

Abstract
Genetic modification of adeno-associated virus (AAV) capsids has previously been exploited to redirect viral tropism. Here we demonstrate that engineering of AAV capsids as scaffolds for antigen display augments antigen-specific immunogenicity. Combining antigen display with vector-mediated overexpression resulted in a single-shot prime-boost vaccine. This new class of vaccines induced immune responses significantly faster and an IgG antibody pool of higher avidity than conventional vectors, highlighting the potency of capsid modification in vaccine development.
TEXT
Adeno-associated virus (AAV) vectors have been shown to exhibit both an excellent safety profile in human clinical trials and therapeutic efficacy in the treatment of rare inherited diseases (11). Recently, their use was expanded to the area of vaccine development by exploiting AAV vectors for in vivo expression of viral antigens (8, 9). Viral capsids are multiprotein complexes that induce immune responses irrespective of viral gene expression. We therefore hypothesized that combining antigen display on the AAV capsid with vector-mediated antigen overexpression would increase the immunogenicity of AAV-based vaccines. In this proof-of-concept study, we made use of AAV serotype 2 vectors, which are the most frequently applied AAV vectors (3), both to display and to express Ag85A, a well-described target antigen of Mycobacterium tuberculosis vaccines (7, 14).
Three viral capsid proteins, VP1, VP2, and VP3, build up the nonenveloped AAV capsid (3). All capsid proteins share the common VP3 region (3). VP2 and VP1 differ from VP3 in N-terminal extensions of 65 amino acids. A further 137 amino acids are unique to VP1 and are required for viral infectivity (3). We and others have recently shown that relatively large proteins can be incorporated into the AAV capsid via N′-terminal fusion to VP2 (1, 10, 12, 17) without compromising viral infectivity or affecting viral tropism (10, 12). Here we inserted the Ag85A gene lacking its mycobacterial leader peptide sequence upstream of the VP2 open reading frame. Expression of native VP2 was ablated by start codon mutation in order to ensure incorporation of Ag85A-VP2 fusion proteins upon capsid assembly (10). For antigen expression, we cloned an AAV2-based vector plasmid encoding Ag85A, including the human tissue plasminogen activator leader peptide and controlled by a human cytomegalovirus promoter.
For packaging of the “prime-boost vaccine” (Ag85A-AAV:Ag85A), which displays Ag85A on the capsid surface and harbors the Ag85A expression cassette, we employed the helper virus-free AAV packaging method (19) followed by iodixanol step gradient and affinity chromatography purification (5). For comparison, we also generated Ag85A-AAV:GFP, which displays Ag85A and encodes enhanced green fluorescent protein (GFP) as a transgene; AAV:Ag85A, a classical DNA vaccine vector that contains the Ag85A expression cassette in a wild-type capsid; and as a negative control, AAV:GFP, which encodes GFP in a wild-type capsid. No significant difference in the capsid-to-genomic particle ratio was observed for capsid-modified versus unmodified vectors (data not shown), indicating that incorporation of Ag85A-VP2 fusion proteins did not adversely affect the packaging efficacy.
We vaccinated BALB/c mice by intramuscular injection with equal amounts of vector particles and adjusted capsid numbers of Ag85A-AAV:Ag85A, AAV:Ag85A, Ag85A-AAV:GFP, and AAV:GFP, respectively (Fig. 1). An additional group of mice was injected subcutaneously with recombinant Ag85A protein (rAg85A) as a positive control.
Fig 1.
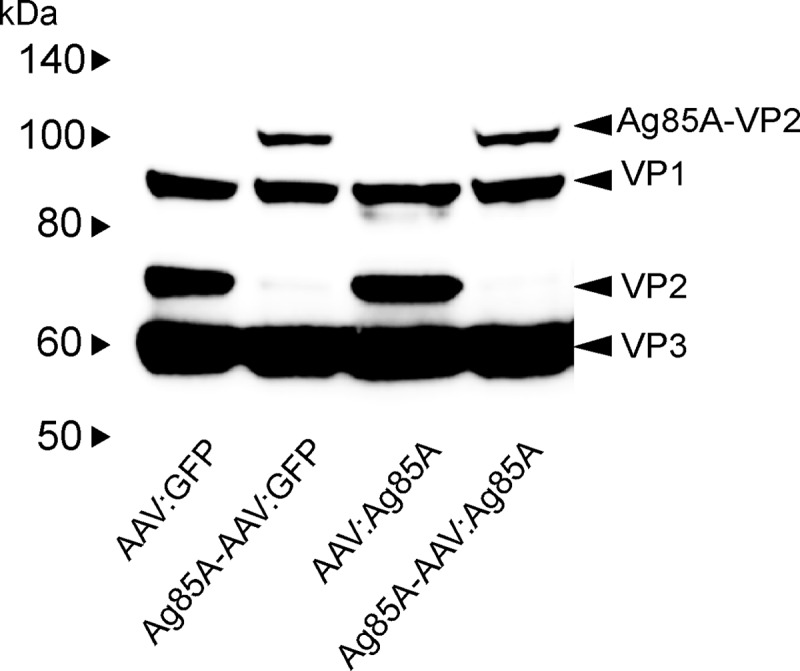
Western blot of AAV preparations. Four different AAV vectors (Ag85A-AAV:Ag85A, Ag85A-AAV:GFP, AAV:Ag85A, and AAV:GFP) were assayed for induction of humoral immune responses. Equal genomic particles of each vector preparation were applied. Since Ag85A antigen is displayed on the capsid of Ag85A-AAV:Ag85A and Ag85A-AAV:GFP, and thus impacts induction of immune responses, capsid numbers were adjusted additionally prior to injection by adding empty Ag85A-AAV capsids to Ag85A-AAV:Ag85A and Ag85A-AAV:GFP and empty wild-type capsid to AAV:Ag85A and AAV:GFP. In order to confirm the success of the adjustment, 3 × 109 genomic particles of each preparation were separated by SDS-PAGE and blotted onto a nitrocellulose membrane followed by incubation with mouse anti-AAV capsid protein antibody B1 (18) and detection by antimouse secondary antibodies (2).
Serum samples were collected from mice prior to vaccination and 2, 4, and 8 weeks postimmunization (p.i.). Serum obtained from mice prior to vaccination was nonreactive to rAg85A (data not shown). The serum of mice treated with AAV:GFP also remained nonreactive throughout the experiments (Fig. 2, 3, 4, and 6). As early as 2 weeks p.i., the prime-boost vaccine, Ag85A-AAV:Ag85A, elicited levels of anti-Ag85A-IgM antibodies as high as the positive control, rAg85A (Fig. 2A). The titer induced by Ag85A-AAV:GFP was lower but significantly higher than that in the group receiving the conventional vector AAV:Ag85A. Remarkably, whereas Ag85A-IgM titers in rAg85A- and Ag85A-AAV:GFP-vaccinated animals declined over time (as expected for antigens delivered as proteins/peptides), the Ag85A-IgM antibody titers in animals that had received the prime-boost vaccine or the conventional vector construct remained high or increased (Fig. 2B and C).
Fig 2.

Anti-Ag85A-specific IgM humoral immune response. Nine- to ten-week-old BALB/c mice had been vaccinated by intramuscular injection (caudal thigh muscle) of 1 × 1010 genomic particles of the indicated vectors (n = 6). Of note, all preparations were adjusted for equal numbers of capsids (Fig. 1). An additional group of mice (n = 6), which had been injected with 10 μg of recombinant Ag85A protein subcutaneously, served as a positive control. Sera of BALB/c mice collected prior to injection (not shown) and 2 (A), 4 (B) and 8 (C) weeks p.i. were assayed for anti-Ag85A antibodies by ELISA. On the y axes, anti-Ag85A IgM levels are displayed as absorbance at 450 nm (sera diluted 1:50). Error bars indicate the standard error of the mean (unpaired two-tailed t test). ***, P ≤ 0.001; **, P = 0.01; *, P < 0.05; NS, not significant.
Fig 3.

Anti-Ag85A-specific IgG humoral immune response. Anti-Ag85A IgG humoral immune responses were determined prior to injection (not shown) and 2 (A), 4 (B), and 8 (C) weeks p.i. by ELISA. On the y axes, anti-Ag85A IgG antibodies are displayed as absorbance at 450 nm. Error bars indicate the standard error of the mean. In comparison to controls, immunization with the Ag85A-AAV:Ag85A, the prime-boost vaccine, led to a significantly stronger humoral immune response at 2 and 4 weeks p.i. (Ag85A-AAV:Ag85A versus AAV:Ag85A, 2 weeks p.i., P < 0.0001, and 4 weeks p.i., P < 0.0001, unpaired two-tailed t test; dilution factor, 1:1,000). At all time points, IgG levels induced by Ag85A-AAV:GFP were significantly higher than those induced by rAg85A (P < 0.005, unpaired two-tailed t test).
Fig 4.

Anti-Ag85A-specific IgG1 and IgG2a humoral immune response. IgG subclasses were determined prior to injection (not shown) and 4 and 8 weeks p.i. by ELISA. On the y axes, anti-Ag85A IgG1 (B) and IgG2a (A) antibodies are displayed as absorbance at 450 nm. Error bars indicate the standard error of the mean. IgG2a (A) was only detected in relevant amounts in the Ag85A-AAV:Ag85A and AAV:Ag85A groups. At 4 and 8 weeks p.i., the prime-boost vaccine evoked a significantly higher IgG2a response than AAV:Ag85A (P = 0.0031 and 0.0483, unpaired two-tailed t test; dilution factor, 1:50). The IgG1 response (B) was similar to that for total IgG (Fig. 3).
Fig 6.

Booster vaccination with rAg85A. Anti-Ag85A IgG titers in sera of mice vaccinated with the vectors described above (Fig. 1) or rAg85A were measured 13 weeks p.i. by ELISA. Next, all mice received a subcutaneous booster vaccination with 10 μg of rAg85A. Two weeks p.i., sera were assayed for anti-Ag85A IgG titers by ELISA. On the y axes, anti-Ag85A IgG antibody titers are displayed as absorbance at 450 nm. Error bars indicate the standard error of the mean. Booster IgG responses were significantly higher for both capsid-modified vectors in comparison with AAV:Ag85A (Ag85A-AAV:Ag85A versus AAV:Ag85A, P = 0.0416, and Ag85A-AAV:GFP versus AAV:Ag85A, P < 0.005, unpaired two-tailed t test; dilution factor, 1:5,000).
Two weeks p.i., high anti-Ag85A-IgG titers were detected only for the prime-boost vaccine. These titers remained significantly higher than in any other treatment group at 4 weeks p.i. (Fig. 3A and B). At 8 weeks p.i., anti-Ag85A IgG titers induced by the Ag85A-expressing vectors were very similar and clearly exceeded those titers of the Ag85A-AAV:GFP and rAg85A groups (Fig. 3C). At all time points, IgG levels induced by Ag85A-AAV:GFP were significantly higher than those induced by rAg85A, indicating that incorporation of the antigen into the capsid as such augments antigen-specific immunogenicity.
Regarding IgG subclasses (Fig. 4), the IgG1 response to the different vectors followed the same pattern as for total IgG, while induction of IgG2a was only detected in relevant amounts in serum from mice that had been vaccinated with the prime-boost vaccine or AAV:Ag85A. Even at 8 weeks p.i., the prime-boost vaccine evoked a significantly higher IgG2a response than AAV:Ag85A.
These data reveal that incorporation of the Ag85A antigen into the AAV capsid is most advantageous with regard to the rapidity of antibody response as total IgM and IgG titers become equivalent for Ag85A-AAV:Ag85A and AAV:Ag85A only at 8 weeks.
IgG avidity is widely used to distinguish between primary and secondary antigenic challenge. After primary antigenic challenge, IgG avidity is low, while antibodies elicited in a secondary response show a substantial higher average avidity (6, 15, 16). We were therefore interested in determining whether the biphasic immune activation strategy of our novel vaccine (i.e., early B-cell activation through Ag85A protein presented by the viral capsid and later B-cell activation upon transgene expression) impacts IgG antibody avidity. To this end, we performed an ammonium thiocyanate (NH4SCN) elution procedure (13) based on a standard enzyme-linked immunosorbent assay (ELISA) with sera obtained from mice vaccinated with Ag85A-AAV:Ag85A and AAV:Ag85A, respectively. We estimated the avidity index as the molar concentration of thiocyanate required to reduce the initial absorbance (in the absence of NH4SCN) by 50% (4). As shown in Fig. 5, the anti-Ag85A IgG antibody pool elicited upon vaccination with the prime-boost vaccine showed significantly higher avidity than IgG antibodies induced by the conventional vector.
Fig 5.

Relative avidity of anti-Ag85A IgG antibody pool elicited upon immunization with prime-boost vaccine compared to a conventional vector-based construct. The ammonium thiocyanate (NH4SCN) elution assay was performed as described previously (13). Specifically, anti-Ag85A IgG antibodies in sera obtained from mice (n = 5) 8 weeks p.i., in a dilution of 1:1,000, were detected by a standard ELISA. The serum dilution of 1:1,000 was carefully chosen at a point at which the absorbance values were linearly proportional to the antibody concentrations (Fig. 3C). After antibody incubation, NH4SCN was added to the appropriate duplicate wells at increasing molarities. The avidity index was calculated by converting absorbance readings in the presence of increasing concentrations of NH4SCN to the appropriate percentage of total bound antibody (absorbance readings in the absence of NH4SCN). The y axis shows the relative avidity index estimated according to Ferreira and Katzin (4) as the molar concentration of thiocyanate required to reduce the initial absorbance by 50%. IgG antibodies elicited upon vaccination with Ag85A-AAV:Ag85A showed significantly higher avidity to the Ag85A antigen than IgG antibodies induced by AAV:Ag85A (P < 0.005).
To further investigate the contribution of capsid modification as such, we checked for memory recall responses as a measure of antigen-specific immunogenicity. Specifically, we performed subcutaneous booster vaccination with rAg85A and determined anti-Ag85A IgG antibody titers 2 weeks later (Fig. 6). As expected, for mice initially injected with Ag85A-AAV:Ag85A or AAV:Ag85A, the Ag85A-encoding vectors, we detected only a moderate increase in anti-Ag85A antibody titers compared to baseline (IgG titer prior to booster vaccination). Of note, overall IgG titers achieved upon booster vaccination were significantly higher for the capsid-modified vectors than for AAV:Ag85A.
The most impressive memory recall response was elicited in mice treated with Ag85A-AAV:GFP, the vector presenting Ag85A as the antigen on the capsid. Compared to the baseline, antibody titers increased more than 10-fold up to the same level as that in mice treated with the prime-boost vaccine. Antibody titers in mice initially vaccinated with rAg85A, however, increased by only 6-fold and thus remained significantly lower than those in all other groups (except for the negative control). Thus, antigen incorporation into AAV capsids augments antigen-specific immunogenicity and is by itself sufficient to induce a memory recall response.
Our findings demonstrate that the combination of antigen incorporation with antigen overexpression after cell transduction can dramatically enhance the antigenic potential of AAV-based vaccines. Humoral immune responses were induced much more rapidly than with conventional vaccination strategies. Due to continuous antigen release, antigen-specific antibody titers remained high even at 13 weeks p.i. in those groups that received antigen-encoding vectors, while levels declined in mice that were vaccinated with Ag85A as protein.
The combination of antigen presentation by the viral capsid with vector-mediated antigen overexpression, as we here demonstrate for AAV2, can easily be translated to more immunogenic serotypes, such as rh32.33 (8), which could further increase the potency of AAV-based vaccines. Based on these findings, a variety of applications for antigen-VP2 fusions are now possible, such as vaccination with multiple antigens using a single construct, for example, where vaccinations require an immunization with antigens from different serotypes, as is the case for dengue virus.
ACKNOWLEDGMENTS
J.R. and P.H. are supported by the German Ministry of Education and Research (Bundesministerium für Bildung und Forschung [BMBF]) grant 01 KI 0771. H.B. is supported by the Collaborative Research Center 670 (SFB670) of the German Research Council (DFG) and the Center for Molecular Medicine of the University of Cologne (B1).
We are much obliged to Edel van Gumpel and Sandra Winter for excellent technical assistance. We thank Oliver Coutelle (University of Cologne) and Elizabeth Schell-Frederick for critically reading the manuscript, Jude Samulski (University of North Carolina at Chapel Hill) for providing pXX6-80, and Jürgen Kleinschmidt (DKFZ, Heidelberg) for providing the anti-capsid protein antibody B1. Polyclonal anti-Mycobacterium tuberculosis antigen 85 complex (anti-serum, rabbit, NR-13800) was obtained through the NIH Biodefense and Emerging Infections Research Resources Repository, NIAID, NIH.
Footnotes
Published ahead of print 3 October 2012
REFERENCES
- 1. Asokan A, Johnson JS, Li C, Samulski RJ. 2008. Bioluminescent virion shells: new tools for quantitation of AAV vector dynamics in cells and live animals. Gene Ther. 15:1618–1622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Boucas J, et al. 2009. Engineering adeno-associated virus serotype 2-based targeting vectors using a new insertion site-position 453- and single point mutations. J. Gene Med. 11:1103–1113 [DOI] [PubMed] [Google Scholar]
- 3. Buning H, Perabo L, Coutelle O, Quadt-Humme S, Hallek M. 2008. Recent developments in adeno-associated virus vector technology. J. Gene Med. 10:717–733 [DOI] [PubMed] [Google Scholar]
- 4. Ferreira MU, Katzin AM. 1995. The assessment of antibody affinity distribution by thiocyanate elution: a simple dose-response approach. J. Immunol. Methods 187:297–305 [DOI] [PubMed] [Google Scholar]
- 5. Hacker UT, et al. 2005. Adeno-associated virus serotypes 1 to 5 mediated tumor cell directed gene transfer and improvement of transduction efficiency. J. Gene Med. 7:1429–1438 [DOI] [PubMed] [Google Scholar]
- 6. Hamkar R, et al. 2005. Assessment of IgM enzyme immunoassay and IgG avidity assay for distinguishing between primary and secondary immune response to rubella vaccine. J. Virol. Methods 130:59–65 [DOI] [PubMed] [Google Scholar]
- 7. Huygen K, et al. 1996. Immunogenicity and protective efficacy of a tuberculosis DNA vaccine. Nat. Med. 2:893–898 [DOI] [PubMed] [Google Scholar]
- 8. Lin J, et al. 2009. A new genetic vaccine platform based on an adeno-associated virus isolated from a rhesus macaque. J. Virol. 83:12738–12750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Logan GJ, Wang L, Zheng M, Coppel RL, Alexander IE. 2010. Antigen fusion with C3d3 augments or inhibits humoral immunity to AAV genetic vaccines in a transgene-dependent manner. Immunol. Cell Biol. 88:228–232 [DOI] [PubMed] [Google Scholar]
- 10. Lux K, et al. 2005. Green fluorescent protein-tagged adeno-associated virus particles allow the study of cytosolic and nuclear trafficking. J. Virol. 79:11776–11787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mingozzi F, High KA. 2011. Therapeutic in vivo gene transfer for genetic disease using AAV: progress and challenges. Nat. Rev. Genet. 12:341–355 [DOI] [PubMed] [Google Scholar]
- 12. Munch RC, et al. 2012. Displaying high-affinity ligands on adeno-associated viral vectors enables tumor cell-specific and safe gene transfer. Mol. Ther. [Epub ahead of print.] doi:10.1038/mt.2012.186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pullen GR, Fitzgerald MG, Hosking CS. 1986. Antibody avidity determination by ELISA using thiocyanate elution. J. Immunol. Methods 86:83–87 [DOI] [PubMed] [Google Scholar]
- 14. Romano M, et al. 2006. Priming but not boosting with plasmid DNA encoding mycolyl-transferase Ag85A from Mycobacterium tuberculosis increases the survival time of Mycobacterium bovis BCG vaccinated mice against low dose intravenous challenge with M. tuberculosis H37Rv. Vaccine 24:3353–3364 [DOI] [PubMed] [Google Scholar]
- 15. Steiner LA, Eisen HN. 1967. The relative affinity of antibodies synthesized in the secondary response. J. Exp. Med. 126:1185–1205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Steiner LA, Eisen HN. 1967. Sequential changes in the relative affinity of antibodies synthesized during the immune response. J. Exp. Med. 126:1161–1183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Warrington KH, Jr, et al. 2004. Adeno-associated virus type 2 VP2 capsid protein is nonessential and can tolerate large peptide insertions at its N terminus. J. Virol. 78:6595–6609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wobus CE, et al. 2000. Monoclonal antibodies against the adeno-associated virus type 2 (AAV-2) capsid: epitope mapping and identification of capsid domains involved in AAV-2-cell interaction and neutralization of AAV-2 infection. J. Virol. 74:9281–9293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Xiao X, Li J, Samulski RJ. 1998. Production of high-titer recombinant adeno-associated virus vectors in the absence of helper adenovirus. J. Virol. 72:2224–2232 [DOI] [PMC free article] [PubMed] [Google Scholar]