

Abstract
Leishmania (Viannia) braziliensis causes three main types of American tegumentary leishmaniasis (ATL), localized cutaneous leishmaniasis (CL), mucosal leishmaniasis (ML), and disseminated leishmaniasis (DL). All forms are observed among individuals of Corte de Pedra, Brazil. We previously used random amplified markers to identify a multiclonal population among L. (V.) braziliensis isolates from ATL patients, defining parasite clades associated with different clinical syndromes. Herein we compared sequences of random amplified markers to identify genotypes of L. (V.) braziliensis recovered from lesions of CL, ML, and DL patients. Six polymorphic genomic loci were sequenced from 35 parasite isolates. Single-nucleotide polymorphisms (SNPs) and insertions-deletions (indels) at each locus allowed us to segregate the L. (V.) braziliensis population according to haplotypes. Several SNPs, indels, and haplotypes were significantly associated with an increased risk of DL. Molecular genotyping may provide markers to identify L. (V.) braziliensis strains likely to cause this emerging, hard-to-treat form of ATL.
INTRODUCTION
Leishmaniasis constitutes a spectrum of human diseases caused by protozoa belonging to the genus Leishmania, divided into two subgenera, Leishmania, which is present in both the Old World and the New World, and Viannia, which is restricted to the New World. Productive infections may either be asymptomatic or lead to different clinical syndromes involving internal organs, skin, and/or mucosal surfaces (3). Leishmania (Viannia) braziliensis is endemic in Latin America and causes at least three distinct clinical types of American tegumentary leishmaniasis (ATL), localized cutaneous leishmaniasis (CL), mucosal leishmaniasis (ML), and disseminated leishmaniasis (DL) (3, 4, 6, 9, 29). The most striking feature that differentiates CL from either ML or DL is the degree of metastasis of lesions from the original inoculation site. CL lesions are restricted to the entry site of the parasites, whereas ML is defined by spreading to mucosal surfaces of the upper digestive and airways tracts. DL is characterized by wide dissemination to distant cutaneous sites. Some individuals with DL develop hundreds of lesions throughout their body surfaces.
Many reports cite ML as the most common metastatic form of disease caused by L. (V.) braziliensis. It was previously more common, but ML currently develops in 4 to 5% of individuals with CL (2, 7, 17). Recently, in our study area, DL has emerged as the most prevalent metastatic form of L. (V.) braziliensis infection (26). The present study is based on the hypotheses that different strains of L. (V.) braziliensis are prone to cause the different forms of ATL, and it is the spread of a DL-predisposing strain that is responsible for the increasing incidence of this new cutaneous syndrome.
Individuals living in the region of Corte de Pedra in the state of Bahia, Northeast Brazil, can be afflicted by any of the three forms of ATL due to L. (V.) braziliensis. However, DL has only recently been recognized in Corte de Pedra (6, 9). Careful longitudinal study of this form of leishmaniasis has revealed a recent increase in the absolute numbers of DL cases, an increase in the relative proportion of DL cases compared to the total number ATL cases, and a pattern of geographic spread of the disease across a wider region of Corte de Pedra over the past 2 decades (16, 26, 29). Indeed, the geographic distribution of DL cases differs from that of ML in Corte de Pedra, which led us to investigate the regional risks of these two forms of L. (V.) braziliensis disease (26).
One of the first clues that different parasite strains may contribute to the different ATL forms was in vitro evidence showing that the total antigen from L. (V.) braziliensis isolated from a DL patient caused peripheral blood mononuclear cells to release larger amounts of the proinflammatory cytokines gamma interferon and tumor necrosis factor alpha than the total antigen of a parasite from a subject with CL (14). Parallel investigations revealed that parasites isolated from ATL patients could be distinguished according to randomly amplified polymorphic genetic markers. These markers were used to define separate subpopulations or clades of L. (V.) braziliensis from Corte de Pedra. Of the five subpopulations described, parasites from individuals with DL were significantly enriched in clades A and D, while clade B contained mostly isolates from CL subjects and isolates from subjects with ML were significantly more frequently associated with clade C (27). These observations led to our hypothesis that genetically distinct strains of L. (V.) braziliensis may determine the clinical outcomes of L. (V.) braziliensis infections.
Here, to strengthen the molecular evidence for the above hypothesis, we compared the sequences of several markers that distinguished clades in our prior study. PCR amplicons generated by random amplification of polymorphic DNA (RAPD) from the genomic DNA of 35 L. (V.) braziliensis isolates from ATL patients in Corte de Pedra were cloned, sequenced, and compared. Using this strategy, we identified six polymorphic loci among parasite strains. Tests for association of single-nucleotide polymorphisms (SNPs), insertion-deletions (indels), and SNP/indel-based haplotypes at those loci with each ATL form were performed. The findings indicated a strong association between DL and particular strains of L. (V.) braziliensis. These data suggest not only that genetically distinct parasite strains are partly responsible for the emerging spread of DL but that the progression of these strains through the region can be tracked by using molecular markers.
MATERIALS AND METHODS
Study area.
Corte de Pedra is composed of 20 municipalities in a rural area located in the southeastern region of the state of Bahia, in the northeast of Brazil. Lutzomyia (Nyssomyia) whitmany and L. (N.) intermedia sandflies that transmit L. (V.) braziliensis are endemic in Corte de Pedra. Residents of this area work mostly in agriculture, which is often carried out in primary or secondary forests. There is little population migration in or out of this region. Study participants' mean time of residence at their addresses at the time of diagnosis and parasite sampling was 17 years; 90% of the study participants lived on farms.
Parasites.
Thirty-five L. (V.) braziliensis isolates were derived from 17 individuals with CL, 9 with ML, and 9 with DL diagnosed in the medical clinic of Corte de Pedra, Bahia, Brazil. The three types of leishmaniasis were defined as follows. CL consisted of an ulcerated skin lesion at a single body site with no more than two secondary or satellite lesions, without clinical evidence of mucosal involvement. ML was defined as the presence of an inflamed or ulcerated mucosal lesion at a site that was noncontiguous with any cutaneous lesion. DL was defined as 10 or more skin lesions of mixed types (acneiform, papular, nodular, and/or ulcerated) located on two or more body parts (head, trunk, arms, and legs). In addition to these clinical criteria, cases had two or three of the following: positive delayed-type hypersensitivity skin response to leishmania antigen (Montenegro test), positive serology, and/or parasites identified in tissue biopsy specimens by histopathology.
The L. (V.) braziliensis isolates used in the present study were cultured from aspirates of the borders of skin or mucosal lesions. Aspirate material was immediately suspended in biphasic liver infusion tryptose-Novy, McNeal, and Nicolle medium and incubated at 26°C for 1 to 2 weeks. The suspension was then transferred to Schneider's medium complemented with 10% heat-inactivated fetal calf serum and 2 mM l-glutamine and incubated at 26°C for up to 2 weeks. Species determination was based upon HSP70 PCR-restriction fragment length polymorphism (11, 18) and later confirmed by a serial real-time quantitative PCR assay system (31). Parasites were frozen without further subculture in 10% dimethyl sulfoxide–90% growth medium in liquid nitrogen and thawed prior to testing for genotypes.
Selection of genomic L. (V.) braziliensis targets.
For sequence-based typing method development, several RAPD-generated bands that had been useful for subtyping of L. (V.) braziliensis isolates from Corte de Pedra were chosen to be analyzed further (see Fig. 1). These loci were reamplified by RAPD from each of nine isolates of L. (V.) braziliensis by using previously described primers and protocols (27). The resulting DNA bands were cloned into the pCR 2.1-TOPO vector (Invitrogen Inc.) according to the manufacturer's instructions. Plasmids were prepared from five representative bacterial clones from each transformation reaction, and then their inserts were sequenced. After alignment of homologous sequences, six polymorphic loci were identified, which were used in the remainder of the study.
Fig 1.
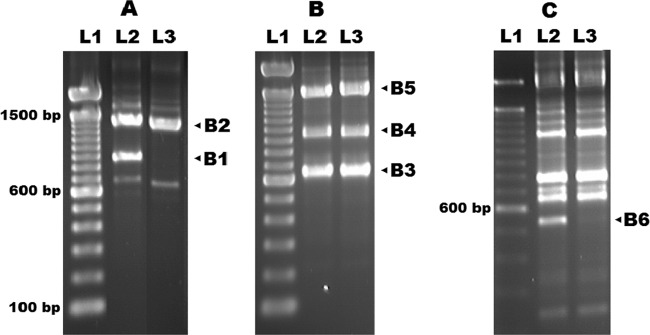
Electrophoretic patterns of L. (V.) braziliensis genomic DNA amplicons generated using three RAPD protocols (separately depicted in panels A, B, and C) previously described (27). Marked bands were excised, cloned, sequenced, and then aligned among nine L. (V.) braziliensis isolates from Corte de Pedra to detect polymorphisms. The polymorphic loci detected among parasites from Corte de Pedra were CHR 24/3074 (chromosome 24, starting at position 3074) (B1), CHR 26/765 (B2), CHR 28/195696 (B3), CHR 28/425451 (B4), CHR 32/1356278 (B5), and CHR 35/335652 (B6). The CHR 26/765 locus was found duplicated on chromosome 33 starting at position 1476284. Lanes: L1, molecular size markers; L2 and L3, examples of L. (V.) braziliensis isolates. Estimated nucleic acid lengths in base pairs are shown.
PCR amplification of selected targets and cloning.
On the basis of the sequences generated with the above-described preliminary assays, six primer pairs were newly designed (Table 1) and used to amplify the selected loci from 35 L. (V.) braziliensis isolates. In these reactions, 3 ng template genomic DNA was incubated with 1 pmol each of the reverse and forward primers. The MgCl2 concentration and annealing temperatures used are listed in Table 1. Amplification conditions were 5 min at 95°C, followed by 35 cycles of 30 s at 95°C, 1 min at the annealing temperature listed in Table 1 (48 to 58°C), and 2 min at 72°C, followed by a final extension at 72°C for 10 min. Amplicons were electrophoresed, and then their corresponding bands were extracted from agarose gels and cloned into pCR 2.1-TOPO. Plasmids containing each of the six amplicons from each of the 35 isolates of L. (V.) braziliensis were transformed into competent Escherichia coli DH5α. Plasmid minipreps were generated from six representative bacterial clones per locus (23).
TABLE 1.
Chromosomal positions of the six polymorphic loci identified in L. (V.) braziliensis from Corte de Pedraa
| Locus (band)b | Primers used for locus amplification by PCR | Chromosome | Location in chromosome | Melting (annealing) temp (°C) | [MgCl2] (mM) | Amplicon size (bp) | Gene product overlapped |
|---|---|---|---|---|---|---|---|
| CHR 24/3074 (B1) | 5′-GGACTGGAGTGATCGAA, 5′-TGGCTCAAGTGTCGCA | 24 | 3074–3848 | 55 | 1.0 | 779 | Pyruvate dehydrogenase kinase |
| CHR 26/765c (B2) | 5′-GAAATAAGAGACGAAAAGGC, 5′-TGACGTAGGGGTAGAGAAAC | 26 | 765–1425 | 58 | 1.0 | 660 | |
| CHR 28/195696 (B3) | 5′-GGACTGGAGTCTGCTTTATTTG, 5′-ACCAGGGTCGTGAACATGCT | 28 | 195696–196300 | 55 | 1.5 | 618 | Nucleobase/nucleoside transporter |
| CHR 28/425451 (B4) | 5′-TAAGGTGAACAAGAAGAATC, 5′-CTGCTCGCTTGCTTTC | 28 | 425451–426067 | 48 | 1.0 | 622 | Long-chain fatty acid-coenzyme A ligase protein |
| CHR 32/1356278 (B5) | 5′-GTACGCACATGAGCCCGGAT, 5′-GCGGCGTATCACGCACTTTT | 32 | 1356278–1356777 | 55 | 2.5 | 498 | |
| CHR 35/335652 (B6) | 5′-TTATAGACGTGACACAGCG, 5′-AGCAGTCCGGAGTTGT | 35 | 335652–336148 | 56 | 1.0 | 497 |
The complete L. (V.) braziliensis MHOM/BR/75/M2904 genome, accessed at the http://blast.ncbi.nlm.nih.gov/Blast.cgi website, was used.
Corresponding band in Fig. 1.
This locus is duplicated in chromosome 33 at position 1476284.
Sequence analysis.
Plasmid inserts were sequenced with a primer complementary to the M13 vector sequence, followed by custom designed primers, which were necessary because of the large sizes of some original amplicons. Sequencing was performed at Macrogen Inc. (Seoul, South Korea). The data were analyzed by the MEGA 4.0 software package (28) to determine the consensus sequence at each locus and to compare alleles at homologous loci from different L. (V.) braziliensis isolates. Sequences from each of the six loci were analyzed for the occurrence of SNPs and/or indels uniquely present in isolates from the CL, ML, or DL phenotype.
Locations of polymorphic loci within L. (V.) braziliensis chromosomes.
In order to locate the detected polymorphic loci within the parasite's genome, the consensus sequence of each locus, obtained as outlined above, was blasted to the sequences of each of the chromosomes listed in the L. (V.) braziliensis MHOM/BR/75/M2904 complete genome, accessed at the website http://blast.ncbi.nlm.nih.gov/Blast.cgi. This allowed first the identification of the chromosome(s) containing the loci and then the determination of the starting point of each locus within these chromosomes.
Human subject approvals.
Studies were approved by Institutional Review Boards of the Federal University of Bahia and the University of Iowa. Protocols were approved by the U.S. NIH and by Comissão Nacional de Ética em Pesquisa in Brazil. The Brazilian Institutional Review Board is registered with the U.S. NIH. Written consent was obtained from all subjects that participated in the study.
Statistical analyses.
Differences in the distribution frequencies of each polymorphism among CL, ML, and DL cases were analyzed by Fisher's exact test, and the relative risk was calculated with a confidence level of 95%. All comparisons were considered significant at P < 0.05. Statistical analyses were performed with the STATA 10.0 software package (StataCorp LP).
RESULTS
Identification of polymorphisms among randomly selected loci of L. (V.) braziliensis from Corte de Pedra.
We previously described the complex population structure of L. (V.) braziliensis obtained from individuals living in Corte de Pedra. Different subpopulations or clades of parasites were identified on the basis of genotypes defined by RAPD data (27). In order to determine specific molecular sequences that would refine our ability to detect different strains among parasites isolated from ATL patients in that region, bands from the RAPD electrophoretic profiles of nine isolates were cloned and sequenced. Polymorphisms were found in six genomic sequences amplified by three RAPD protocols from the L. (V.) braziliensis isolates. The electrophoretic patterns of the sequences amplified with these three protocols are shown in Fig. 1A, B, and C. The loci generating the six polymorphic amplicons fell into distinct chromosomes at diverse locations within these chromosomes. Their distributions and genes contained within or overlapping with amplicons are shown in Table 1.
In order to amplify the targeted loci more specifically than by the RAPD strategy, new PCR primer sets were designed that were complementary to sequences flanking the polymorphic stretches found at each locus (Table 1). Using these primer pairs, each locus was reamplified from genomic DNA of L. (V.) braziliensis isolates, yielding amplicons that ranged from 400 to 800 bp in length. These amplicons were cloned into pCR2.1-TOPO, and the sequences of six clones representing each amplicon (i.e., locus) from each Leishmania isolate were determined.
The analysis of the 35 L. (V.) braziliensis isolates from different forms of ATL revealed a total of 27 SNPs and indels within the six polymorphic loci. Because some lesions may have harbored two or more parasite strains, we could not formally determine the allele frequencies. However, on the basis of the number of samples from which alleles were identified, we were able to determine major and minor alleles for each polymorphic chromosomal position (Table 2). Usually a predominant nucleotide could be found in the sample with substitutions or indels occurring in a smaller subset of the isolates. The strategy of sequencing cloned amplicons allowed us to identify haplotypes containing SNPs and indels at each locus. The frequencies of the most common haplotypes found are indicated in Table 3. We identified 17 different haplotypes distributed in the six loci, which could be detected in at least 10% of the L. (V.) braziliensis isolates.
TABLE 2.
SNPs/indels found per L. (V.) braziliensis locus studied and frequencies of their alleles in the sample
| Locus (band)a and polymorphism | Chromosomal position | Alleles (proportion of L. [V.] braziliensis isolates with each allele)b |
|---|---|---|
| CHR 24/3074 (B1) | ||
| SNP24/3099ag | 3099 | A/G (0.23/1) |
| SNP24/3120ga | 3120 | G/A (0.23/1) |
| SNP24/3136ta | 3136 | T/A (0.23/1) |
| SNP24/3197ag | 3197 | A/G (0.23/1) |
| Indel24/3207a- | 3207 | A/indel (0.23/1) |
| SNP24/3537ct | 3537 | C/T (0.23/1) |
| SNP24/3672tc | 3672 | T/C (0.23/1) |
| CHR 26/765c (B2) | ||
| SNP26/808ca | 808 | C/A (0.90/0.42) |
| Indel26/815-c | 815 | Indel/C (1/0.17) |
| SNP26/909ga | 909 | G/A (0.77/0.77) |
| SNP26/953tg | 953 | T/G (0.77/0.81) |
| SNP26/1007ga | 1007 | G/A (0.77/0.81) |
| SNP26/1050ac | 1050 | A/C (0.77/0.81) |
| CHR 28/195696 (B3) | ||
| SNP28/195926gc | 195926 | G/C (0.77/0.83) |
| SNP28/196016cg | 196016 | C/G (0.77/0.83) |
| SNP28/196100ga | 196100 | G/A (0.77/0.86) |
| CHR 28/425451 (B4) | ||
| SNP28/425481tc | 425481 | T/C (0.8/0.74) |
| SNP28/425737ct | 425737 | C/T (0.83/0.71) |
| Indel28/425996t- | 425996 | T/indel (0.69/0.83) |
| CHR 32/1356278 (B5) | ||
| SNP32/1356321gt | 1356321 | G/T (0.97/0.31) |
| SNP32/1356500ca | 1356500 | C/A (0.94/0.61) |
| Indel32/1356597c- | 1356597 | C/indel (1/0.12) |
| SNP32/1356598ca | 1356598 | C/A (0.94/0.53) |
| SNP32/1356599ca | 1356599 | C/A (1/0.12) |
| CHR 35/335652 (B6) | ||
| SNP35/335735ca | 335735 | C/A (1/0.23) |
| SNP35/336018tc | 336018 | T/C (1/0.08) |
| SNP35/336094ca | 336094 | C/A (1/0.06) |
Corresponding band in Fig. 1.
Proportion of isolates presenting the particular allele at the specified locus.
This locus is duplicated in chromosome 33 at position 1476284.
TABLE 3.
Haplotypes found in at least 10% of the parasite isolates for each L. (V.) braziliensis locus studied
| Locus | Haplotypes (proportions of L. [V.] braziliensis isolates with haplotypes) |
|---|---|
| CHR 24/3074 | gaag-tc (0.97), agtaact (0.23) |
| CHR 26/765a | c-agac (0.75), c-gtga (0.44), a-gtga (0.37) |
| CHR 28/195696 | cga (0.83), gcg (0.66) |
| CHR 28/425451 | cct (0.71), cc- (0.11), tt- (0.8) |
| CHR 32/1356278 | gcccc (0.82), tcccc (0.12), gacac (0.37), gaccc (0.21), tacac (0.19) |
| CHR 35/335652 | ctc (1.0), cta (0.23) |
This locus is duplicated in chromosome 33 at position 1476284.
Association of SNPs, indels, and haplotypes with forms of ATL.
The sequence differences identified at those loci enabled us to more precisely define parasite strains and re-examine our hypothesis that genetically distinct L. (V.) braziliensis strains are significantly associated with different disease syndromes. The frequencies of CL, ML, and rapidly emerging DL were determined among individuals infected with parasites containing each of the SNPs, indels, or haplotypes. The risk of having a particular clinical disease type given a particular genotype of the parasite (expressed as a risk ratio [RR]) was evaluated (see Tables 4 to 7).
TABLE 4.
Association between SNP/indel haplotypes in L. (V.) braziliensis loci investigated and DL phenotype
| Locus or loci [haplotype(s)]a | f (count)b | f′ (count)c | RR | Pd | 95% CI | AFe |
|---|---|---|---|---|---|---|
| CHR 28/425451 [cc- (+)] | 0.44 (4/9) | 0 (0/26) | 6.2 | 0.002 | 2.8–13.8 | 0.8 |
| CHR 28/425451 [cct (+)] | 1 (9/9) | 0.62 (16/26) | 1.6 | 0.03 | 1.2–2.2 | 0.8 |
| CHR 28/425451 [cct (+), cc- (+)] | 0.44 (4/9) | 0 (0/26) | 6.2 | 0.002 | 2.8–13.8 | 0.8 |
| CHR 28/425451 [cct (+), tt- (+)] | 0.89 (8/9) | 0.38 (10/26) | 7.5 | 0.01 | 1.0–54.2 | 0.9 |
| CHR 24/3074 [agtaact (−)], CHR 28/425451 [cct (+)] | 1 (9/9) | 0.38 (10/26) | 8.6 | 0.004 | 1.2–60.7 | 0.9 |
| CHR 24/3074 [agtaact (−)], CHR 32/1356278 [gacac (+)], CHR 28/425451 [cct (+)] | 0.50 (4/8) | 0.17 (4/24) | 3.0 | 0.08 | 1.0–9.3 | 0.7 |
| CHR 24/3074 [agtaact (−)], CHR 28/195696 [cga (+)], CHR 28/425451 [cct (+)] | 1 (9/9) | 0.35 (9/26) | 9.5 | 0.002 | 1.3–67.3 | 0.9 |
(+) indicates that the presence of the haplotype in L. (V.) braziliensis isolates was evaluated for association with DL. (−) indicates that the absence of the haplotype in L. (V.) braziliensis isolates was evaluated for association with DL.
f, frequency of DL isolates with the polymorphism. In parentheses is the number of DL cases presenting the L. (V.) braziliensis haplotype(s) divided by the total number of DL cases that had isolates evaluated in that particular analysis.
f ′, frequency of ML and CL isolates with the polymorphism. In parentheses is the number of CL and ML cases presenting the L. (V.) braziliensis haplotype(s) divided by the total number of CL and ML cases that had isolates evaluated in that particular analysis.
Fisher's exact test.
AF, fraction of DL cases attributable to the polymorphism.
TABLE 7.
Association between combined SNPs in L. (V.) braziliensis and risk of DL phenotype
| Combined SNP polymorphisms | f (count)a | f′ (count)b | RR | Pc | 95% CI | AFd |
|---|---|---|---|---|---|---|
| SNP24/3672c only, SNP28/196100ga, SNP28/425737ct, SNP32/1356598ca | 0.50 (4/8) | 0 (0/24) | 7 | 0.002 | 2.8–17.3 | 0.9 |
| SNP24/3672c only, SNP28/425737ct, SNP32/1356598ca | 0.75 (6/8) | 0.08 (2/24) | 9 | 0.0008 | 2.2–36.0 | 0.9 |
| SNP24/3672c only, SNP28/196100ga, SNP28/425737ct | 0.67 (6/9) | 0.19 (5/26) | 4.4 | 0.01 | 1.3–14.3 | 0.8 |
| SNP24/3672c only, SNP28/425737ct | 0.89 (8/9) | 0.27 (7/26) | 10.7 | 0.002 | 1.5–76.3 | 0.9 |
| SNP24/3672c only, SNP32/1356598ca | 0.75 (6/8) | 0.29 (7/24) | 4.4 | 0.03 | 1.0–18.4 | 0.8 |
| SNP28/196100ga, SNP28/425737ct | 0.67 (6/9) | 0.19 (5/26) | 4.4 | 0.01 | 1.3–14.3 | 0.8 |
| SNP28/425737ct, SNP32/1356598ca | 0.75 (6/8) | 0.12 (3/24) | 7.7 | 0.002 | 1.9–31.2 | 0.9 |
| SNP32/1356598ca, SNP26/909g only | 0.37 (3/8) | 0 (0/22) | 5.4 | 0.01 | 2.4–11.9 | 0.8 |
f, frequency of DL isolates with the polymorphism. In parentheses is the number of DL cases presenting the L. (V.) braziliensis SNPs divided by the total number of DL cases that had isolates evaluated in that particular analysis.
f′, frequency of ML and CL isolates with the polymorphism. In parentheses is the number of CL and ML cases presenting the L. (V.) braziliensis SNPs divided by the total number of CL and ML cases that had isolates evaluated in that particular analysis.
Fisher's exact test.
AF, fraction of DL attributable to that combination of polymorphisms.
Several SNPs, indels, and haplotypes were significantly associated with DL. Six of the 17 haplotypes that were present in at least 10% of the L. (V.) braziliensis isolates in the sample associated with DL (Table 4). The locus most often associated with DL was CHR28/425451. Isolates with haplotypes cc-, cct, and tt- at that locus were associated with RRs between 1.6 and 7.5 for DL. Isolates containing CHR28/425451 cct combined with CHR28/195696 cga but lacking CHR24/3074 agtaact comprised 100% of the DL cases and had a RR of 9.5 for this form of ATL. Further strengthening the link between the parasite genetic background and the form of disease, the estimated fraction of the ATL patients who developed DL associated with these haplotypes was above 80%. Interestingly, some of the haplotype combinations that showed a significantly increased risk of DL (Table 4) conversely displayed significantly decreased risks (i.e., RRs of <1.0) of ML and CL outcomes (Table 5). This suggests a high specificity of these molecular markers of disseminated leishmaniasis.
TABLE 5.
Levels of association between the ML and CL phenotypes and the L. (V.) braziliensis SNP/indel haplotypes that showed an association with DLa
| Locus or loci [haplotype(s)]b | ML |
CL |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| f (count)c | f′ (count)d | RR | Pe | 95% CI | f (count)f | f′ (count)g | RR | Pe | 95% CI | |
| CHR 28/425451 [cc- (+)] | 0 (0/9) | 0.15 (4/26) | 0 | 0.28 | — | 0 (0/17) | 0.22 (4/18) | 0 | 0.058 | — |
| CHR 28/425451 [cct (+)] | 0.67 (6/9) | 0.73 (19/26) | 0.8 | 0.51 | 0.2–2.6 | 0.59 (10/17) | 0.83 (15/18) | 0.6 | 0.11 | 0.3–1.1 |
| CHR 28/425451 [cct (+), cc- (+)] | 0 (0/9) | 0.15 (4/26) | 0 | 0.28 | — | 0 (0/17) | 0.22 (4/18) | 0 | 0.058 | — |
| CHR 28/425451 [cct (+), tt- (+)] | 0.33 (3/9) | 0.58 (15/26) | 0.5 | 0.19 | 0.1–1.6 | 0.41 (7/17) | 0.61 (11/18) | 0.7 | 0.20 | 0.3–1.3 |
| CHR 24/3074 [agtaact (−)], CHR 28/425451 [cct (+)] | 0.22 (2/9) | 0.65 (17/26) | 0.2 | 0.03 | 0.06–1 | 0.47 (8/17) | 0.61 (11/18) | 0.7 | 0.31 | 0.4–1.5 |
| CHR 24/3074 [agtaact (−)], CHR 32/1356278 [gacac (+)], CHR 28/425451 [cct (+)] | 0.11 (1/9) | 0.30 (7/23) | 0.4 | 0.26 | 0.05–2.5 | 0.2 (3/15) | 0.29 (5/17) | 0.7 | 0.42 | 0.3–2 |
| CHR 24/3074 [agtaact (−)], CHR 28/195696 [cga (+)], CHR 28/425451 [cct (+)] | 0.22 (2/9) | 0.61 (16/26) | 0.3 | 0.049 | 0.06–1.1 | 0.41 (7/17) | 0.61 (11/18) | 0.7 | 0.20 | 0.3–1.3 |
See Table 4.
(+) indicates that the presence of the haplotype in L. (V.) braziliensis isolates was evaluated for associations; (−) indicates that the absence of the haplotype in L. (V.) braziliensis isolates was evaluated for associations.
f, frequency of ML isolates with the polymorphism. In parentheses is the number of ML cases presenting the L. (V.) braziliensis haplotype(s) divided by the total number of ML cases that had isolates evaluated in that particular analysis.
f′, frequency of DL and CL isolates with the polymorphism. In parentheses is the number of DL and CL cases presenting the L. (V.) braziliensis haplotype(s) divided by the total number of DL and CL cases that had isolates evaluated in that particular analysis.
Fisher's exact test.
f, frequency of CL isolates with the polymorphism. In parentheses is the number of CL cases presenting the L. (V.) braziliensis haplotype(s) divided by the total number of CL cases that had isolates evaluated in that particular analysis.
f′, frequency of DL and ML isolates with the polymorphism. In parentheses is the number of DL and ML cases presenting the L. (V.) braziliensis haplotype(s) divided by the total number of DL and ML cases that had isolates evaluated in that particular analysis.
Associations between parasite genotypes and increased risks of ML and CL did not reach significance in the study sample. The associations that were closest to significance are displayed in Table 6. The SNP that approached significance with the highest RR for ML was SNP32/1356500c, in isolates where no other allele could be found for that position (i.e., c-only isolates). This SNP revealed an RR of 3.1 (confidence interval [CI], 0.9 to 10.2; P = 0.06) for this form of ATL (Table 6). The only haplotype that approached a significant risk of ML was CHR 24/3074 agtaact (RR of 2.7; CI, 0.9 to 7.7; P = 0.09).
TABLE 6.
Association between SNPs and combinations of SNPs in L. (V.) braziliensis and risk of CL or ML phenotypes
| Combined SNP polymorphisms | ATL form | f (count)a | f′ (count)b | RR | Pc | 95% CI | AFd |
|---|---|---|---|---|---|---|---|
| SNP24/3672tc | ML | 0.44 (4/9) | 0.15 (4/26) | 2.7 | 0.09 | 0.9–7.7 | 0.6 |
| SNP32/1356500c only | ML | 0.67 (6/9) | 0.29 (7/24) | 3.1 | 0.06 | 0.9–10.2 | 0.7 |
| SNP32/1356598c only | ML | 0.67 (6/9) | 0.39 (9/23) | 2.3 | 0.1 | 0.7–7.5 | 0.5 |
| SNP32/1356598c only, SNP24/3672tc | ML | 0.33 (3/9) | 0.08 (2/26) | 3.0 | 0.09 | 1.1–8.2 | 0.7 |
| SNP26/953g only | CL | 0.93 (13/14) | 0.70 (12/17) | 3.1 | 0.1 | 0.5–19.4 | 0.7 |
f, frequency of either ML or CL isolates with the polymorphism. In parentheses is the number of ML or CL cases presenting the L. (V.) braziliensis SNPs divided by the total number of ML or CL cases that had isolates evaluated in that particular analysis.
f′, frequency of either non-ML or non-CL isolates with the polymorphism. In parentheses is the number of either non-ML or non-CL cases presenting the L. (V.) braziliensis SNPs divided by the total number of either non-ML or non-CL cases that had isolates evaluated in that particular analysis.
Fisher's exact test.
AF, fraction of ML or CL attributable to that combination of polymorphisms.
Given the emerging nature of DL and the fact that it is more refractory to treatment than other forms of tegumentary leishmaniasis in the study area, combinations of SNPs from different loci were tested for increased association with disseminated leishmaniasis. Only combinations of SNPs and indels that approached significant (P < 0.1) RRs of greater than 2 when tested individually were used in this analysis. Eight combinations of polymorphisms that were significantly (0.03 > P > 0.0008) associated with DL were detected. These were associated with RRs between 4.4 and 10.7 (Table 7). These associations of SNPs may be candidates to be explored in diagnostic tools for identifying patients with an increased risk of developing this severe and difficult-to-treat form of leishmaniasis.
DISCUSSION
Previous studies have directly or indirectly underscored the influences of both the parasite and the host in the clinical manifestations of disease caused by L. (V.) braziliensis (5, 7, 8, 12, 22, 24–27). In the present report, we tested whether genotypes of L. (V.) braziliensis isolated from ATL patients would associate with a form of disease, indicating a cause-effect relationship between the strain of the infecting parasite and the outcome of the infection. We analyzed the nucleotide sequences of polymorphic loci discovered by RAPD in our previous study (27). We compared these sequences from isolates that were derived from individuals from Corte de Pedra, Brazil, with the three common forms of disease caused by L. (V.) braziliensis. Our study revealed SNPs and indels within six polymorphic loci in the parasite's genome that are associated with DL. Strong associations indicating increased risks of CL and ML were not detected.
DL has been rapidly emerging, with increasing spread and incidence in the vicinity of Corte de Pedra over the past 2 decades (6, 9, 16, 26, 29). We previously noted differences between the geographic distributions of DL versus that of ML, the other disseminated form of ATL due to L. (V.) braziliensis (26). We also reported that new DL cases tend to occur in clusters with recent cases of the disease (26). These observations suggest that DL may result predominantly from infection with a particular strain or group of related strains of the parasite. The large number of SNPs, indels, and haplotypes found associated with DL in the present study reinforces this hypothesis and suggests that this parasite subpopulation may have recently emerged in the Corte de Pedra region.
ML has long been described as a disease determined by several factors, among which are the duration of CL (15), nutritional status (15), and genetic background of the human host (8, 20). The detection of few near-significant associations between L. (V.) braziliensis genotypes and ML likely reflects its multifactorial nature, as well as the small sample size, which decreased the power of the present study to detect such associations. ML has been diagnosed in Corte de Pedra for many decades, and it is plausible that the population of strains that predispose to this form of disseminated disease is more diverse than that for recently emerged DL. Thus, a greater number of ML-derived parasite isolates may be needed to detect strains of parasites that carry an increased risk of this disease.
Localized skin ulcers constitute the most common presentation of ATL caused by any strain of L. (V.) braziliensis. Their long-standing presence in the region may reflect a diverse group of parasites causing CL. Furthermore, given that a single or a few skin ulcers usually precede the more severe metastatic ATL outcomes, early diagnosis of infection may cause some patients infected with strains associated with either DL or ML to be treated before dissemination has occurred. Thus, parasite isolates and strains from the subgroup of patients classified as CL are expected to be more heterogeneous than those labeled as ML and DL. As discussed above for ML, a greater number of CL-derived parasite isolates may be needed to detect associations.
It should be noted that each L. (V.) braziliensis isolate used in this study consisted of a first-pass culture of promastigotes isolated from patients' specimens. The Leishmania spp., and particularly the visceralizing species, notoriously lose virulence characteristics rapidly in culture (13, 21). In order to preserve parasites as close as possible to the true infection-induced parasites, there was no attempt to clone the isolates. Because some lesions may have harbored two or more parasite strains, this strategy would minimize difficulties due to the differential growth of one isolate over others and minimize the chromosomal rearrangements that can occur during Leishmania culture (10). Recent experimental and natural evidence has shown that there is genetic exchange in the Leishmania species, and this most likely occurs within the sand fly vector (1, 19).
Although it is advisable to minimize artifacts, the characterization of whole isolates rather than clones complicates the interpretation of the number of alleles identified in each parasite. Many isolates presented with two alleles at single loci. This could indicate heterozygosity of a single strain or the presence of two or more homozygous or heterozygous strains within the isolates. There were only seven instances in which more than two alleles could be detected. This affected seven different isolates (i.e., 20%) in the study sample. In six of these isolates, there were three different haplotypes either in locus CHR 28/425451 or in locus CHR 32/1356278. In only one isolate, four haplotypes could be found in locus CHR 32/1356278. Such findings indicate coinfections with multiple strains, the presence of a single strain that was aneuploid within these isolates, or both (30). Hence the data in Table 2 are presented as the frequency of alleles in cultured isolates and cannot imply the true allele frequencies in the total population.
Nonetheless, it is noteworthy that in the vast majority of the isolates tested, only one or two haplotypes could be found at each locus. We speculate that this may indicate that most human infections are caused either by single strains of L. (V.) braziliensis or by combinations of the parasite within which there may be dominant strains.
Although several significant associations between genotypes of L. (V.) braziliensis and DL could be detected, no single SNP displayed a significant RR for any form of ATL. This probably reflects the small sample employed in the study, which lacked power to discriminate the nucleotides that would be more common at single polymorphic sites among different clinical phenotypes. Nevertheless, the significantly increased risk of DL that several haplotypes presented helps shed light on the still elusive role of the parasite strain in determining the outcome of ATL, while the detected associations between combinations of as few as two SNPs at different loci and DL offer targets that may be tested with simple PCR-based tools that may prove useful in the diagnostic management of ATL.
Our previous studies of tegumentary leishmaniasis in the Corte de Pedra region revealed that (i) different subpopulations or clades of L. (V.) braziliensis determined by RAPD were associated with distinct clinical outcomes (27); (ii) whole antigen of L. (V.) braziliensis isolated from distinct ATL forms, namely, CL and DL, caused distinct in vitro reactivity from peripheral blood mononuclear cells of ATL patients (14); and (iii) different forms of severe ATL (i.e., ML and DL) displayed distinct geographic distributions (26). The present findings reflect these previous studies and suggest that certain strains in that complex L. (V.) braziliensis population may constitute major risks of DL.
Future studies will focus on the stability of the detected SNPs/indels, haplotypes, and association with the outcome of ATL in a replication sample of L. (V.) braziliensis from Corte de Pedra. Genotypes that show consistent and stable associations with the more severe forms of the disease may be good candidates to be explored in the development of tools for the improved diagnosis and management of human leishmaniasis cases.
ACKNOWLEDGMENTS
We deeply thank all personnel in the health post of Corte de Pedra, Bahia, Brazil, for their careful help with patient management and Kátia Salgado for laboratory support with parasite isolation and management.
This work was supported by the Brazilian Council for Scientific and Technological Development (CNPq), through the action plan Brazil-Switzerland, process number 590016/2010-5; by the U.S. National Institutes of Health (NIH), through grants NIH P50-AI30639-16 and NIH R03 A167663-01; and by the Fogarty International Clinical Research Scholars and Fellows (FICRS-F) Program, through National Institutes of Health/Fogarty International Center grant R24 TW007988. A.Q. was the recipient of a CAPES Ph.D. scholarship.
We do not have any conflicts of interest, commercial or otherwise, that might interfere with this study, its results, or its analyses.
Footnotes
Published ahead of print 3 October 2012
REFERENCES
- 1. Akopyants NS, et al. 2009. Demonstration of genetic exchange during cyclical development of Leishmania in the sand fly vector. Science 324:265–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Amato VS, Tuon FF, Bacha HA, Neto VA, Nicodemo AC. 2008. Mucosal leishmaniasis. Current scenario and prospects for treatment. Acta Trop. 105:1–9 [DOI] [PubMed] [Google Scholar]
- 3. Azulay RD, Azulay DR., Junior 1995. Immune-clinical-pathologic spectrum of leishmaniasis. Int. J. Dermatol. 34:303–307 [DOI] [PubMed] [Google Scholar]
- 4. Bacellar O, et al. 2002. Up-regulation of Th1-type responses in mucosal leishmaniasis patients. Infect. Immun. 70:6734–6740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cabrera M, et al. 1995. Polymorphism in tumor necrosis factor genes associated with mucocutaneous leishmaniasis. J. Exp. Med. 182:1259–1264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Carvalho EM, Barral A, Costa JM, Bittencourt A, Marsden P. 1994. Clinical and immunopathological aspects of disseminated cutaneous leishmaniasis. Acta Trop. 56:315–325 [DOI] [PubMed] [Google Scholar]
- 7. Castellucci L, et al. 2005. Familial aggregation of mucosal leishmaniasis in northeast Brazil. Am. J. Trop. Med. Hyg. 73:69–73 [PubMed] [Google Scholar]
- 8. Castellucci L, et al. 2006. IL6-174 G/C promoter polymorphism influences susceptibility to mucosal but not localized cutaneous leishmaniasis in Brazil. J. Infect. Dis. 194:519–527 [DOI] [PubMed] [Google Scholar]
- 9. Costa JM, et al. 1986. Disseminated cutaneous leishmaniasis in a field clinic in Bahia, Brazil: a report of eight cases. J. Trop. Med. Hyg. 89:319–323 [PubMed] [Google Scholar]
- 10. Cruz AK, Titus R, Beverley SM. 1993. Plasticity in chromosome number and testing of essential genes in Leishmania by targeting. Proc. Natl. Acad. Sci. U. S. A. 90:1599–1603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Garcia L, et al. 2004. Culture-independent species typing of neotropical Leishmania for clinical validation of a PCR-based assay targeting heat shock protein 70 genes. J. Clin. Microbiol. 42:2294–2297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kahl LP, Byram JE, David JR, Comerford SA, Von Lichtenberg F. 1991. Leishmania (Viannia) braziliensis: comparative pathology of golden hamsters infected with isolates from cutaneous and mucosal lesions of patients residing in Tres Bracos, Bahia, Brazil. Am. J. Trop. Med. Hyg. 44:218–232 [DOI] [PubMed] [Google Scholar]
- 13. Katakura K, Kobayashi A. 1985. Enhancement of infectivity of Leishmania donovani promastigotes by serial mouse passages. J. Parasitol. 71:393–394 [PubMed] [Google Scholar]
- 14. Leopoldo PT, et al. 2006. Differential effects of antigens from L. braziliensis isolates from disseminated and cutaneous leishmaniasis on in vitro cytokine production. BMC Infect. Dis. 6:75 doi:10.1186/1471-2334-6-75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Machado-Coelho GL, Caiaffa WT, Genaro O, Magalhaes PA, Mayrink W. 2005. Risk factors for mucosal manifestation of American cutaneous leishmaniasis. Trans. R. Soc. Trop. Med. Hyg. 99:55–61 [DOI] [PubMed] [Google Scholar]
- 16. Machado PR, et al. 2011. Reappraisal of the immunopathogenesis of disseminated leishmaniasis: in situ and systemic immune response. Trans. R. Soc. Trop. Med. Hyg. 105:438–444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Marsden PD. 1994. Mucosal leishmaniasis due to Leishmania (Viannia) braziliensis L(V)b in Tres Bracos, Bahia-Brazil. Rev. Soc. Bras. Med. Trop. 27:93–101 [DOI] [PubMed] [Google Scholar]
- 18. Montalvo AM, et al. 2010. Heat-shock protein 70 PCR-RFLP: a universal simple tool for Leishmania species discrimination in the New and Old World. Parasitology 137:1159–1168 [DOI] [PubMed] [Google Scholar]
- 19. Nolder D, Roncal N, Davies CR, Llanos-Cuentas A, Miles MA. 2007. Multiple hybrid genotypes of Leishmania (viannia) in a focus of mucocutaneous leishmaniasis. Am. J. Trop. Med. Hyg. 76:573–578 [PubMed] [Google Scholar]
- 20. Ramasawmy R, et al. 2010. The −2518bp promoter polymorphism at CCL2/MCP1 influences susceptibility to mucosal but not localized cutaneous leishmaniasis in Brazil. Infect. Genet. Evol. 10:607–613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Roberts SC, Wilson ME, Donelson JE. 1995. Developmentally regulated expression of a novel 59-kDa product of the major surface protease (Msp or gp63) gene family of Leishmania chagasi. J. Biol. Chem. 270:8884–8892 [DOI] [PubMed] [Google Scholar]
- 22. Salhi A, et al. 2008. Immunological and genetic evidence for a crucial role of IL-10 in cutaneous lesions in humans infected with Leishmania braziliensis. J. Immunol. 180:6139–6148 [DOI] [PubMed] [Google Scholar]
- 23. Sambrook J, Fritsch EF, Maniatis T. 1989. Molecular cloning: a laboratory manual, 2nd ed Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 24. Saravia NG, et al. 1998. Epidemiologic, genetic, and clinical associations among phenotypically distinct populations of Leishmania (Viannia) in Colombia. Am. J. Trop. Med. Hyg. 59:86–94 [DOI] [PubMed] [Google Scholar]
- 25. Saravia NG, et al. 2002. Heterogeneity, geographic distribution, and pathogenicity of serodemes of Leishmania viannia in Colombia. Am. J. Trop. Med. Hyg. 66:738–744 [DOI] [PubMed] [Google Scholar]
- 26. Schriefer A, et al. 2009. Geographic clustering of leishmaniasis in northeastern Brazil. Emerg. Infect. Dis. 15:871–876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Schriefer A, et al. 2004. Multiclonal Leishmania braziliensis population structure and its clinical implication in a region of endemicity for American tegumentary leishmaniasis. Infect. Immun. 72:508–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tamura K, Dudley J, Nei M, Kumar S. 2007. MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol. Biol. Evol. 24:1596–1599 [DOI] [PubMed] [Google Scholar]
- 29. Turetz ML, et al. 2002. Disseminated leishmaniasis: a new and emerging form of leishmaniasis observed in northeastern Brazil. J. Infect. Dis. 186:1829–1834 [DOI] [PubMed] [Google Scholar]
- 30. Ubeda JM, et al. 2008. Modulation of gene expression in drug resistant Leishmania is associated with gene amplification, gene deletion and chromosome aneuploidy. Genome Biol. 9:R115 doi:10.1186/gb-2008-9-7-r115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Weirather JL, et al. 2011. Serial quantitative PCR assay for detection, species-discrimination, and quantification of Leishmania spp. in human samples. J. Clin. Microbiol. 49:3892–3904 [DOI] [PMC free article] [PubMed] [Google Scholar]