

Abstract
Mycobacterium bovis isolates carry restricted allelic variation yet exhibit a range of disease phenotypes and host preferences. Conventional genotyping methods target small hypervariable regions of the M. bovis genome and provide anonymous biallelic information that is insufficient to develop phylogeny. To resolve phylogeny and establish trait-allele associations, we interrogated 75 M. bovis and 61 M. tuberculosis genomes for single nucleotide polymorphisms (SNPs), using iPLEX MassArray (Sequenom Inc., CA) technology. We indexed nucleotide variations in 306 genic and 44 intergenic loci among isolates derived from outbreaks in the United States from 1991 to 2010 and isolated from a variety of mammalian hosts. Two hundred six variant SNPs classified the 136 isolates and 4 previously sequenced strains (AF2122/97, BCG Pasteur, H37Rv, and CDC1551) into 5 major “SNP cluster groups.” M. bovis isolates clustered into three major lineages based on 118 variant SNPs, while 84 SNPs differentiated the M. bovis BCG lineage from the virulent isolates. Forty-nine of the 51 human M. tuberculosis isolates were identical at all 350 loci studied. Thus, SNP-based analyses resolved the genotypic differences within M. bovis strains and differentiated these strains from M. tuberculosis strains representing diversity in time and space, providing population genetic frameworks that may aid in identifying factors responsible for the wide host range and disease phenotypes of M. bovis.
INTRODUCTION
Bovine tuberculosis is a disease of significant economic importance in the developed world, affecting animal productivity and trade of animal products (12). The introduction of milk pasteurization and “test and slaughter” cattle control programs in the early 1900s was successful in eradicating bovine tuberculosis in most developed nations (12). In some countries, such as Ireland, the United States, and New Zealand, Mycobacterium bovis infections in wildlife serve as a reservoir of the pathogen, with severe consequences for livestock in those countries. Bovine tuberculosis in wildlife poses serious difficulties for control and eradication of this insidious infection and contributes to the maintenance of the infection and its periodic spillover to domesticated animals (19, 20). M. bovis infection is a zoonosis and is a major concern in pastoral settings of the developing world where the animal-human interface is close and HIV prevalence is high. A recent study of all human tuberculosis cases in the United States from 1995 through 2005 estimated that only 1.4% of cases were caused by M. bovis (15). In San Diego, CA, over 45% of all culture-confirmed tuberculosis cases in children and 8% of all tuberculosis cases were caused by M. bovis (15). M. bovis is unable to utilize glycerol as a carbon source, and because this carbon source is commonly used in culture media for M. tuberculosis, M. bovis needs supplementation with pyruvate. Thus, it is likely that M. bovis infections in humans are underreported. This implies that the true prevalence of M. bovis infections in humans is unknown, especially in developing countries where the animal-human interface is close. It is therefore important for public health policy makers to be able to differentiate human infections caused by M. bovis from those due to M. tuberculosis.
Differentiation of genetic variants has become an indispensable tool to study the evolution, epidemiology, and ecology of pathogenic organisms and to gain insights into host-pathogen interactions (3, 14). M. bovis belongs to the Mycobacterium tuberculosis complex (MTC) group of organisms, which are characterized by 99.9% nucleotide sequence identity and carry identical 16S rRNA genes and very restricted allelic variation in their structural genes (16, 17). In the postgenomic era, single nucleotide polymorphisms (SNPs) have emerged as a robust tool for delineating phylogenetic relationships between closely related strains of pathogenic bacteria, including M. tuberculosis (7, 10, 11). Besides being a rich primary source of genetic variation, SNPs are easy to assay and provide for large-scale population genetic studies (10, 11). A study by Garcia Pelayo et al. (9) discovered over 700 SNPs, by comparative genomic analysis of the virulent M. bovis strain AF2122/97 (from the United Kingdom) and the vaccine strain M. bovis BCG Pasteur (from the parent strain M. bovis Nocard, originally obtained from a cow with tuberculosis mastitis in France), that redefined the global BCG strain genealogy and distinguished between M. bovis isolates of French and British lineages.
In the present study, we used SNP genotyping analysis based on a subset of 350 SNP loci from these previously identified SNPs (9) to derive a population genetic framework for 75 M. bovis and 61 M. tuberculosis isolates from the United States, isolated from a wide range of host species from diverse geographic locations.
MATERIALS AND METHODS
Bacterial isolates.
A collection of 75 M. bovis isolates associated with bovine tuberculosis outbreaks in the United States from 1990 to 2009 and isolated from a variety of hosts—cattle (n = 25), deer (n = 6), elk (n = 10), elephants (n = 2), swine (n = 7), humans (n = 24), and the environment (n = 1)—were used for the study. Sixty-one M. tuberculosis isolates, from humans (n = 51), primates (n = 7), a bird (n = 1), and elephants (n = 2), were also included in the analysis. The 75 M. bovis strains and 61 M. tuberculosis strains are shown in Table 2, along with brief epidemiological information about these isolates. Some of the M. bovis isolates were derived from slaughterhouse surveillance cases within the United States known to trace back to various states in Mexico. All of these isolates have been characterized by spoligotyping and were made available from the APHIS-USDA culture collections (isolates 1 to 67) and the Public Health Research Institute Center (PHRI), Newark, NJ (isolates 68 to 136). The DNAs for these strains were isolated at APHIS-USDA, IA, and PHRI, Newark, NJ, using standard DNA extraction protocols for mycobacteria (1), and then were shipped to our lab. The whole-genome DNA samples were amplified in our lab by use of a Qiagen repli-G kit (Qiagen Inc., Valencia, CA) and were stored at −80°C until further use.
TABLE 2.
Metadata on the isolates used for SNP analysis
| Isolate group and no. | Isolate IDa | Host—yr of isolation | State, city, or countryd | Spoligotype | VNTR profile | No. of IS6110 bands |
|---|---|---|---|---|---|---|
| M. bovis isolates from APHIS-USDA, Ames, IA (n = 57) | ||||||
| 1 | HC2045T | Cattle | TX | SB0673 | 25237452534 | |
| 2 | 08-5055 | Cattle | CA | SB0140 | 25215452534 | |
| 3 | 08-4513 | Cattle | TX | SB0971 | 25237452534 | |
| 4 | 08-2906 | Cattle | TX | SB0121 | 23326442232 | |
| 5 | 08-2630 | Cattle | MN | SB0271 | 25237452534 | |
| 6 | 08-2431 | Cattle | CA | SB0121 | 23326442232 | |
| 7 | 08-0955 | Cattle | MI | SB0815 | 23237552533 | |
| 8 | 08-0168 | Cattle | OK | SB0673 | 25237452534 | |
| 9 | 07-6182 | Cattle | SD | SB0152 | 25336442635 | |
| 10 | 07-5545 | Cattle | NM | SB0673 | 25237452534 | |
| 11 | 07-3557 | Cattle | MI | SB0145 | 23237552533 | |
| 12 | 07-3280 | Deer | MN | SB0271 | 25237452534 | |
| 13 | 07-1437 | Cattle | OK | SB0327 | 25134452323 | |
| 14 | 07-0608 | Cattle | MN | SB0271 | 25237452534 | |
| 15 | 06-8471 | Cattle | TX | SB0121 | 23326442232 | |
| 16 | 06-6855 | Cattle | MI | SB0145 | 23237552533 | |
| 17 | 06-3641 | Deer | MN | SB0271 | 25237452534 | |
| 18 | 06-4034 | Cattle | MI | SB0145 | 23237572533 | |
| 19 | 06-2501 | Cattle | TX | SB0265 | 23335432534 | |
| 20 | 04-0901 | Cattle | MX | SB0673 | 25245452534 | |
| 21 | 04-3121 | Cattle | TX | SB1040 | 25237552533 | |
| 22 | 03-5025 | Cattle | TX | SB0140 | 25234452534 | |
| 23 | 03-2620 | Cattle | CA | SB1345 | 25336442542 | |
| 24 | 03-0196 | Cattle | CA | SB0673 | 25237452432 | |
| 25 | 95-1315 | Deer | MI | SB0145 | 23237552533 | |
| 26 | 91-2299 | Deer | NY | SB1069 | 25337441535 | |
| 27 | 09-4591 | Deer | MN | SB0271 | 25237452534 | |
| 28 | Hbo-5 | Environment | CA | SB1040 | 25237552533 | |
| 29 | Hbo-7 | Human | CA | SB0145 | 25237472533 | |
| 30 | Hbo-11 | Human | CA | SB1040 | 25238352533 | |
| 31 | Hbo-13 | Human | CA | Unregisteredb | 25336442642 | |
| 32 | 92-3043 | Elk | NY | SB0265 | 23335432534 | |
| 33 | 94-0704 | Elk | MT | SB0265 | 23335432534 | |
| 34 | 94-2161 | Elk | MT | SB0265 | 23335432534 | |
| 35 | 95-0059 | Elk | MO | SB1069 | 25337441535 | |
| 36 | 97-2516 | Feral swine | HI | SB0145 | 25247542533 | |
| 37 | 97-3839 | Elk | WI | SB0265 | 23335432534 | |
| 38 | 98-1511 | Elk | KS | SB0265 | 23335432534 | |
| 39 | 99-3877 | Feral swine | HI | SB0815 | 25247542533 | |
| 40 | 00-0121 | Elk | WI | SB0265 | 23335432534 | |
| 41 | 00-2550 | Elk | WI | SB0265 | 23335432534 | |
| 42 | 00-5477 | Elephant | DC | SB0134 | 25432422535 | |
| 43 | 00-5480 | Elephant | DC | SB0134 | 25435422535 | |
| 44 | 02-1372 | Feral swine | HI | SB0145 | 25247542533 | |
| 45 | 03-5734 | Feral swine | HI | SB0145 | 25247542533 | |
| 46 | 05-5341 | Human | NY | SB0673 | 25237442534 | |
| 47 | 05-5354 | Human | NY | SB0673 | 25237442534 | |
| 48 | 06-4387 | Feral swine | HI | SB0145 | 25247542533 | |
| 49 | 07-6292 | Cattle | MX | SB0673 | 25237452534 | |
| 50 | 09-3461 | Elk | NE | SB0265 | 23335432534 | |
| 51 | 09-6071 | Elk | NE | SB0265 | 23335432534 | |
| 52 | 07-6293 | Cattle | MX | SB0121 | 23336442535 | |
| 53 | 07-7253 | Cattle | MX | SB0145 | 25237551533 | |
| 54 | 07-7901 | Human | MX | SB1828 | 26336442635 | |
| 55 | 07-11680 | Feral swine | HI | SB0145 | 25247542533 | |
| 56 | 08-5155 | Feral swine | HI | SB0145 | 25247542533 | |
| 57 | 08-8559 | Deer | NY | SB1069 | 25337441534 | |
| M. tuberculosis sensu stricto isolates from APHIS-USDA, Ames, IA (n = 10) | ||||||
| 58 | 09-0453 | Primate | PA | SB1622 | 24438452534 | |
| 59 | 09-0454 | Primate | PA | SB1622 | 24438452534 | |
| 60 | 09-0455 | Primate | PA | SB1622 | 24438452534 | |
| 61 | 09-3381 | Avian | TX | Unregisteredb | 44344221637 | |
| 62 | 06-8534 | Monkey | WI | Unregisteredb | 74354421658 | |
| 63 | 09-4348 | Primate | NV | Unregisteredb | 24257242256 | |
| 64 | 05-4400 | Elephant | TX | Unregisteredb | 34242121527 | |
| 65 | 09-8103 | Primate | SC | Unregisteredb | 54343421858 | |
| 66 | 09-7906 | Primate | NV | Unregisteredb | 44332221537 | |
| 67 | 97-0352 | Elephant | IL | Unregisteredb | 34314221639 | |
| M. bovis isolates from PHRI, NJ (n = 9) | ||||||
| 68 | 21540 | Human—2006 | NYC | SB0173 | 1 | |
| 69 | 24489 | Human—2009 | NYC | SB1157 | 1 | |
| 70 | 20701 | Human—2006 | NYC | SB0242 | 1 | |
| 71 | 23244 | Human—2008 | NYC | SB0172 | 1 | |
| 72 | 23396 | Human—2008 | NJ | SB0333 | 2 | |
| 73 | 26515 | Human—2009 | NYC | SB0509 | 1 | |
| 74 | 16862 | Human—2003 | NYC | SB0846 | 1 | |
| 75 | 23217 | Human—2008 | NYC | SB1847 | 1 | |
| 76 | 16158 | Human—2002 | Egyptc | SB1160 | 2 | |
| Isolates of M. bovis from PHRI, NJ, typed as strain BCG by SNP analysis (n = 9) | ||||||
| 77 | 20658 | Human—2005 | NYC | SB0025 | 1 | |
| 78 | 21068 | Human—2006 | NYC | SB0025 | 2 | |
| 79 | 24644 | Human—2009 | NYC | SB0025 | 1 | |
| 80 | 20051 | Human—2005 | NY | SB0025 | 1 | |
| 81 | 9682 | Human—1999 | Russiac | SB0025 | 2 | |
| 82 | 9680 | Human—1999 | Russiac | SB0025 | 2 | |
| 83 | 7768 | Human—1997 | NH | SB0025 | 1 | |
| 84 | 22666 | Human—2007 | NYC | SB0025 | 1 | |
| 85 | 20502 | Human—2005 | NYC | SB0025 | 1 | |
| M. tuberculosis isolates from PHRI, NJ, typed by SNP analysis and previously identified as M. bovis (n = 2) | ||||||
| 86 | 24282 | Human—2008 | NYC | SB0228 | 3 | |
| 87 | 18463 | Human—2003 | NYC | SB0242 | 3 | |
| M. tuberculosis sensu stricto isolates from PHRI, NJ (n = 49) | ||||||
| 88 | 6401 | Human—1997 | NJ | SB0075 | 1 | |
| 89 | 6519 | Human—1997 | NJ | SB0075 | 1 | |
| 90 | 7396 | Human—1997 | NYC | SB0075 | 1 | |
| 91 | 8072 | Human—1998 | NJ | SB0075 | 1 | |
| 92 | 9723 | Human—1999 | NJ | SB0075 | 1 | |
| 93 | 10225 | Human—1999 | NJ | SB0075 | 1 | |
| 94 | 10425 | Human—1999 | NJ | SB0075 | 1 | |
| 95 | 13260 | Human—2001 | NJ | SB0075 | 1 | |
| 96 | 14435 | Human—2002 | NYC | SB0075 | 1 | |
| 97 | 17147 | Human—2003 | NYC | SB0075 | 1 | |
| 98 | 17781 | Human—2003 | NYC | SB0075 | 1 | |
| 99 | 17996 | Human—2003 | NYC | SB0075 | 1 | |
| 100 | 22813 | Human—2007 | NYC | SB0075 | 1 | |
| 101 | 23257 | Human—2008 | NYC | SB0075 | 1 | |
| 102 | 24091 | Human—2008 | NJ | SB0075 | 1 | |
| 103 | 18928 | Human—2004 | NYC | SB0030 | 1 | |
| 104 | 6365 | Human—1997 | NY | SB0030 | 3 | |
| 105 | 8423 | Human—1998 | NYC | SB0030 | 3 | |
| 106 | 9688 | Human—1999 | NYC | SB0030 | 3 | |
| 107 | 13602 | Human—2001 | NYC | SB0030 | 3 | |
| 108 | 19733 | Human—2005 | NYC | SB0030 | 3 | |
| 109 | 21946 | Human—2007 | NYC | SB0030 | 3 | |
| 110 | 23771 | Human—2008 | NYC | SB0030 | 3 | |
| 111 | 25703 | Human—2009 | NYC | SB0030 | 3 | |
| 112 | 913 | Human—1992 | NYC | SB0030 | 3 | |
| 113 | 5401 | Human—1996 | NJ | SB0009 | 3 | |
| 114 | 9319 | Human—1998 | NJ | SB0075 | 3 | |
| 115 | 9904 | Human—1999 | NJ | SB0075 | 3 | |
| 116 | 6478 | Human—1997 | NJ | SB0009 | 2 | |
| 117 | 9136 | Human—1998 | NYC | SB0009 | 2 | |
| 118 | 12721 | Human—2000 | NJ | SB0009 | 2 | |
| 119 | 13571 | Human—2001 | NYC | SB0009 | 2 | |
| 120 | 18104 | Human—2003 | NYC | SB0009 | 2 | |
| 121 | 19711 | Human—2005 | NYC | SB0009 | 2 | |
| 122 | 22665 | Human—2007 | NYC | SB0009 | 2 | |
| 123 | 26033 | Human—2010 | NYC | SB0009 | 2 | |
| 124 | 11064 | Human—1997 | NJ | SB0030 | 2 | |
| 125 | 24991 | Human—2009 | NYC | SB0075 | 2 | |
| 126 | 5855 | Human—1997 | NJ | SB0075 | 2 | |
| 127 | 7061 | Human—1997 | NJ | SB0075 | 2 | |
| 128 | 8433 | Human—1998 | NJ | SB0075 | 2 | |
| 129 | 9140 | Human—1998 | NJ | SB0075 | 2 | |
| 130 | 9898 | Human—1999 | NJ | SB0075 | 2 | |
| 131 | 10296 | Human—1999 | NJ | SB0075 | 2 | |
| 132 | 10443 | Human—1999 | NJ | SB0075 | 2 | |
| 133 | 11055 | Human—1999 | NJ | SB0075 | 2 | |
| 134 | 21307 | Human—2006 | NYC | SB0075 | 2 | |
| 135 | 24810 | Human—2009 | NJ | SB0075 | 2 | |
| 136 | 15069 | Human—2002 | NJ | SB0075 | 2 |
For isolates 1 to 67, the first two digits represent the year of isolation, except for isolate 1 (early 1990s) and isolates 28 to 31 (not known).
For isolates with newly identified, unregistered spoligotypes, the octal codes are (in order of appearance in the table) 676713676777600, 000000000003771, 000000000003771, 777777774413771, 777774077560731, 000000000003761, 777717607760771, and 776377777760771.
One of three isolates from locations outside the United States.
NYC, New York City.
SNP selection and identification.
Based on a recent genomewide analysis of the sequenced M. bovis AF2122/97 and M. bovis BCG Pasteur strains, a total of 782 SNPs were identified by Garcia Pelayo et al. (9). These 782 sites included transitions, transversions, insertions or deletions, and block substitutions (where a block of >1 bp replaces another). We selected a set of 350 target loci from this data set, including SNPs in genic (n = 44) and intergenic (n = 306) regions, choosing loci that showed diversity among the M. bovis isolates associated with outbreaks in the United Kingdom and France (see the supplemental material for a list of the target loci). Selected SNP sites were representative of the whole M. bovis genome (Fig. 1A). The information on these SNP positions, as it occurs in the M. bovis BCG genome, is available through the study of Garcia Pelayo et al., with genomic position, locus, and gene/intergenic presence identified. Using this information, we located and verified each of the SNPs in the genome sequences of M. bovis strain AF2122/97 and the M. tuberculosis strains H37Rv and CDC1551, available freely through the public database of the National Center for Biotechnology Information (NCBI) (www.ncbi.nlm.nih.gov).
Fig 1.

(A) Genomewide distribution of 350 target SNP loci across the 4.3-Mb M. bovis genome. (B) Genomewide distribution of 206 variant SNPs across the 4.3-Mb M. bovis genome. The 59 synonymous substitutions are shown in blue, the 120 nonsynonymous changes are shown in red, and the 27 intergenic SNPs are shown in green. Both panels were generated using the DNAPlotter tool from the Artemis genome browser and annotation tool.
Single nucleotide polymorphism-based genotyping.
Genotyping was performed using iPLEX chemistry on the MassArray genotyping platform (Sequenom Inc., San Diego, CA) available at the BioMedical Genomics Center, University of Minnesota. During the iPLEX reaction, oligonucleotide primers anneal directly adjacent to the SNP of interest. SNPs were queried using oligonucleotides that annealed at position −1 relative to the base of interest; allele-specific extension products were then analyzed via matrix-assisted laser desorption ionization mass spectrometry to identify the base at each SNP position across the panel of strains. Allele-specific extension products were then produced by single-base extension of the oligonucleotide with terminator nucleotides, each of unique mass. Multiplexed iPLEX assays comprising 1 to 8 assays per iPLEX reaction were designed to detect 350 single nucleotide base changes, using the Sequenom Assay Design v.3.0.2.0 package. Allele-specific products resulting from the iPLEX reaction were desalted through the addition of an anion-exchange resin and then analyzed by matrix-assisted laser desorption ionization–time of flight (MALDI-TOF) mass spectrometry. Genotypes were assigned in real time and then evaluated using SpectroCALLER and SpectroACQUIRE software (Sequenom Inc., San Diego, CA).
Phylogenetic analysis.
The 206 variant SNPs were concatenated into a string of single characters, resulting in a single 206-bp sequence for each strain. Sequence alignment and phylogenetic analysis were carried out using MEGA 4.1 software (20; http://www.megasoftware.net/).
RESULTS
SNP diversity analysis.
We genotyped 350 loci on the M. bovis genome and identified 206 (Fig. 1A and B) to be variable among 75 M. bovis and 61 M. tuberculosis isolates. Information on these 350 loci was also obtained for four previously sequenced strains, including M. bovis AF2122/97, M. bovis BCG Pasteur, M. tuberculosis H37Rv, and M. tuberculosis CDC1551. Between the 75 M. bovis isolates alone, 202 SNPs were identified, among which 118 SNPs (Table 1) were observed between the disease-associated M. bovis isolates. Of these 118 variant SNPs, 91 were genic SNPs and 27 were in the intergenic region. A second set of 84 genic SNPs (Fig. 2) was able to distinguish isolates of the attenuated vaccine lineage of M. bovis strain BCG from the virulent isolates. A set of 9 isolates previously genotyped as M. bovis by IS6110 profiling and spoligotyping was submitted to the study. However, SNP analysis identified these isolates as the BCG Pasteur vaccine strain. Further analysis for region of difference 1 (RD1) among these 9 isolates (19) confirmed them to be M. bovis BCG strains.
TABLE 1.
Variant SNPs among 67 virulent Mycobacterium bovis isolates representing three cluster groups on the phylogenetic treef
| Isolate no. | SNP locus | Nucleotide at: |
||
|---|---|---|---|---|
| SCG-1 | SCG-2 | SCG-3 | ||
| 1 | atpH | C | C | G |
| 2 | corA | C | T | T |
| 3 | dha | A | A | G |
| 4 | fadD28 | T | T | G |
| 5 | fadD9-1 | A | A | G |
| 6 | fadD9-2 | A | G | G |
| 7 | fadE20 | C | C | G |
| 8 | fadE27 | A | G | G |
| 9 | galT | G | G | C |
| 10 | glmU | C | C | T |
| 11 | glnA3 | A | A | G |
| 12 | glnB | A | G | G |
| 13 | glnD | C | C | A |
| 14 | glpKb | G | C | C |
| 15 | hisD | G | A | A |
| 16 | ispD | C | C | T |
| 17 | lpqB | G | G | C |
| 18 | lpqF | A | A | G |
| 19 | mmpL12 | C | C | T |
| 20 | mmsA | C | C | A |
| 21 | narL | G | G | C |
| 22 | narU | T | T | C |
| 23 | nuoB | C | C | A |
| 24 | PE31 | T | T | C |
| 25 | pks12 | T | T | C |
| 26 | pks6b | G | T | T |
| 27 | pks7 | A | A | G |
| 28 | PPE21 | G | A | A |
| 29 | recBb | G | A | A |
| 30 | rhlE | C | T | T |
| 31 | sodC | A | A | G |
| 32 | speE | A | G | G |
| 33 | sseA | A | G | G |
| 34 | thioA | T | T | C |
| 35 | Mb0085 | T | T | C |
| 36 | Mb0139 | Deletion | Deletion | G |
| 37 | Mb0228c | T | T | C |
| 38 | Mb0278c | T | T | C |
| 39 | Mb0353 | Deletion | Deletion | A |
| 40 | Mb0378c | A | G | G |
| 41 | Mb0393 | C | A | A |
| 42 | Mb0458c | A | G | G |
| 43 | Mb0849 | G | A | A |
| 44 | Mb0899c | C | T | T |
| 45 | Mb0937 | T | T | C |
| 46 | Mb0963 | T | T | C |
| 47 | Mb1013 | A | G | G |
| 48 | Mb1150c | C | G | G |
| 49 | Mb1365c | A | G | G |
| 50 | Mb1427 | G | A | A |
| 51 | Mb1707 | G | C | C |
| 52 | Mb1885c | T | C | C |
| 53 | Mb1904 | A | G | G |
| 54 | Mb2029 | C | T | T |
| 55 | Mb2204c | G | T | T |
| 56 | Mb2381c | T | C | C |
| 57 | Mb2410c | C | T | T |
| 58 | Mb2441c | T | T | C |
| 59 | Mb2492c | G | G | A |
| 60 | Mb2501c | T | T | C |
| 61 | Mb2507c | G | G | A |
| 62 | Mb2512c | T | C | C |
| 63 | Mb2550 | A | G | G |
| 64 | Mb2596 | T | T | C |
| 65 | Mb2661 | G | C | C |
| 66 | Mb2996 | T | C | C |
| 67 | Mb3193 | C | T | T |
| 68 | Mb3328 | A | G | G |
| 69 | Mb3421c | T | T | C |
| 70 | Mb3478 | A | C | C |
| 71 | Mb3619c | C | C | T |
| 72 | Mb3718c | T | C | C |
| 73 | Tb39.8-1 | C | C | G |
| 74 | Tb39.8-2 | C | C | T |
| 75 | cysN | T | T | T/Ca |
| 76 | dacB1 | A | A | A/Ga |
| 77 | fusA2b | A | A | A/Ga |
| 78 | PPE31 | T | T | C/Tb |
| 79 | typA | T | T | C/Tb |
| 80 | Mb0007 | G | G | A/Gb |
| 81 | Mb0244 | T | T | C/Tb |
| 82 | Mb1072c | T | T | T/Ga |
| 83 | Mb1404 | A | A | A/Gc |
| 84 | Mb1495 | C | C | C/Td |
| 85 | Mb1794c-1 | G | G | G/Aa |
| 86 | Mb1794c-2 | T | T | T/Ca |
| 87 | Mb1860 | T | T | T/Ca |
| 88 | Mb2067c | A | A | A/Ga |
| 89 | Mb2261 | A | A | A/Ga |
| 90 | Mb2439c | C | C | T/Cb |
| 91 | Mb2558 | A | A | G/Ae |
Allele observed in only two isolates (16158 and 23217).
Allele observed in only five isolates (95-0059, 08-8559, 91-2299, 00-5480, and 00-5477).
Allele observed only in isolate 08-2906.
Allele observed only in isolate 16158.
Alllele observed in only four isolates (08-8559, 91-2299, 00-5480, and 00-5477).
Isolates 92 to 118 had SNPs in intergenic regions IGR1, IGR2, IGR3, IGR4, IGR5, IGR6, IGR7, IGR8, IGR9, IGR10, IGR11, IGR12, IGR13, IGR14, IGR15, IGR16, IGR17, IGR18, IGR19, IGR20, IGR21, IGR22, IGR23, IGR24, IGR25, IGR26, and IGR27, respectively. No SNP cluster group-specific distribution was observed for the 27 SNPs in intergenic regions.
Fig 2.
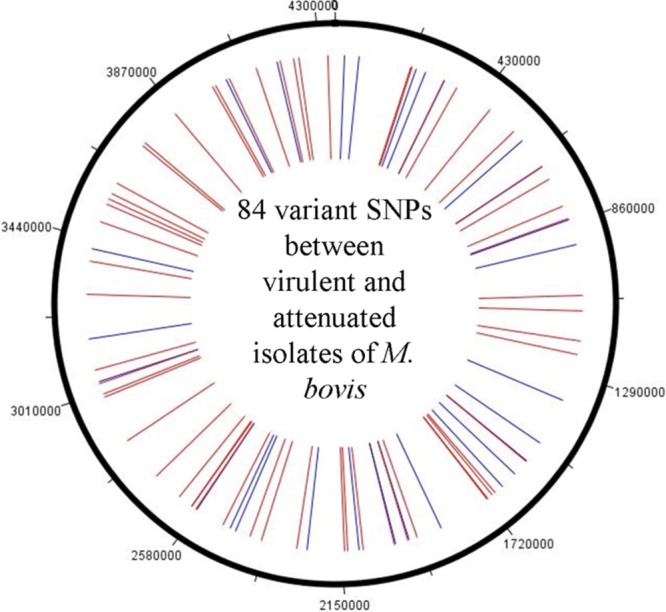
Genomewide distribution of the 84 genic SNPs that separate the 67 virulent M. bovis isolates from the 10 attenuated BCG lineage isolates. The synonymous changes are shown in blue, and the nonsynonymous changes are shown in red.
Forty-nine of the 51 M. tuberculosis isolates from human hosts were identical at all 350 loci examined and clustered in a single clade. The two variant human M. tuberculosis isolates (Table 2) used in the study (isolates 86 [18463] and 87 [24282]) were submitted as human M. bovis strains, classified as having spoligotypes SB0228 and SB0242, respectively, and carried 3 copies of IS6110. These 2 isolates varied from the other human M. tuberculosis isolates at 11 of the 350 typed loci, including the genic SNPs at katG codon 463 and Mb1794c (n = 2) codons 72 and 132 and eight SNPs that were in the intergenic region (IGR1, IGR14, IGR15, and IGR17 to IGR21). These two isolates also lacked the M. bovis signature SNP at pncA codon 57. Further probing for the presence of RD9 (RD9 loci included Rv2073c) confirmed the isolates as M. tuberculosis, not M. bovis. Ten M. tuberculosis isolates derived from animal hosts had nearly identical SNP profiles to those of the human isolates, except at 19 loci. These included 5 genic SNPs, at katG codon 463, oxyR codon 78, fadD9 codon 600, and Mb1794 (n = 2) codons 72 and 132, and 14 intergenic SNPs (IGR1, IGR14, IGR15, and IGR17 to IGR27).
M. bovis phylogeny.
A consensus phylogenetic tree was derived using the maximum parsimony algorithm with 1,000 bootstrap replicates. The 206 variant SNPs resolved the 136 isolates used in this study, as well as 4 sequenced strains (AF2122/97, BCG Pasteur, H37Rv, and CDC1551), into 5 major genetic clusters, or “SNP cluster groups”: 4 groups of M. bovis isolates and 1 cluster that included all M. tuberculosis isolates (Fig. 3). Based on 118 SNPs, M. bovis isolates were differentiated into three principal SNP cluster groups. These included isolates from both animal and human hosts. However, variations observed in the intergenic SNPs were not lineage specific. The fourth group, which exclusively clustered 9 human M. bovis isolates along with the vaccine strain BCG Pasteur, differed at 84 genic loci from the virulent isolates (Fig. 2). Strain AF2122/97 (M. bovis strain from the United Kingdom) clustered with M. bovis isolates in SNP cluster group 1. M. tuberculosis strains CDC1551 and H37Rv clustered with cluster group 5, which included all M. tuberculosis isolates used in our analysis. Isolates from Michigan (n = 5), Minnesota (n = 5), and Hawaii (n = 7) clustered within their respective SNP cluster groups. Isolates from states other than Michigan, Minnesota, and Hawaii carried diverse genetic profiles, as evidenced by their distribution across all 3 M. bovis SNP cluster groups. All elk isolates (n = 10) from a variety of geographic locations, including Missouri, Montana, Nebraska, New York, Wisconsin, and Kansas, and isolated from 1992 to 2009, clustered in SNP cluster group 3. The fourth SNP cluster group of M. bovis isolates was unique in that it included only BCG strains from humans. These isolates shared the SNP genotype of BCG Pasteur. This unique SNP signature permits differentiation of BCG from virulent M. bovis isolates.
Fig 3.
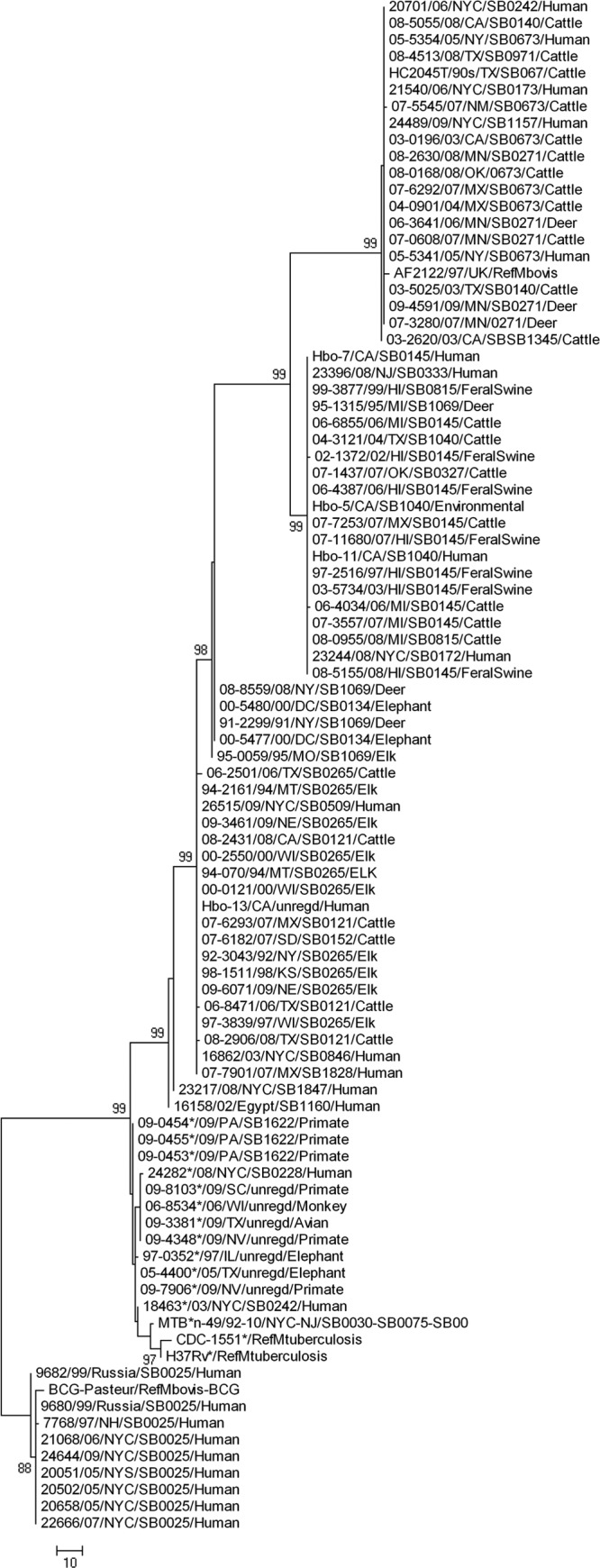
Consensus linear phylogenetic tree generated using the maximum parsimony algorithm with 1,000 bootstrap replicates, using MEGA4.1 software. The tree represents the SNP genotypes of 75 M. bovis (confirmed by our SNP analysis) and 61 M. tuberculosis (includes 2 isolates previously identified as M. bovis) isolates, along with the sequences of virulent M. bovis strain AF2122/97, M. bovis vaccine strain BCG Pasteur, and two M. tuberculosis strains (H37Rv and CDC1551). The tree is rooted to the isolates of the M. bovis BCG strain. Five major SNP cluster groups, i.e., cluster groups 1 through 5 (top to bottom), indicative of the five “SNP genotypes,” were identified. The first 3 cluster groups are the major M. bovis SNP cluster groups, which include 66 virulent isolates from various hosts and geographic locations. Cluster group 1 (the first 20 isolates, along with strain AF2122/97) has all the isolates from Minnesota, cluster group 2 (20 isolates) includes all the isolates from Michigan and Hawaii, and cluster group 3 (26 isolates) has all the elk isolates, which vary in time and geographic origin. Cluster group 5 (at the bottom of the tree) includes the 9 human M. bovis isolates, which cluster together with the attenuated BCG Pasteur strain. Cluster group 4 (isolates marked with asterisks) includes all the M. tuberculosis isolates from animal and human hosts, including the two sequenced strains. The details of the isolates that represent the five SNP cluster groups are listed in Table 2.
Analysis of synonymous, nonsynonymous, and intergenic SNPs.
Among the 206 SNPs, we identified both intergenic (n = 27) and genic (n = 179) SNPs that were distributed evenly around the genome (Fig. 1B). Of the 179 genic SNPs, 59 were synonymous changes and 120 were nonsynonymous mutations. The ratio of synonymous SNPs to nonsynonymous SNPs was 1:2.
Variations in spoligotyping, VNTR, and IS6110 RFLP profiles of strains.
All isolates were previously characterized (Table 2) by spoligotyping and variable-number tandem-repeat (VNTR) profiling (APHIS-USDA culture collections) or by IS6110 restriction fragment length polymorphism (RFLP) profiling and spoligotyping (PHRI culture collections). We examined the relationship between phylogenetic lineages of these isolates and their spoligotyping/VNTR/RFLP profiles. M. bovis isolates with common spoligotype patterns or VNTR/RFLP profiles clustered together. However, each of the 3 SNP cluster groups was represented by more than one spoligotype or VNTR/RFLP profile. Similarly, the 49 M. tuberculosis isolates from humans that were identical by their SNP profiles had diverse IS6110 and spoligotype profiles. The human M. tuberculosis sensu stricto isolates that had identical SNP genotypes in this study were isolated from 1992 to 2010, mainly from the New York City and New Jersey areas. Seven of the 10 M. tuberculosis isolates from animal hosts had unique, unregistered spoligotypes and variant VNTR profiles.
DISCUSSION
Genomewide SNPs of M. bovis differentiate between isolates.
In a 2009 study by Garcia Pelayo et al. (9), 782 SNPs were identified across the entire genomes of M. bovis and M. bovis BCG. We derived information from their study on a subset of 350 SNPs and used this information to generate a population genetic framework for outbreak-associated isolates from the United States. Molecular variation and outbreak tracking of M. tuberculosis complex isolates typically employs IS6110 profiling, spoligotyping, or mycobacterial interspersed repetitive unit-VNTR (MIRU-VNTR) analysis. While these targets and tools are considered sufficient for molecular epidemiology, they are unable to sufficiently index the population genetic structure of this genus, as they represent small hypervariable regions within the genome that generally evolve at higher rates than the rest of the genome. Thus, SNPs have been used to define the extent of genetic diversity in M. tuberculosis and other pathogenic mycobacteria, providing insights into the evolution, pathogenicity, and molecular epidemiology of tuberculosis globally. A previous study identified 782 SNPs between the virulent M. bovis strain AF2122/97 and the vaccine strain BCG Pasteur, among which 158 SNPs separated all the M. bovis strains of French lineage from the M. bovis strains of British lineage. This may also be a reflection of the fact that all M. bovis BCG strains originated from a French strain of M. bovis, while the sequenced strain AF2122 is of British origin. These findings further suggest that the attenuation of M. bovis BCG may go beyond large sequence polymorphisms and that examining the functional consequences of variant SNPs may aid in understanding the shortcomings of BCG as a vaccine (3–6).
The current study documents 206 SNPs across the genome that are sufficient to resolve M. bovis phylogeny and genetic relatedness into three major lineages and sets the platform for downstream studies involving phenotypic characterization of factors affecting virulence and pathogenesis. Furthermore, among the 206 SNPs, we noted a 2:1 ratio of nonsynonymous SNPs to synonymous SNPs, similar to the case in genomewide SNP studies of M. tuberculosis (10, 11) which have indicated recent emergence of these strains resulting from a population bottleneck.
SNPs differentiate lineages of M. bovis and M. tuberculosis.
In the current study, SNP-based phylogenetic analysis was able to differentiate M. bovis strains—both virulent strains and the attenuated BCG strains that conventional genotyping techniques fail to resolve. This is important in clinical diagnosis of tuberculosis, because the BCG vaccine, although considered safe, is known to cause disease in immunocompromised hosts (2, 18, 21, 22).
Furthermore, SNP genotyping resolved misclassifications of 2 M. tuberculosis isolates as M. bovis and of 9 M. bovis BCG isolates identified as virulent M. bovis by previous typing techniques. SNPs in oxyR codon 78, katG codon 463, and pncA codon 57 identified isolates as either M. tuberculosis or M. bovis, and 206 SNP profiles differentiated M. tuberculosis from M. bovis BCG. Thus, M. bovis infections and outbreaks in the United States, documented for humans by use of conventional methods, have a tendency toward misclassification. This further implies that genomewide SNP sets may serve as powerful markers for the differentiation of biotypes within the M. tuberculosis complex. Furthermore, unambiguous classification would be useful for indexing zoonotic transmission of M. bovis in rural areas of the developing world, where the animal-human interface is intensifying as land use patterns are changing.
SNP-based spatial and host associations.
Bovine tuberculosis is a reemerging infectious disease in the United States, where the deer population has been identified as a potential reservoir for M. bovis infections (13, 14). Within the United States, the state of Michigan has had one of the longest ongoing bovine tuberculosis epidemics. Our deer and cattle tuberculosis isolates from Michigan (n = 5), collected between 1995 and 2008, and from Minnesota (n = 5), isolated between 2006 and 2009, clustered in distinct lineages specific to geographic origin. The spatial specificity of lineages is suggestive of a founder effect where, upon introduction, the strains evolved independently in the deer and cattle populations. Evidence suggests that the Michigan strain of M. bovis spilled over into the white-tailed deer population in the 1930s and has since been maintained in that population (14).
A significant observation in our study was that Hawaiian isolates shared their SNP genotype with isolates from other geographic locations, despite little or no epidemiological linkage. Despite depopulation and restocking of cattle on the islands of Hawaii in an attempt to eradicate bovine tuberculosis, periodic cattle infections have been detected. Epidemiological studies suggest that feral pigs serve as a reservoir of infection in that state (8). The fact that the feral swine isolates from Hawaii share a SNP genotype with cattle and deer isolates from other geographic locations, such as Michigan, Texas, California, New York, Oklahoma, and Mexico, suggests that the organism was introduced into that swine population by infected deer or cattle relocated from other states, leading to its rapid spread and maintenance within the new feral hosts.
All animal isolates identified as M. tuberculosis were identical to the human counterparts at all loci examined, except for 19 loci. This was likely due to intrahost adaptive changes that may have occurred in the animal hosts after transmission from humans or suggests that animal species are susceptible only to some subtypes of M. tuberculosis. Our data also provide robust information on diversity among M. bovis isolates and documents loci that can be used to differentiate M. tuberculosis from M. bovis within animals, between animals and humans, and between M. bovis and M. bovis BCG.
Elk M. bovis isolates from 6 states of the United States and representing the period from 1992 to 2009 were the only strains to cluster in a single clade, suggesting a degree of host specificity for this genotype. These isolates also showed identical spoligotypes and VNTR profiles, suggesting a clonal spread of a single strain in this host, despite geographic and temporal distance. It is likely that the particular SNP genotype is elk adapted and highly virulent for this host, or elk may be exclusively highly susceptible to this genotype of M. bovis. The presence of several SNP genotypes among isolates from cattle, deer, and humans suggests multiple sources of introduction of infection in these host species. The identification of SNP genotypes from Mexico in every clade suggests a high level of diversity in and interspecies transmission of isolates from that location.
We conclude that SNP-based genotyping is able to resolve misclassification of the infecting species, to identify patterns of host or spatial associations, and to differentiate lineages and phylogenetic structures among M. bovis strains. With the increasing availability of multiple whole-genome sequences, SNP identification will add considerably to phylogenetic analysis and evolutionary studies. We present a snapshot of the diversity and structure of strains, using 206 “informative” SNPs; further investigations should derive from comparisons of whole-genome sequences of isolates from diverse geographic locations. We propose that the SNP cluster groups identified in this study should facilitate investigations of functional and biological variation between and within the isolates of these five phylogenetic lineages.
Supplementary Material
ACKNOWLEDGMENTS
We thank the Biomedical Genomics Center at the University of Minnesota for providing the MassArray SNP typing service. We thank the Minnesota Supercomputing Institute for access to supercomputing resources and genetic analysis software.
This study was supported by the Rapid Agricultural Response Fund (Agriculture Experiment Station) and by a USDA-Specials grant awarded to S.S.
Footnotes
Published ahead of print 19 September 2012
Supplemental material for this article may be found at http://jcm.asm.org/.
REFERENCES
- 1. Amaro A, Duarte E, Amado A, Ferronha H, Botelho A. 2008. Comparison of three DNA extraction methods for Mycobacterium bovis, Mycobacterium tuberculosis and Mycobacterium avium subsp. avium. Lett. Appl. Microbiol. 47:8–11 [DOI] [PubMed] [Google Scholar]
- 2. Azzopardi P, Bennett CM, Graham SM, Duke T. 2009. Bacille Calmette-Guerin vaccine-related disease in HIV-infected children: a systematic review. Int. J. Tuberc. Lung Dis. 13:1331–1344 [PubMed] [Google Scholar]
- 3. Behr MA. 2002. BCG—different strains, different vaccines? Lancet Infect. Dis. 2:86–92 [DOI] [PubMed] [Google Scholar]
- 4. Behr MA. 2001. Comparative genomics of BCG vaccines. Tuberculosis (Edinb.) 81:165–168 [DOI] [PubMed] [Google Scholar]
- 5. Behr MA, Small PM. 1997. Has BCG attenuated to impotence? Nature 389:133–134 [DOI] [PubMed] [Google Scholar]
- 6. Behr MA, Small PM. 1999. A historical and molecular phylogeny of BCG strains. Vaccine 17:915–922 [DOI] [PubMed] [Google Scholar]
- 7. Filliol I, et al. 2006. Global phylogeny of Mycobacterium tuberculosis based on single nucleotide polymorphism (SNP) analysis: insights into tuberculosis evolution, phylogenetic accuracy of other DNA fingerprinting systems, and recommendations for a minimal standard SNP set. J. Bacteriol. 188:759–772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Freier JE, Miller RS, Geter KD. 2007. Geospatial analysis and modelling in the prevention and control of animal diseases in the United States. Vet. Ital. 43:549–557 [PubMed] [Google Scholar]
- 9. Garcia Pelayo MC, et al. 2009. A comprehensive survey of single nucleotide polymorphisms (SNPs) across Mycobacterium bovis strains and M. bovis BCG vaccine strains refines the genealogy and defines a minimal set of SNPs that separate virulent M. bovis strains and M. bovis BCG strains. Infect. Immun. 77:2230–2238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gutacker MM, et al. 2006. Single-nucleotide polymorphism-based population genetic analysis of Mycobacterium tuberculosis strains from 4 geographic sites. J. Infect. Dis. 193:121–128 [DOI] [PubMed] [Google Scholar]
- 11. Gutacker MM, et al. 2002. Genome-wide analysis of synonymous single nucleotide polymorphisms in Mycobacterium tuberculosis complex organisms: resolution of genetic relationships among closely related microbial strains. Genetics 162:1533–1543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Michel AL, Muller B, van Helden PD. 2010. Mycobacterium bovis at the animal-human interface: a problem, or not? Vet. Microbiol. 140:371–381 [DOI] [PubMed] [Google Scholar]
- 13. O'Brien DJ, et al. 2001. Tuberculous lesions in free-ranging white-tailed deer in Michigan. J. Wildl. Dis. 37:608–613 [DOI] [PubMed] [Google Scholar]
- 14. O'Brien DJ, et al. 2002. Epidemiology of Mycobacterium bovis in free-ranging white-tailed deer, Michigan, USA, 1995–2000. Prev. Vet. Med. 54:47–63 [DOI] [PubMed] [Google Scholar]
- 15. Rodwell TC, et al. 2010. Tracing the origins of Mycobacterium bovis tuberculosis in humans in the USA to cattle in Mexico using spoligotyping. Int. J. Infect. Dis. 14(Suppl 3):e129–e135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Scorpio A, et al. 1997. Rapid differentiation of bovine and human tubercle bacilli based on a characteristic mutation in the bovine pyrazinamidase gene. J. Clin. Microbiol. 35:106–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sreevatsan S, et al. 1997. Restricted structural gene polymorphism in the Mycobacterium tuberculosis complex indicates evolutionarily recent global dissemination. Proc. Natl. Acad. Sci. U. S. A. 94:9869–9874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Talbot EA, Perkins MD, Silva SF, Frothingham R. 1997. Disseminated bacille Calmette-Guerin disease after vaccination: case report and review. Clin. Infect. Dis. 24:1139–1146 [DOI] [PubMed] [Google Scholar]
- 19. Talbot EA, Williams DL, Frothingham R. 1997. PCR identification of Mycobacterium bovis BCG. J. Clin. Microbiol. 35:566–569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tamura K, Dudley J, Nei M, Kumar S. 2007. MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol. Biol. Evol. 24:1596–1599 [DOI] [PubMed] [Google Scholar]
- 21. Toida I. 2011. BCG, a tuberculosis vaccine—Japanese contribution. Kekkaku 86:603–606 [PubMed] [Google Scholar]
- 22. Waddell RD, et al. 2001. Bacteremia due to Mycobacterium tuberculosis or M. bovis, bacille Calmette-Guerin (BCG) among HIV-positive children and adults in Zambia. AIDS 15:55–60 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.