

Abstract
Mammalian reoviruses display serotype-specific patterns of tropism and disease in the murine central nervous system (CNS) attributable to polymorphisms in viral attachment protein σ1. While all reovirus serotypes use junctional adhesion molecule-A as a cellular receptor, they differ in their utilization of carbohydrate coreceptors. This observation raises the possibility that carbohydrate binding by σ1 influences reovirus pathology in the CNS. In this study, we sought to define the function of carbohydrate binding in reovirus neuropathogenesis. Newborn mice were inoculated intramuscularly with wild-type strain type 3 Dearing (T3D) and T3D-σ1R202W, a point mutant T3D derivative that does not bind sialic acid (SA). Infected mice were monitored for survival, and viral loads at the sites of primary and secondary replication were quantified. Fewer mice inoculated with the wild-type virus survived in comparison to those inoculated with the mutant virus. The wild-type virus also produced higher titers in the spinal cord and brain at late times postinoculation but lower titers in the liver in comparison to those produced by the mutant virus. In addition, the wild-type virus was more virulent and produced higher titers in the brain than the mutant following intracranial inoculation. These animal infectivity studies suggest that T3D-σ1R202W harbors a defect in neural growth. Concordantly, compared with the wild-type virus, the mutant virus displayed a decreased capacity to infect and replicate in primary cultures of cortical neurons, a property dependent on cell surface SA. These results suggest that SA binding enhances the kinetics of reovirus replication in neural tissues and highlight a functional role for sialylated glycans as reovirus coreceptors in the CNS.
INTRODUCTION
Attachment to specific cell surface molecules is the initial step in viral infection. As such, receptor engagement can influence many aspects of viral pathogenesis, perhaps most prominently systemic dissemination and tropism for distinct host cells and tissues. Tropism of human immunodeficiency virus (HIV) (27), poliovirus (32), and severe acute respiratory syndrome (SARS) coronavirus (15), among others, is strongly influenced by the capacity of the virus to bind specific receptors. Infection of discrete regions within the central nervous system (CNS) by strains of adeno-associated virus (AAV) is dependent on carbohydrate-binding specificity. AAV4 infects ependymal cells and utilizes α(2,3)-linked sialic acid (SA) as a receptor. In contrast, AAV5 uses both α(2,3)- and α(2,6)-linked SA to infect neurons (22). While much is known about the importance of these receptor-mediated functions in viral spread and tropism, the precise mechanisms by which receptor utilization regulates viral infection in the CNS are not fully understood.
Mammalian orthoreoviruses (reoviruses), members of the Reoviridae, are nonenveloped viruses comprising two concentric protein shells that enclose the 10 segments of the viral double-stranded RNA genome (37). These viruses are genetically tractable tools for use in studies of virus-receptor interactions and viral pathogenesis. In neonatal mice, reovirus serotypes 1 and 3 disseminate from sites of primary replication to the CNS. However, tropism for neural cells and resulting disease outcomes vary in a serotype-specific manner. Type 1 reovirus strains spread to the CNS via hematogenous routes and infect ependymal cells in the brain, which leads to a nonlethal hydrocephalus (40, 43, 44). In contrast, type 3 reovirus strains spread to the CNS using neural and hematogenous routes and target neurons in the brain, which results in lethal encephalitis (1, 6, 28, 40). These serotype-specific differences in tropism and disease segregate genetically with the viral S1 gene (43, 44), which encodes attachment protein σ1 (26, 45) and nonstructural protein σ1s (9, 17, 35). Both S1 gene products influence reovirus pathogenesis (1, 5–7, 23), with the σ1 protein thought to target the virus to discrete cell types in the CNS (14, 39).
Reovirus attachment protein σ1 is a filamentous trimer with three discrete domains: the tail, the body, and the head (11, 19, 31). Residues 1 to 160 encompass the tail domain, which inserts into the virion capsid (11, 20). This region of the molecule is predicted to form an α-helical coiled coil (16, 29). The body domain encompasses residues 170 to 309 and comprises a β-spiral repeat motif interrupted by a short α-helical coiled coil (31). The globular head domain incorporates residues 310 to 455 and folds into an 8-stranded β-barrel (11, 24, 36).
Receptors for reoviruses are engaged by a multistep adhesion-strengthening process in which σ1 first binds to an abundant cell surface carbohydrate with low affinity (3), which is followed by high-affinity interactions with junctional adhesion molecule-A (JAM-A) (2, 4). Sequences in the σ1 body domain mediate SA binding by type 3 strains (10, 31), whereas sequences in the σ1 head domain engage JAM-A (24). Interactions between σ1 and SA involve residues at the N-terminal portion of the body domain, between β-spiral repeats 2 and 3, where the SA moiety docks into a shallow pocket that is formed mainly by residues in the third β-spiral repeat (31).
While all reovirus serotypes engage JAM-A (8, 30), they differ in carbohydrate coreceptor utilization, suggesting that carbohydrate binding by σ1 influences reovirus pathology in the CNS. Studies in newborn mice comparing the pathogenesis of reovirus strains that differ in glycan utilization revealed that SA binding enhances spread from the intestine to peripheral sites of replication such as the liver, spleen, and brain (5). In addition, the capacity to bind SA confers reovirus tropism for bile duct epithelia (5). However, it is not known whether SA binding influences reovirus dissemination, tropism, and replication within the CNS.
In this study, we sought to define the function of carbohydrate binding in the neuropathogenesis of reovirus. We monitored the survival of newborn mice inoculated with wild-type type 3 Dearing (T3D) and T3D-σ1R202W, a T3D point mutant virus that does not bind SA (31), and quantified the viral loads in various organs of infected animals. We found that the wild-type virus was more virulent and produced higher titers in neural tissues than the mutant virus following intramuscular or intracranial inoculation. Furthermore, we found that the wild-type virus displayed an increased capacity to infect and replicate in primary cultures of cortical neurons in an SA-dependent manner. Collectively, these results suggest that binding to SA enhances the kinetics of reovirus replication in neural tissues and point to a functional role for sialylated glycan engagement in reovirus neurovirulence.
MATERIALS AND METHODS
Cells and viruses.
L929 cells (34) were maintained in Joklik's minimum essential medium supplemented to contain 5% fetal bovine serum (FBS), 2 mM l-glutamine, 100 U/ml of penicillin, 100 μg/ml of streptomycin, and 25 ng/ml of amphotericin B (Invitrogen).
Strains T3D (25) and T3D-σ1R202W (31) were generated by plasmid-based reverse genetics. Purified reovirus virions were obtained using second- or third-passage L929 cell lysate stocks of twice-plaque-purified reovirus, as described previously (20). Viral particles were Freon extracted from infected cell lysates, layered onto 1.2 to 1.4 g/cm3 CsCl gradients, and centrifuged at 62,000 × g for 18 h. Bands corresponding to virions (1.36 g/cm3) (38) were collected and dialyzed in virion storage buffer (150 mM NaCl, 15 mM MgCl2, 10 mM Tris-HCl [pH 7.4]). The concentration of reovirus virions in purified preparations was determined from an equivalence of 1 optical density (OD) unit at 260 nm equaling 2.1 × 1012 virions (38). The viral titers were determined by plaque assay using L929 cells (42). The average particle-to-PFU ratios for T3D and T3D-σ1R202W were 893 (n = 4) and 1,050 (n = 5), respectively. These values do not differ statistically.
Infection of mice.
C57BL/6J mice were obtained from the Jackson Laboratory to establish breeding colonies. At 2 to 3 days of age, newborn mice were inoculated intramuscularly or intracranially with purified reovirus diluted in phosphate-buffered saline (PBS). Intramuscular (IM) inoculations (5 μl) were delivered into the right hind limb, and intracranial (IC) inoculations (5 μl) were delivered into the left cerebral hemisphere; in both cases, Hamilton syringes with 30-gauge needles were used. For the analysis of viral virulence, the mice were monitored for clinical signs of disease for 21 days postinoculation. For survival experiments, mice were euthanized when found to be moribund (defined by rapid or shallow breathing, lethargy, or paralysis). Death was not used as an endpoint. For the analysis of virus replication, mice were euthanized at various intervals following inoculation, and their organs were collected into 1 ml of PBS and homogenized by freezing, thawing, and sonicating. For the analysis of viremia, mice were euthanized and decapitated at various intervals following inoculation, and whole blood was collected from their necks into a 1-ml syringe containing 100 μl Alsever's solution (Sigma). The viral titers in organ homogenates were determined by plaque assay using L929 cells (42).
For experiments in which viral titers in an organ or blood were determined, the Mann-Whitney test was used to calculate two-tailed P values. This test is appropriate for experimental data that display a non-Gaussian distribution (33). P values of less than 0.05 were considered statistically significant. When all values are less than the limit of detection, a Mann-Whitney test P value cannot be calculated. Statistical analyses were performed using Prism software (GraphPad Software, Inc.).
All animal husbandry and experimental procedures were performed in accordance with Public Health Service policy and approved by the Vanderbilt University School of Medicine Institutional Animal Care and Use Committee.
Histology and immunohistochemical staining for reovirus antigen.
At 2 to 3 days of age, newborn mice were inoculated intramuscularly or intracranially with reovirus diluted in PBS. The mice were euthanized 8 days postinoculation, and their organs were resected and incubated in 10% formalin at room temperature for 24 to 48 h. Fixed organs were embedded in paraffin, and consecutive 6-mm sections were stained with hematoxylin and eosin (H&E) for the evaluation of histopathologic changes or processed for immunohistochemical detection of reovirus proteins (5) or the activated form of caspase-3 (Cell Signaling Technology, Inc.) (12).
Quantification of viral RNA using RT-qPCR.
Total RNA was extracted from 200 μl of whole blood-Alsever's mixture using the High Pure Viral RNA kit (Roche). RNA was eluted into a final volume of 40 μl. Reverse transcription-quantitative PCR (RT-qPCR) was performed using an ABI 7000 sequence detection system (Applied Biosystems) and EZ RT-PCR (Roche) according to the manufacturer's instructions with minor modifications. Reovirus RNA was quantified using 10 μl of RNA extract. Forward (S4 83F, 5′-CGCTTTTGAAGGTCGTGTATCA-3′) and reverse (S4 153R, 5′-CTGGCTGTGCTGAGATTGTTTT-3′) primers corresponding to the viral S4 gene were used for RT and qPCR amplification. The S4-specific fluorogenic probe used was 5′-dFAM-AGCGCGCAAGAGGGATGGGA-BHQ-1 (where dFAM is 6-carboxyfluorescein and BHQ is black hole quencher) to 3′ (Biosearch Technologies). RT was performed at 50°C for 2 min, followed by incubation at 60°C for 30 min. The reaction was terminated by incubation at 95°C for 5 min. Subsequently, 40 cycles of qPCR were performed at 95°C for 15 s, followed by incubation at 60°C for 30 s. Standard curves relating threshold cycle values to copies of plasmid DNA template were generated using 10-fold dilutions of a T3D S4-encoding plasmid (pT7-T3D S4) (25). The concentration of viral RNA in each sample was extrapolated from standard curves. The final S4 RNA copy number was calculated by multiplying the copy number obtained by extrapolation from the standard curve by 4 to account for using one-quarter of the extracted RNA as a template.
Preparation of cortical neuron cultures from embryonic mice.
Primary cultures of mouse cortical neurons were established using the cerebral cortices of C57/BL6 embryos at developmental day 15.5 (1). Fetuses were decapitated, the brains were removed, and the cortical lobes were dissected and submerged in Hanks' balanced salt solution (Gibco) on ice. The cortices were incubated in 0.6 mg/ml trypsin solution at room temperature for 30 min, washed twice, and manually dissociated twice using a Pasteur pipette. Viable cells were plated at a density of 2.5 × 105 cells/ml in 24-well plates. The wells were treated prior to plating with a 10 μg/ml poly-d-lysine solution (BD Biosciences) and a 1.64 μg/ml laminin solution (BD Biosciences). Cultures were incubated for the first 24 h in Neurobasal medium (Gibco) supplemented to contain 10% FBS (Gibco), 0.6 mM l-glutamine, 50 U/ml penicillin, and 50 μg/ml streptomycin. Cultures were thereafter maintained in Neurobasal medium supplemented to contain B27 supplement (Gibco), 50 U/ml penicillin, and 50 μg/ml streptomycin. One-half of the medium was replaced with fresh medium every 3 to 4 days. Neurons were cultivated for 7 days prior to use.
Assessment of reovirus infectivity by indirect immunofluorescence.
Monolayers of murine cortical neurons (2.5 × 105 cells/well) seeded in 24-well plates were adsorbed with reovirus at a multiplicity of infection (MOI) of 1,000 PFU/cell at room temperature for 1 h. Cells were incubated with either PBS or 40 mU Arthrobacter ureafaciens neuraminidase at 37°C for 1 h and washed three times with PBS prior to virus adsorption. Following removal of the virus inoculum, the cells were washed with PBS and incubated in complete medium at 37°C for 21 h to permit completion of a single cycle of viral replication. Monolayers were fixed with 1 ml of methanol at −20°C for at least 30 min, washed twice with PBS, and blocked with 0.1% Tween 20 (Sigma) and 20% normal goat serum (Vector Laboratories) in PBS. The cells were washed once with PBS and stained with polyclonal rabbit anti-reovirus serum at a 1:1,000 dilution in PBS-0.5% Triton X-100 at room temperature for 1 h. Neurons were visualized after washing three times with PBS and staining with a monoclonal mouse anti-β-tubulin antibody (TUJ1; Covance) at a 1:1,000 dilution in PBS-0.5% Triton X-100 at room temperature for 1 h. Monolayers were washed twice with PBS-0.5% Triton X-100 and incubated with a 1:1,000 dilution of Alexa 488-labeled anti-rabbit and Alexa 546-labeled anti-mouse IgG (Invitrogen). Nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI) (Invitrogen) at a 1:10,000 dilution. Monolayers were washed with PBS, and infected cells were visualized by indirect immunofluorescence using an Axiovert 200 fluorescence microscope (Carl Zeiss).
Assessment of reovirus replication by plaque assay.
Murine primary cortical neurons (105 cells/well) were adsorbed with reovirus strains at an MOI of 1 PFU/cell. Cells were incubated with either PBS or 40 mU A. ureafaciens neuraminidase at 37°C for 1 h and washed three times with PBS prior to virus adsorption. Following incubation with the virus at room temperature for 1 h, the cells were washed three times with PBS and incubated at 37°C for various intervals. Samples were frozen and thawed twice, and viral titers were determined by plaque assay using L929 cells (42). For each experiment, the samples were infected in triplicate. The viral yields were calculated according to the following formula: log10 yieldtx = log10(PFU/ml)tx − log10(PFU/ml)t0, where tx is the time postinfection. The mean values from two independent experiments were compared using an unpaired Student's t test (applied in Microsoft Excel). P values of less than 0.05 were considered statistically significant.
RESULTS
SA binding enhances reovirus neurovirulence and spinal cord injury.
To determine whether SA binding influences reovirus neuropathogenesis, we monitored survival and clinical signs of disease in 2-day-old mice following IM inoculation with 5 × 106 PFU of either wild-type T3D or mutant T3D-σ1R202W (Fig. 1A). Reovirus strain T3D-σ1R202W contains a point mutation in σ1 that ablates the SA-binding capacity (31). Mice were observed for 21 days after inoculation and euthanized when found to be moribund. Significantly fewer mice infected with strain T3D survived than those infected with strain T3D-σ1R202W. The survival frequency of mice infected with T3D was 25.9% compared with 63.6% for the mice infected with T3D-σ1R202W (P < 0.005). These results suggest that SA binding enhances reovirus virulence.
Fig 1.

SA-binding capacity enhances reovirus virulence and paralysis following IM inoculation. Newborn C57/BL6 mice were inoculated in the right hind limb with 5 × 106 PFU of either T3D or T3D-σ1R202W (R202W). (A) Mice (n = 22 to 27 for each virus strain) were monitored for survival for 21 days. The asterisk indicates a P value of <0.005, as determined by log-rank test in comparison to strain T3D. (B) Mice were monitored for the development of reovirus-induced acute flaccid paralysis on day 8 postinoculation (n = 32 to 44 mice for each virus strain). Complete paralysis was first observed at this time point; survival frequencies of wild-type and mutant-infected mice at this time point were comparable.
Following IM inoculation of newborn mice, serotype 3 reovirus causes acute flaccid paralysis (AFP) between 8 and 10 days postinoculation (21). Infected mice exhibit ipsilateral, followed by contralateral, hind-limb paralysis as a consequence of injury within the anterior horn of the spinal cord associated with motor neuron loss and the spread of viral antigen (21). We observed differences in the development of AFP in mice infected with the wild-type and the mutant viruses. On day 8 postinoculation, mice infected with the wild-type virus displayed hind-limb paralysis at a 5-fold greater frequency than those infected with the mutant virus (33% versus 6%, respectively) (Fig. 1B). Furthermore, the percentage of mice infected with the wild-type virus exhibiting bilateral hind-limb paralysis was almost four times greater than that of mice infected with the mutant virus (11% versus 3%, respectively) (Fig. 1B). These data are concordant with results gathered from the survival experiments and provide further evidence that SA binding enhances reovirus virulence.
To investigate whether enhanced virulence and AFP development in mice infected with the wild-type virus compared with the mutant virus correspond with more extensive reovirus infection and tissue damage in the spinal cord, we compared histologic sections of spinal cords resected from mice inoculated intramuscularly with 5 × 106 PFU of either T3D or T3D-σ1R202W (Fig. 2). Transverse sections of the inferior spinal cord were sectioned and processed for H&E staining, staining for reovirus antigen, or staining for the activated form of caspase-3, a marker of apoptosis. Following H&E staining, spinal cords of the mice infected with the wild-type virus displayed more widespread injury than that observed in mutant-infected mice, as indicated by increased apoptotic bodies, reactive endothelia, inflammatory infiltrates, and neurons with eosinophilic cytoplasmic inclusions. Immunohistochemistry for reovirus antigen showed correspondingly more abundant and intense staining in mice infected with the wild-type virus than in spinal cords from the mice infected with the mutant virus. Staining for the activated form of caspase-3 demonstrated more apoptotic neurons in the spinal cord tissues of mice infected with the wild-type virus in comparison to those from mutant-infected mice. Quantification of caspase-3-positive cells revealed that mice infected with the wild-type virus contained a significantly higher number of apoptotic cells in the spinal cord sections (P < 0.005) (Fig. 3). These findings suggest that binding to SA leads to increased reovirus infection and apoptotic injury to motor neurons in the spinal cord.
Fig 2.

SA-binding capacity enhances reovirus-induced injury in the spinal cord following IM inoculation. Newborn C57/BL6 mice were inoculated in the right hind limb with 5 × 106 PFU of either T3D or T3D-σ1R202W (R202W). The inferior spinal cord was resected on day 8 postinoculation, and consecutive lumbosacral sections were stained with hematoxylin and eosin (H&E), polyclonal reovirus antiserum, or antibodies specific for the cleaved (active) form of caspase-3. Representative sections of spinal cord matched for lumbar depth are shown. Boxes indicate areas of enlargement.
Fig 3.
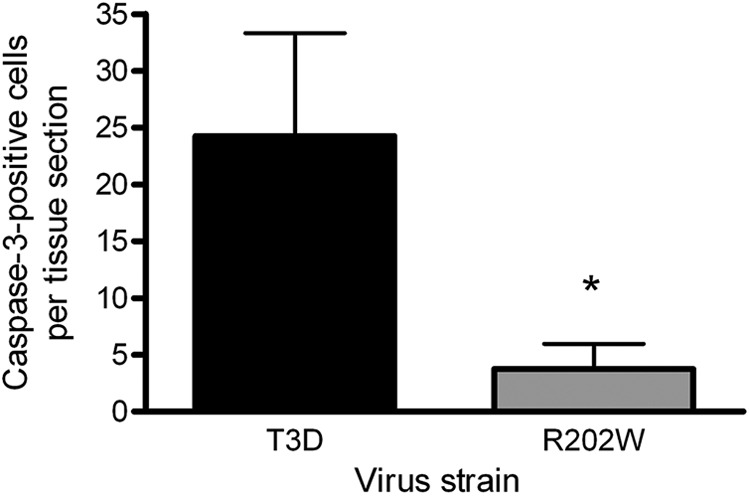
Quantification of apoptotic cells in the spinal cord of reovirus-infected mice. Lumbosacral sections of the inferior spinal cord resected from newborn mice inoculated intramuscularly in the right hind limb with 5 × 106 PFU of either T3D or T3D-σ1 R202W (R202W) were stained for the cleaved form of caspase-3. Cells staining positive for caspase-3 were enumerated in individual sections obtained from four animals infected with each virus strain. Results are expressed as the mean caspase-3-positive cells per tissue section. Error bars represent the SD. The asterisk indicates a P value of <0.005, as determined by Student's t test, in comparison to strain T3D.
SA binding influences reovirus replication at sites of secondary replication.
To better understand how SA-binding capacity influences reovirus virulence, we inoculated 2-day-old mice intramuscularly with 5 × 106 PFU of either T3D or T3D-σ1R202W and quantified viral titers in the hind limb, spinal cord, brain, and liver on days 2, 4, 8, and 10 postinoculation (Fig. 4). In the hind-limb muscle, the titers of both viruses were comparable, except on day 4 when the mutant virus produced higher titers than the wild-type virus (P < 0.005). In the spinal cord and brain, the mutant virus produced significantly higher titers than the wild-type virus at day 4 (P < 0.05). However, on days 8 and 10, titers of the wild-type virus were significantly higher than those of the mutant (P < 0.05). Interestingly, titers in the liver of mice infected with the wild-type virus were significantly lower than those in mice infected with the mutant virus throughout the experimental time course of infection (P < 0.05 on day 2 and P < 0.005 on days 4 and 8).
Fig 4.
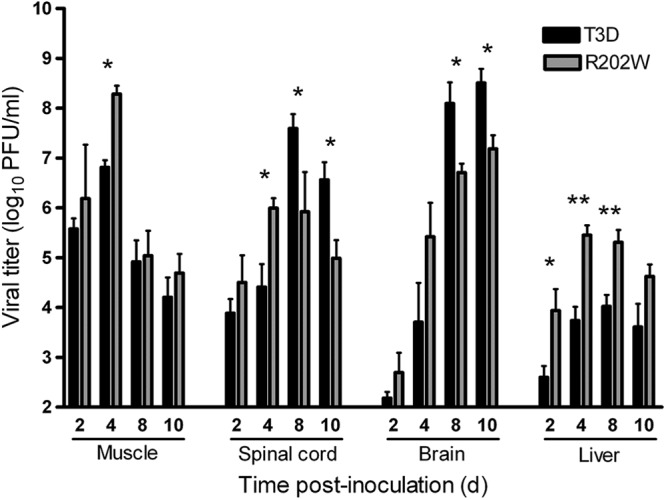
SA binding is associated with higher peak titers of reovirus in neural tissues. Newborn C57/BL6 mice were inoculated in the right hind limb with 5 × 106 PFU of either T3D or T3D-σ1R202W (R202W). At days 2, 4, 8, and 10 postinoculation, the mice were euthanized, hind-limb muscle, spinal cord, brain, and liver were resected, and the viral titers in organ homogenates were determined by plaque assay. Results are expressed as mean viral titers for 6 to 9 animals for each time point. Error bars indicate the SEM. One asterisk indicates P values of <0.05, and two asterisks indicate P values of <0.005, as determined by the Mann-Whitney test, in comparison to strain T3D.
To determine whether SA-binding capacity influences the kinetics of hematogenous spread, we again inoculated 2-day-old mice intramuscularly with 5 × 106 PFU of either T3D or T3D-σ1R202W. In this experiment, we quantified viral titers in the heart and spleen (Fig. 5A) and monitored viral genome copies in the serum (Fig. 5B). There were statistically significant differences in the titers produced by the wild-type and mutant viruses in the heart on day 4 postinoculation and in the spleen on days 4 and 10 postinoculation. On day 4 postinoculation, the wild-type virus produced a higher titer in the heart but a lower titer in the spleen than did the mutant virus. On day 10 postinoculation, the wild-type virus produced a higher titer than the mutant virus in the spleen. Although these differences are statistically significant, because of the modest magnitude of the differences, we do not think that they are biologically relevant. Furthermore, the number of viral genome copies detected in the serum of mice infected with either virus was comparable. Thus, SA-binding capacity enhances overall peak titers in the murine CNS. However, utilization of SA is not required for infection of the liver and does not appear to influence hematogenous dissemination in mice.
Fig 5.
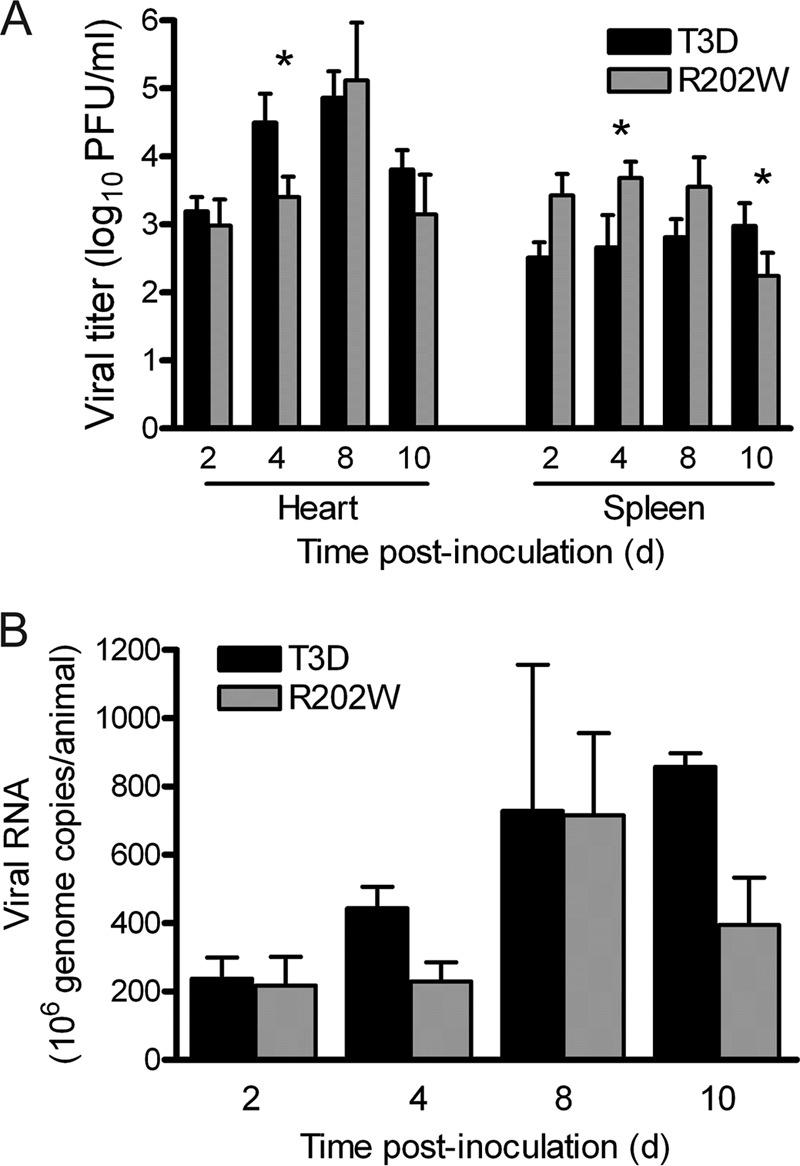
Binding to SA is not required for dissemination by hematogenous routes. Newborn C57/BL6 mice were inoculated in the right hind limb with 5 × 106 PFU of either T3D or T3D-σ1R202W (R202W). (A) At days 2, 4, 8, and 10 postinoculation, the mice were euthanized, and their hearts and spleens were resected. Viral titers in the organ homogenates were determined by plaque assay. Results are expressed as mean viral titers for 7 to 10 animals for each time point. Error bars indicate the SEM. The asterisks indicate P values of <0.05, as determined by the Mann-Whitney test, in comparison to strain T3D. (B) At days 2, 4, 8, and 10 postinoculation, the mice were euthanized, their blood was collected, and the viral genome copies in serum were determined by RT-qPCR. Results are expressed as the mean viral genome copies per animal for 3 to 5 animals at each time point. Error bars indicate the SEM.
SA binding enhances reovirus neurovirulence and replication in the brain.
Differences in titers produced by strains T3D and T3D-σ1R202W in the spinal cord and brain could result from more efficient spread of the wild-type virus from the muscle to the CNS, from enhanced replication of the wild-type virus in the CNS, or from both effects. To distinguish between these possibilities, we inoculated 3-day-old mice intracranially with 2 PFU of either the wild-type or the mutant virus. We monitored infected mice for survival and clinical signs of disease for 21 days and quantified viral loads in the brain at days 4, 8, and 12 following inoculation (Fig. 6). We again observed that mice infected with the wild-type virus displayed a lower frequency of survival (33%) compared with those infected with the mutant virus (85%) (P < 0.001) (Fig. 6A). In addition, the wild-type virus produced higher titers in the brain than did the mutant following IC inoculation (Fig. 6B), and this difference reached statistical significance on day 8 postinoculation (P < 0.05). Taken together, these findings suggest that SA binding enhances reovirus virulence by augmenting viral replication in the CNS.
Fig 6.
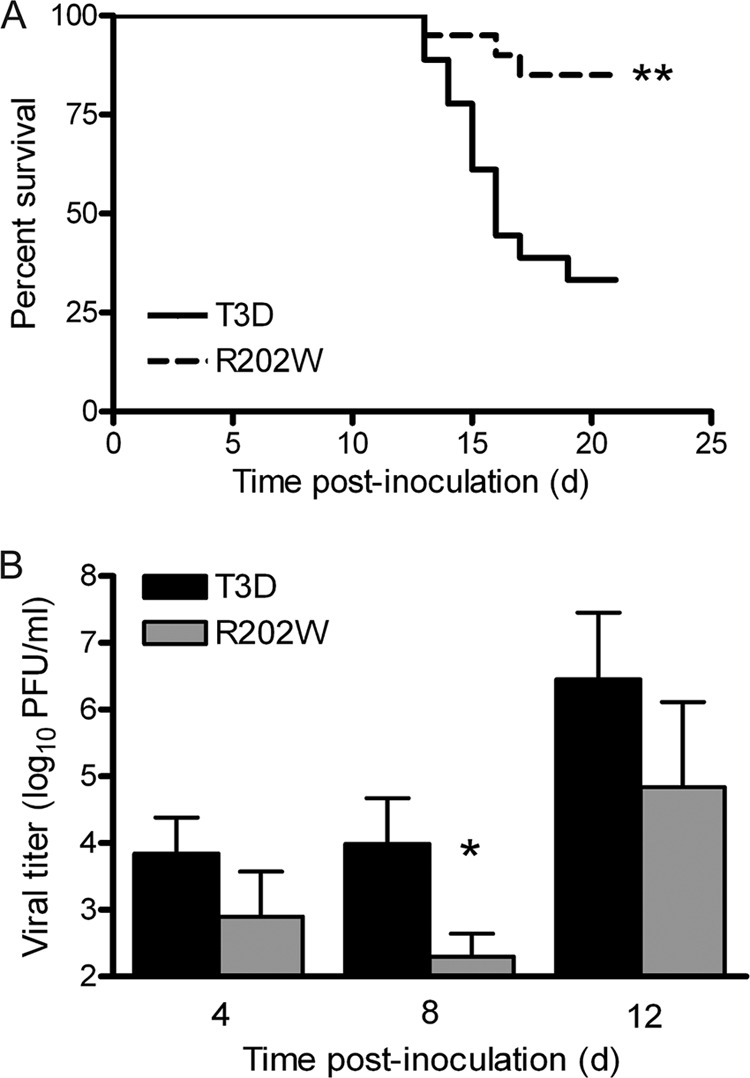
SA-binding capacity enhances reovirus neurovirulence and replicative capacity following IC inoculation. Newborn C57/BL6 mice were inoculated intracranially with 2 PFU of either T3D or T3D-σ1R202W (R202W). (A) Mice (n = 18 to 21 for each virus strain) were monitored for survival for 21 days. Two asterisks indicate a P value of <0.001, as determined by the log-rank test, in comparison to strain T3D. (B) At days 4, 8, and 12 postinoculation, the viral titers in brain homogenates were determined by plaque assay. Results are expressed as the mean viral titers for 4 to 7 animals for each time point. Error bars indicate the SEM. The asterisk indicates a P value of <0.05, as determined by the Mann-Whitney test, in comparison to strain T3D.
SA binding does not alter reovirus tropism in the brain.
To test whether σ1-SA interactions are required for neural tropism of the type 3 reovirus, we compared histologic sections of brains resected from mice inoculated intracranially with 100 PFU of either T3D or T3D-σ1R202W (Fig. 7). Brains chosen for the histologic analysis were matched anatomically based on the sizes and shapes of landmarks such as the hippocampus as well as the display of comparable viral titers. The intensity of reovirus antigen staining, and the cellular distribution of reovirus antigen, was similar in brain sections from the mice infected with either T3D or T3D-σ1R202W. Characteristic reovirus tropism involving several areas the brain, including the cortex, the C2-C4 region of the hippocampus, and the thalamus, was apparent in mice infected with either virus (Fig. 7A). In addition, involvement of the cerebellum, specifically infection of the Purkinje cells, was observed in brain sections of mice infected with either virus (Fig. 7B). Staining for the activated form of caspase-3 demonstrated apoptotic foci in sections of brain tissue from the animals infected with either T3D or T3D-σ1R202W. However, the number of apoptotic cells did not appear to differ in these sections (data not shown), perhaps due to the inoculum dose used in this experiment. Therefore, the SA-binding capacity of type 3 reovirus does not influence targeting of reovirus to specific regions within the brain but, rather, appears to influence replication efficiency in brain tissue.
Fig 7.

Binding to SA does not alter reovirus tropism in the brain. Newborn C57/BL6 mice were inoculated intracranially with 100 PFU of either T3D or T3D-σ1R202W (R202W). On day 8 postinoculation, brains of the infected mice were resected. The brains were bisected, the left hemispheres were processed for histopathology, and the right hemispheres were prepared for viral titer determination by plaque assay. Consecutive coronal sections of the brain were stained with hematoxylin and eosin (H&E) or polyclonal reovirus antiserum. Representative sections of brain hemisphere matched for hippocampal depth (A) and cerebellum (B) are shown. Strain T3D-infected brain sections are from the left hemisphere of a brain with a titer of 8 × 108 PFU, and strain T3D-σ1R202W-infected brain sections are from the left hemisphere of a brain with a titer of 3.5 × 108 PFU.
SA binding enhances infection of murine primary cortical neurons.
Results gathered thus far reveal that SA binding enhances the capacity of reovirus to replicate in the brain following IM or IC inoculation. We hypothesized that this difference is attributable to enhanced infection and replication in neurons by SA-binding reovirus strains. To test this hypothesis, we quantified infection of primary cortical neuron cultures using an immunofluorescence assay (Fig. 8A and B) and compared the titers of strains T3D and T3D-σ1R202W following replication in primary cortical neuron cultures at 24, 48, and 72 h postinfection (Fig. 8C). Primary cultures of mouse cortical neurons were established using cerebral cortices of C57/BL6 embryos at developmental day 15.5 and cultured for 7 days prior to infection (1). The wild-type virus infected a significantly higher percentage of cortical neurons (5%) than did the mutant virus (1%) (P < 0.0001) (Fig. 8B). At 24 h postinfection, yields of the wild-type and mutant viruses were comparable (Fig. 8C). However, by 48 and 72 h postinfection, the wild-type virus produced significantly higher yields than those produced by the mutant virus (P < 0.05) (Fig. 8C). These findings suggest that the engagement of SA by reovirus enhances infection and replication efficiency in neurons.
Fig 8.

SA-binding capacity increases the efficiency of reovirus infection of primary cortical neurons. Cortical neurons were harvested from C57/BL6 mouse embryos at developmental day 15.5 and cultured for 7 days prior to infection. (A) Neurons were adsorbed with either T3D or T3D-σ1R202W (R202W) at an MOI of 1,000 PFU/cell following pretreatment with either PBS or 40 mU A. ureafaciens neuraminidase and incubated for 21 h. Cells were stained with TUJ1 neural-specific marker to detect neurons (red), DAPI to detect nuclei (blue), and polyclonal reovirus antiserum to detect reovirus antigen (green) and visualized using indirect immunofluorescence microscopy. Representative wells from duplicate experiments are shown. (B) The percentage of infected cells was quantified by dividing the number of neurons exhibiting reovirus staining by the total number of cell nuclei exhibiting DAPI staining in three fields of ×400 view in triplicate wells (n = 2). The fields of view contained between 200 and 600 nuclei. Error bars indicate the SEM. The asterisk indicates a P value of <0.0001, as determined by Student's t test, in comparison to strain T3D. (C) Neurons were adsorbed with either T3D or T3D-σ1R202W at an MOI of 1 PFU/cell. Titers of the virus in cell lysates were determined by plaque assay at the indicated times postadsorption. Results are expressed as the mean viral yield for duplicate samples. Error bars indicate the SD. The asterisk indicates a P value of <0.05, as determined by Student's t test, in comparison to strain T3D.
To confirm the importance of SA binding for infectivity and replication of primary cultures of neurons, cells were treated with A. ureafaciens neuraminidase to remove cell surface SA. A. ureafaciens neuraminidase is a broad-spectrum enzyme that cleaves α(2,3)-, α(2,6)-, and α(2,8)-terminally linked SA present on glycan chains (41). Neuraminidase treatment diminished the infectivity and replication of the wild-type virus to the levels observed for the mutant virus (Fig. 8A to C). These findings demonstrate that both infectivity and replication in primary neuronal cultures by type 3 reovirus are enhanced by SA binding. Collectively, these results suggest that the capacity of reovirus to bind cell surface SA enhances the efficiency of infection of cells in the murine CNS.
DISCUSSION
In this study, we found that the capacity to bind cell surface SA enhances the neurovirulence of type 3 reovirus. Following either IM or IC inoculation of newborn mice, the wild-type strain T3D was more virulent and produced higher titers in neural tissues than the non-SA-binding mutant virus T3D-σ1R202W. Furthermore, mice infected with the wild-type virus displayed a higher frequency of AFP and enhanced tissue injury than the mutant-infected mice. Injury in the spinal cord and the brain is associated with apoptosis, with the number of apoptotic neurons in the spinal cord correlating with the extent of neurological symptoms. The key finding of this study is that SA-binding capacity dictates increased reovirus infectivity and replicative capacity in murine neurons. These data suggest that SA binding enhances the neurovirulence of type 3 reovirus by mediating the efficient infection of target cells in the CNS, resulting in increased replication and pathology.
We found that wild-type T3D displays increased infectivity and replicative capacity in primary cultures of murine cortical neurons, a phenotype that is dependent on cell surface SA. Binding of type 3 reovirus strains to SA facilitates viral attachment through low-affinity adhesion that places the virus on the cell surface, where access to the higher-affinity but lower-abundance proteinaceous receptor is thermodynamically favored (3). This adhesion-strengthening mechanism may be the basis for the enhanced replication and neurovirulence of T3D. Although the proteinaceous receptor on neurons for type 3 reovirus is not known, our findings suggest that its optimal engagement likely requires initial low-affinity glycan binding to anchor the virus to the cell surface. We favor this scenario because neuraminidase treatment of primary murine cortical neurons decreases but does not abolish infection or replication (Fig. 8), consistent with the existence of other host mediators of infection.
We previously identified residues in the type 3 σ1 molecule that are required for functional binding of reovirus to SA, i.e., binding that results in productive infection (31). Using reverse genetics, we introduced structure-guided mutations in the type 3 σ1 molecule and characterized the resultant mutant viruses using assays to quantify SA-binding capacity. This analysis highlights the importance of Arg202 in the interaction of the type 3 σ1 molecule with SA. The Arg202 side chain forms a bidentate salt bridge with the carboxylate moiety of SA, which is an important electrostatic contact between the protein and its carbohydrate ligand. The replacement of Arg202 with a tryptophan in T3D-σ1R202W would remove the electrostatic interaction between the arginine and SA and introduce a structural clash with the glycan (Fig. 9). It is therefore unlikely that the mutant can bind to SA, even in the improbable case that the overall structure of the binding site is not altered by the mutation. This model provides a structural framework for understanding the decreased virulence displayed by strain T3D-σ1R202W.
Fig 9.

Comparison of T3D and T3D-σ1R202W σ1 interactions with the terminal SA of α(2,3) sialyllactose. (A) Ribbon drawing of the T3D σ1 body and head domains in complex with α(2,3) sialyllactose. The σ1 monomers are shown in red, blue, and yellow. The body domain consists of seven triple β-spiral repeats (β1–β7) and an α-helical coiled-coil domain that is inserted between β-spiral repeats β4 and β5. The bound α(2,3) sialyllactose is shown in stick representation and colored in orange (enclosed by the red box). (B) An enlarged view of the T3D σ1 SA-binding domain. The σ1 residue 202 of a single σ1 monomer is drawn in stick representation, while the remainder of the monomer is shown as a ribbon tracing. The other monomers are shown in a surface-shaded representation. The SA moiety of α(2,3) sialyllactose is shown in stick representation, with carbons colored orange, oxygens colored red, and nitrogens colored blue. Residue Arg202 (cyan) of wild-type T3D forms a salt bridge with the carboxylate group of SA (31). This bond is represented by a dashed red line, illustrating that it is likely lost when Arg202 is replaced with a tryptophan (modeled in yellow) in T3D-σ1R202W.
Following IM inoculation, the wild-type T3D strain produced lower titers in the livers of infected mice than did the non-SA binding T3D-σ1R202W strain (Fig. 4). This is an interesting result, because previous studies from our laboratory revealed that SA binding confers tropism of reovirus for bile duct epithelia and reovirus-induced injury to the liver (5). The SA-binding strain used in that study, T3SA+, produces higher titers in liver tissue than its non-SA-binding counterpart, T3SA−, at early times after infection. However, SA-binding capacity is not strictly required for infection and replication in the liver, as strain T3SA− produces titers in the liver that approximate those of T3SA+ at late times of infection (1, 5). It is possible that the genetic backgrounds of the strains used by Barton et al. (5) (T1L) and here (T3D) account for the observed differences. Alternatively, it is possible that the routes of inoculation, peroral (5) versus IM (current study), influence hepatic tropism. We also noted that the mutant T3D-σ1R202W produced higher titers than the wild-type virus in the spinal cord and brain of infected mice on day 4 postinoculation. Since reovirus is delivered initially to the CNS by hematogenous routes (1, 6), it is possible that bloodstream dissemination of the mutant virus is subtly enhanced in comparison to the wild-type virus, although if so, this difference was not sufficient to permit detection in our assays.
Data presented in this and another study (5) indicate that the capacity of type 3 reovirus to bind cell surface SA can target the virus to specific sites of replication and lead to enhanced replication and disease. These observations suggest that the SA-binding capacity confers a fitness advantage for reovirus. For example, it is possible that an SA-mediated replication enhancement leads to increased viral shedding and transmission of the virus from host to host. However, there are naturally circulating strains of reovirus that do not bind to SA (13), suggesting that SA-binding capacity is a balanced polymorphism in nature that also imposes a fitness cost. For example, SA-binding capacity diminishes reovirus infection of polarized human airway epithelial cells (18). Strain T3SA+ does not efficiently infect these cells, whereas T3SA− does. Moreover, neuraminidase pretreatment to remove cell surface SA enhances T3SA+ infection of these cells (18). Determination of the precise contribution of SA binding to viral fitness will require competition studies using strains that vary solely in their SA-binding capacity and populations of susceptible mice.
For many viruses, including several important human pathogens, glycan engagement plays an important role in infection and pathogenesis. Here, we demonstrate that utilization of sialylated glycans as coreceptors enhances the neurovirulence of type 3 reovirus. This study broadens our understanding of the mechanisms of reovirus attachment to neuronal cells and supports the hypothesis that type 3 reovirus-glycan interactions are key determinants of reovirus virulence in the CNS.
ACKNOWLEDGMENTS
We thank Jim Chappell and members of our laboratory for many helpful suggestions and useful discussions. We thank Karl Boehme and Bernardo Mainou for careful review of the manuscript. We are grateful to Laura Ooms for assistance with the PCR assays.
This work was supported by Public Health Service awards T32 AI07611 (J.M.F.), R01 AI76983 (T.S.D. and T. S.), and R37 AI38296 (T.S.D.) and the Elizabeth B. Lamb Center for Pediatric Research. Additional support was provided by Public Health Service awards P30 CA68485 for the Vanderbilt-Ingram Cancer Center and P60 DK20593 for the Vanderbilt Diabetes Research and Training Center. We have no financial conflicts of interest.
Footnotes
Published ahead of print 3 October 2012
REFERENCES
- 1. Antar AAR, et al. 2009. Junctional adhesion molecule-A is required for hematogenous dissemination of reovirus. Cell Host Microbe 5:59–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Barton ES, Chappell JD, Connolly JL, Forrest JC, Dermody TS. 2001. Reovirus receptors and apoptosis. Virology 290:173–180 [DOI] [PubMed] [Google Scholar]
- 3. Barton ES, Connolly JL, Forrest JC, Chappell JD, Dermody TS. 2001. Utilization of sialic acid as a coreceptor enhances reovirus attachment by multistep adhesion strengthening. J. Biol. Chem. 276:2200–2211 [DOI] [PubMed] [Google Scholar]
- 4. Barton ES, et al. 2001. Junction adhesion molecule is a receptor for reovirus. Cell 104:441–451 [DOI] [PubMed] [Google Scholar]
- 5. Barton ES, et al. 2003. Utilization of sialic acid as a coreceptor is required for reovirus-induced biliary disease. J. Clin. Invest. 111:1823–1833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Boehme KW, Frierson JM, Konopka JL, Kobayashi T, Dermody TS. 2011. The reovirus sigma1s protein is a determinant of hematogenous but not neural virus dissemination in mice. J. Virol. 85:11781–11790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Boehme KW, Guglielmi KM, Dermody TS. 2009. Reovirus nonstructural protein σ1s is required for establishment of viremia and systemic dissemination. Proc. Natl. Acad. Sci. U. S. A. 106:19986–19991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Campbell JA, et al. 2005. Junctional adhesion molecule-A serves as a receptor for prototype and field-isolate strains of mammalian reovirus. J. Virol. 79:7967–7978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cashdollar LW, Chmelo RA, Wiener JR, Joklik WK. 1985. Sequences of the S1 genes of the three serotypes of reovirus. Proc. Natl. Acad. Sci. U. S. A. 82:24–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chappell JD, Duong JL, Wright BW, Dermody TS. 2000. Identification of carbohydrate-binding domains in the attachment proteins of type 1 and type 3 reoviruses. J. Virol. 74:8472–8479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chappell JD, Prota A, Dermody TS, Stehle T. 2002. Crystal structure of reovirus attachment protein σ1 reveals evolutionary relationship to adenovirus fiber. EMBO J. 21:1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Danthi P, et al. 2008. Reovirus apoptosis and virulence are regulated by host cell membrane-penetration efficiency. J. Virol. 82:161–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dermody TS, Nibert ML, Bassel-Duby R, Fields BN. 1990. Sequence diversity in S1 genes and S1 translation products of 11 serotype 3 reovirus strains. J. Virol. 64:4842–4850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dichter MA, Weiner HL. 1984. Infection of neuronal cell cultures with reovirus mimics in vitro patterns of neurotropism. Ann. Neurol. 16:603–610 [DOI] [PubMed] [Google Scholar]
- 15. Ding Y, et al. 2004. Organ distribution of severe acute respiratory syndrome (SARS) associated coronavirus (SARS-CoV) in SARS patients: implications for pathogenesis and virus transmission pathways. J. Pathol. 203:622–630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Duncan R, Horne D, Cashdollar LW, Joklik WK, Lee PWK. 1990. Identification of conserved domains in the cell attachment proteins of the three serotypes of reovirus. Virology 174:399–409 [DOI] [PubMed] [Google Scholar]
- 17. Ernst H, Shatkin AJ. 1985. Reovirus hemagglutinin mRNA codes for two polypeptides in overlapping reading frames. Proc. Natl. Acad. Sci. U. S. A. 82:48–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Excoffon KJDA, et al. 2008. Reovirus preferentially infects the basolateral surface and is released from the apical surface of polarized human respiratory epithelial cells. J. Infect. Dis. 197:1189–1197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fraser RDB, et al. 1990. Molecular structure of the cell-attachment protein of reovirus: correlation of computer-processed electron micrographs with sequence-based predictions. J. Virol. 64:2990–3000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Furlong DB, Nibert ML, Fields BN. 1988. Sigma 1 protein of mammalian reoviruses extends from the surfaces of viral particles. J. Virol. 62:246–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Goody RJ, Schittone SA, Tyler KL. 2008. Experimental reovirus-induced acute flaccid paralysis and spinal motor neuron cell death. J. Neuropathol. Exp. Neurol. 67:231–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kaludov N, Brown KE, Walters RW, Zabner J, Chiorini JA. 2001. Adeno-associated virus serotype 4 (AAV4) and AAV5 both require sialic acid binding for hemagglutination and efficient transduction but differ in sialic acid linkage specificity. J. Virol. 75:6884–6893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kaye KM, Spriggs DR, Bassel-Duby R, Fields BN, Tyler KL. 1986. Genetic basis for altered pathogenesis of an immune-selected antigenic variant of reovirus type 3 Dearing. J. Virol. 59:90–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kirchner E, Guglielmi KM, Strauss HM, Dermody TS, Stehle T. 2008. Structure of reovirus σ1 in complex with its receptor junctional adhesion molecule-A. PLoS Pathog. 4:e1000235 doi:10.1371/journal.ppat.1000235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kobayashi T, et al. 2007. A plasmid-based reverse genetics system for animal double-stranded RNA viruses. Cell Host Microbe 1:147–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lee PWK, Hayes EC, Joklik WK. 1981. Protein σ1 is the reovirus cell attachment protein. Virology 108:156–163 [DOI] [PubMed] [Google Scholar]
- 27. Maddon PJ, et al. 1986. The T4 gene encodes the AIDS virus receptor and is expressed in the immune system and the brain. Cell 47:333–348 [DOI] [PubMed] [Google Scholar]
- 28. Morrison LA, Sidman RL, Fields BN. 1991. Direct spread of reovirus from the intestinal lumen to the central nervous system through vagal autonomic nerve fibers. Proc. Natl. Acad. Sci. U. S. A. 88:3852–3856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nibert ML, Dermody TS, Fields BN. 1990. Structure of the reovirus cell-attachment protein: a model for the domain organization of σ1. J. Virol. 64:2976–2989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Prota AE, et al. 2003. Crystal structure of human junctional adhesion molecule 1: implications for reovirus binding. Proc. Natl. Acad. Sci. U. S. A. 100:5366–5371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Reiter DM, et al. 2011. Crystal structure of reovirus attachment protein σ1 in complex with sialylated oligosaccharides. PLoS Pathog. 7:e1002166 doi:10.1371/journal.ppat.1002166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ren R, Costantini FC, Gorgacz EJ, Lee JJ, Racaniello VR. 1990. Transgenic mice expressing a human poliovirus receptor: a new model for poliomyelitis. Cell 63:353–362 [DOI] [PubMed] [Google Scholar]
- 33. Richardson BA, Overbaugh J. 2005. Basic statistical considerations in virological experiments. J. Virol. 79:669–676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sanford KK, Earle WR, Likely GD. 1948. The growth in vitro of single isolated tissue cells. J. Natl. Cancer Inst. 9:229–246 [PubMed] [Google Scholar]
- 35. Sarkar G, et al. 1985. Identification of a new polypeptide coded by reovirus gene S1. J. Virol. 54:720–725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Schelling P, et al. 2007. The reovirus σ1 aspartic acid sandwich: a trimerization motif poised for conformational change. J. Biol. Chem. 282:11582–11589 [DOI] [PubMed] [Google Scholar]
- 37. Schiff LA, Nibert ML, Tyler KL. 2007. Orthoreoviruses and their replication, p 1853–1915 In Knipe DM, Howley PM. (ed), Fields virology, 5th ed, vol 2 Lippincott, Williams & Wilkins, Philadelphia [Google Scholar]
- 38. Smith RE, Zweerink HJ, Joklik WK. 1969. Polypeptide components of virions, top component and cores of reovirus type 3. Virology 39:791–810 [DOI] [PubMed] [Google Scholar]
- 39. Tardieu M, Weiner HL. 1982. Viral receptors on isolated murine and human ependymal cells. Science 215:419–421 [DOI] [PubMed] [Google Scholar]
- 40. Tyler KL, McPhee DA, Fields BN. 1986. Distinct pathways of viral spread in the host determined by reovirus S1 gene segment. Science 233:770–774 [DOI] [PubMed] [Google Scholar]
- 41. Uchida Y, Tsukada Y, Sugimori T. 1979. Enzymatic properties of neuraminidases from Arthrobacter ureafaciens. J. Biochem. 86:1573–1585 [DOI] [PubMed] [Google Scholar]
- 42. Virgin HW, Bassel-Duby IVR, Fields BN, Tyler KL. 1988. Antibody protects against lethal infection with the neurally spreading reovirus type 3 (Dearing). J. Virol. 62:4594–4604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Weiner HL, Drayna D, Averill DR, Jr., Fields BN. 1977. Molecular basis of reovirus virulence: role of the S1 gene. Proc. Natl. Acad. Sci. U. S. A. 74:5744–5748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Weiner HL, Powers ML, Fields BN. 1980. Absolute linkage of virulence and central nervous system tropism of reoviruses to viral hemagglutinin. J. Infect. Dis. 141:609–616 [DOI] [PubMed] [Google Scholar]
- 45. Weiner HL, Ramig RF, Mustoe TA, Fields BN. 1978. Identification of the gene coding for the hemagglutinin of reovirus. Virology 86:581–584 [DOI] [PubMed] [Google Scholar]