

Abstract
Several studies have previously defined host-derived signaling events capable of driving lytic gammaherpesvirus replication or enhancing immediate-early viral gene expression. Yet signaling pathways that regulate later stages of the productive gammaherpesvirus replication cycle are still poorly defined. In this study, we utilized a mass spectrometric approach to identify c-Jun as an abundant cellular phosphoprotein present in late stages of lytic murine gammaherpesvirus 68 (MHV68) infection. Kinetically, c-Jun phosphorylation was enhanced as infection progressed, and this correlated with enhanced phosphorylation of the c-Jun amino-terminal kinases JNK1 and JNK2 and activation of AP-1 transcription. These events were dependent on progression beyond viral immediate-early gene expression, but not dependent on viral DNA replication. Both pharmacologic and dominant-negative blockade of JNK1/2 activity inhibited viral replication, and this correlated with inhibition of viral DNA synthesis and reduced viral gene expression. These data suggest a model in which MHV68 by necessity amplifies and usurps JNK/c-Jun signaling as infection progresses in order to facilitate late stages of the MHV68 lytic infection cycle.
INTRODUCTION
Gammaherpesviruses (GHVs) are DNA tumor viruses defined by their genetic similarity and the capacity to establish life-long latent infection in lymphocytes, primarily B cells. The GHV subfamily of herpesviruses includes Epstein-Barr virus (EBV), Kaposi sarcoma-associated herpesvirus (KSHV), herpesvirus Saimiri (HVS), rhesus rhadinovirus (RRV), murine gammaherpesvirus 68 (MHV68 [also referred to as γHV68]), and two recently isolated rodent viruses, wood mouse herpesvirus and herpesvirus Peru (22, 29). Infections by the human pathogens EBV and KSHV are etiologically associated with numerous diseases, including a staggering list of cancers and tumors (10, 31).
Like all herpesviruses, GHVs exhibit two distinct phases of the infectious cycle. The lytic, or productive, replication cycle is characterized by temporally regulated viral gene expression that culminates in viral DNA replication and packaging into infectious virions. In contrast, during the latent phase of the infectious cycle, often referred to as latency, viral gene expression is exquisitely restricted, and the viral genome is maintained as a circular episome that replicates concurrent to cellular DNA replication. Latent GHVs can reinitiate the lytic cycle in a process known as reactivation in response to mitogenic signals, host cell stress, or terminal differentiation of B cells to plasma cells (15, 26, 28, 40, 46, 57).
In vivo studies utilizing MHV68, a natural rodent pathogen genetically and pathogenetically related to EBV and KSHV, have indicated that lytic viral replication is necessary for dissemination from epithelial surfaces to distal latency reservoirs (33). Furthermore, ongoing productive viral replication is necessary for KSHV persistence in cultured endothelial cells (18), which correlates with clinical data that suggest that ongoing lytic replication, or at least failure of the immune system to control KSHV lytic gene expression, contributes to the pathogenesis of AIDS-associated Kaposi sarcoma (12, 31). While roles for many viral proteins in productive GHV replication have been established (13, 40, 44), much less is known about the cellular signaling events that facilitate the lytic infection process, especially those signals that are actively triggered by GHVs and then usurped for replication. This knowledge gap is in part due to the lack of robust lytic replication systems available for studying EBV and KSHV replication in culture. MHV68, on the other hand, replicates to high titers upon de novo infection of numerous cell types in culture and thus provides an important experimental system for defining biochemical pathways necessary for efficient GHV replication.
In this report, we describe experiments that identify the JNK/c-Jun signaling pathway as a critical host pathway regulating MHV68 lytic replication. JNK was activated and c-Jun phosphorylated at time points coinciding with the early-to-late transition in viral gene expression. In agreement, robust JNK and c-Jun phosphorylation and activation of an AP-1 reporter construct did not occur following infection with UV-inactivated or replication-deficient MHV68, and robust phosphorylation and activation by wild-type (WT) MHV68 did not require viral DNA synthesis. Viral gene expression and replication also were inhibited by dominant-negative JNK1 (DN-JNK1) and DN-JNK2 expression and by treatment with the pharmacologic JNK inhibitor SP600125. However, JNK inhibition did not globally repress viral gene expression as has been previously suggested in reactivation-based analyses (55), but rather blocked viral DNA replication and complete expression of viral lytic antigens. Further, while early viral gene expression was not suppressed following JNK inhibition, viral replication remained sensitive to JNK inhibition for several hours after infection of target cells. Our data support a model in which JNK signaling, possibly through c-Jun activation, regulates late events in the lytic replication cycle that are necessary for the production of progeny virus.
MATERIALS AND METHODS
Cell culture and viruses.
NIH 3T12, NIH 3T3-SA (referred to as 3T3 fibroblasts throughout), and 293T cells were purchased from ATCC. C57BL/6 mouse embryonic fibroblasts (MEFs) were prepared from day 13.5 embryos as described previously (53). All cells were cultured in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% fetal calf serum (FCS), 100 units/ml penicillin, 100 μg/ml streptomycin, and 2 mM l-glutamine (cMEM). Serum starvation involved culturing of cells in medium containing 1% FCS for 18 to 24 h prior to infection or treatment. Cells were cultured at 37°C in an atmosphere containing 5% CO2 and ∼99% humidity. Wild-type MHV68 was strain WUMS (ATCC VR1465) or WT bacterial artificial chromosome (BAC)-derived MHV68 (1). ORF50-null (50.STOP) MHV68 was previously described (39). UV inactivation of WT BAC-derived MHV68 was accomplished by diluting stock to 1 × 107 PFU/ml and auto-cross-linking viruses plated on a 60-mm plate in a Stratalinker prior to infection.
Viral infections.
Viral stocks were diluted in cMEM and adsorbed to monolayers of cells plated the previous day. The time of adsorption was considered time zero. After low-volume adsorption, inocula were removed for all high-multiplicity infections. After adsorption, cells were incubated in a normal culture volume of cMEM for the indicated times at 37°C. Cells were harvested by direct lysis for immunoblot assays or by freezing at −80°C for titer determinations. Progeny virions were liberated by freeze-thaw lysis, lysates were serially diluted, and titers were determined by MHV68 plaque assay essentially as described previously (47), except cells were overlaid with a 1.5% methyl cellulose solution in DMEM containing 2.5% FCS.
To determine the 50% inhibitory concentrations (IC50), SP600125 titration data were transformed according to the following parameters: X = log(X), Y = Y · K, where K = 1, X = [SP600125], and Y = viral yield. Viral yields were calculated by dividing output titers at either 24 or 48 h by the amount of virus present after adsorption (see the hour data in the graph shown in Fig. 4, below). Transformed data were then analyzed using a 4-parameter nonlinear regression analysis. Analyses were performed using GraphPad Prism software. Note that for log transformations, the no-treatment control concentration was arbitrarily set at 0.01.
Fig 4.

Pharmacologic inhibition of JNK activity with SP600125 blocks MHV68 replication. (A) Serum-starved 3T3 fibroblasts were treated with vehicle (DMSO) or SP600125 at the indicated concentrations 1 h prior to infection with MHV68 at an MOI of 5 PFU/cell. Cells were harvested at 0, 24, and 48 h postinfection and subjected to freeze-thaw lysis, and viral titers were determined by plaque assay. (B) Data from the 48-h time point in panel A were transformed, and a four-parameter nonlinear regression analysis was performed to determine the IC50 of SP600125 against MHV68 replication in culture. For all experiments described, results are means of triplicate samples. Error bars represent standard deviations.
Mass spectrometry.
Peptide samples from infected or mock-infected cell lysates with 1 μg of protein in a final concentration of 0.1% formic acid (Fluka) were analyzed by the nano-liquid chromatography–tandem mass spectrometry (LC/MS/MS) technique on an ion trap tandem mass spectrometer. An autosampler was used for automatic injection of tryptic peptides from a 96-well plate to the NanoLC 2D system (Eksigent). The peptides were separated by reverse-phase HPLC (high-performance LC) by using a 10-cm-long analytical column (C12 resin; Phenomenex). The chromatographic eluate thus obtained was ionized by electrospray ionization (ESI), followed by MS/MS analysis using collision-induced dissociation (CID) on an LTQ Orbitrap hybrid MS apparatus (Thermo Finnigan, San Jose, CA) with two mass analyzers, a linear ion trap (LTQ) and an Orbitrap. One MS scan by the Orbitrap was followed by 7 MS/MS scans by the LTQ. Other relevant parameters included the following: spray voltage, 2.0 kV; m/z range, 350 to 1,500; isolation width (m/z), 2.5; normalized collision energy, 35%. The MS data were acquired using XCalibur 2.0 software in RAW file format.
Antibodies, drug treatments, and vectors.
Polyclonal MHV68 antiserum was obtained from C57BL/6 mice 6 weeks postinfection (16). ORF59-directed chicken IgY was previously described (48). Phospho-c-Jun (S63-#9261 or S73-#9164), total c-Jun (9615), phospho-JNK1/2 (9251), and total JNK1/2 (9252) antibodies were purchased from Cell Signaling Technology. β-Actin mouse monoclonal antibody was purchased from Sigma (A2228). Horseradish peroxidase (HRP)-conjugated secondary antibodies for immunoblot analyses were purchased from Jackson ImmunoResearch. Alexa 568-conjugated secondary antibodies for immunofluorescence analyses were purchased from Invitrogen. Phosphono-acetic acid was added to culture media at 200 μg/ml following viral adsorption. SP600125 was dissolved in dimethyl sulfoxide (DMSO) and diluted to the indicated concentrations in growth media. For pretreatments, cells were treated with SP600125 for 1 h prior to infection. Otherwise, SP600125 was added at the indicated time postinfection. Cells were treated with DMSO as a negative control. Homo sapiens dominant-negative JNK1 (13846) and dominant-negative JNK2 (13761) in pcDNA3 were purchased from AddGene. The empty vector control for dominant-negative experiments was pcDNA3.1+ (Invitrogen).
Immunoblot analyses.
Cells were lysed with alternative RIPA buffer (150 mM NaCl, 20 mM Tris, 2 mM EDTA, 1% NP-40, 0.25% deoxycholate, supplemented with Complete mini-EDTA-free protease inhibitor tablet [Roche] and phosphatase inhibitor cocktail 2 [Sigma]) and quantified using the Bio-Rad DC or Thermo bicinchoninic acid protein assay prior to resuspending in Laemmli sample buffer, or equivalent numbers of cells (1 × 105 to 2 × 105) were directly lysed with 100 μl Laemmli sample buffer (25). Samples were heated to 100°C for 10 min and resolved by SDS-PAGE. Resolved proteins were transferred to nitrocellulose and identified with the indicated antibodies. Immobilized antigens and antibodies were detected with HRP-conjugated secondary antibodies and SuperSignal Pico West enhanced chemiluminescence reagent (Thermo Scientific) and exposed to film.
Immunofluorescence assays.
Immunofluorescence analyses were performed as previously described (14). Briefly, cells plated on glass coverslips were fixed with 10% formalin 18 h postinfection, washed with phosphate-buffered saline (PBS), permeabilized and blocked with 0.1% Triton X-100 (TX100) in PBS containing 5% bovine serum albumin (blocking buffer). Infected cells were stained with mouse polyclonal MHV68 antiserum (1:1,000 dilution in blocking buffer plus 1% normal donkey serum) for 2 h at room temperature. Cells were washed 3 times with PBS-TX100 and incubated with Alexa 568 goat anti-mouse secondary antibody (1:8,000 dilution in blocking buffer). Cells were washed 3 times in PBS-TX100, once with PBS, and once with deionized H2O and mounted on slides using 4′,6-diamidino-2-phenylindole (DAPI)-containing ProLong Anti-Fade Gold reagent. Images were captured using a Zeiss Axio A1 imager fluorescence microscope.
Reporter assays.
Cells in 10-cm dishes were transfected with the pAP-1-luc luciferase reporter plasmid (Stratagene) using Superfect (Qiagen). The following day, cells were trypsinized and replated in 24-well plates. Approximately 48 h after transfection, cells were infected with the indicated viruses or treatments. Cells were lysed using Promega passive lysis buffer 18 h postinfection. Luciferase activity was determined using luciferase reporter assay kits (Promega). Luciferase activity was measured using a Turner Designs 2000 luminometer.
Quantitative PCR.
Cells were infected with MHV68 at a multiplicity of infection (MOI) of 5 PFU/cell. After adsorption, cells were washed 3 times with PBS to remove cell-free virus. Cells were harvested, and DNA was isolated using the Qiagen DNeasy kit at 6 h and 18 h after adsorption. The 6-h time point represented the “eclipse” phase following viral genome deposition in the nucleus and preceding de novo viral DNA synthesis. This time point thus provides an input control upon which amplification by 18 h can be quantified. A 200-μg aliquot of total DNA was diluted in iQ SYBR green supermix (Bio-Rad) and analyzed by quantitative real-time PCR using primers corresponding to ORF25 in the MHV68 genome or the cellular GAPDH gene (15). The experiment was performed in biological triplicates, and duplicate wells were analyzed for each biological replicate. Data were analyzed using the ΔΔCT method, where ΔΔCT = [CT(ORF25) − CT(GAPDH)6 h] − [CT(ORF25) − CT(GAPDH)18 h]. Data are presented as the evaluation 2−ΔΔCT for each treatment, represented as the fold change in MHV68 DNA at 18 h relative to 6 h and normalized to the GAPDH results.
RESULTS
A JNK/c-Jun signaling axis is induced during MHV68 lytic replication.
To better understand the signaling events that regulate lytic GHV replication, we performed phosphoproteomic analyses comparing mock-infected to MHV68-infected NIH 3T3 fibroblasts. From two independent experiments, each analyzed in triplicate, we detected c-Jun peptides phosphorylated on S63 and S73 in analytes from infected cells but not mock-infected cells (Fig. 1A). These events, depicted in the c-Jun sequence in Fig. 1C, represent known activating posttranslation modifications of the c-Jun proto-oncogene (51). Immunoblot analyses comparing mock-infected to MHV68-infected cells confirmed infection-associated c-Jun phosphorylation on S63 and S73 (Fig. 1B).
Fig 1.

Identification of c-Jun as an MHV68-induced phosphoprotein. (A) Phosphopeptide enrichment was performed for NIH 3T3 fibroblasts infected with MHV68 18 h postinfection. LC/MS/MS analyses identified two unique phosphopeptides corresponding to the consensus c-Jun sequence phosphorylated by JNK isoforms. Representative peptide spectra are shown for each unique peptide. (B) Serum-starved 3T3 fibroblasts were mock infected (−) or infected (+) with MHV68 at an MOI of 5 PFU/cell. Cells were harvested at the indicated times postinfection, and proteins were resolved by SDS-PAGE. Resolved proteins were detected by immunoblot analyses with antibodies that recognize the indicated proteins. (C) Unique peptides are highlighted in yellow on the murine c-Jun sequence. Posttranslationally modified amino acids are highlighted in green.
c-Jun is a classic target of the c-Jun amino-terminal kinases JNK1 and JNK2. The finding that c-Jun was phosphorylated upon MHV68 infection suggested that JNK1 and/or JNK2 would also be activated during MHV68 infection. Induction of JNK1 and JNK2 kinase activity is triggered by phosphorylation of conserved Tyr and Thr residues within the activation loop by the dual specificity mitogen-activated protein kinase kinases (MAPKKs) MKK4 and MKK7 (41). Thus, we hypothesized that JNK1/2 phosphorylation also would occur during lytic MHV68 infection. Indeed, immunoblot analyses revealed increased JNK1/2 phosphorylation following MHV68 infection compared to results with mock-infected cells (Fig. 2A). Further, we also detected enhanced JNK1/2 kinase activity against a c-Jun substrate in MHV68-infected fibroblasts compared to mock-infected cells, and JNK/c-Jun phosphorylation also occurred following infection of primary murine embryonic fibroblasts, murine endothelial cells, and 3T3 fibroblasts regardless of serum concentration (data not shown). Interestingly, while phosphorylated JNK was detectable immediately after viral adsorption (see the 1-h time point), this analysis suggested more robust JNK phosphorylation (and also c-Jun phosphorylation, particularly on S73) as infection progressed. While comparisons of diverse antigens by immunoblot analyses cannot be directly quantitated due to the potential influences of antibody affinities and other variables, it is interesting that robust JNK and c-Jun phosphorylation became apparent at time points coinciding to robust expression of the viral DNA polymerase processivity factor ORF59 and viral lytic antigens detected with MHV68 antiserum. These data demonstrated JNK1/2 activation as a consequence of lytic MHV68 infection and suggested that activation of JNK/c-Jun signaling is triggered through the expression of particular viral genes.
Fig 2.
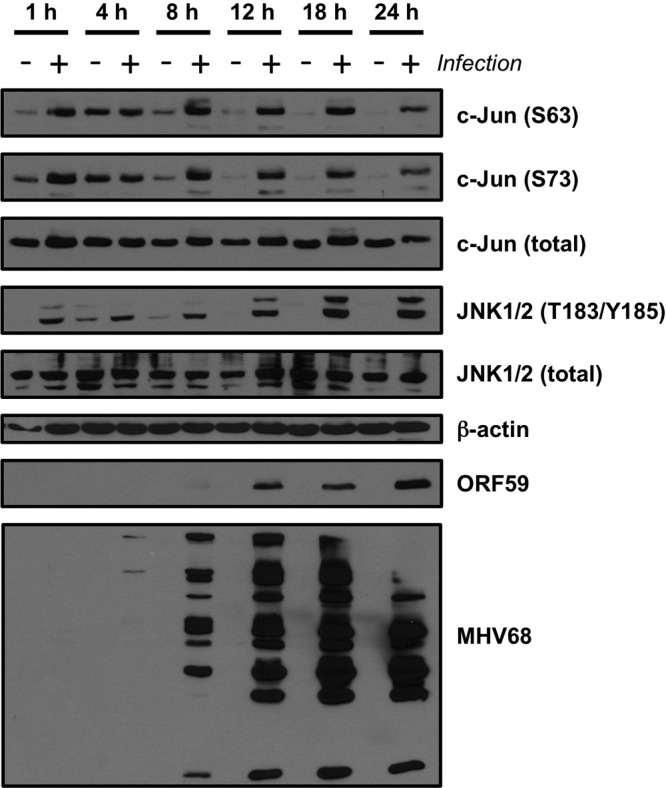
JNK phosphorylation is induced during MHV68 infection. Serum-starved 3T3 fibroblasts were mock infected (−) or infected (+) with MHV68 at an MOI of 5 PFU/cell. Cells were harvested at the indicated times postinfection, and proteins were resolved by SDS-PAGE. Resolved proteins were detected by immunoblot analyses using antibodies that recognize the indicated proteins.
Robust JNK/c-Jun phosphorylation and AP-1 transcriptional activation require viral gene expression, but not DNA replication.
To gain better insight into the kinetics of JNK1/2 and c-Jun phosphorylation induced by MHV68 infection, especially as it relates to phase of the viral infectious cycle, we infected fibroblasts with UV-inactivated (UVI) virus, replication-defective ORF50-null (50.Stop) virus (39), and WT MHV68 in the presence or absence of phosphono-acetic acid (PAA), which blocks viral DNA replication. Again, we detected robust c-Jun and JNK1/2 phosphorylation in immunoblot analyses performed 18 h after infection with WT MHV68 (Fig. 3A). Comparatively, neither UVI nor ORF50-null MHV68 elicited robust phosphorylation of c-Jun or JNK, although a slight increase in c-Jun S73 and JNK phosphorylation was observed relative to mock-infected lysates in replicate experiments. Importantly, MHV68 antigens were not detectable following infection by either UVI or ORF50-null MHV68, which confirmed their replication incompetence and blockade to viral gene expression. Thus, these data suggest that replication-incompetent MHV68 particles in and of themselves elicit a modicum of JNK/c-Jun phosphorylation. Such activity may account for the early JNK1/2 and c-Jun phosphorylation observed in time course analyses (Fig. 1 and 2). Further, since ORF50-null MHV68 is incapable of progressing beyond immediate-early (IE) gene expression, these findings suggest that events coinciding with either RTA expression, early viral gene expression, or both are necessary for robust JNK/c-Jun-related signaling events during lytic MHV68 replication. Finally, c-Jun and JNK phosphorylation were not inhibited by PAA treatment (Fig. 3A), suggesting that viral DNA synthesis does not influence JNK/c-Jun activation.
Fig 3.
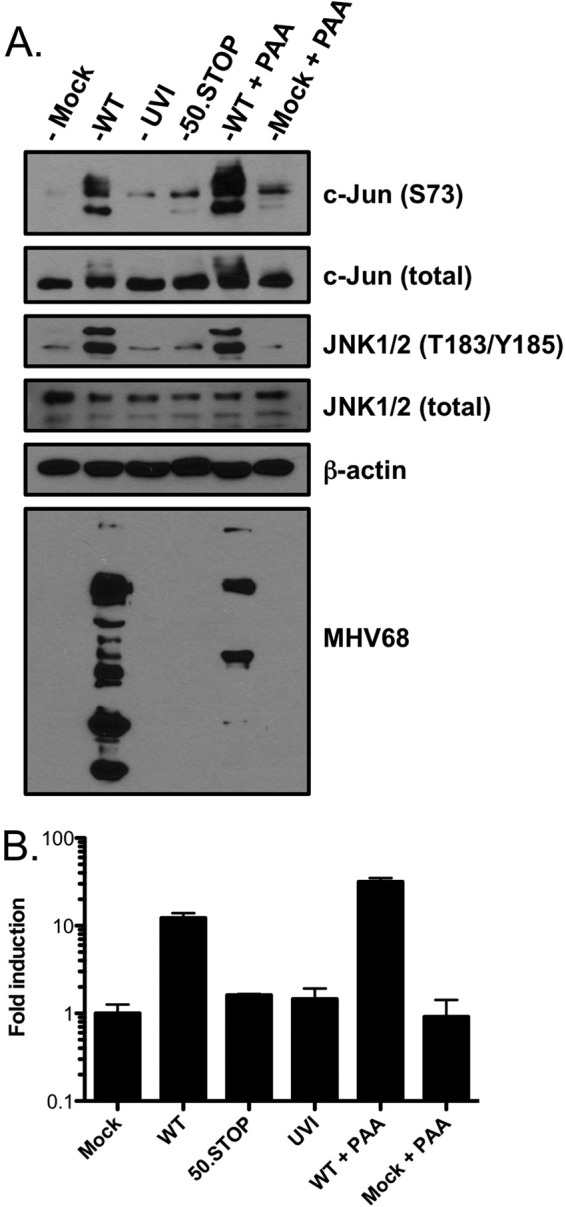
JNK/c-Jun phosphorylation and AP-1 reporter activity are amplified as infection progresses. (A) Serum-starved 3T3 fibroblasts were either mock infected or infected at an MOI of 5 PFU/cell with WT MHV68, UVI MHV68, ORF50-null MHV68 (50.STOP), mock infected, or infected with WT MHV68 in the presence of PAA (200 μg/ml). Cells were harvested 18 h postinfection, and proteins were resolved by SDS-PAGE. Resolved proteins were detected by immunoblot analyses using antibodies that recognize the indicated proteins. (B) 3T3 fibroblasts were transiently transfected with an AP-1 reporter plasmid to evaluate JNK/c-Jun transcriptional activity. At 24 h posttransfection, cells were replated and infected as described for panel A. Cells were harvested 18 h postinfection, and luciferase activity was quantified. Results are means of triplicate samples. Error bars represent standard deviations.
Once activated by JNK-mediated phosphorylation, c-Jun contributes to the transcriptional activity of an AP-1 transcription factor complex comprised of either c-Jun homodimers or heterodimers of c-Jun and various other transcription factors, including c-Fos (reviewed in reference 51). The findings that JNK is phosphorylated and active and c-Jun is phosphorylated suggest that AP-1 transcription factor activity is induced in response to MHV68 infection. To test this hypothesis, we transiently transfected NIH 3T3 fibroblasts with an AP-1 luciferase reporter construct prior to infection with WT MHV68, UVI MHV68, ORF50-null MHV68, or WT MHV68 in the presence of PAA (Fig. 3B). While, UVI and ORF50-null MHV68 only slightly enhanced AP-1 reporter activity compared to mock infection, both WT MHV68 and MHV68 in the presence of PAA elicited greater-than-10-fold induction of the AP-1 reporter relative to mock infection. The results thus corroborated the analogous immunoblot experiment. Together, these findings strongly suggest that a JNK/c-Jun signaling cascade capable of driving AP-1 transcriptional activity is triggered by MHV68 particles and amplified coincidental to viral gene expression, but prior to viral DNA synthesis, during MHV68 infection.
JNK activity is necessary for MHV68 replication.
As JNK/c-Jun activation appeared to be elicited by viral gene expression, we next sought to determine the necessity for JNK signaling in MHV68 replication. Thus, we tested the capacity of the pharmacologic JNK inhibitor SP600125 to block MHV68 replication. SP600125 is an anthrapyrazole that specifically targets and inhibits the kinase domain of JNK isoforms (3). We tested a range of SP600125 concentrations for the capacity to inhibit MHV68 replication at 24 and 48 h postinfection. While concentrations of SP600125 ranging from 0.5 to 2.5 μM did not inhibit MHV68 replication, SP600125 concentrations greater than 5 μM potently inhibited MHV68 replication, with no increase in viral titers over time occurring at 20 and 50 μM concentrations (Fig. 4A). These doses were in good agreement with previous data demonstrating SP600125 inhibition of JNK activity in cultured cells (3). It should be noted that 50 μM SP600125 did exhibit toxicity to the monolayer, while lower concentrations did not. The calculated IC50 for SP600125 against MHV68 48 h after infection was 2.24 μM (95% confidence interval, 0.963 to 5.20 μM) (Fig. 4B). These data demonstrate that JNK can be pharmacologically targeted to inhibit MHV68 replication in culture.
As a further and complementary test of the hypothesis that JNK activation is required for MHV68 replication, we evaluated the capacity of dominant-negative JNK1 (DN-JNK1) and DN-JNK2 expression to inhibit MHV68 replication. For this experiment, 293T cells were transfected with MHV68 BAC in combination with empty vector, DN-JNK1, DN-JNK2, or both DN-JNK1 and DN-JNK2. Cells were harvested at 48, 72, and 96 h posttransfection, and immunoblot analyses were performed using polyclonal MHV68 antiserum to evaluate the expression of MHV68 lytic antigens over time. While MHV68 antiserum readily detected viral antigens in cells cotransfected with MHV68 BAC and empty vector, expression of either DN-JNK1 or DN-JNK2 resulted in reduced detection of MHV68 proteins. The reduction in MHV68 proteins detected was even more dramatic in cells transfected with both DN-JNK1 and DN-JNK2 (Fig. 5). Although reproducible, it was not immediately clear why DN-JNK2 expression was reduced when coexpressed with DN-JNK1. In agreement with the observed reduction in viral antigen detectability, MHV68 titers in the supernatants of cells transfected with DN-JNK constructs were markedly reduced at each time point, especially when both DN-JNK1 and DN-JNK2 were coexpressed, compared to empty vector transfections (Table 1). Importantly, neither of the DN-JNK constructs negatively impacted transfection of the MHV68 BAC, as evidenced by detection of green fluorescent protein (GFP) expressed from the MHV68 BAC 24 h after transfection (data not shown). These findings provided an important specificity control for inhibition of viral replication by SP600125 and further demonstrated the importance of JNK signaling to MHV68 replication. Moreover, since the effects of expressing DN-JNK1 and DN-JNK2 were additive, these findings suggest that both JNK1 and JNK2 may be utilized by MHV68 to facilitate lytic replication.
Fig 5.
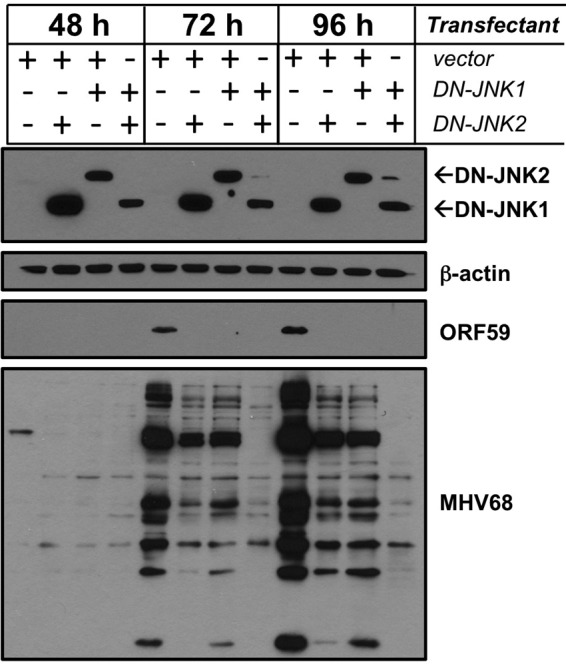
Dominant-negative JNK expression blocks MHV68 gene expression. HEK293T cells were transfected with infectious MHV68 BAC DNA in combination with empty vector, DN-JNK1, DN-JNK2, or both DN-JNK1 and DN-JNK2. Cells were harvested 48, 72, and 96 h posttransfection, and lysates were resolved by SDS-PAGE. Resolved proteins were detected by immunoblot analyses using antibodies that recognize the indicated proteins. Note that DN-JNK constructs encode a FLAG epitope tag. DN-JNK1 and DN-JNK2 proteins were detected using FLAG-specific antibodies.
TABLE 1.
MHV68 titers from cells expressing DN-JNK1 and DN-JNK2
| Vector | MHV68 titer (PFU/ml) at: |
||
|---|---|---|---|
| 48 h | 72 h | 96 h | |
| Empty vector | 1.8 × 106 | 6.0 × 106 | 1.1 × 107 |
| DN-JNK1 | 1.5 × 105 | 4.9 × 105 | 1.5 × 106 |
| DN-JNK2 | 1.2 × 105 | 7.7 × 105 | 4.8 × 105 |
| DN-JNK1 and DN-JNK2 | 4.6 × 102 | 4.7 × 103 | 1.9 × 104 |
JNK signaling regulates late steps in the MHV68 lytic cycle.
Data presented thus far suggest that JNK signaling is amplified and required for MHV68 replication. To begin to understand what roles JNK activity might play in MHV68 productive infection, we performed immunoblot analyses to determine what effect SP600125 treatment exerts on MHV68 gene expression (Fig. 6A). When cells were pretreated with vehicle, MHV68 infection once again elicited robust c-Jun and JNK phosphorylation in comparison to mock infection. Treatment with SP600125, however, inhibited virus-related c-Jun phosphorylation, which was consistent with the drug blocking JNK phosphorylation of downstream targets and indicated that c-Jun phosphorylation during late-stage MHV68 infection is largely due to JNK activity, as opposed to other MAPKs or viral kinases. Interestingly, pharmacologic inhibition of JNK activity correlated with reduced expression of MHV68 lytic antigens detected using MHV68-directed antiserum. In parallel immunofluorescence analyses, SP600125-treated cells exhibited altered viral protein staining patterns, most notably an apparent exclusion or absence of viral proteins from the host cell nucleus, 18 h postinfection (Fig. 6B). These images also illustrated that SP600125 treatment does not simply block or reduce MHV68 infection, as similar numbers of cells stained positive for viral proteins.
Fig 6.

JNK activity is necessary for MHV68 genome replication. (A) 3T3 fibroblasts were treated with vehicle (DMSO) or SP600125 (20 μM) 1 h prior to infection with MHV68 at an MOI of 5 PFU/cell. Cells were harvested 18 h postinfection, and lysates were resolved by SDS-PAGE. Resolved proteins were detected by immunoblot analyses using antibodies that recognize the indicated proteins. (B) Cells were treated as for panel A, except cells were fixed in 10% formalin 18 h postinfection for indirect immunofluorescence analyses. Fixed and permeabilized cells were stained with MHV68 antiserum to detect lytic viral antigens. DNA was stained with DAPI. (C) 3T3 fibroblasts were treated with DMSO, 20 μM SP600125 (SP6), or 200 μg/ml PAA 1 h prior to infection with MHV68 at an MOI of 5 PFU/cell. Cells were harvested at 6 and 18 h postinfection, and total cellular and viral DNAs were isolated. Quantitative PCR was performed to detect viral genomes, and values were normalized to total cellular DNA at each time point. Data represent the change in viral DNA copy number between 6 and 18 h postinfection as determined using the ΔΔCT method. Results are means of triplicate samples. Error bars represent standard deviations.
Since herpesvirus late gene expression by and large coincides with viral DNA replication, we performed quantitative PCR analyses to test the hypothesis that JNK activity is necessary for viral DNA replication (Fig. 6C). While vehicle-treated cells exhibited robust viral DNA replication by 18 h postinfection, treatment of cells with SP600125 inhibited viral DNA synthesis to almost the same extent as the known viral DNA polymerase inhibitor PAA. For both SP600125 and PAA, this equated to a greater-than-1,000-fold inhibition of viral DNA synthesis. These data demonstrate that MHV68 utilizes JNK activity to facilitate viral DNA replication, which also may account for the reduced expression of late viral antigens recognized by MHV68 antiserum.
Together, the findings that JNK and c-Jun phosphorylation progressively increased as infection proceeded and that JNK activity was necessary for viral DNA replication suggest that amplification of JNK signaling occurs to regulate late steps in the viral replication cycle. To better define when in the viral replication cycle JNK signaling is critical for viral replication, we performed kinetic analyses in which infected cells were treated with vehicle or SP600125 at specific times after viral adsorption. In immunoblot analyses of samples harvested 8 h postinfection, both pretreatment and treatment with SP600125 4 h postinfection effectively reduced virus-induced c-Jun phosphorylation compared to vehicle controls. However, neither treatment appeared to inhibit expression of the early gene ORF59 or of any viral proteins detected by MHV68 antiserum (note that some were reduced) (Fig. 7A). These data indicate that JNK activity, likely through c-Jun, is not a critical requirement for immediate-early and early viral gene expression. In contrast, pretreatment and addition of SP600125 at 4 or 8 h postinfection effectively reduced c-Jun phosphorylation and reduced the relative expression levels of ORF59 and viral antigens detected with MHV68 antiserum in immunoblot analyses of samples harvested 18 h postinfection (Fig. 7B). These data demonstrated that infected cells remain sensitive to JNK inhibition up to several hours after initial infection and that JNK inhibition reduces the accumulation of viral antigens within infected cells. In related experiments testing the effects of delayed JNK inhibition on viral replication, SP600125 substantially suppressed viral replication when added at 4 and 8 h postinfection (Fig. 7C). Sensitivity to SP600125 treatment was progressively lost when added at later times postinfection. Collectively, these data are consistent with observations from immunoblot analyses demonstrating that potent JNK and c-Jun phosphorylation occurs between 8 and 12 h postinfection. These data strongly suggest that JNK controls steps in the viral replication cycle downstream of immediate-early and early viral gene expression that are necessary for viral DNA replication.
Fig 7.

JNK activity regulates late phases of the MHV68 replication cycle. (A and B) 3T3 fibroblasts were treated with vehicle (DMSO) or SP600125 (20 μM) 1 h prior to infection or at 4 h or 8 h (B only) postinfection with MHV68 at an MOI of 5 PFU/cell. Cells were harvested 8 h (A) or 18 h (B) postinfection, and lysates were resolved by SDS-PAGE. Resolved proteins were detected by immunoblot analyses using antibodies that recognize the indicated proteins. (C) 3T3 fibroblasts were treated with 20 μM SP600125 at the indicated times postinfection with MHV68 at an MOI of 5 PFU/cell. Cells were harvested 24 h postinfection and subjected to freeze-thaw lysis, and viral titers were determined by plaque assay. Results are means of triplicate samples. Error bars represent standard deviations.
DISCUSSION
In this report, we validated the mass spectrometric identification of c-Jun as a cellular phosphoprotein induced during productive MHV68 infection. We extended these findings to demonstrate infection-associated activation of JNK1/2 signaling pathways, leading to c-Jun phosphorylation, and we further demonstrated that JNK1 and JNK2 are necessary for efficient MHV68 replication. By utilizing UV-inactivated virus, replication-deficient virus, and inhibitors of viral DNA replication, we found that JNK/c-Jun activation likely occurs in conjunction with early viral gene expression. These findings were supported by kinetic experiments in which pharmacologic inhibition of JNK activity most efficiently blocks viral replication when added prior to time points coinciding with robust late gene synthesis (6, 32). Finally, we demonstrated that pharmacologic JNK inhibition does not simply globally block viral gene expression but apparently suppresses late MHV68 gene expression, which correlated with altered localization of detectable viral antigens within infected cells and blockade of viral genome replication. These findings suggest a model in which JNK1/2 and possibly c-Jun activity are functionally amplified as productive viral replication proceeds, in order to facilitate DNA replication and/or viral gene expression toward completing the lytic replication cycle (Fig. 8).
Fig 8.
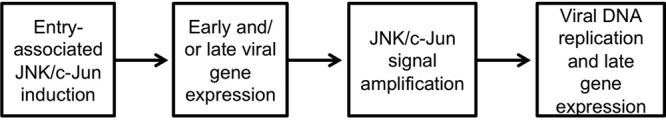
Model of JNK/c-Jun functions in productive MHV68 replication.
Our data demonstrate that JNK/c-Jun activation occurs early after infection, presumably as a consequence of viral attachment or entry, and increases as the infection process progresses (Fig. 1, 2, and 3). The modest activation coincident with viral engagement of the host cell is consistent with data published for KSHV. These previous studies suggested that MAPK activation (p38, extracellular signal-regulated kinase, and JNK) in general facilitates or enhances immediate-early viral gene expression (37, 43, 56). Furthermore, MAPK activation in general also regulates KSHV reactivation following induction with chemical stimuli (17, 55). These reports also suggested that MAPKs facilitate activation of the promoter for ORF50, which encodes the RTA protein (37, 43). As RTA expression is an absolute requirement for rhadinovirus replication (11, 39, 45), and presumably the expression of all non-IE lytic genes, it stands to reason that inhibition of factors involved in driving ORF50 transcription would block GHV reactivation. Thus, we were surprised that pretreatment with SP600125 did not globally block viral gene expression, as evidenced by the detection of MHV68 lytic antigens using MHV68 antiserum in immunoblotting and immunofluorescence analyses (Fig. 6). In fact, infected cells were sensitive to JNK inhibition several hours postinfection, by which time ORF50 transcription and subsequent early gene expression are well under way (6, 24), and remained modestly sensitive to SP600125 treatment up to 12 h postinfection (Fig. 7). Our findings suggest that JNK/c-Jun pathways are not absolute requirements for GHV IE gene expression upon de novo infection but are essential to a later stage in the replication process.
Although the mechanism by which the JNK/c-Jun signaling axis is amplified as infection progresses is not immediately clear, our data support two likely hypotheses. First, it is conceivable that JNK activation and amplification are simply passive processes due to infection-associated stress on host cells. JNK1 and JNK2 are also known as stress-associated protein kinases (SAPKs), and JNK activation is induced by numerous external and internal cellular stressors, including cytokine signaling, UV exposure, ionizing radiation, ribosome inhibition, and nutrient deprivation, to name a few (41). As a host cell response to infection by diverse pathogens, JNK activation is associated with infection-induced apoptosis (2, 8, 30) and contributes to host innate immune responses (7, 35, 42, 49). In the case of MHV68, our data imply that amplification of the JNK signal occurs concurrently with progression through the lytic gene expression cascade but prior to viral DNA replication and presumably late gene expression. The signal would then be usurped, perhaps acting as a switch that controls completion of the replication process. Second, JNK may be actively triggered (or amplified) by specific viral gene products to facilitate distinct aspects of MHV68 replication. For KSHV, both ORF36 and ORF49 are capable of stimulating JNK activation in overexpression systems (17, 20). Likewise, the EBV Na protein, the lymphocryptovirus homolog of rhadinovirus ORF49, also is linked to JNK phosphorylation (19). The expression kinetics of both ORF36 and ORF49 (6, 24), which also are MHV68 tegument proteins (4, 36), would fit with the kinetics of JNK/c-Jun phosphorylation and AP-1 reporter activity we observed. However, an ORF36 transposon mutant (44) elicited JNK/c-Jun phosphorylation comparable to WT MHV68 in our hands (J. A. Stahl and J. C. Forrest, unpublished observation). Moreover, ORF36 is not required for MHV68 replication, whereas JNK activity is. A similar analysis for ORF49 is complicated by a severe replication defect (36, 44). It would be of interest to determine if exogenous stimulation of JNK/c-Jun activity could serve as a host-cell complement to ORF49 deficiency. Suffice it to say, additional tests are necessary to define the mechanism of JNK/c-Jun induction during de novo infection by MHV68.
Whether specific cellular signaling cascades are necessary for JNK/c-Jun amplification to facilitate completion of MHV68 replication also is not clear. A recent study from the Sun laboratory found that several MAPK family members, including MAP2K3, MAP3K2, MAP3K5, MAP3K8/Tpl2/COT, and MAP3K11, were capable of enhancing MHV68 replication when overexpressed in 293T cells (27). In the context of our data, these upstream MAPKKs and MAPKKKs may serve as activators of JNK concurrent to viral entry or may function in our proposed signal amplification event that facilitates late steps in the infectious cycle. Importantly, those authors demonstrated that Tpl2 enhanced activation of promoters for ORF50/RTA and ORF57, as well as promoting increased c-Fos expression immediately following MHV68 infection (27). These findings suggest that Tpl2 may influence immediate-early gene expression. Further, those authors demonstrated that overexpression of dominant-negative c-Jun also inhibited ORF50 promoter activity and MHV68 replication in general (27). The promoter inhibition data seem consistent with the modest entry-associated JNK/c-Jun phosphorylation and AP-1 reporter activity described here (Fig. 3). Thus, the current data indicate that Tpl2 regulates immediate-early AP-1 activity during infection, but whether Tpl2 is responsible for amplification of JNK signaling as infection progresses is not yet clear.
Interestingly, varicella-zoster virus (VZV) also requires JNK activity for productive infection (58). And, for VZV, MKK4 and MKK7, which are dual-specificity upstream MAPKKs that are potently capable of activating JNK1/2 (41), are activated during productive replication. Again, MKK4/7 activation also was demonstrated for KSHV ORF36 (20), and we cannot completely rule out a similar function for the MHV68 ORF36 homolog. It will be of interest to determine if Tpl2 or other MAPKKKs that enhance MHV68 replication (27) function to activate MKK4 or MKK7, leading to JNK/c-Jun amplification as MHV68 infection progresses.
The mechanism by which JNK activity regulates MHV68 DNA synthesis and possibly late gene expression remains to be determined. Since treatment with SP600125 also reduced virus-related c-Jun phosphorylation, a role for JNK-mediated c-Jun activation currently seems the most likely interpretation. Searching for the AP-1 consensus binding site, TGA(G/C)TCA, within the MHV68 genome revealed 31 potential AP-1 binding elements (Table 2). Of these, 11 are situated less than 500 nucleotides upstream of an annotated open reading frame (50), which suggests a capacity to function in transcriptional activation of the adjacent viral gene. This includes elements within previously defined AP-1-responsive promoters for ORF50 and ORF57 (27) which, remarkably, are conserved in KSHV (27, 43, 52). It seems reasonable to suggest that entry-associated JNK/c-Jun activity facilitates transactivation of the ORF50 promoter, thus potentiating entry into the lytic replication cycle. Indeed, expression of dominant-negative c-Jun inhibited Tpl2-related ORF50 promoter activity (27). However, mutagenesis of the ORF50 promoter AP-1 response elements within the MHV68 genome did not yield a replication-incompetent virus, although replication was attenuated (27). Thus, while our studies demonstrated that JNK signaling is necessary for completion of the lytic replication cycle, AP-1/c-Jun-driven RTA expression is not an absolute requirement for progression through the lytic cycle.
TABLE 2.
Presence of AP-1 consensus sites within the MHV68 genome
| Genomic position (bp) | Gene namea |
|---|---|
| 7470 | IG M3-M4 |
| 11080 | IG ORF4-6 (ORF6 pro?) |
| 19067 | IG ORF8-9 (ORF9 pro?) |
| 19726 | ORF9 |
| 29711 | ORF17 |
| 32397 | ORF20 |
| 37831 | ORF23 |
| 38410 | ORF24 |
| 42009 | ORF25 |
| 42920 | ORF25 |
| 44804 | ORF26 |
| 46967 | ORF29b |
| 51067 | ORF29a |
| 52659 | ORF35 |
| 53890 | ORF36 |
| 57393 | ORF40 |
| 60148 | ORF43 |
| 63872 | ORF45 |
| 65370 | ORF47 (ORF46 pro?) |
| 66659 | ORF50 pro-1 |
| 71610 | ORF53 (ORF52 pro?) |
| 72796 | ORF55 |
| 75634 | ORF57 pro |
| 75724 | ORF57 pro |
| 83200 | ORF62 (ORF61 pro?) |
| 83298 | ORF62 (ORF61 pro?) |
| 96675 | ORF68 (ORF67 pro?) |
| 103420 | M11 (ORF72 pro?) |
| 109773 | ORF75c |
| 111044 | ORF75b |
| 111140 | ORF75b |
IG, intergenic region. The AP-1 sites sit between ORFs 4 and 6 and ORFs 8 and 9. Upstream and downstream genes are listed. Hypothetical promoters are shown in parentheses.
ORF57 is an interesting potential target, as the ORF57 protein product (also known as the multifunctional transcription activator [MTA]) facilitates the export of unspliced late viral transcripts from the nucleus for translation in the cytoplasm (reviewed in reference 23). ORF57 also enhances translation of viral proteins (5). Thus, inhibition of AP-1-driven ORF57 transcription may result in a late blockade to viral replication. Testing such a hypothesis would require direct mutagenesis of AP-1 elements in the ORF57 promoter, as ORF57 transcription also is highly responsive to RTA (38, 54). It is also interesting to note the presence of AP-1 elements upstream of ORF9, which encodes the viral DNA polymerase. It is tempting to speculate that SP600125-mediated inhibition of viral DNA synthesis results from failed expression of the viral DNA polymerase.
Finally, it also is possible that c-Jun activity is dispensable for transcriptional events leading to viral DNA synthesis and production of progeny virions. While c-Jun is a classic target of JNK activity, JNK is capable of targeting numerous proteins within a cell. According to information on the website PhosphoSite.org (21), there are 69 JNK1 and 33 JNK2 substrates currently defined. Clearly, an attempt to determine whether all of these JNK1/2 targets regulate discrete steps in the MHV68 replication cycle is a large undertaking, but it is worth noting that our phosphoproteomic analysis identified numerous proteins—both cellular and viral—in addition to c-Jun that were phosphorylated on MAPK target motifs. Hence, potential alternative targets for JNK activity likely are present within MHV68-infected cells. Clearly, there are numerous testable hypotheses to explore in future experiments.
ACKNOWLEDGMENTS
We are indebted to Eric Siegel for help with IC50 calculations. We thank members of the Forrest and Speck laboratories for helpful comments and discussions.
This work was supported by a pilot grant from the UAMS Translational Research Institute (UL1TR000039) and startup funds from the Arkansas Biosciences Institute and UAMS College of Medicine to J.C.F. and by Public Health Service award R01CA052004 from the National Cancer Institute to S.H.S.
Footnotes
Published ahead of print 26 September 2012
REFERENCES
- 1. Adler H, Messerle M, Wagner M, Koszinowski UH. 2000. Cloning and mutagenesis of the murine gammaherpesvirus 68 genome as an infectious bacterial artificial chromosome. J. Virol. 74:6964–6974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Autret A, et al. 2007. Poliovirus induces Bax-dependent cell death mediated by c-Jun NH2-terminal kinase. J. Virol. 81:7504–7516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bennett BL, et al. 2001. SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc. Natl. Acad. Sci. U. S. A. 98:13681–13686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bortz E, et al. 2003. Identification of proteins associated with murine gammaherpesvirus 68 virions. J. Virol. 77:13425–13432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Boyne JR, Jackson BR, Taylor A, Macnab SA, Whitehouse A. 2010. Kaposi's sarcoma-associated herpesvirus ORF57 protein interacts with PYM to enhance translation of viral intronless mRNAs. EMBO J. 29:1851–1864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cheng BY, et al. 2012. Tiled microarray identification of novel viral transcript structures and distinct transcriptional profiles during two modes of productive murine gammaherpesvirus 68 infection. J. Virol. 86:4340–4357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chu WM, et al. 1999. JNK2 and IKKβ are required for activating the innate response to viral infection. Immunity 11:721–731 [DOI] [PubMed] [Google Scholar]
- 8. Clarke P, et al. 2004. JNK regulates the release of proapoptotic mitochondrial factors in reovirus-infected cells. J. Virol. 78:13132–13138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Reference deleted.
- 10. Damania B. 2007. DNA tumor viruses and human cancer. Trends Microbiol. 15:38–44 [DOI] [PubMed] [Google Scholar]
- 11. Deng H, Liang Y, Sun R. 2007. Regulation of KSHV lytic gene expression. Curr. Top. Microbiol. Immunol. 312:157–183 [DOI] [PubMed] [Google Scholar]
- 12. Dittmer DP, Richards KL, Damania B. 2012. Treatment of Kaposi sarcoma-associated herpesvirus-associated cancers. Front. Microbiol. 3:141 doi:10.3389/fmicb.2012.00141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fields BN, Knipe DM, Howley PM. 2007. Fields virology, 5th ed Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 14. Forrest JC, Paden CR, Allen RD, III, Collins J, Speck SH. 2007. ORF73-null murine gammaherpesvirus 68 reveals roles for mLANA and p53 in virus replication. J. Virol. 81:11957–11971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Forrest JC, Speck SH. 2008. Establishment of B-cell lines latently infected with reactivation-competent murine gammaherpesvirus 68 provides evidence for viral alteration of a DNA damage-signaling cascade. J. Virol. 82:7688–7699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gargano LM, Moser JM, Speck SH. 2008. Role for MyD88 signaling in murine gammaherpesvirus 68 latency. J. Virol. 82:3853–3863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gonzalez CM, et al. 2006. Identification and characterization of the Orf49 protein of Kaposi's sarcoma-associated herpesvirus. J. Virol. 80:3062–3070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Grundhoff A, Ganem D. 2004. Inefficient establishment of KSHV latency suggests an additional role for continued lytic replication in Kaposi sarcoma pathogenesis. J. Clin. Invest. 113:124–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hagemeier SR, Barlow EA, Kleman AA, Kenney SC. 2011. The Epstein-Barr virus BRRF1 protein, Na, induces lytic infection in a TRAF2- and p53-dependent manner. J. Virol. 85:4318–4329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hamza MS, et al. 2004. ORF36 protein kinase of Kaposi's sarcoma herpesvirus activates the c-Jun N-terminal kinase signaling pathway. J. Biol. Chem. 279:38325–38330 [DOI] [PubMed] [Google Scholar]
- 21. Hornbeck PV, Chabra I, Kornhauser JM, Skrzypek E, Zhang B. 2004. PhosphoSite: a bioinformatics resource dedicated to physiological protein phosphorylation. Proteomics 4:1551–1561 [DOI] [PubMed] [Google Scholar]
- 22. Hughes DJ, et al. 2010. Characterization of a novel wood mouse virus related to murid herpesvirus 4. J. Gen. Virol. 91:867–879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jackson BR, Noerenberg M, Whitehouse A. 2012. The Kaposi's sarcoma-associated herpesvirus ORF57 protein and its multiple roles in mRNA biogenesis. Front. Microbiol. 3:59 doi:10.3389/fmicb.2012.00059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Johnson LS, Willert EK, Virgin HW. 2010. Redefining the genetics of murine gammaherpesvirus 68 via transcriptome-based annotation. Cell Host Microbe 7:516–526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Laemmli UK. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685 [DOI] [PubMed] [Google Scholar]
- 26. Laichalk LL, Thorley-Lawson DA. 2005. Terminal differentiation into plasma cells initiates the replicative cycle of Epstein-Barr virus in vivo. J. Virol. 79:1296–1307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Li X, et al. 2010. Tpl2/AP-1 enhances murine gammaherpesvirus 68 lytic replication. J. Virol. 84:1881–1890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Liang X, Collins CM, Mendel JB, Iwakoshi NN, Speck SH. 2009. Gammaherpesvirus-driven plasma cell differentiation regulates virus reactivation from latently infected B lymphocytes. PLoS Pathog. 5:e1000677 doi:10.1371/journal.ppat.1000677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Loh J, et al. 2011. Identification and sequencing of a novel rodent gammaherpesvirus that establishes acute and latent infection in laboratory mice. J. Virol. 85:2642–2656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lu X, et al. 2010. The PI3K/Akt pathway inhibits influenza A virus-induced Bax-mediated apoptosis by negatively regulating the JNK pathway via ASK1. J. Gen. Virol. 91:1439–1449 [DOI] [PubMed] [Google Scholar]
- 31. Martin D, Gutkind JS. 2008. Human tumor-associated viruses and new insights into the molecular mechanisms of cancer. Oncogene 27(Suppl. 2):S31–S42 [DOI] [PubMed] [Google Scholar]
- 32. Martinez-Guzman D, et al. 2003. Transcription program of murine gammaherpesvirus 68. J. Virol. 77:10488–10503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Moser JM, Farrell ML, Krug LT, Upton JW, Speck SH. 2006. A gammaherpesvirus 68 gene 50 null mutant establishes long-term latency in the lung but fails to vaccinate against a wild-type virus challenge. J. Virol. 80:1592–1598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Reference deleted.
- 35. Nociari M, Ocheretina O, Murphy M, Falck-Pedersen E. 2009. Adenovirus induction of IRF3 occurs through a binary trigger targeting Jun N-terminal kinase and TBK1 kinase cascades and type I interferon autocrine signaling. J. Virol. 83:4081–4091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Noh CW, et al. 2012. The virion-associated open reading frame 49 of murine gammaherpesvirus 68 promotes viral replication both in vitro and in vivo as a derepressor of RTA. J. Virol. 86:1109–1118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pan H, Xie J, Ye F, Gao SJ. 2006. Modulation of Kaposi's sarcoma-associated herpesvirus infection and replication by MEK/ERK, JNK, and p38 multiple mitogen-activated protein kinase pathways during primary infection. J. Virol. 80:5371–5382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pavlova I, Lin CY, Speck SH. 2005. Murine gammaherpesvirus 68 Rta-dependent activation of the gene 57 promoter. Virology 333:169–179 [DOI] [PubMed] [Google Scholar]
- 39. Pavlova IV, Virgin HW, IV, Speck SH. 2003. Disruption of gammaherpesvirus 68 gene 50 demonstrates that Rta is essential for virus replication. J. Virol. 77:5731–5739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pipas J, Damania B. 2009. DNA tumor viruses. Springer-Verlag, New York, NY [Google Scholar]
- 41. Raman M, Chen W, Cobb MH. 2007. Differential regulation and properties of MAPKs. Oncogene 26:3100–3112 [DOI] [PubMed] [Google Scholar]
- 42. Rincon M, Davis RJ. 2009. Regulation of the immune response by stress-activated protein kinases. Immunol. Rev. 228:212–224 [DOI] [PubMed] [Google Scholar]
- 43. Sharma-Walia N, et al. 2005. ERK1/2 and MEK1/2 induced by Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) early during infection of target cells are essential for expression of viral genes and for establishment of infection. J. Virol. 79:10308–10329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Song MJ, et al. 2005. Identification of viral genes essential for replication of murine gamma-herpesvirus 68 using signature-tagged mutagenesis. Proc. Natl. Acad. Sci. U. S. A. 102:3805–3810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Staudt MR, Dittmer DP. 2007. The Rta/Orf50 transactivator proteins of the gamma-herpesviridae. Curr. Top. Microbiol. Immunol. 312:71–100 [DOI] [PubMed] [Google Scholar]
- 46. Sun CC, Thorley-Lawson DA. 2007. Plasma cell-specific transcription factor XBP-1s binds to and transactivates the Epstein-Barr virus BZLF1 promoter. J. Virol. 81:13566–13577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Upton JW, Speck SH. 2006. Evidence for CDK-dependent and CDK-independent functions of the murine gammaherpesvirus 68 v-cyclin. J. Virol. 80:11946–11959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Upton JW, van Dyk LF, Speck SH. 2005. Characterization of murine gammaherpesvirus 68 v-cyclin interactions with cellular CDKs. Virology 341:271–283 [DOI] [PubMed] [Google Scholar]
- 49. Van der Velden J, et al. 2012. Differential requirement for c-Jun N-terminal kinase 1 in lung inflammation and host defense. PLoS One 7:e34638.doi:10.1371/journal.pone.0034638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Virgin HW, IV, et al. 1997. Complete sequence and genomic analysis of murine gammaherpesvirus 68. J. Virol. 71:5894–5904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Vogt PK. 2001. Jun, the oncoprotein. Oncogene 20:2365–2377 [DOI] [PubMed] [Google Scholar]
- 52. Wang SE, et al. 2004. Early activation of the Kaposi's sarcoma-associated herpesvirus RTA, RAP, and MTA promoters by the tetradecanoyl phorbol acetate-induced AP1 pathway. J. Virol. 78:4248–4267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Weck KE, Barkon ML, Yoo LI, Speck SH, Virgin HI. 1996. Mature B cells are required for acute splenic infection, but not for establishment of latency, by murine gammaherpesvirus 68. J. Virol. 70:6775–6780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wu TT, Tong L, Rickabaugh T, Speck S, Sun R. 2001. Function of Rta is essential for lytic replication of murine gammaherpesvirus 68. J. Virol. 75:9262–9273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Xie J, Ajibade AO, Ye F, Kuhne K, Gao SJ. 2008. Reactivation of Kaposi's sarcoma-associated herpesvirus from latency requires MEK/ERK, JNK and p38 multiple mitogen-activated protein kinase pathways. Virology 371:139–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Xie J, Pan H, Yoo S, Gao SJ. 2005. Kaposi's sarcoma-associated herpesvirus induction of AP-1 and interleukin 6 during primary infection mediated by multiple mitogen-activated protein kinase pathways. J. Virol. 79:15027–15037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Yu F, et al. 2007. B cell terminal differentiation factor XBP-1 induces reactivation of Kaposi's sarcoma-associated herpesvirus. FEBS Lett. 581:3485–3488 [DOI] [PubMed] [Google Scholar]
- 58. Zapata HJ, Nakatsugawa M, Moffat JF. 2007. Varicella-zoster virus infection of human fibroblast cells activates the c-Jun N-terminal kinase pathway. J. Virol. 81:977–990 [DOI] [PMC free article] [PubMed] [Google Scholar]