

Abstract
Theiler's virus-induced demyelinating disease has been extensively investigated as a model for persistent viral infection and multiple sclerosis (MS). However, the role of CD8+ T cells in the development of disease remains unclear. To assess the role of virus-specific CD8+ T cells in the pathogenesis of demyelinating disease, a single amino acid substitution was introduced into the predominant viral epitope (VP3 from residues 159 to 166 [VP3159-166]) and/or a subdominant viral epitope (VP3173-181) of susceptible SJL/J mice by site-directed mutagenesis. The resulting variant viruses (N160V, P179A, and N160V/P179A) failed to induce CD8+ T cell responses to the respective epitopes. Surprisingly, mice infected with N160V or N160V/P179A virus, which lacks CD8+ T cells against VP3159-166, did not develop demyelinating disease, in contrast to wild-type virus or P179A virus lacking VP3173-181-specific CD8+ T cells. Our findings clearly show that the presence of VP3159-166-specific CD8+ T cells, rather than viral persistence itself, is strongly correlated with disease development. VP3173-181-specific CD8+ T cells in the central nervous system (CNS) of these virus-infected mice expressed higher levels of transforming growth factor β, forkhead box P3, interleukin-22 (IL-22), and IL-17 mRNA but caused minimal cytotoxicity compared to that caused by VP3159-166-specific CD8+ T cells. VP3159-166-specific CD8+ T cells exhibited high functional avidity for gamma interferon production, whereas VP3173-181-specific CD8+ T cells showed low avidity. To our knowledge, this is the first report indicating that the induction of the IL-17-producing CD8+ T cell type is largely epitope specific and that this specificity apparently plays a differential role in the pathogenicity of virus-induced demyelinating disease. These results strongly advocate for the careful consideration of CD8+ T cell-mediated intervention of virus-induced inflammatory diseases.
INTRODUCTION
It has been well established that virus-specific CD8+ T cells play the most efficient role in eradicating viral persistence from the infected host. These antiviral CD8+ T cells typically produce gamma interferon (IFN-γ) and/or tumor necrosis factor alpha (TNF-α) upon activation and exhibit strong cytolytic function via the granzyme/perforin and the Fas/FasL systems (8, 16). However, the presence of intereukin-17 (IL-17)-producing CD8+ T cells, termed Tc17 cells (versus the conventional IFN-γ-producing Tc1 cells), has recently been detected following viral infection (14, 20), some autoimmune disease lesions (41), and cytokine-derived in vitro differentiation (19). Interestingly, Tc17 cells produce a distinct set of cytokines, including IL-17, and exhibit a low cytotoxic function. Recent studies indicated that the environments associated with tumor and chronic inflammation promote the induction of abundant IL-17-producing CD8+ cells with reduced cytolytic function (5, 29). However, the induction mechanisms and roles of the Tc1 and Tc17 subpopulations in protection or pathogenesis following viral infection remain unclear. Therefore, it would be important to investigate the roles of these CD8+ T cell subtypes in the protection/pathogenesis of virus-induced chronic inflammatory disease and whether induction is dependent on the cognate epitopes following viral infection. Furthermore, there is growing evidence to suggest the potential involvement of CD8+ T cells in the pathogenesis of multiple sclerosis (MS), including the presence of greater numbers of CD8+ T cells than CD4+ T cells in MS lesions (12, 34, 51). In this study, we examine these questions using the Theiler's murine encephalomyelitis virus (TMEV)-induced demyelinating disease model of MS.
SJL/J (SJL) mice infected with TMEV reproducibly develop chronic progressive demyelinating disease, displaying histopathological similarities to human MS (7, 26, 28). Interestingly, the resistance of C57BL/6 (B6; H-2b) mice to TMEV-induced demyelinating disease (TMEV-IDD) is genetically linked to the H-2D major histocompatibility complex (MHC) class I locus (31, 47). In addition, perforin-deficient (43) or β2-microglobin-deficient (44) mice with a resistant B6 background develop demyelinating disease, suggesting the possibility that CD8+ T cells may be important in maintaining resistance to TMEV-IDD. More recently, it was shown that transgenic (Tg) expression of H-2Db in susceptible FVB mice (H-2q) rendered them resistant (35). Furthermore, when the predominant H-2Db-restricted CD8+ T cells specific for the VP2 epitope from residues 121 to 130 (VP2121-130) were tolerized by infusion with the epitope peptide, resistant H-Db-transgenic FVB mice developed demyelinating disease. Therefore, immunodominant VP2121-130-specific CD8+ T cells appear to be responsible for conferring resistance in B6 mice.
In contrast, little is known about the role of CD8+ T cells in the pathogenesis of TMEV-IDD in susceptible strains, such as SJL (H-2s) mice. Existing evidence, however, suggests that CD8+ T cells in susceptible SJL mice play an important role in the protection against TMEV-IDD: class I-deficient SJL mice succumbed to higher persisting viral titers and developed exacerbated demyelination and clinical disease (2). Epitope-specific CD8+ T cells in BeAn strain TMEV-infected SJL mice account for up to 70% of central nervous system (CNS)-infiltrating CD8+ T cells (24). Furthermore, virus-specific CD8+ T cells from susceptible SJL mice showed activation markers and levels of IFN-γ production similar to those for resistant B6 mice. However, the number of virus-specific IFN-γ-producing CD8+ T cells in susceptible SJL mice was roughly 3-fold lower than that in resistant B6 mice (32). Therefore, the overall levels of initial virus-specific IFN-γ-producing CD8+ T cells seem to be critical for maintaining resistance to TMEV-IDD.
Despite the protective role of TMEV-specific CD8+ T cells, a pathogenic role for virus-specific CD8+ T cells remains possible. A group of investigators previously reported that perforin is necessary for the development of clinical symptoms but not demyelination following infection of resistant B6 mice with the DA strain of TMEV (37). These results suggest that perforin-mediated cytotoxicity by virus-specific CD8+ T cells may play a role in the pathogenesis of TMEV-induced clinical disease. However, these studies utilized mice with a resistant genetic background, and conflicting results were reported, depending on the viral strains and/or investigators (43, 44). We previously showed that transgenic SJL mice expressing the P1 region of TMEV-encoding capsid proteins are unresponsive to the predominant VP3159-166 epitope but have intact CD8+ T cell responses to subdominant epitopes (40). Interestingly, P1-transgenic SJL mice lacking CD8+ T cell responses to the predominant epitope develop less severe delayed clinical disease. These results suggest an interesting possibility that VP3159-166-specific CD8+ T cells, and not subdominant epitope-reactive CD8+ T cells, may play a pathogenic role in the development of TMEV-induced demyelinating disease.
To further analyze the role of epitope-specific CD8+ T cells, we utilized mutant viruses containing a single or double substitution in the CD8+ T cell epitope regions of the TMEV genome. We chose to generate epitope-null viruses rather than to induce immune tolerance to the epitopes, as an immune tolerance often accompanies the generation of regulatory cells and/or deviations of immune cells that may complicate the interpretation. SJL mice infected with the N160V virus, which lacks the CD8+ T cell responses to the predominant VP3159-166, remained free of clinical symptoms. In contrast, mice infected with the P179A virus, which lacks the CD8+ T cell responses to the subdominant VP3173-181, displayed clinical signs comparable to those of wild-type (WT) virus-infected mice. These results strongly suggest that individual epitope-specific CD8+ T cells play different roles in pathogenesis or protection during the development of TMEV-IDD. Surprisingly, CNS-infiltrating CD8+ T cells specific to VP3173-181 expressed high levels of forkhead box P3 (FoxP3), IL-17, and transforming growth factor β1 (TGF-β1) mRNA. Furthermore, our data show that Tc1-like VP3159-166-specific CD8+ T cells, which exhibit superior cytolytic function and IFN-γ production, promote the development of disease, whereas Tc17-like VP3173-181-specific CD8+ T cells, which display low cytolytic function, do not. These results strongly suggest that cytolytic Tc1-like CD8+ T cells participate, perhaps together with Th17 cells, in the pathogenesis of virally induced demyelinating disease. Thus, our current observation reveals an important function of virus-specific CD8+ T cell subpopulations associated with protection versus pathogenesis, which is a critical consideration for CD8+ T cell-mediated immune intervention.
MATERIALS AND METHODS
Animals.
SJL/J mice were purchased from Harlan Sprague-Dawley and housed in the Animal Care Facility of Northwestern University. Six- to 8-week-old female mice were used for all experiments.
Synthetic peptides and antibodies.
All peptides used were purchased from GeneMed (GeneMed Synthesis Inc., CA) and used as described previously (24). All antibodies were purchased from BD Pharmingen.
Viruses and cell lines.
The wild-type and variant viruses of the BeAn strain of TMEV were generated by site-directed mutagenesis (Fig. 1). These viruses were propagated and titers were determined in BHK-21 cells grown in Dulbecco's modified Eagle medium supplemented with 7.5% donor calf serum. For intracerebral (i.c.) infection, 30 μl virus solution, containing 30 × 106 PFU, was injected into the right cerebral hemisphere of 6- to 8-week-old mice anesthetized with isoflurane. Clinical symptoms of disease were assessed weekly on the following grading scale: grade 0, no clinical signs; grade 1, mild waddling gait; grade 2, severe waddling gait; grade 3, moderate hind limb paralysis; grade 4, severe hind limb paralysis.
Fig 1.

Generation and growth properties of cytotoxic T lymphocyte epitope variant viruses. (A) Diagram depicting the site-directed mutagenesis of the predominant and subdominant TMEV BeAn strain CD8+ T cell epitopes. To eliminate the ability to stimulate epitope-specific CD8+ T cells, a series of site-specific mutations was introduced into the cytotoxic T lymphocyte epitope regions (VP3159-166 and/or VP3173-181) of the TMEV BeAn genome using a QuikChange kit. (B) Variant viruses replicated comparably to the WT virus in BHK-21 cells and primary mouse astrocytes. BHK-21 cells or primary mouse astrocytes were infected with wild-type and variant viruses harboring a single amino acid substitution (N160V or P179A) or double amino acid substitutions (N160V/P179A). Infectious TMEV titers were assessed at the indicated time points from the viral lysates of BHK-21 cell or primary astrocyte cultures by plaque assay. The results of a representative of two separate experiments are shown. (C) Virus binding and infection of bone-marrow-derived DCs were determined by flow cytometry using anti-TMEV capsid antibody. The results of one of two similar experiments are shown.
Reverse transcriptase PCR and real-time PCR.
Total cellular RNA was isolated from the brain and spinal cord of infected SJL/J mice using TRIzol reagents (Invitrogen, Carlsbad, CA). First-strand cDNA was synthesized from 1 μg total RNA utilizing SuperScript III first-strand synthesis Supermix or Moloney murine leukemia virus reverse transcriptase (Invitrogen, Carlsbad, CA). Primers for control GAPDH (glyceraldehyde-3-phosphate dehydrogenase) and cytokine genes were purchased from Integrated DNA Technologies, as follows: for GAPDH, forward primer AACTTTGGCATTGTGGAAGG and reverse primer ACACATTGGGGGTAGGAACA; for TGF-β1, ACAGAGAAGAACTGCTGTGTGC and GTTGTGTTGGTTGTAGAGGGCAA; for IFN-γ, ACTGGCAAAAGGATGGTGAC and TGAGCTCATTGAATGCTTGG; for IFN-β, CCCTATGGAGATGACGGAGA and CTGTCTGCTGGTGGAGTTCA; for IL-10, GCCAAGCCTTATCGGAAATGATCC and AGACACCTTGGTCTTGGAGCTT; for IL-18, ACAACTTTGGCCGACTTCAC and GGGTTCACTGGCACTTTGAT; for IL-17A, CTCCAGAAGGCCCTCAGACTAC and AGCTTTCCCTCCGCATTGACACAG; for IL-23, CAAGCGGGACATATGAATCT and CATGGGGCTATCAGGGAGTA; for IL-6, AGTTGCCTTCTTGGGACTGA and TCCACGATTTCCCAGAGAAC; for TNF-α, GGTCACTGTCCCAGCATCTT and CTGTGAAGGGAATGGGTGTT; for FoxP3, TTGGTTTACTCGCATGTTCGCC and TGGAGGTCAAGGGCAGGGATTG; and for VP1, TGACTAAGCAGGACTATGCCTTCC and CAACGAGCCACATATGCGGATTAC. GAPDH expression served as an internal reference for normalization. Real-time PCR was performed in triplicate.
Plaque assay.
Viral titers in infected CNS tissues were enumerated by a standard plaque assay on BHK-21 cell monolayers (27). After methanol fixation, 0.1% crystal violet was used to visualize plaques on the monolayer.
Viral replication assays.
Wild-type or mutant virus was infected at a multiplicity of infection of 10 into BHK-21 cells or primary mouse astrocytes with rocking and resting for 1 h followed by an extensive wash. Cells were then cultured for various time periods at 33°C as previously described (3). Both cells and supernatants were harvested and kept at −80°C until use. Primary mouse astrocytes were prepared as described elsewhere (42). Briefly, single-cell suspensions were obtained from 0- to 3-day-old neonatal brains and seeded on flasks coated with poly-l-lysine (25 μg/ml) (48). After differential shakings at 200 and 250 rpm, adherent astrocytes were collected as previously described (42). The purity of the cell preparations (>95% pure) was routinely confirmed by staining with antibodies specific for an astrocyte marker, glial fibrillary acidic protein (Dako, CA).
Isolation of CNS-infiltrating MNCs.
Mice were perfused with sterile Hanks' balanced salt solution (HBSS). The excised brains and spinal cords were forced through steel mesh to prepare single-cell suspensions, which were incubated at 37°C for 45 min in 250 mg/ml collagenase type 4 (Worthington Biochemical Corp., Lakewood, NJ). CNS-infiltrating mononuclear cells (MNCs) were then enriched in the 1/3 bottom fraction of a continuous Percoll (Pharmacia, Piscataway, NJ) gradient after centrifugation for 30 min at 27,000 × g as described previously (10).
Intracellular staining of cytokine production.
Freshly isolated CNS-infiltrating MNCs were cultured in 96-well round-bottom plates in the presence of relevant or control peptide. Allophycocyanin-conjugated anti-CD8 (clone Ly2) or anti-CD4 (clone L3T4) antibody and phycoerythrin (PE)-labeled rat monoclonal anti-IFN-γ (XMG1.2) antibody were used for intracellular cytokine staining. Cells were analyzed on a Becton Dickinson FACSCalibur or LSRII cytometer.
Generation of H-2Ks tetramers.
H-2Ks tetramers were generated as previously described (1). Briefly, H-2Ks and human β2-microglobulin genes were subcloned into a pET28 bacterial expression vector. Escherichia coli BL21(DE3) was transformed, and protein expression was induced with IPTG (isopropyl-β-d-thiogalactopyranoside) for 4 to 5 h. The inclusion body was purified and refolded in the presence of peptides. The soluble monomeric form of peptide-MHC was biotinylated with BirA at room temperature and tetramerized with streptavidin-PE (Invitrogen, Carlsbad, CA).
T cell proliferation assay.
Spleen cells from SJL/J mice infected with wild-type or variant viruses were cultured in the presence of peptides for 3 days and then pulsed with 1 μCi [3H]thymidine deoxyribose ([3H]TdR) for 18 h. Cells were harvested, and the levels of [3H]TdR incorporated were measured using a TopCount liquid scintillation counter (Perkin-Elmer, San Jose, CA). Data are expressed as the mean ± standard deviation (SD) of triplicate samples.
Histopathological analyses.
At day 120 postinfection, mice were anesthetized and perfused with 0.1% cold glutaraldehyde in phosphate-buffered saline (PBS). Spinal cords were excised, fixed in 1% OsO4, and embedded in Epon. Sections were cut at a 1-μm thickness, stained with toluidine blue, and analyzed by microscopy (44). Ten different sections of the lumbar region of the spinal cord of individual mice (two mice per experimental group) were assessed. For Luxol Fast Blue (LFB) staining for axonal demyelination and Bielschowsky silver staining for axon loss and damage, spinal cords from mice infected with WT or N160V virus were dissected and fixed in 4% formalin in PBS for 24 h. The tissues were embedded in paraffin and sectioned at 6 μm. Adjacent sets of 3 sections from each animal were deparaffinized, rehydrated, and stained. Slides were examined using a Leica DMR light microscope, and images were captured using AxioCam MRc camera and AxioVision imaging software.
Statistical analysis.
Data are presented as the mean ± SD of either two to three independent experiments or triplicates of one representative from at least three separate experiments. The significance of the differences in the mean values was determined by Student's t test. The statistical significance of disease incidence was tested by Fisher's exact test. Clinical scores were analyzed by the Mann-Whitney U test. P values of <0.05 were considered statistically significant.
RESULTS
TMEV variants of the CD8+ T cell epitopes replicate similarly to WT virus in BHK-21 cells and mouse astrocytes.
We previously showed that CD8+ T cells reactive to the predominant (VP3159-166) and subdominant (VP3173-181) epitopes of the TMEV BeAn strain do not recognize the equivalent epitopes of the closely related TMEV DA strain due to a single amino acid substitution in each (23). To investigate the role of epitope-specific CD8+ T cells in the development of TMEV-IDD, mutant viruses containing a single amino acid substitution or double amino acid substitutions in the CD8+ T cell epitopes of the BeAn VP3 protein with the DA residues were generated by site-directed mutagenesis of an expression cDNA clone (pSBW) of the BeAn strain (Fig. 1A). Therefore, it was expected that the variant viruses, encoding a single substitution or double substitutions in the epitope regions of the VP3 protein, would be deficient in the corresponding epitope-specific CD8+ T cell responses in the CNS and periphery of virus-infected SJL mice.
The levels of WT and variant virus replication were first assessed using BHK-21 cells and primary mouse astrocytes (Fig. 1B). The WT and variant viruses replicated similarly in BHK-21 cells that were deficient in type I interferon production (Fig. 1B, left). This result suggests that no stage of the virus life cycle, including virus binding on a cellular receptor(s), viral entry, uncoating of the viral genome, RNA replication, viral coat protein translation, and virus assembly, was significantly affected by the introduction of mutations. To further investigate if these viruses replicate similarly in mouse cells, primary SJL astrocytes were infected with WT and variant viruses. Similar viral replication levels were also observed in astrocytes infected with WT and variant viruses (Fig. 1B, right). In addition, these viruses displayed no significant differences in their ability to bind to mouse cells, as shown with dendritic cells (DCs) (Fig. 1C). Therefore, it is likely that WT and variant viruses have comparable infection and growth rates in vitro.
Variant viruses do not induce CD8+ T cell responses to the corresponding epitopes.
To assess the CD8+ T cell responses to the corresponding epitopes, susceptible SJL/J mice were infected with WT and variant viruses. Levels of epitope-specific CD8+ T cells in the CNS of infected mice were determined at day 8 postinfection by assessing the levels of binding of VP3159-166- and VP3173-181-loaded H-2Ks tetramers to CD8+ T cells (Fig. 2A and B). Of the CD8+ T cells from WT virus-infected mice, 32% were VP3159-166 specific and 11% were VP3173-181 specific. In contrast, less than 3% of the CD8+ T cells from N160V virus-infected mice were VP3159-166 specific but 24% were VP3173-181 specific. These results suggest that unresponsiveness to the predominant epitope is partly compensated for by an increase in the subdominant VP3173-181-specific CD8+ T cell response. However, the lack (0.5%) of a VP3173-181-specific CD8+ T cell response in mice infected with the P179A virus was not accompanied by such an increase in the dominant VP3159-166-specific CD8+ T cell response. Mice infected with the doubly substituted (N160V/P179A) virus did not induce the CD8+ T cells reactive to any of these epitopes (Table 1).
Fig 2.

Epitope-specific CD8+ T cell responses in mice infected with WT and variant viruses. (A) On the indicated days postinfection, SJL mice (3 mice per group) infected with WT or variant viruses were sacrificed, and isolated CNS-infiltrating MNCs were stimulated with PBS or VP3159-166, VP3173-181, or VP111-20 peptide for 6 h. The proportion of CNS CD8+ T cells was analyzed using flow cytometry after staining with H-2Ks–VP3159-166 and H-2Ks–VP3173-181 tetramers. (B) The proportion of epitope-specific tetramer-positive CD8+ T cells (top) and the number of epitope-specific tetramer-positive CD8+ T cells in the CNS (bottom) are shown. *, P < 0.05; **, P < 0.01. (C) The percentage of IFN-γ-producing CD8+ T cells of the total CD8+ T cells is shown in the upper right quadrant of each plot. Cells were stained for both CD8 and intracellular IFN-γ for flow cytometric analysis. A representative fluorescence-activated cell sorter analysis plot of three independent experiments is shown. (D) The proportion (top) and number (bottom) of epitope-specific IFN-γ-producing CD8+ T cells in the CNS of SJL mice during viral infection are shown. The values given are the means of the percentages or numbers of IFN-γ-producing CD8+ T cells from four independent experiments (mean ± SD).
TABLE 1.
Mutant viruses and their properties
| Virus | Reactivity of cytotoxic T lymphocytec | Presence ofa: |
||
|---|---|---|---|---|
| Clinical symptoms | Demyelination | Virus persistenceb | ||
| WT | VP3159-166, VP3173-181, VP111-20 | +++ | +++ | + |
| N160V | VP3173-181, VP111-20 (VP3159-166) | ± | ± | ++ |
| P179A | VP3159-166, VP111-20 (VP3173-181) | + | +++ | + |
| N160V/P179A | VP111-20 (VP3159-166, VP3173-181) | ± | ± | ++ |
+++, very high; ++, high; +, moderate; ±, low.
Based on viral message levels in the spinal cords at 8, 21, and 120 days postinfection.
Parentheses indicate absence.
To further correlate the CD8+ T cell response with the production of key T cell cytokines, IFN-γ production in response to the epitopes was analyzed by flow cytometry (Fig. 2C). Variant viruses were similarly unable to induce the corresponding epitope-specific CD8+ T cells producing IFN-γ. For example, the N160V virus was unable to induce VP3159-166-specific IFN-γ-producing CD8+ T cells, in contrast to WT virus. N160V/P179A virus-infected animals did not induce a CD8+ T cell response to either of the respective epitopes in the CNS. In addition, the loss of these epitope-specific CD8+ T cell responses was not compensated for by an increase of VP111-20-specific CD8+ T cells. These patterns of epitope-specific CD8+ T cell responses remained at day 120 postinfection (Fig. 2D).
N160V and N160V/P179A viruses, deficient in the predominant CD8+ T cell epitope, do not induce demyelinating disease.
To further investigate how differences in virus-specific CD8+ T cell responses in the CNS affect the development of TMEV-IDD, SJL mice (9 to 10 mice/group) were infected with WT or variant viruses. Mice infected with the WT or P179A virus (>75%) developed clinical disease by 50 days postinfection (Fig. 3A). In contrast, the majority of the N160V or N160V/P179A virus-infected animals (>80%) did not develop disease. In addition, clinically affected mice infected with the N160V or N160V/P179A virus showed only minimal disease severity (P < 0.01 at 42 days postinfection and thereafter) compared to the mice infected with the WT or P179A virus. These studies suggest that VP3159-166-specific CD8+ T cell responses are critical in disease development (Table 1).
Fig 3.
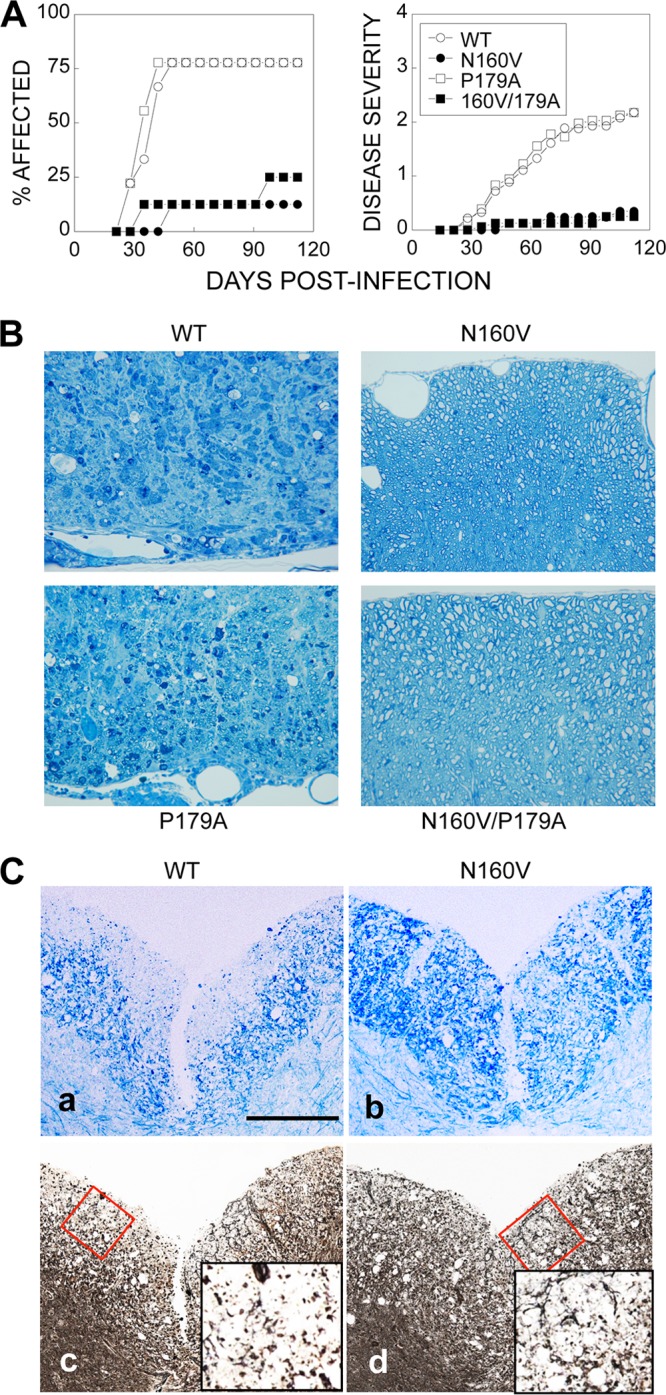
Comparison of the clinical disease course in mice infected with either WT or variant viruses. (A) SJL mice (n = 9 to 10) were infected with WT or variant TMEVs (30 × 106 PFU/mouse). Animals were graded for clinical signs as described in Materials and Methods. The results are expressed as the percentage (left) or mean (right) clinical score of affected animals on the indicated days postinfection. The statistical significance of disease incidence was tested by Fisher's exact test. The clinical scores were analyzed by the Mann-Whitney U test. For WT-infected versus N160V- or N160V/P179A-infected mice, Pwas <0.05 at day 35 postinfection and thereafter; for WT-infected versus P179A-infected mice, P was <0.05 at day 63 and thereafter; for N160V-infected versus P179A-infected mice, P was <0.05 at day 49 and thereafter; for N160V- or P179A-infected versus N160V/P179A-infected mice, P was not significant at all time points. (B) Spinal cords from SJL mice infected with WT or variant viruses for 120 days were obtained (two mice per group). One-micron-thick sections were prepared and stained with toluidine blue. Ten different sections of the lumbar region of the spinal cord of each individual mouse were graded for demyelination by microscopy. Results of a representative of three different experiments are shown. (C) Spinal cords of virus-infected mice were stained with Luxol Fast Blue (a, b) or Bielschowsky (c, d). The red boxes (c and d) indicate the areas of the insets. Density comparisons of 10 random areas stained with Luxol Fast Blue showed values of 40.8 ± 5.0 for WT virus-infected mice and 117.9 ± 32.6 for N160V-infected mice. Density comparisons of 10 random areas stained with Bielschowsky showed values of 54.2 ± 25.4 for WT virus-infected mice and 133.9 ± 28.6 for N160V-infected mice.
The histopathology of the spinal cord was further examined at 120 days after viral infection and was correlated with disease development by the epitope mutant viruses. The WT and P179A virus-infected animals exhibited similar severe demyelination in the spinal cord, whereas the N160V and N160V/P179A virus-infected animals remained free of demyelinating lesions (Fig. 3B). These results are consistent with the clinical disease levels induced by the WT and mutant viruses. To further determine if the level of axon damages reflects the pathogenic function of VP3159-166-specific CD8+ T cells, the integrity of axons in the spinal cord of mice infected with the WT and N160V virus were compared on the basis of the density of the stains after Bielschowsky silver staining (Fig. 3C). The levels of axonal loss and damage were more extensive in the spinal cord of the WT virus-infected mice (54.2 ± 25.4) than that of the N160V virus-infected mice (133.9 ± 28.6) lacking the VP3159-166-specific CD8+ T cell response. These data strongly suggest that the CD8+ T cells specific for the predominant VP3159-166 epitope but not those specific for the subdominant P179A epitope play an important role in the pathogenesis of demyelination in the spinal cord and consequent clinical disease.
Mice infected with the N160V or N160V/P179A virus display higher viral loads in the CNS.
To determine if the reduced disease development in mice infected with viruses to which CD8+ T cell responses to the predominant epitope are deficient is associated with lower levels of viral replication, viral RNA levels in the CNS of infected mice were assessed using real-time PCR (Fig. 4A). Interestingly, the N160V and N160V/P179A virus-infected mice lacking VP3159-166-specific CD8+ T cells displayed significantly higher levels of viral messages in the spinal cord at 8 days postinfection. In contrast, viral message levels in the CNS of the P179A virus-infected mice were significantly lower than those in the CNS of the WT virus-infected mice. Levels of infectious virus in the CNS of these mice were further determined by plaque assay (Fig. 4B). Roughly 2-log-unit higher infectious viral titers were detected in the brain and spinal cord of N160V virus-infected mice than WT-infected mice (P < 0.0005). The N160V/P179A virus-infected mice similarly displayed more infectious virus in the brain at day 8 postinfection (P < 0.017). In contrast, 10-fold fewer infectious viruses were detected in the brain of the P179A virus-infected mice, which induce a significantly higher level of VP3159-166-specific CD8+ T cells in the CNS than the WT virus-infected mice (P < 0.001). The levels of viral load remained high in both the N160V and N160V/P179A virus-infected mice at day 21 postinfection. However, at day 120 postinfection, infectious viruses were not detectable in the spinal cord of the N160V or P179A virus-infected mice, whereas the levels of viral load in the brain were largely comparable in all groups (Fig. 4B). These results suggest that VP3159-166-specific CD8+ T cells are most efficient in viral clearance and that the loss of the CD8+ T cell response results in an elevated viral load at the early stages of viral infection.
Fig 4.

Viral persistence in mice infected with WT and variant viruses. Brain and spinal cord homogenates were prepared from SJL mice (n = 3) infected with WT and variant TMEV. (A) Viral RNA levels in the brain and spinal cord homogenates from mice infected with viruses for 8 days were compared with the fold VP1 expression using real-time PCR. (B) Infectious titers in the tissue homogenates of mice were assessed by plaque assay on days 8, 21, and 120 (d8, d21, and d120, respectively) postinfection. The values given are the mean numbers from two to three independent experiments (mean ± SD). *, P < 0.05; **, P < 0.01; ***, P < 0.001; ND, not detectable (below the detection level).
Levels of IFN-γ-producing TMEV-specific CD4+ T cells are comparable between WT and variant virus-infected mice.
Epitope-specific CD4+ T cell levels were also investigated to examine if the mutations in the CD8+ T cell epitope regions of the TMEV genome affect the virus-specific CD4+ T cell responses in the CNS (Fig. 5). Proliferative responses of splenic CD4+ T cells to viral epitopes were not significantly different between WT and variant virus-infected groups throughout the course of viral infection (Fig. 5A). Similarly, the levels of virus-specific (combined capsid and noncapsid epitopes) CD4+ T cells in the CNS measured by IFN-γ production were comparable among all experimental groups, as shown for a representative experiment at 8 days postinfection (Fig. 5B). The proportions and numbers of CNS-infiltrating CD4+ T cells at 8 days postinfection were further assessed, and the combined results of 3 separate experiments are shown in Fig. 5C. These results suggest that there are no significant alterations in the CD4+ T cell responses to the variant viruses during the course of infection. Therefore, the differences in virus-specific CD4+ T cell responses unlikely resulted in the altered clinical outcome following infection with VP3159-166-modified viruses (Fig. 3).
Fig 5.

Reactivity of CD4+ T cells in mice infected with WT and variant viruses. (A) Splenic cells from mice infected with WT and variant viruses at 8 days postinfection were stimulated for 3 days in the presence of PBS or a structural CD4+ T cell epitope mix (S; VP1233-250, VP274-86, and VP324-37) and the predominant nonstructural epitope (NS; 3D21-36). Proliferation levels were assessed by [3H]TdR uptake. (B) CNS-infiltrating MNCs were stimulated with PBS or a CD4+ T cell epitope mix (VP1233-250, VP274-86, VP324-37, and 3D21-36). After 6 h of stimulation, cells were stained for CD4 and intracellular IFN-γ. The percentage of CD4+ IFN-γ-producing cells is shown in the upper right corner of each plot. The data are representative of three independent experiments. (C) Epitope-specific IFN-γ production by CNS-infiltrating CD4+ T cells. The proportion of epitope-specific CD4+ T cells in the CNS (left) and the number of epitope-specific CD4+ T cells (right) are shown. The values given are the means of the percentages or numbers from 3 independent experiments (mean ± SD).
Distinct cytokine genes are expressed in CD8+ T cells reactive to the dominant VP3159-166 epitope versus the subdominant VP3173-181epitope.
It is conceivable that CD8+ T cells produce a different set of cytokines depending on epitope recognition, which would consequently result in differential viral clearance and pathogenesis. To examine this possibility, CNS cells from mice infected with the WT virus were restimulated in vitro with dominant and subdominant epitope peptides (Fig. 6A and B). Because it is difficult to isolate the individual epitope-specific CD8+ T cells from the CNS, we have determined the levels of cytokine messages induced after stimulation with the cognate epitopes compared to those in the unstimulated controls. In contrast to the minimal level of IFN-γ in the control cultures with PBS, similarly high levels of IFN-γ mRNAs were detected upon cognate stimulation with the VP3159-166 and VP3173-181 peptides. Therefore, the level of IFN-γ mRNA appears to reflect the cytokine message levels produced by the respective epitope-specific CD8+ T cells. Interestingly, the VP3173-181-specific CD8+ T cells expressed significantly higher levels of IL-10, IL-17, IL-22, and TGF-β1 genes than the VP3159-166-specific CD8+ T cells. These results suggest that the major T cell populations specific for these two epitopes represent distinct CD8+ T cell types. The predominant VP3159-166-specific CD8+ T cells appear to represent the typical antiviral CD8+ T cells, whereas the subdominant VP3173-181-specific CD8+ T cells may represent the recently described Tc17 cell type (14, 19, 20).
Fig 6.

Distinct cytokine profiles of different epitope-specific CD8+ T cells. CNS-infiltrating MNCs from SJL mice infected with WT virus were restimulated in vitro at 8 days postinfection with PBS, VP3159-166, or VP3173-181 for 6 h. The mRNA levels of various cytokines in these cultures were analyzed using either conventional RT-PCR (A) or quantitative real-time PCR (B). The RNA levels of TMEV, FoxP3, and various cytokines in CNS-infiltrating cells of mice infected with either WT or variant viruses were assessed using conventional RT-PCR (C) or real-time PCR (D). Fold increase indicates the relative fold increases after normalization on the basis of GAPDH levels. One representative set from three separate experiments is shown. *, P < 0.05; **, P < 0.01.
To further delineate the potential mechanism(s) underlying the resistance to the development of demyelinating disease in the absence of VP3159-166-specific CD8+ T cells, the levels of T cell-associated cytokine gene expression in the CNS of WT- and variant virus-infected mice were analyzed at 8 days postinfection using PCR (Fig. 6C and D). The expression of IL-10, IL-17, TGF-β1, and FoxP3 mRNA was noticeably higher in the CNS of the VP3159-166-deficient N160V virus-infected mice, which have increased levels of VP3173-181-specific CD8+ T cells, than in the CNS of either the WT virus-infected or VP3173-181-deficient P179A virus-infected mice displaying high levels of VP3159-166-specific CD8+ T cells. These results clearly demonstrate that viral RNA and IL-17, TGF-β1, and FoxP3 mRNA levels are highly elevated in the absence of VP3159-166-specific CD8+ T cells. Because the number of VP3173-181-specific CD8+ T cells is increased in the absence of VP3159-166-specific CD8+ T cells, the elevated cytokine gene expression in N160V virus-infected mice likely reflects the cytokine profile of VP3173-181-specific CD8+ T cells. However, the presence of intracellular IL-17 was undetectable by flow cytometry, suggesting that the IL-17 levels produced in these CD8+ T cells are relatively low (not shown). Nevertheless, the differences in T cell cytokine levels suggest that the predominant virus-specific CD8+ T cells in the CNS recognize VP3159-166 and that the deletion of this population results in a drastically altered cytokine profile of CD8+ T cells. These results are consistent with the cytokine production levels obtained upon stimulation with the respective epitope peptides (Fig. 6A and B).
VP3159-166- and VP3173-181-specific CD8+ T cells differ in their cytolytic function.
Because SJL mice infected with the N160V virus deficient in the VP3159-166 epitope do not develop demyelinating disease, we further examined if differences in the epitope-dependent cytolytic function of CD8+ T cells are associated with disease development. The cytolytic activity of CD8+ T cells from SJL mice infected with the WT or the N160V or P179A mutant virus was assessed in vitro at various effector-to-target cell ratios (Fig. 7A). The CD8+ T cells from the WT virus-infected mice failed to lyse VP3173-181-loaded target cells at an effector-to-target cell ratio as high as 100:1, whereas these CD8+ T cells effectively lysed VP3159-166-loaded target cells at an effector-to-target cell ratio as low as 10:1. Furthermore, the CD8+ T cells from the N160V virus-infected mice lacking VP3159-166-specific CD8+ T cells could not lyse the target cells. In contrast, the CD8+ T cells from the P179A virus-infected mice deficient in VP3173-181-specific T cells effectively lysed the target cells. These results clearly indicate that VP3159-166-specific CD8+ T cells, but not VP3173-181-specific CD8+ T cells, exhibit strong cytolytic activity in vitro.
Fig 7.

Different cytolytic functions of epitope-specific CD8+ T cells. (A) Splenocytes from naive SJL mice were loaded with control ovalbumin from residues 323 to 339 (OVA323-339) and viral (VP3159-166 or VP3173-181) peptides and labeled with a high and a low concentration of CFSE, respectively. A fixed number of CFSE-labeled target cells and various numbers of splenic effector cells from mice at 8 days postinfection with WT or variant viruses were cocultured for 60 h at the indicated effector-to-target cell ratios. The numbers in each histogram represent the percentages of cells labeled with low (VP3159-166 or VP3173-181) and high (OVA323-339) concentrations of CFSE. (B) At 8 days after infection, in vivo lysis activity was evaluated in virus- and mock-infected mice (n = 3) that received the peptide-loaded CFSE-labeled target cells. OVA323-339 and viral (both VP3159-166 and VP3173-181) peptides were used to load the target cells. Mixtures of control and viral peptide-loaded target cells were injected into SJL mice (n = 3) infected with WT, N160V, or P179A virus and mock infected with PBS. Cytolytic profiles of three individual mice are shown. (C) SJL mice were treated with either the isotype (n = 3) or anti-IL-17 (n = 3) antibody at days 0 and 7 relative to the time of TMEV infection. At 8 days postinfection, in vivo cytolysis activity was evaluated in virus- and mock-infected mice (n = 3) that received the target cells. (D) CNS-infiltrating cells from the SJL mice infected with TMEV were stimulated with VP3159-166 or VP3173-181 at different concentrations for 6 h, and intracellular IFN-γ production was assessed by flow cytometry. The numbers of IFN-γ-producing CD8+ T cells after stimulation with 2 μM peptide were considered to be 100% of the epitope-specific IFN-γ-producing cells. (E) CNS-infiltrating cells (1.5 × 106) from SJL mice infected with the TMEV BeAn strain were stimulated with various concentrations of VP3159-166 or VP3173-181 peptide for 72 h. The production of IFN-γ and IL-17 in the culture supernatants was measured using the respective cytokine-specific ELISA.
Despite the lack of significant cytolytic function by VP3173-181-specific CD8+ T cells in vitro (Fig. 7A), it is conceivable that in vitro cytotoxicity may not reflect cytolytic function in virus-infected mice. To explore this possibility, we further examined the potential differences in cytolytic functions of CD8+ T cells in virus-infected mice using target cells loaded with an equal mixture of the VP3159-166 and VP3173-181 epitopes (Fig. 7B). Control and cognate peptide-loaded target cells labeled with two different intensities of carboxyfluorescein succinimidyl ester (CFSE; high and low) were administered to virus-infected mice (3 mice per viral group) at 8 days postinfection, as previously described (17). The N160V virus-infected mice (3/3) lacking VP3159-166-specific CD8+ T cells failed to lyse the target cells. In contrast, the WT virus-infected mice and P179A virus-infected mice deficient in VP3173-181-specific CD8+ T cells efficiently lysed the target cells. These results clearly indicate that WT virus-infected SJL mice are able to efficiently clear VP3159-166- and VP3173-181-bearing cells, suggesting that only VP3159-166-specific CD8+ T cells are capable of effectively clearing the target cells in infected animals, which is consistent with the in vitro cytotoxicity (Fig. 7A).
It has recently been shown that IL-17 inhibits the cytolytic function of CD8+ T cells by protecting virus-infected target cells from apoptosis (17) and that IL-17-producing CD8+ T cells display deficient cytolytic function in vitro (14). Despite the relatively higher level of IL-17 mRNA in VP3173-181-specific CD8+ T cells, we failed to detect intracellular IL-17 by flow cytometry. Therefore, it is conceivable that a relatively small amount of IL-17 in CD8+ T cells is sufficient for blocking the cytolysis of target cells. To examine this possibility, either isotype or anti-IL-17 antibody-treated SJL mice were infected with WT TMEV. Eight days after infection, in vivo cytolytic activity was evaluated in the virus- and mock-infected mice (Fig. 7C). Interestingly, both the VP3159-166- and VP3173-181-specific CD8+ T cells in the anti-IL-17 antibody-treated mice significantly gained cytolytic function. In particular, VP3173-181-specific CD8+ T cells in the anti-IL-17 antibody-treated mice displayed strong cytolysis (>50%) of target cells, whereas there was a lack of detectable cytolysis in the isotype antibody-treated mice. A similar improvement in the cytolytic activity of the VP3173-181-specific CD8+ T cells was also detected in vitro in the presence of anti-IL-17 antibody (not shown). Therefore, these results suggest that a small amount of IL-17 production by CD8+ T cells may efficiently block target cell cytolysis in cell-cell contact interactions. It is also noteworthy that the cytolytic levels in virus-infected mice (Fig. 7C) were much lower than those assessed with in vitro cytotoxic assays (Fig. 7A). This may be attributable to the presence of higher IL-17 levels in virus-infected mice. Taken together, these results strongly suggest that CD8+ T cells with strong cytolytic function in vivo are essential for the pathogenesis of demyelinating disease, as mice lacking this CD8+ T cell population do not develop the disease.
VP3159-166- and VP3173-181-specific CD8+ T cells display different avidities for their epitopes and produce distinct cytokines.
It has recently been shown that T cell activation signals affect the development of CD4+ T cell subtypes (11, 30). To further understand the potential underlying mechanisms of the differences in cytokine production and cytolytic function between VP3159-166- and VP3173-181-specific CD8+ T cells, we determined the functional avidity of T cells to the epitopes by measuring the levels of IFN-γ and/or IL-17A production upon stimulation with various concentrations of the cognate peptides (Fig. 7D and E), as recently described (25). Interestingly, the concentrations of VP3159-166 peptide required to stimulate intracellular IFN-γ production over 6 h to the levels similar to those of the VP3173-181 peptide were 30- to 100-fold lower (Fig. 7D). For example, more than 50 nM VP3173-181 peptide versus less than 2 nM VP3159-166 peptide was required to achieve 50% of the maximum IFN-γ production. These results indicate that the functional avidity of VP3159-166-specific CD8+ T cells to the cognate epitope required to stimulate the initial production of IFN-γ is at least 30-fold higher than that of VP3173-181-specific CD8+ T cells. However, intracellular IL-17 production after 6 h of in vitro stimulation was not detectable by flow cytometry (not shown), although the increase in IL-17 mRNA production was observed by reverse transcription-PCR (RT-PCR) (Fig. 6). To further determine the production of IL-17 by the epitope-specific CD8+ T cells over a longer time period, we incubated 1.5 × 106 CNS-infiltrating cells from TMEV-infected SJL mice for 72 h in the presence of the epitope peptides. The levels of IFN-γ and IL-17 that accumulated in the culture supernatants were assessed using cytokine-specific enzyme-linked immunosorbent assays (ELISAs). Although the production of IFN-γ was detected in the cultures stimulated with either the VP3159-166 or the VP3173-181 peptide, IL-17 production was detectable only after VP3173-181 peptide stimulation (Fig. 7D). Therefore, it is conceivable that differences in the functional avidity toward their cognate epitopes and/or the type of cytokines produced may affect the consequent cytolytic function of CD8+ T cell populations in an epitope-dependent manner.
DISCUSSION
TMEV infection in susceptible strains of mice induces chronic demyelinating disease mediated by CD4+ T cells and macrophages (17, 26, 28). However, epitope-specific CD4+ T cells can be protective, depending on the time of availability in conjunction with viral infection (36). In addition, the pathogenicity of CD4+ T cells may also be dependent on the recognizing viral epitopes (21, 54). Therefore, epitope-specific cytotoxic CD8+ T cells may similarly play a protective or pathogenic role depending on the type and/or reactivity of epitopes. We previously demonstrated that the level of the VP3159-166-specific CD8+ T cell response in susceptible SJL mice is inversely correlated with viral persistence in P1-transgenic (P1-Tg) SJL mice (40), highlighting the importance of VP3159-166-specific CD8+ T cells in limiting the viral load in vivo. Surprisingly, however, higher viral persistence in P1-Tg mice did not lead to a higher disease incidence and/or severity. These results posed an interesting possibility that VP3159-166-specific CD8+ T cells may be associated with the pathogenesis of TMEV BeAn-induced demyelinating disease. Because the TMEV DA strain, which induces a similar chronic demyelinating disease, does not contain this epitope (23), the DA virus may utilize CD8+ T cells recognizing a different epitope for its pathogenesis.
To further address the possibility that VP3159-166-specific CD8+ T cells play a pathogenic role in disease development, a series of mutant viruses containing a single amino acid substitution or double amino acid substitutions in the predominant and/or subdominant CD8+ T cell epitopes of TMEV was generated (Fig. 1). These variant viruses failed to mount the corresponding epitope-specific CD8+ T cell responses (Fig. 2) without affecting CD4+ T cell responses (Fig. 5). Interestingly, the viral load was higher in the CNS of the VP3159-166-deficient N160V virus-infected mice during the early stages of infection, suggesting that VP3159-166-specific CD8+ T cells are critical for viral clearance (Fig. 3). The high viral load is unlikely to reflect more rapid viral replication, as these variant viruses did not replicate differently in BHK-21 cells and mouse astrocytes or bone marrow-derived dendritic cells (Fig. 1) and in the CNS of Rag1−/− mice (not shown). Since these cell populations are the major cell populations infected by the WT virus, it is unlikely that the mutant viruses infect different cell types. In addition, similar viral loads in the CNS of Rag1−/− mice, which do not have adaptive immune responses, would reflect the comparable tropism of these variant viruses. However, differences in the infectivity to minor cell populations or viral trafficking to different CNS compartments would be possible.
Interestingly, the loss of a VP3159-166-specific CD8+ T cell response in the N160V virus-infected mice was compensated for by an increase in CD8+ T cells reactive to the subdominant VP3173-181 epitope (Fig. 2). However, the VP3173-181-specific CD8+ T cells whose levels were elevated in the N160V virus-infected mice were apparently unable to efficiently limit the viral load (Fig. 4). The inefficient control of viral load was due to the poor cytolytic function of VP3173-181-specific CD8+ T cells compared to VP3159-166-specific CD8+ T cells (Fig. 7). In contrast, the VP3173-181-deficient P179A virus-infected mice, which mount a greater VP3159-166-specific CD8+ T cell response, efficiently controlled the viral load in the CNS. Interestingly, however, the virus level in the CNS did not correlate with the development of clinical disease (Fig. 4), which is consistent with the previous observation in P1-Tg mice (40). Therefore, viral persistence alone may not be sufficient for the induction of demyelinating disease, and CD8+ T cell responses to certain viral epitopes may be an integral component of the pathogenesis, as previously suggested (34).
It is known that the presence of a strong CD8+ T cell response is essential for protection from viral diseases. However, the majority of the N160V virus-infected animals (>90%), which lacked the predominant cytolytic VP3159-166-reactive CD8+ T cells bearing high functional avidity (Fig. 7), remained free of clinical signs (Fig. 3). In contrast, the majority of the P179A virus-infected animals (80%), which had high levels of VP3159-166-reactive CD8+ T cells in the absence of VP3173-181-reactive CD8+ T cells, developed the full-blown disease (Fig. 3). In addition, the N160V/P179A virus-infected SJL mice, which lacked both VP3159-166- and VP3173-181-reactive CD8+ T cells, were also largely free of demyelinating disease. The observation that a highly cytolytic CD8+ T cell response in the CNS is associated with the development of TMEV-induced demyelinating disease in SJL mice was unexpected (Fig. 3 and 7). Taken together, these results strongly suggest that cytolytic CD8+ T cells with high avidity (Fig. 7) may cause an initial infliction of CNS damage leading to the development of demyelinating disease.
It is not clear at this time whether CD4+ T cells and CD8+ T cells independently or cooperatively induce clinical disease. It has previously been assumed that CD4+ T cells alone induce TMEV-induced demyelinating disease, based on the amelioration of disease development after treatment with antibodies against CD4+ T cells (17, 46, 53) and the development of severe demyelinating disease in β2-microglobulin-deficient SJL mice (2). However, β2-microglobulin is critical for all types of T cells restricted with class I and class I-like molecules, including NK T cells and other nonclassical class I-restricted T cells. Therefore, it is difficult to discern the potential role and function of these T cell populations in the pathogenesis and/or protection from viral disease development. It is interesting to note that CD4-deficient susceptible SJL and PLJ mice display more severe demyelination than CD8-deficient counterpart mice (38). These results suggest that subpopulations of both CD4+ and CD8+ T cell types could be cooperatively involved in the pathogenesis of demyelinating disease.
The mechanisms underlying the differences in viral clearance and cytotoxic function among epitope-specific CD8+ T cells are unknown. Although CD8+ T cells recognizing the VP3159-166 and VP3173-181 epitopes can produce relatively high levels of IFN-γ, the efficiency of the VP3173-181-specific CD8+ T cells to induce IFN-γ production was as much as 100-fold lower than that of the VP3159-166-specific CD8+ T cells (Fig. 7D). In contrast, the VP3173-181-specific CD8+ T cells induced significantly higher levels of IL-10, IL-17, IL-22, and TGF-β1 mRNA expression than VP3159-166-specific CD8+ T cells (Fig. 6). This CD8+ T cell population appears to be similar to the IL-17-producing CD8+ T cell population recently named Tc17 (14), including the expression of FoxP3 (Fig. 6D) and retinoic acid-related orphan nuclear receptor gamma t (ROR-γt) transcription factors (not shown). It is interesting to note that the Tc17-type response is largely epitope dependent (Fig. 6 and 7E). As far as we know, this is the first report indicating that the induction of a Tc17 response is epitope dependent. The drastic difference in functional avidity between the VP3159-166- and VP3173-181-specific CD8+ T cells (Fig. 7) may lead to the differential induction of CD8+ T cell types. In addition, it is possible that various cytokines favoring the development of pathogenic Th17 cells in TMEV-infected mice (17) may also promote the epitope-dependent development of Tc17 cells. For example, TGF-β and IL-6 genes are highly expressed in various glial cells and infiltrating antigen-presenting cells in the CNS of TMEV-infected SJL mice (4, 18, 22, 50). Therefore, viral infection-induced TGF-β, together with IL-6, may also participate in the development of IL-17-producing CD8+ T cells in the presence of a low level of IFN-γ (14, 19), similar to the development of Th17 cells (15, 17, 33, 52).
The VP3173-181-specific CD8+ T cells producing IL-17 also expressed TGF-β1 mRNA in response to their cognate epitope (Fig. 6). Furthermore, these CD8+ T cells upregulated the surface expression of CD103 (not shown), which is a marker for the recent expression of TGF-β (9, 39). TGF-β interferes with the effector functions of CD8+ T cells by inhibiting the expression of perforin and FasL on T cells, both of which are important for the cytotoxic function of CD8+ T cells (6, 13, 45, 49). In addition, IL-17 inhibits the cytolytic function of CD8+ T cells by protecting the virus-infected target cells from apoptosis (17). Consequently, IL-17-producing CD8+ T cells may exhibit deficient cytolytic function in vitro (Fig. 7A) and in vivo (Fig. 7B), as shown previously (14, 19). Although the presence of anti-IL-17 antibody enhanced cytolytic function (Fig. 7C), the IL-17 level produced by VP3173-181-specific CD8+ T cells appears to be too low to detect using intracellular cytokine staining to assess the amount of intracellular IL-17 produced in 6 h (not shown). However, these CD8+ T cells produced a significant level of IL-17 in 3 days (Fig. 7E), suggesting that the amount of IL-17 produced by the epitope-specific CD8+ T cells is significant, although smaller than that produced by CD4+ T cells. Therefore, a relatively small amount of IL-17 locally produced by CD8+ T cells may be sufficient for blocking cytolysis of target cells during the cognate cell-cell interaction.
The role of the IL-17-producing CD8+ T cell subtype in the protection or pathogenesis of demyelinating disease is unclear. A protective role of Tc17 in influenza viral infection has been proposed, as mice treated with anti-IL-17 antibody showed higher mortality (14). Perhaps the increased mortality after treatment with the anti-IL-17 antibody reflects extensive tissue damage in the absence of IL-17, which prevents apoptotic cell death (17). However, mice with high levels of Tc17 cells harbor higher viral loads in the CNS (Fig. 4), suggesting less efficient viral clearance by the cells. Interestingly, this CD8+ T cell population failed to result in the development of TMEV-induced demyelinating disease, in contrast to conventional IFN-γ-producing Tc1 CD8+ T cells (Fig. 2 and 3). It is surprising to observe that the highly cytolytic VP3159-166-specific Tc1 population is associated with the pathogenesis of demyelination and axon damage, whereas the noncytolytic VP3173-181-specific Tc17 population is not (Fig. 3). This is unexpected, in light of our recent findings that IL-17-producing CD4+ T cells (Th17) play a critical pathogenic role in the development of virus-induced demyelinating disease (17). Therefore, it is conceivable that IFN-γ-producing VP3159-166-specific CD8+ T cells with high cytolytic function are required to initiate the pathogenic process by destroying infected neurons and/or releasing sequestered autoantigens, whereas IL-17-producing low-cytolytic VP3173-181-specific CD8+ T cells fail to do so. Because Th17 cells secreting high levels of IL-17 play a critical role in the pathogenesis of TMEV-induced demyelinating disease (17), virus-specific cytolytic CD8+ T cells may initiate the process, followed by Th17-mediated inhibition of cytolytic function promoting viral persistence and subsequent extensive tissue damage. Thus, both Th17 and Tc1 populations reactive to viral epitopes may cooperatively be involved in the pathogenesis of virally induced demyelinating disease.
ACKNOWLEDGMENTS
This work was supported by United States Public Health Service grants (RO1 NS28752 and RO1 NS33008) and by a grant from the National Multiple Sclerosis Society (RG 4001-A6).
Footnotes
Published ahead of print 10 October 2012
REFERENCES
- 1. Altman JD, et al. 1996. Phenotypic analysis of antigen-specific T lymphocytes. Science 274:94–96 [PubMed] [Google Scholar]
- 2. Begolka WS, et al. 2001. CD8-deficient SJL mice display enhanced susceptibility to Theiler's virus infection and increased demyelinating pathology. J. Neurovirol. 7:409–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Calenoff MA, Faaberg KS, Lipton HL. 1990. Genomic regions of neurovirulence and attenuation in Theiler murine encephalomyelitis virus. Proc. Natl. Acad. Sci. U. S. A. 87:978–982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chang JR, Zaczynska E, Katsetos CD, Platsoucas CD, Oleszak EL. 2000. Differential expression of TGF-beta, IL-2, and other cytokines in the CNS of Theiler's murine encephalomyelitis virus-infected susceptible and resistant strains of mice. Virology 278:346–360 [DOI] [PubMed] [Google Scholar]
- 5. Chang Y, et al. 2011. CD8 positive T cells express IL-17 in patients with chronic obstructive pulmonary disease. Respir. Res. 12:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chen ML, et al. 2005. Regulatory T cells suppress tumor-specific CD8 T cell cytotoxicity through TGF-beta signals in vivo. Proc. Natl. Acad. Sci. U. S. A. 102:419–424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dal Canto MC, Kim BS, Miller SD, Melvold RW. 1996. Theiler's murine encephalomyelitis virus (TMEV)-induced demyelination: a model for human multiple sclerosis. Methods 10:453–461 [DOI] [PubMed] [Google Scholar]
- 8. Doherty PC, et al. 1997. Effector CD4+ and CD8+ T-cell mechanisms in the control of respiratory virus infections. Immunol. Rev. 159:105–117 [DOI] [PubMed] [Google Scholar]
- 9. Dubois CM, Laprise MH, Blanchette F, Gentry LE, Leduc R. 1995. Processing of transforming growth factor beta 1 precursor by human furin convertase. J. Biol. Chem. 270:10618–10624 [DOI] [PubMed] [Google Scholar]
- 10. Fuller AC, et al. 2005. Gender bias in Theiler's virus-induced demyelinating disease correlates with the level of antiviral immune responses. J. Immunol. 175:3955–3963 [DOI] [PubMed] [Google Scholar]
- 11. Gabrysova L, et al. 2011. Integrated T-cell receptor and costimulatory signals determine TGF-beta-dependent differentiation and maintenance of Foxp3+ regulatory T cells. Eur. J. Immunol. 41:1242–1248 [DOI] [PubMed] [Google Scholar]
- 12. Gay FW, Drye TJ, Dick GW, Esiri MM. 1997. The application of multifactorial cluster analysis in the staging of plaques in early multiple sclerosis. Identification and characterization of the primary demyelinating lesion. Brain 120:1461–1483 [DOI] [PubMed] [Google Scholar]
- 13. Genestier L, Kasibhatla S, Brunner T, Green DR. 1999. Transforming growth factor beta1 inhibits Fas ligand expression and subsequent activation-induced cell death in T cells via downregulation of c-Myc. J. Exp. Med. 189:231–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hamada H, et al. 2009. Tc17, a unique subset of CD8 T cells that can protect against lethal influenza challenge. J. Immunol. 182:3469–3481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Harrington LE, et al. 2005. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat. Immunol. 6:1123–1132 [DOI] [PubMed] [Google Scholar]
- 16. Harty JT, Tvinnereim AR, White DW. 2000. CD8+ T cell effector mechanisms in resistance to infection. Annu. Rev. Immunol. 18:275–308 [DOI] [PubMed] [Google Scholar]
- 17. Hou W, Kang HS, Kim BS. 2009. Th17 cells enhance viral persistence and inhibit T cell cytotoxicity in a model of chronic virus infection. J. Exp. Med. 206:313–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hou W, So EY, Kim BS. 2007. Role of dendritic cells in differential susceptibility to viral demyelinating disease. PLoS Pathog. 3:e124 doi:10.1371/journal.ppat.0030124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Huber M, et al. 2009. A Th17-like developmental process leads to CD8+ Tc17 cells with reduced cytotoxic activity. Eur. J. Immunol. 39:1716–1725 [DOI] [PubMed] [Google Scholar]
- 20. Intlekofer AM, et al. 2008. Anomalous type 17 response to viral infection by CD8+ T cells lacking T-bet and eomesodermin. Science 321:408–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jin YH, Kang B, Kim BS. 2009. Theiler's virus infection induces a predominant pathogenic CD4+ T cell response to RNA polymerase in susceptible SJL/J mice. J. Virol. 83:10981–10992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jin YH, et al. 2007. Differential virus replication, cytokine production, and antigen-presenting function by microglia from susceptible and resistant mice infected with Theiler's virus. J. Virol. 81:11690–11702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kang BS, Lyman MA, Kim BS. 2002. Differences in avidity and epitope recognition of CD8(+) T cells infiltrating the central nervous systems of SJL/J mice infected with BeAn and DA strains of Theiler's murine encephalomyelitis virus. J. Virol. 76:11780–11784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kang BS, Lyman MA, Kim BS. 2002. The majority of infiltrating CD8+ T cells in the central nervous system of susceptible SJL/J mice infected with Theiler's virus are virus specific and fully functional. J. Virol. 76:6577–6585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kang HS, Kim BS. 2010. Predominant clonal accumulation of CD8+ T cells with moderate avidity in the central nervous systems of Theiler's virus-infected C57BL/6 mice. J. Virol. 84:2774–2786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kim BS, et al. 2001. Pathogenesis of virus-induced immune-mediated demyelination. Immunol. Res. 24:121–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kim BS, Mohindru M, Kang B, Kang HS, Palma JP. 2005. Effects of the major histocompatibility complex loci and T-cell receptor beta-chain repertoire on Theiler's virus-induced demyelinating disease. J. Neurosci. Res. 81:846–856 [DOI] [PubMed] [Google Scholar]
- 28. Kim BS, Palma JP, Inoue A, Koh CS. 2000. Pathogenic immunity in Theiler's virus-induced demyelinating disease: a viral model for multiple sclerosis. Arch. Immunol. Ther. Exp. (Warsz.) 48:373–379 [PubMed] [Google Scholar]
- 29. Kuang DM, et al. 2010. Tumor-activated monocytes promote expansion of IL-17-producing CD8+ T cells in hepatocellular carcinoma patients. J. Immunol. 185:1544–1549 [DOI] [PubMed] [Google Scholar]
- 30. Leitenberg D, Bottomly K. 1999. Regulation of naive T cell differentiation by varying the potency of TCR signal transduction. Semin. Immunol. 11:283–292 [DOI] [PubMed] [Google Scholar]
- 31. Lipton HL, Melvold R. 1984. Genetic analysis of susceptibility to Theiler's virus-induced demyelinating disease in mice. J. Immunol. 132:1821–1825 [PubMed] [Google Scholar]
- 32. Lyman MA, Myoung J, Mohindru M, Kim BS. 2004. Quantitative, not qualitative, differences in CD8+ T cell responses to Theiler's murine encephalomyelitis virus between resistant C57BL/6 and susceptible SJL/J mice. Eur. J. Immunol. 34:2730–2739 [DOI] [PubMed] [Google Scholar]
- 33. Mangan PR, et al. 2006. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature 441:231–234 [DOI] [PubMed] [Google Scholar]
- 34. McDole J, Johnson AJ, Pirko I. 2006. The role of CD8+ T-cells in lesion formation and axonal dysfunction in multiple sclerosis. Neurol. Res. 28:256–261 [DOI] [PubMed] [Google Scholar]
- 35. Mendez-Fernandez YV, Johnson AJ, Rodriguez M, Pease LR. 2003. Clearance of Theiler's virus infection depends on the ability to generate a CD8+ T cell response against a single immunodominant viral peptide. Eur. J. Immunol. 33:2501–2510 [DOI] [PubMed] [Google Scholar]
- 36. Mohindru M, Kang B, Kim BS. 2006. Initial capsid-specific CD4+ T cell responses protect against Theiler's murine encephalomyelitisvirus-induced demyelinating disease. Eur. J. Immunol. 36:2106–2115 [DOI] [PubMed] [Google Scholar]
- 37. Murray PD, et al. 1998. Perforin-dependent neurologic injury in a viral model of multiple sclerosis. J. Neurosci. 18:7306–7314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Murray PD, Pavelko KD, Leibowitz J, Lin X, Rodriguez M. 1998. CD4(+) and CD8(+) T cells make discrete contributions to demyelination and neurologic disease in a viral model of multiple sclerosis. J. Virol. 72:7320–7329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Myers L, Croft M, Kwon BS, Mittler RS, Vella AT. 2005. Peptide-specific CD8 T regulatory cells use IFN-gamma to elaborate TGF-beta-based suppression. J. Immunol. 174:7625–7632 [DOI] [PubMed] [Google Scholar]
- 40. Myoung J, Il Bahk Y, Kang HS, Dal Canto MC, Kim BS. 2008. Anti-capsid immunity level, not viral persistence level, correlates with the progression of Theiler's virus-induced demyelinating disease in viral P1-transgenic mice. J. Virol. 82:5606–5617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ortega C, et al. 2009. IL-17-producing CD8+ T lymphocytes from psoriasis skin plaques are cytotoxic effector cells that secrete Th17-related cytokines. J. Leukoc. Biol. 86:435–443 [DOI] [PubMed] [Google Scholar]
- 42. Palma JP, Kim BS. 2004. The scope and activation mechanisms of chemokine gene expression in primary astrocytes following infection with Theiler's virus. J. Neuroimmunol. 149:121–129 [DOI] [PubMed] [Google Scholar]
- 43. Palma JP, et al. 2001. Enhanced susceptibility to Theiler's virus-induced demyelinating disease in perforin-deficient mice. J. Neuroimmunol. 116:125–135 [DOI] [PubMed] [Google Scholar]
- 44. Pullen LC, Miller SD, Dal Canto MC, Kim BS. 1993. Class I-deficient resistant mice intracerebrally inoculated with Theiler's virus show an increased T cell response to viral antigens and susceptibility to demyelination. Eur. J. Immunol. 23:2287–2293 [DOI] [PubMed] [Google Scholar]
- 45. Ranges GE, Figari IS, Espevik T, Palladino MA., Jr 1987. Inhibition of cytotoxic T cell development by transforming growth factor beta and reversal by recombinant tumor necrosis factor alpha. J. Exp. Med. 166:991–998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rodriguez M, Lafuse WP, Leibowitz J, David CS. 1986. Partial suppression of Theiler's virus-induced demyelination in vivo by administration of monoclonal antibodies to immune-response gene products (Ia antigens). Neurology 36:964–970 [DOI] [PubMed] [Google Scholar]
- 47. Rodriguez M, Leibowitz J, David CS. 1986. Susceptibility to Theiler's virus-induced demyelination. Mapping of the gene within the H-2D region. J. Exp. Med. 163:620–631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Skias DD, et al. 1987. Susceptibility of astrocytes to class I MHC antigen-specific cytotoxicity. J. Immunol. 138:3254–3258 [PubMed] [Google Scholar]
- 49. Smyth MJ, Strobl SL, Young HA, Ortaldo JR, Ochoa AC. 1991. Regulation of lymphokine-activated killer activity and pore-forming protein gene expression in human peripheral blood CD8+ T lymphocytes. Inhibition by transforming growth factor-beta. J. Immunol. 146:3289–3297 [PubMed] [Google Scholar]
- 50. So EY, Kang MH, Kim BS. 2006. Induction of chemokine and cytokine genes in astrocytes following infection with Theiler's murine encephalomyelitis virus is mediated by the Toll-like receptor 3. Glia 53:858–867 [DOI] [PubMed] [Google Scholar]
- 51. Sospedra M, Martin R. 2005. Immunology of multiple sclerosis. Annu. Rev. Immunol. 23:683–747 [DOI] [PubMed] [Google Scholar]
- 52. Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. 2006. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity 24:179–189 [DOI] [PubMed] [Google Scholar]
- 53. Welsh CJ, Tonks P, Nash AA, Blakemore WF. 1987. The effect of L3T4 T cell depletion on the pathogenesis of Theiler's murine encephalomyelitis virus infection in CBA mice. J. Gen. Virol. 68:1659–1667 [DOI] [PubMed] [Google Scholar]
- 54. Yauch RL, Palma JP, Yahikozawa H, Koh CS, Kim BS. 1998. Role of individual T-cell epitopes of Theiler's virus in the pathogenesis of demyelination correlates with the ability to induce a Th1 response. J. Virol. 72:6169–6174 [DOI] [PMC free article] [PubMed] [Google Scholar]