- 10. Chevallet M, et al. 2008. Sweet silver: a formaldehyde-free silver staining using aldoses as developing agents, with enhanced compatibility with mass spectrometry. Proteomics 8:4853–4861 [DOI] [PubMed] [Google Scholar]
- 11. Child J, Willetts A. 1978. Microbial metabolism of aliphatic glycols. Bacterial metabolism of ethylene glycol. Biochim. Biophys. Acta 538:316–327 [DOI] [PubMed] [Google Scholar]
- 12. Duine JA, Jongejan JA. 1989. Quinoproteins, enzymes with pyrrolo-quinoline quinone as cofactor. Annu. Rev. Biochem. 58:403–426 [DOI] [PubMed] [Google Scholar]
- 13. Fincher EL, Payne WJ. 1962. Bacterial utilization of ether glycols. Appl. Environ. Microbiol. 10:542–547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gaston LW, Stadtman ER. 1962. Fermentation of ethylene glycol by Clostridium glycolicum, sp. n. J. Bacteriol. 85:356–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gavagan JE, et al. 1995. Glyoxylic acid production using microbial transformant catalysts. J. Org. Chem. 60:3957–3963 [Google Scholar]
- 16. Gliese N. 2004. AgmR controls transcription of a regulon with several operons essential for ethanol oxidation in Pseudomonas aeruginosa ATCC 17933. Microbiology 150:1851–1857 [DOI] [PubMed] [Google Scholar]
- 17. Gonzalez CF, Taber WA, Zeitoun MA. 1972. Biodegradation of ethylene glycol by a salt-requiring bacterium. Appl. Microbiol. 24:911–919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Görisch H. 2003. The ethanol oxidation system and its regulation in Pseudomonas aeruginosa. Biochim. Biophys. Acta 1647:98–102 [DOI] [PubMed] [Google Scholar]
- 19. Gould TA, van de Langemheen H, Muñoz-Elías EJ, McKinney JD, Sacchettini JC. 2006. Dual role of isocitrate lyase 1 in the glyoxylate and methylcitrate cycles in Mycobacterium tuberculosis. Mol. Microbiol. 61:940–947 [DOI] [PubMed] [Google Scholar]
- 20. Graf N, Altenbuchner J. 2011. Development of a method for markerless gene deletion in Pseudomonas putida. Appl. Environ. Microbiol. 77:5549–5552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Isobe K. 1995. Oxidation of ethylene glycol and glycolic acid by glycerol oxidase. Biosci. Biotechnol. Biochem. 59:576–581 [DOI] [PubMed] [Google Scholar]
- 22. Isobe K, Nishise H. 1995. A new enzymatic method for glycolaldehyde production from ethylene glycol. J. Mol. Catal. B Enzym. 1:37–43 [Google Scholar]
- 23. Kataoka M, Sasaki M, Hidalgo AR, Nakano M, Shimizu S. 2001. Glycolic acid production using ethylene glycol-oxidizing microorganisms. Biosci. Biotechnol. Biochem. 65:2265–2270 [DOI] [PubMed] [Google Scholar]
- 24. Kornberg HL, Gotto AM. 1961. The metabolism of C2 compounds in microorganisms. 6. Synthesis of cell constituents from glycollate by Pseudomonas sp. Biochem. J. 78:69–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kornberg HL, Morris JG. 1965. The utilization of glycolate by Micrococcus denitrificans: the β-hydroxyaspartate pathway. Biochem. J. 95:577–586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kornberg HL. 1961. Metabolism of C2 compounds in microorganisms. 8. A dicarboxylic acid cycle as a route for the oxidation of glycollate by Escherichia coli. Biochem. J. 81:503–513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kretzschmar U, Khodaverdi V, Jeoung JH, Görisch H. 2008. Function and transcriptional regulation of the isocitrate lyase in Pseudomonas aeruginosa. Arch. Microbiol. 190:151–158 [DOI] [PubMed] [Google Scholar]
- 28. Li XZ, Klebensberger J, Rosche B. 2010. Effect of gcl, glcB and aceA disruption on glyoxylate conversion by Pseudomonas putida JM37. J. Microbiol. Biotechnol. 20:1006–1010 [DOI] [PubMed] [Google Scholar]
- 29. Mattioda G, Christidis Y. 2000. Glyoxylic acid, p 89–92 In Bohnet M, Brinker CG, Cornils B. (ed), Ullmann's encyclopedia of industrial chemistry. Wiley-VCH Verlag GmbH & Co KGaA, Weinheim, Germany: doi:10.1002/14356007.a12_495 [Google Scholar]
- 30. Mern DS, Ha SW, Khodaverdi V, Gliese N, Gorisch H. 2010. A complex regulatory network controls aerobic ethanol oxidation in Pseudomonas aeruginosa: indication of four levels of sensor kinases and response regulators. Microbiology 156:1505–1516 [DOI] [PubMed] [Google Scholar]
- 31. Pfennig N, Lippert KD. 1966. Über das Vitamin B12-Bedürfnis phototropher Schwefelbakterien. Arch. Microbiol. 55:245–256 [Google Scholar]
- 32. Promden W, Vangnai AS, Toyama H, Matsushita K, Pongsawasdi P. 2009. Analysis of the promoter activities of the genes encoding three quinoprotein alcohol dehydrogenases in Pseudomonas putida HK5. Microbiology 155:594–603 [DOI] [PubMed] [Google Scholar]
- 33. Rupp M, Görisch H. 1988. Purification, crystallisation and characterization of quinoprotein ethanol dehydrogenase from Pseudomonas aeruginosa. Biol. Chem. Hoppe-Seyler 369:431–439 [DOI] [PubMed] [Google Scholar]
- 34. Sajtos A. May 1991. Process for the preparation of glyoxylic acid and glyoxylic acid derivates. US patent 5,015,760
- 35. Sambrook J, Fritsch EF, Maniatis T. 1989. Molecular cloning: a laboratory manual, 2nd ed Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 36. Schobert M, Görisch H. 2001. A soluble two-component regulatory system controls expression of quinoprotein ethanol dehydrogenase (QEDH) but not expression of cytochrome c550 of the ethanol-oxidation system in Pseudomonas aeruginosa. Microbiology 147(Pt 2):363–372 [DOI] [PubMed] [Google Scholar]
- 37. Shevchenko A, Tomas H, Havlis J, Olsen JV, Mann M. 2007. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc. 1:2856–2860 [DOI] [PubMed] [Google Scholar]
- 38. Wiegant W, de Bont J. 1980. A new route for ethylene glycol metabolism in Mycobacterium E44. Microbiology 120:325–331 [Google Scholar]
- 39. Yadav G, Gupta V. 2000. Synthesis of glyoxalic acid from glyoxal. Process Biochem. 36:73–78 [Google Scholar]
- 40. Yanisch-Perron C, Vieira J, Messing J. 1985. Improved M13 phage cloning vectors and host strains: nucleotide sequences of the M13mp18 and pUC19 vectors. Gene 33:103–119 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.