

Abstract
Interferon-inducible transmembrane (IFITM) protein family members IFITM1, -2, and -3 restrict the infection of multiple enveloped viruses. Significant enrichment of a minor IFITM3 allele was recently reported for patients who were hospitalized for seasonal and 2009 H1N1 pandemic flu. This IFITM3 allele lacks the region corresponding to the first amino-terminal 21 amino acids and is unable to inhibit influenza A virus. In this study, we found that deleting this 21-amino-acid region relocates IFITM3 from the endosomal compartments to the cell periphery. This finding likely underlies the lost inhibition of influenza A virus that completes its entry exclusively within endosomes at low pH. Yet, wild-type IFITM3 and the mutant with the 21-amino-acid deletion inhibit HIV-1 replication equally well. Given the pH-independent nature of HIV-1 entry, our results suggest that IFITM3 can inhibit viruses that enter cells via different routes and that its N-terminal region is specifically required for controlling pH-dependent viruses.
INTRODUCTION
The interferon-inducible transmembrane (IFITM) proteins comprise a small protein family that is conserved across many eukaryotic species (16, 27, 30). Although only about 130 amino acids in length, they all have two putative transmembrane domains interspersed by a conserved cytoplasmic region. Humans have five ifitm genes, including ifitm1, ifitm2, ifitm3, ifitm5, and ifitm10. They are all located on chromosome 11, with ifitm1, ifitm2, ifitm3, and ifitm5 clustered within a 26.5-kb region and ifitm10 located 1.4 Mb away (16). Very little is known about the function of IFITM10 despite the fact that it is the most conserved of all IFITMs among different species (16). IFITM5 is expressed strictly in osteoblasts and is involved in bone mineralization and maturation (26). Expression of IFITM1, -2, and -3 is stimulated by interferon (21), which suggests their role in interferon-mediated antiviral innate immunity.
Indeed, IFITM1, -2, and -3 inhibit multiple important human-pathogenic viruses. A functional genomic small interfering RNA (siRNA) screen led to the finding that IFITM1, -2, and -3 potently inhibit infection by influenza A H1N1 virus, West Nile virus (WNV), and dengue virus (4). Subsequently, more viruses were reported to be subject to IFITM restriction. These include yellow fever virus (YFV), vesicular stomatitis virus (VSV), Marburg virus (MARV), Ebola virus (EBOV), SARS coronavirus (SARS-CoV) and human immunodeficiency virus type 1 (HIV-1) (4, 6, 17, 18, 22, 28, 35). It is noted that these viruses are inhibited to different extents by different IFITM proteins. For example, influenza A virus is more sensitive to IFITM3, whereas MARV and EBOV are more readily restricted by IFITM1 (17). This is likely due to the sequence divergence that has occurred between IFITM proteins. In contrast to the high homology shared by IFITM2 and IFITM3, IFITM1 has a shorter N-terminal region and a relatively longer C-terminal region (22, 30).
IFITM proteins restrict viral infection by interfering with virus entry (4). This inhibition mechanism was first revealed by studies using murine leukemia virus (MLV) that was pseudotyped with different viral envelope proteins (4). Inhibition was observed for MLV pseudovirus bearing envelope proteins from influenza A virus, WNV, YFV, EBOV, and SARS-CoV but not from lymphocytic choriomeningitis virus, Lassa virus, Machupo virus, or amphotropic MLV (4, 17). More direct evidence was reported in studies using a BlaM-Vpr-based HIV-1 virion fusion assay to demonstrate that IFITM3 inhibits entry mediated by HIV-1 envelope protein, influenza A virus hemagglutinin, and VSV-G protein (12, 22). When the entry of influenza A virus particles was monitored in cells expressing IFITM3 by microscopy, virions were seen to accumulate in the IFITM3-positive acidic membrane compartments and were found to fail completion of cytosolic entry (12, 17). This suggests that IFITM3 blocks the final escape of virus particles from late endosomes rather than affecting the earlier steps of influenza A virus entry, such as binding to the sialic acid receptor, endocytosis, and trafficking to the late endosomes. In support of the site of action of IFITM3 being at these acidic compartments, IFITM3 restriction of SARS-CoV S protein-mediated entry was bypassed when trypsin digestion was used to trigger the membrane fusion at or near the plasma membrane instead of within low-pH cellular compartments (17).
An in vivo role of IFITM3 in limiting virus infection has been reported for both mice and humans. First, ifitm3 knockout mice were found to develop fulminant viral pneumonia when challenged with an otherwise-low-pathogenicity H3N2 influenza A virus (10). Second, hospitalized patients who were severely ill from seasonal flu or 2009 H1N1 pandemic flu exhibited an enrichment of a minor ifitm3 allele (SNP rs12252-C) that lacks the region corresponding to the first amino-terminal 21 amino acids due to the alteration of a splice acceptor site (10). Results of in vitro studies further showed that IFITM3 encoded by this allele is unable to inhibit influenza virus (10). In line with the latter observation, removing these 21 amino acids abrogates IFITM3 inhibition of VSV infection (35), further highlighting the importance of this region in the antiviral function of IFITM3. We now report that the N-terminal region of IFITM3 contributes to antiviral action by regulating its localization to endosomes.
MATERIALS AND METHODS
Plasmid DNA and antibodies.
The Tet-IFITM3 DNA construct was described previously (22). The N-terminal region of IFITM3 was mutated by PCR with the indicated primers to generate the following mutations (Fig. 1A): Δ(1-21), 5′-GCACGGATCCATGGATTACAAGGATGACGACGATAAGATGCTCAAGGAGGAGCACGAG-3′ and 5′-GCACGAATTCCTATCCATAGGCCTGGAAG-3′; Δ(17-20), 5′-CTCTCCTGTCAACAGTGGCCAGGAGATGCTCAAGGAGGAGCAC-3′ and 5′-GTGCTCCTCCTTGAGCATCTCCTGGCCACTGTTGACAGGAGAG-3′; Δ1718, 5′-CCTGTCAACAGTGGCCAGAACTATGAGATGCTCAAG-3′ and 5′-CTTGAGCATCTCATAGTTCTGGCCACTGTTGACAGG-3′; ΔY20, 5′-CAGTGGCCAGCCCCCCAACGAGATGCTCAAGGAGGAGC-3′ and 5′-GCTCCTCCTTGAGCATCTCGTTGGGGGGCTGGCCACTG-3′; 17PTAP, 5′-GTCAACAGTGGCCAGCCCACCGCCCCTGAGATGCTCAAGGAGGAGC-3′ and 5′-GCTCCTCCTTGAGCATCTCAGGGGCGGTGGGCTGGCCACTGTTGAC-3′; 17YPDL, 5′-CCTGTCAACAGTGGCCAGTACCCCGACCTGGAGATGCTCAAGGAGGAGC-3′ and 5′-GCTCCTCCTTGAGCATCTCCAGGTCGGGGTACTGGCCACTGTTGACAGG-3′; K24R, 5′-CAACTATGAGATGCTCAGGGAGGAGCACGAGGTG-3′ and 5′-CACCTCGTGCTCCTCCCTGAGCATCTCATAGTTG-3′; Y20F, 5′-GTGGCCAGCCCCCCAACTTCGAGATGCTCAAGGAGG-3′ and 5′-CCTCCTTGAGCATCTCGAAGTTGGGGGGCTGGCCAC-3′; and Y20A, 5′-GTGGCCAGCCCCCCAACGCTGAGATGCTCAAGGAGG-3′ and 5′-CCTCCTTGAGCATCTCAGCGTTGGGGGGCTGGCCAC-3′.
Fig 1.

IFITM3 inhibits infection by VSV-G protein-pseudotyped viruses. (A) Illustration of IFITM3 and its N-terminal mutations. The two transmembrane domains (TM1 and TM2) and the N-terminal sequence of IFITM3 are depicted. Sequences that are mutated in each mutant are also shown. (B) Western blotting to show the expression of IFITM3 and its mutants in stably transduced HEK293 cells. Tubulin was probed as an internal control. (C) Flow cytometry data of one representative infection experiment showing the effect of IFITM3 and its mutants on infection by VSV-G protein-pseudotyped MLV reporter viruses. Control cells were stably transduced with the pQCXIP empty retroviral vector. The percentages of infected cells (GFP positive) are indicated. (D) The relative infectivity was calculated by dividing the percentages of GFP-positive cells from IFITM3-expressing cell lines by the percentage from the control cell line (pQCXIP). The results are the averages of three independent infection experiments. The error bars indicate standard deviations.
IFITM3 and its mutants have a Flag tag attached to their N termini to facilitate their detection by Western blotting or immunofluorescence microscopy.
The pQCXIP retroviral vector was purchased from Clontech. The infectious HIV-1 proviral DNA clone BH10 was obtained from the NIH AIDS Research and Reference Reagent Program. Plasmid NLENY1-ES-IRES was a gift of David Levy (20); the pCMV-BlaM-Vpr DNA was provided by Warner Greene (5).
The human Fyn cDNA construct was purchased from Origene (SC118862). Three Fyn mutants, the GGAA, E314A, and E314Q mutants, were generated by site-directed mutagenesis with the following primers: for GGAA, 5′-CCCTGCAGTTGATCAAGAGACTGGCCAATGCCCAGTTTGGGGAAGTATGGATG-3′ and 5′-CATCCATACTTCCCCAAACTGGGCATTGGCCAGTCTCTTGATCAACTGCAGGG-3′; for E314A, 5′-TGTCCCCCGAATCATTCCTTGAGGCTGCGCAGATCATGAAGAAGCTGAAGCAC-3′ and 5′-GTGCTTCAGCTTCTTCATGATCTGCGCAGCCTCAAGGAATGATTCGGGGGACA-3′; and for E314Q, 5′-TGTCCCCCGAATCATTCCTTGAGCAGGCGCAGATCATGAAGAAGCTGAAGCAC-3′ and 5′-GTGCTTCAGCTTCTTCATGATCTGCGCCTGCTCAAGGAATGATTCGGGGGACA-3′.
Anti-Src (catalogue number NB110-57592), anti-Hck (catalogue number NB100-79963), anti-Lyn (catalogue number NB100-92043), and anti-Fyn (catalogue number NB500-517) antibodies were purchased from Novus Biologicals, anti-Fgr (catalogue number H00002268-M03) and anti-IFITM3 (mouse) (catalogue number H00010410-B02) antibodies from Abnova, anti-phosphotyrosine (pY) antibody from Abcam (catalogue number ab10321), and anti-IFITM3 (rabbit) (catalogue number 11714-1-AP) antibody from ProteinTech. U0126 (catalogue number U120), PP2 (catalogue number P0042), genistein (catalogue number G6649), sodium orthovanadate, and a phosphatase inhibitor cocktail (catalogue number P5726) were purchased from Sigma.
Stable cell lines.
Tetracycline-inducible IFITM3 SupT1 cell lines were produced as previously described (22). We also generated human embryonic kidney HEK293 stable cell lines using pQCXIP retroviral vectors that express IFITM3 and its various mutants. Briefly, HEK293T cells were transfected with 2 μg pQCXIP, 2 μg pCMV-gag-pol-MLV, and 0.5 μg pVSV-G. The retroviral particles were then used to infect HEK293 cells in the presence of Polybrene (5 μg/ml) by spinoculation at 1,800 × g for 45 min at room temperature. The stably transduced HEK293 cells were selected with 2 μg/ml puromycin. Expression of IFITM3 and its mutants was assessed by Western blotting.
siRNA knockdown of Fyn.
The following siRNA oligonucleotides targeting human Fyn were purchased from Qiagen: siRNA1 (catalogue number SI00605451; AAGACATGTGGTGTATATAAA), siRNA2 (catalogue number SI02654729; GTGGCCCTTTATGACTATGAA), and siRNA3 (catalogue number SI02659545; AAGAAGCAGGATGCTGATCTA). HEK293T cells (0.3 × 106/well) were seeded in 6-well plates 1 day before transfection with 40 nM siRNA using Lipofectamine 2000 (Invitrogen). After two sequential siRNA transfections, cells were transfected with the pQCXIP plasmids expressing IFITM3. In order to detect tyrosine phosphorylation of IFITM3, cells were treated with 1 mM Na3VO4 for 1 h to block the activity of phosphatases before being harvested for immunoprecipitation and Western blotting.
Immunoprecipitation and Western blotting.
Cells (2 × 106) were lysed in a buffer containing 20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 5 mM EDTA, 1% Triton X-100, 1 mM Na3VO4, and protease inhibitors (Roche). Following clarification by centrifugation, the lysates were incubated with anti-Flag M2 affinity agarose (Sigma) overnight at 4°C. Resins were washed 3 times with the aforementioned lysis buffer, and the bound proteins were eluted with 3× Flag solution (Sigma) on ice for 20 min. To detect the phosphorylation of endogenous IFITM3, HeLa cells (8 × 107) were first treated with Na3VO4 (10 mM) for 1 h and then lysed in the aforementioned lysis buffer. The cell lysates were incubated with control IgG or anti-IFITM3 antibody, followed by absorption onto protein A-agarose (Millipore). After 3 washes, the precipitated materials were extracted from the agarose with 2× SDS loading buffer at 90°C for 15 min. The protein samples were resolved in 1% sodium dodecyl sulfate-12% polyacrylamide gel electrophoresis (SDS-PAGE) gels and then transferred onto polyvinylidene difluoride (PVDF) membranes (Roche). The membranes were blocked with 5% skim milk and further probed with the following antibodies: anti-Flag (1:5,000), anti-pY (1:1,000), anti-Fyn (1:1,000), anti-Fgr (1:1,000), anti-Src (1:1,000), anti-Lyn (1:1,000), anti-Hck (1:1,000), and anti-IFITM3 (1:1,000). Following a further incubation with horseradish peroxidase (HRP)-conjugated secondary antibodies, the protein bands were visualized by exposure to X-ray films following a brief treatment of the membranes with enhanced chemiluminescence (ECL) reagents.
Virus infections.
VSV-G protein-pseudotyped MLV-green fluorescent protein (MLV-GFP) reporter viruses were produced by transfecting HEK293T cells with plasmid DNA pCMV-gag-pol-MLV, pCMV-GFP-MLV, and pVSV-G as described previously (9). The titer of the produced viruses was determined by infecting HEK293 cells. Infected cells were scored as GFP positive by flow cytometry, and an amount of viruses that infected 15% to 20% of HEK293 cells was used in the following infection studies. Briefly, HEK293 cell lines that expressed IFITM3 and its various mutants were exposed to VSV-G protein-pseudotyped MLV-GFP reporter viruses for 3 h before the inoculated viruses were washed off. Forty hours later, cells were detached from the plates by trypsin digestion and fixed with 1% paraformaldehyde. GFP-positive cells were scored by flow cytometry.
The HIV-1 BH10 virus was produced by transfecting HEK293T cells. Virus amounts were then determined by enzyme-linked immunosorbent assay (ELISA) for viral p24. BH10 virus (equivalent to 20 ng viral p24) was used to infect SupT1 cells (2 × 106) that express IFITM3 or its mutants. Virus production was then determined at various time intervals by measuring viral reverse transcriptase (RT) activity in the supernatants (22).
Virion fusion assay.
The virion fusion assay was performed as previously described (5, 22). BlaM-Vpr-labeled virus particles were prepared by transfecting HEK293T cells with plasmid DNA BH10 or NLENY1-ES-IRES and pVSV-G together with pCMV-BlaM-Vpr. The viruses were concentrated by ultracentrifugation and quantified by ELISA to measure viral p24 amounts. SupT1 cells were then exposed to virus BH10/BlaM-Vpr (200 ng p24) or NLENY1-ES-IRES/VSV-G/BlaM-Vpr (50 ng p24) for 2 h before cells were washed with CO2-independent medium (Invitrogen). After incubation with CCF2/AM substrates at room temperature for 1 h in darkness, the cells were then maintained in development medium for 16 h at room temperature before being fixed in 1% paraformaldehyde and analyzed by flow cytometry to measure the cleavage of CCF2/AM.
Immunofluorescence microscopy.
HEK293 cells were seeded in a 4-chamber slide 1 day prior to transfection with pTet-IFITM3 or pTet-Δ(1–21) together pTet-ON plasmid DNA. In order to determine the localization of Fyn and its mutants, HEK293 cells were also transfected with Fyn plasmid DNA clones. Expression of IFITM3 and the Δ(1–21) mutant was induced by doxycycline (500 ng/ml). Both IFITM3 and the Δ(1–21) mutant had a Flag tag attached to their N termini. Cells were fixed with 4% paraformaldehyde (in 1× phosphate-buffered saline) for 10 min at room temperature and permeabilized with 0.1% Triton X-100 for 10 min at room temperature. Cells were then stained for 2 h at room temperature with antibodies against Flag (1:500 dilution, mouse or rabbit), EEA1 (1:500 dilution, rabbit), CD63 (1:200 dilution, rabbit), TGN46 (1:200 dilution, rabbit), or Fyn (1:100 dilution, mouse). After being washed with 1× phosphate-buffered saline, cells were incubated with either Alexa Fluor 488-conjugated secondary anti-mouse antibody (1:2,000 dilution; Invitrogen) or Alexa Fluor 594-conjugated secondary anti-rabbit antibody (1:2,000 dilution; Invitrogen). Images were recorded using the Zeiss Pascal laser scanning confocal microscope.
RESULTS
IFITM3 mutated at tyrosine residue 20 (Y20) does not inhibit VSV-G protein-mediated virus infection.
It was previously reported that deleting the first 21 amino acids renders IFITM3 unable to inhibit VSV (35). We reproduced this observation using VSV-G protein-pseudotyped MLV-GFP reporter viruses (Fig. 1). IFITM3 and its Δ(1–21) mutant were first stably transduced into HEK293 cells (Fig. 1A and B), followed by infection of these cell lines with VSV-G protein-pseudotyped MLV-GFP viruses (Fig. 1C). In order to summarize the data of different infection experiments, the virus infection rate of the control cell line (named pQCXIP) was arbitrarily set as 1. Virus infection rates of IFITM3-expressing cell lines were calculated accordingly and are presented in Fig. 1D. The results showed that IFITM3 reduced the infection by 2-fold, whereas the Δ(1–21) mutant exhibited no effect (Fig. 1C and D). We subsequently used this pseudovirus infection assay to investigate how these 21 amino acids regulate the antiviral function of IFITM3.
We first asked which of these 21 amino acids are the key determinants of IFITM3 antiviral activity. We noted that this protein region has a 17-PPNY-20 motif that represents one of the three viral late domains that enveloped viruses exploit to promote virus budding (7, 13). The PPxY late domain (“x” represents any of the 20 amino acids) functions through recruiting the Nedd4 E3 ubiquitin ligase, which ubiquitinates viral proteins. The ubiquitinated proteins are then recognized by the ESCRT (endosomal sorting complex required for transport) complexes and sorted into multivesicular bodies (MVB) (15). We initially speculated that by recruiting Nedd4, the 17-PPNY-20 motif may direct the location of IFITM3 to the endosomal compartments. To test this hypothesis, we generated three mutations, Δ(17–20), Δ1718, and ΔY20, in which the PPNY sequence was completely or partially deleted (Fig. 1A). We also mutated K24 to R, as K24 is the lysine residue closest to PPNY and if PPNY recruits Nedd4, K24 is most likely ubiquitinated. In addition to PPxY, there are two other types of viral late domains, P(T/S)AP and YPxL, that can functionally replace PPxY in recruiting ESCRT and promoting virus budding (13). We therefore also changed 17-PPNY-20 to 17PTAP or 17YPDL (Fig. 1A). All mutants were expressed at levels similar to that of wild-type IFITM3 in stably transduced HEK293 cells except for the Δ(17–20) and 17YPDL mutants, which were poorly expressed (Fig. 1B). We also noticed that the ΔY20, Δ(17–20), and 17PTAP mutants exhibited slower mobility than wild-type IFITM3 (Fig. 1B). This may have resulted from alterations in protein local conformation or in SDS-binding capacity due to the mutations inserted.
We then infected these different cell lines with VSV-G protein-pseudotyped MLV-GFP reporter viruses and measured virus infection by scoring the rate of GFP-positive cells (Fig. 1C). Compared to wild-type IFITM3, the Δ(17–20) mutant lost its ability to inhibit viral infection (Fig. 1C and D). In order to test whether the lost inhibition of virus infection is due to the low expression level of the Δ(17–20) mutant compared to that of wild-type IFITM3, we generated two mutants, the Δ1718 and ΔY20 mutants, that also had altered 17-PPNY-20 motifs but were expressed at levels similar to that of wild-type IFITM3 (Fig. 1B). The results of infection studies showed that deletion of the Y20 residue, but not 17-PP-18, abrogated the antiviral activity of IFITM3 (Fig. 1C and D). Considering the essential role of the two prolines in the function of PPxY as a viral late domain (13), this result also suggests that Y20 itself, independent of the PPNY motif, enables IFITM3 to inhibit VSV-G protein-mediated virus infection. Supporting this conclusion, replacing 17-PPNY-20 with 17PTAP or 17YPDL did not preserve the antiviral activity of IFITM3 (Fig. 1C and D). It is noted that although 17YPDL was poorly expressed, the expression level of the 17PTAP mutant was similar to that of wild-type IFITM3 (Fig. 1B). We also observed that the K24R mutant inhibited virus infection no less than wild-type IFITM3 (Fig. 1C and D). In order to further confirm the key role of Y20 in IFITM3 restriction of VSV, we generated two more mutations, Y20F and Y20A. As expected, neither of these two mutants inhibited VSV-G protein-pseudotyped reporter virus (Fig. 2). Together, these results demonstrate that the Y20 residue itself governs the ability of IFITM3 to restrict VSV-G protein-mediated virus infection.
Fig 2.
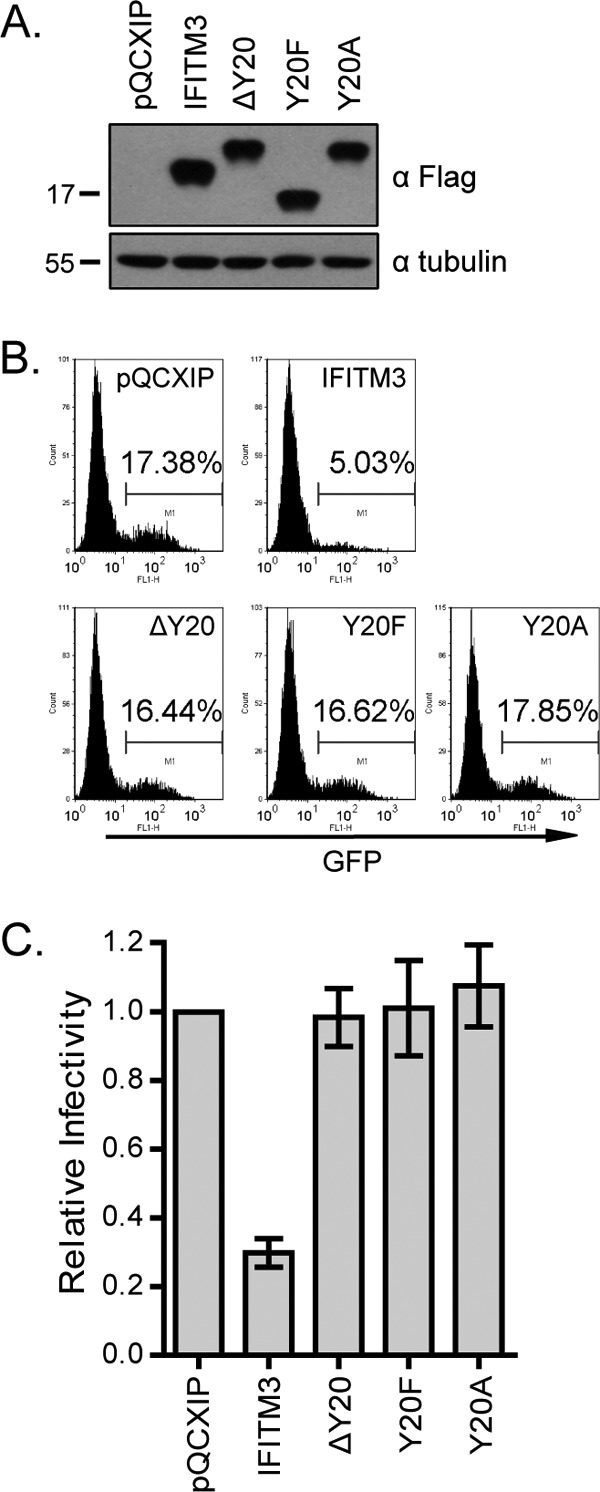
Mutating Y20 abolishes IFITM3 inhibition of infection by VSV-G protein-pseudotyped virus. (A) Expression of the Y20A and Y20F mutants in stably transduced HEK293 cells. (B) Infection of HEK293 cell lines by VSV-G protein-pseudotyped MLV reporter viruses. Results shown are from one representative infection. (C) Summary of three independent infections. The infection efficiency of the control cells is arbitrarily set as 1.
The IFITM3 Δ(1–21) mutant profoundly suppresses HIV-1 replication.
We then asked whether the N-terminal region of IFITM3 is required for inhibiting other viruses, such as HIV-1. To answer this question, some of the aforementioned IFITM3 mutants were cloned into the pRetroX-Tight-Pur retroviral vector and stably transduced into SupT1 cells. Expression of IFITM3 and its mutants was induced by doxycycline (Fig. 3A). We then challenged these cell lines with HIV-1. In the absence of doxycycline (i.e., without induction of IFITM3 expression), virus showed similar replication profiles in the different cell lines (Fig. 3B), although moderate differences in the levels of viral RT activity were noted for different cell lines. This may result from the nonsynchronized growth between cell lines. Given this observation, we compared the replication of HIV-1 under doxycycline treatment to that without doxycycline in the context of the same cell line. The results showed that induction of IFITM3 expression by doxycycline markedly suppressed HIV-1 replication (Fig. 3B). This inhibition was not alleviated by deleting the first N-terminal 21 amino acids or by mutating the 17-PPNY-20 motif (Fig. 3B). In fact, some of the mutants, including the Δ(17–20), ΔY20, 17PTAP, and 17YPDL mutants, inhibited HIV-1 replication more profoundly than wild-type IFITM3 (Fig. 3B). Therefore, as opposed to their essential role in IFITM3 inhibition of VSV and influenza A virus (10, 35), the first 21 amino acids of IFITM3 are not required for inhibiting HIV-1 replication.
Fig 3.

IFITM3 and its N-terminal mutants suppress HIV-1 replication. (A) IFITM3 and its various mutants were cloned into the pRetro-X-Tight vector and stably transduced into SupT1 cells. Expression of IFITM3 and its mutants was induced by doxycycline (Dox) and examined by Western blotting. (B) HIV-1 replication. SupT1 cells were infected with HIV-1. Virus growth was monitored over various time periods through measuring viral reverse transcriptase activity in the culture supernatants.
The IFITM3 Δ(1–21) mutant inhibits virus entry mediated by the HIV-1 envelope protein but not the VSV-G envelope protein.
We next performed the BlaM-Vpr-based HIV-1 virion fusion assay to directly measure the effect of IFITM3 and Δ(1–21) mutant expression on virus entry mediated by either the HIV-1 envelope or the VSV-G protein. We first transfected HEK293T cells with pCMV-BlaM-Vpr plasmid DNA and infectious HIV-1 BH10 proviral DNA to produce BH10/BlaM-Vpr viruses that bear HIV-1 envelope proteins. In order to generate VSV-G protein-pseudotyped BlaM-Vpr-labeled HIV-1 particles, transfections were conducted with pCMV-BlaM-Vpr, pVSV-G, and NLENY1-ES-IRES DNA. NLENY1-ES-IRES expresses all HIV-1 proteins except for viral envelope protein (20). The viruses were concentrated by ultracentrifugation, quantified by HIV-1 p24 ELISA, and then used to infect SupT1 cells expressing wild-type IFITM3 or the Δ(1–21) mutant. Two hours after virus exposure, cells were processed and the number of BlaM-Vpr-positive cells was determined by flow cytometry. The results showed that IFITM3 and its Δ(1–21) mutant markedly decreased the entry of BH10/BlaM-Vpr viruses into target cells (Fig. 4A). In contrast, when the same experiments were performed with the VSV-G/NLENY1-ES-IRES/BlaM-Vpr viruses, inhibition of virus entry was observed only for wild-type IFITM3 and not for the Δ(1–21) mutant (Fig. 4B). Therefore, the N-terminal region of IFITM3 is required for inhibiting VSV G protein-mediated, but not HIV-1 envelope protein-mediated, virus entry.
Fig 4.
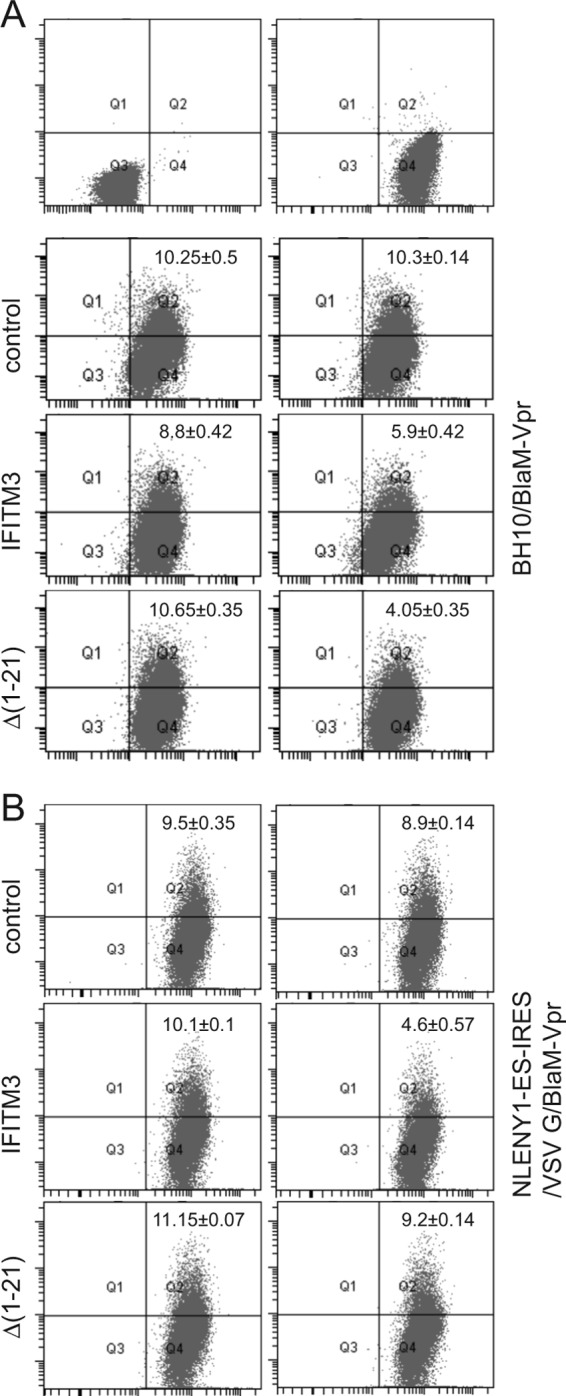
Effects of IFITM3 and the Δ(1–21) mutant on virus entry. (A) BlaM-Vpr-labeled BH10 viruses were used to infect SupT1 cells that were stably transduced with the pRetro-X-Tight vector itself (control) or the vector expressing IFITM3 or the Δ(1–21) mutant. The number of infected cells was determined by monitoring the cleavage of the CCF2/AM substrate by BlaM-Vpr. (B) Results of similar experiments performed with BlaM-Vpr labeled NLENY1-ES-IRES viruses that were pseudotyped with VSV-G protein. Data shown are the averages of three independent infections.
The N-terminal region of IFITM3 determines its cellular location.
In order to understand why the IFITM3 Δ(1–21) mutant inhibits HIV-1 envelope-mediated virus entry but not entry mediated by the VSV-G protein, we studied the cellular locations of both mutant and wild-type IFITM3 by fluorescence microscopy. In contrast to wild-type IFITM3, which colocalized with endocytosed transferrin (Fig. 5A), the Δ(1–21) mutant was mostly seen at the cell periphery and barely exhibited colocalization with endocytosed transferrin molecules (Fig. 5A). This suggests that the 21-amino-acid deletion causes a relocation of IFITM3 from the endocytic compartments; therefore, IFITM3 may miss the opportunity to encounter the viruses that enter cells via endocytosis. Since HIV-1 entry occurs at or near the plasma membrane at neutral pH (25, 31), the Δ(1–21) mutant, now located at the cell periphery, is still well positioned to inhibit HIV-1 entry and viral replication.
Fig 5.
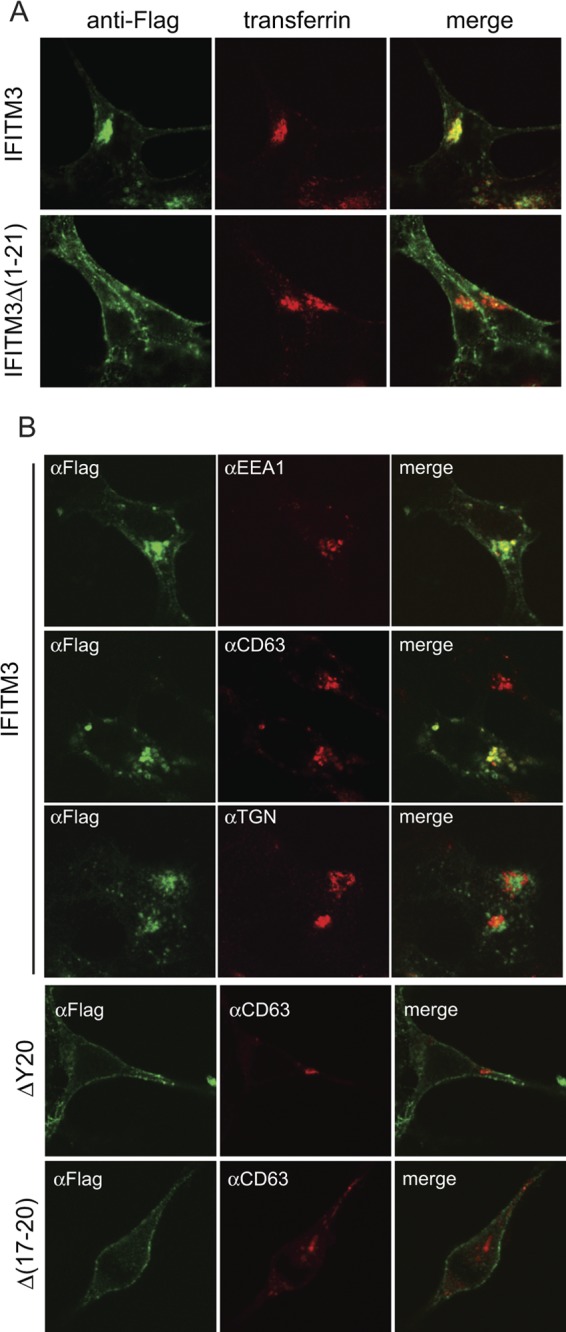
Cellular location of IFITM3 and its mutants. (A) IFITM3 and Δ(1–21) mutant DNAs were transfected into HEK293 cells. Prior to fixation, cells were incubated with Alexa Fluor 555-conjugated transferrin (pseudocolored in red) (5 μg/ml in serum-free Dulbecco modified Eagle medium) for 10 min at 37°C. IFITM3 and the Δ(1–21) mutant were detected by immunostaining with anti-Flag antibodies (pseudocolored in green). (B) IFITM3 and Δ(17–20) and ΔY20 mutant DNAs were transfected into HEK293 cells. Their expression was detected by immunostaining with anti-Flag antibodies (pseudocolored in green). The locations of EEA1, CD63, and TGN46 were visualized by immunostaining with respective antibodies (pseudocolored in red). Representative images are shown.
We then further characterized the cellular location of IFITM3 through immunostaining for EEA1, CD63, and TGN46, which are marker proteins of early endosomes, late endosomes, and the trans-Golgi network, respectively (19, 24, 32). IFITM3 colocalized with EEA1 and CD63 but not with TGN46 (Fig. 5B), suggesting that IFITM3 is mainly located at the early and late endosomes. This observed cellular location enables IFITM3 to inhibit pH-dependent viruses such as VSV and influenza virus that complete their entry within endosomes at low pH; i.e., VSV requires a pH of 6 for entry (within early endosomes), whereas influenza A virus needs a pH of 5 for entry (within late endosomes) (23). We also investigated the cellular location of the ΔY20 and Δ(17-20) mutants. Similar to the Δ(1-21) mutant, the ΔY20 and Δ(17-20) mutants were mainly seen at the cell periphery and barely exhibited colocalization with the late endosome marker CD63 (Fig. 5B). Together, these data suggest that the N-terminal region of IFITM3, specifically the Y20 residue, regulates IFITM3 cellular location and thus determines the inhibition of pH-dependent viruses.
IFITM3 tyrosine residue (Y20) is phosphorylated.
Tyrosine is subject to phosphorylation by kinases. This represents an important mechanism to regulate protein-protein interaction and relay signals along various biological pathways. Given the importance of Y20 in IFITM3 inhibition of VSV-G protein-mediated virus infection, we further studied whether Y20 is phosphorylated and, if so, how this modification contributes to the antiviral function of IFITM3. In addition to IFITM3, we also included IFITM1 and IFITM2 in the assays to detect tyrosine phosphorylation. We first expressed IFITM1, -2, and -3 proteins in HEK293T cells. Following treatment with sodium orthovanadate or a phosphatase inhibitor cocktail for 1 h to block the activity of phosphatases, cells were harvested and IFITM1, -2, and -3 proteins were immunoprecipitated. The precipitated materials were then probed by Western blotting with antibodies specifically recognizing phosphorylated tyrosine (pY). Only IFITM3, not IFITM1 or IFITM2, was recognized by pY antibodies, suggesting that only the tyrosines of IFITM3 are phosphorylated (Fig. 6B).
Fig 6.

IFITM3 is phosphorylated at Y20. (A) Illustration of IFITM3 sequence and the location of tyrosine residues. (B) Detection of IFITM3 phosphorylation using antibodies recognizing phosphorylated tyrosine (pY). DNA vectors expressing IFITM1, IFITM2, and IFITM3 were transfected into HEK293T cells. Na3VO4 or phosphatase inhibitor cocktail was used to treat cells before cell lysates were harvested. The IFITM1, IFITM2, and IFITM3 proteins were precipitated (IP) with anti-Flag antibodies and then tested by Western blotting with anti-pY antibodies. (C) Phosphorylation of endogenous IFITM3. HeLa cells were treated with 500 U/ml IFN-α for 24 h, followed by incubation with 10 mM Na3VO4 for 1 h prior to harvest. The endogenous IFITM3 was precipitated and subject to Western blotting with anti-pY antibody. (D) Detection of phosphorylation of wild-type IFITM3 and the ΔY20, Y80A, Y99A, and Y132A mutants in Western blotting with anti-pY antibodies. (E) Tyrosine phosphorylation of wild-type IFITM3 and theΔY20, Y20F, and Y20A mutants. (F) Tyrosine phosphorylation of IFITM3 and the Δ(1–21), Δ1718, ΔY20, Δ(17–20), 17PTAP, 17YPDL, and K24R mutants. Positions of the IgG light chain (Ig-L) on the gels are indicated by arrows.
We next asked whether the endogenous IFITM3 is phosphorylated at the tyrosine residue. To explore this, we treated HeLa cells with alpha interferon (IFN-α) (500 U/ml) for 24 h, followed by 1 h of incubation with sodium orthovanadate (10 mM). The endogenous IFITM3 was precipitated using the anti-IFITM3 antibody and examined by Western blotting with anti-pY antibody. Indeed, the endogenous IFITM3 was recognized by the anti-pY antibody, suggesting its phosphorylation by tyrosine kinases (Fig. 6C).
There are four tyrosines in IFITM3: Y20, Y80, Y99, and Y132 (Fig. 6A). In order to determine which tyrosines are phosphorylated, in addition to ΔY20, we created three other mutations: Y80A, Y99A, and Y132A. The anti-pY antibodies recognized the Y80A, Y99A, and Y132A mutants but not the ΔY20 mutant (Fig. 6D). To further demonstrate that Y20 is phosphorylated, we generated two more mutants, the Y20F and Y20A mutants, and examined their phosphorylation status. The results showed that neither of these two mutants was recognized by the anti-pY antibody (Fig. 6E). Together, these data suggest that Y20 is the major tyrosine phosphorylation site in IFITM3. In support of this observation, IFITM3 mutants, including the Δ(1–21), Δ(17–20), and 17PTAP mutants, that lack Y20 were also not detected by the anti-pY antibody (Fig. 6F). The 17YPDL mutant has a tyrosine at position 17 but was not phosphorylated, likely due to the loss of the sequence context that is required for recognition by tyrosine kinases (Fig. 6F).
The tyrosine kinase Fyn phosphorylates IFITM3.
We next tried to determine which tyrosine kinase phosphorylates IFITM3. To this end, we first tested the effect of tyrosine kinase inhibitors genistein, PP2, and U0126 on IFITM3 phosphorylation. Genistein is a general tyrosine kinase inhibitor, PP2 selectively inhibits Src family kinases, and U0126 inhibits mitogen-activated protein/extracellular signal-regulated kinase (MAPK/ERK) kinases (1, 11, 14). Both genistein and PP2, but not U0126, effectively diminished IFITM3 phosphorylation (Fig. 7A), suggesting that a member(s) of the Src kinase family phosphorylates IFITM3. We next performed immunoprecipitation experiments to determine which Src kinases interact with IFITM3. We probed the immunoprecipitated IFITM3 samples in Western blots using antibodies against Fyn, Fgr, Src, Lyn, and Hck. A measurable association of Fyn with IFITM3 was detected (Fig. 7B). Deletion of the Y20 residue diminished this interaction (Fig. 7B). This result is consistent with a previous observation showing that rat IFITM3/Rat8 is associated with Fyn (42). In order to further demonstrate the role of Fyn in phosphorylating IFITM3, we used siRNA to knock down endogenous Fyn in HEK293T cells and then measured the level of tyrosine phosphorylation of IFITM3. Among the three siFyn oligonucleotides tested, siFyn1 was the most effective in both depleting endogenous Fyn and diminishing IFITM3 phosphorylation (Fig. 7C). In support of the siRNA knockdown data, overexpression of Fyn substantially increased the level of IFITM3 phosphorylation (Fig. 7D). Moreover, IFITM3 phosphorylation was markedly impeded by the expression of Fyn GGAA, E314A, and E314Q mutants, which were mutated in the ATP-binding region (G278 and G280) or the catalytic site (E314) and thus lost the kinase activity (Fig. 7D) (37, 38). Similar to wild-type Fyn, these mutants also associated with IFITM3, as shown by the results of coimmunoprecipitation and immunofluorescence staining experiments (Fig. 7D and E), which suggests that these ectopically expressed Fyn mutants compete with the endogenous Fyn for binding to IFITM3 and, as a result, diminish IFITM3 phosphorylation. Together, these data indicate that Fyn, which is also membrane associated (2), phosphorylates IFITM3.
Fig 7.

Identification of tyrosine kinase that phosphorylates IFITM3. (A) Effects of kinase inhibitors on IFITM3 phosphorylation. Cells were treated with genistein, PP2, or U0126 to inhibit Src family kinases or the ERK family. Phosphorylation of IFITM3 was detected with antibodies against pY. (B) Interaction of IFITM3 with Src family members, including Fyn, Fgr, Src, Lyn, and Hck. Wild-type IFITM3 or its ΔY20 mutant was precipitated with anti-Flag antibodies and then assessed by Western blotting with antibodies recognizing Fyn, Fgr, Src, Lyn, or Hck. (C) Effect of Fyn knockdown on IFITM3 phosphorylation. HEK293T cells were transfected with siRNA oligonucleotides targeting Fyn and IFITM3 mRNAs. Phosphorylation of IFITM3 was then evaluated by Western blotting using anti-pY antibodies. (D) Effects of Fyn and its GGAA, E314A, and E314Q mutants on IFITM3 phosphorylation. Wild-type Fyn or these mutants were transiently expressed in HEK293T cells that had also been transfected with IFITM3 plasmid DNA. The ectopically expressed IFITM3 was immunoprecipitated with anti-Flag antibody and then subjected to Western blotting for the detection of Fyn and its mutants, as well as tyrosine-phosphorylated IFITM3. The position of the IgG heavy chain (Ig-H) on the gel is indicated by an arrow. (E) Colocalization of Fyn with IFITM3. IFITM3, wild-type Fyn, and Fyn mutants were transiently expressed in HEK293 cells. Flag-tagged IFITM3 was detected by immunofluorescence staining with anti-Flag antibody (pseudocolored in green). Fyn and its mutants were detected with anti-Fyn antibody (pseudocolored in red). Representative images are shown.
DISCUSSION
We report several novel findings from this study. First, the N-terminal 21 amino acids of IFITM3 are dispensable for suppressing HIV-1 replication in vitro (Fig. 3B), although they are essential for IFITM3 to inhibit VSV-G-bearing MLV pseudovirus (Fig. 1), as has been observed for both influenza A virus and VSV (10, 35). Second, deleting the first 21 amino acids changes the location of IFITM3 from endosomal compartments to the cellular periphery (Fig. 5A), which suggests that one function of the N-terminal region of IFITM3 is to target the protein to endosomes where IFITM3 can encounter invading viruses and block their entry. Third, mutagenesis studies reveal Y20 as the key residue that determines IFITM3 localization in cells.
It is intriguing that IFITM3 inhibits virus entry mediated by both HIV-1 Env and VSV-G protein, given that the entry of VSV and that of HIV-1 have different requirement for pH (Fig. 4) (12, 22). VSV, like influenza A virus, needs a low pH to trigger the conformational change of its G protein in order to initiate membrane fusion, whereas HIV-1 entry is pH independent and takes place at the plasma membrane or early endocytic vesicles. In order to inhibit both pH-dependent and pH-independent viruses, IFITM3 is expected to be present at the cellular compartments where these different viruses complete their entry. Indeed, IFITM3 has been observed to colocalize with several marker proteins of endosomes/lysosomes, including Rab5, Rab7, lamp1, and LysoTracker (12, 17, 39). Results of this study further showed colocalization of IFITM3 with endocytosed transferrin and the endosomal markers EEA1 and CD63 (Fig. 5).
The localization of IFITM3 to endosomes depends on the Y20 amino acid. Deletion of Y20 led to relocation of IFITM3 from endosomes to the cell periphery (Fig. 5B). Since Y20 does not function in the context of the 17-PPNY-20 motif, we propose a role of Y20 in the 20-YEML-23 sequence that conforms to a tyrosine-based sorting signal with a consensus sequence, YxxΦ (Φ represents residues with bulky hydrophobic side chains) (3). YxxΦ motifs are involved in rapid internalization of proteins from the plasma membrane and targeting proteins to lysosomes (3). It is thus likely that the 20-YEML-23 motif serves as a protein sorting signal and allows rapid removal of IFITM3 from the plasma membrane. In line with this hypothesis, mutation of Y20 causes accumulation of IFITM3 on the plasma membrane, likely as a result of the impaired internalization of IFITM3.
Then what is the function of Y20 phosphorylation? This modification may present a mechanism to regulate the interaction of the 20-YEML-23 motif with the μ subunit of the AP complex and thereby modulate IFITM3 internalization. This form of regulatory mechanism has been demonstrated with the T cell costimulatory receptor CTLA-4. CTKA-4 has a YVKM sequence that can be phosphorylated at the Y residue when a T cell is activated. The phosphorylated Y blocks the interaction of YVKM motif with μ2 of the AP2 complex and thereby leads to the retention of CTLA-4 on the surface of activated T cells (8, 29, 41). We noted that the phosphorylated IFITM3 represents a minor fraction of the total IFITM3 in cells (Fig. 6), which explains the endosomal localization of the majority of IFITM3 that is suspected not to be phosphorylated at Y20 (Fig. 5). It is conceivable that the extent of Y20 phosphorylation affects IFITM3 distribution in different cellular membrane compartments.
We identified Fyn as one tyrosine kinase that phosphorylates Y20 of IFITM3 (Fig. 7). Fyn is a member of the Src kinase family. Fyn's association with the plasma membrane is dependent on myristoylation and palmitoylation at its N terminus (33, 36). Mutating the palmitoylation site, cysteine 3, abrogates Fyn localization to the plasma membrane. It is notable that IFITM3 is also palmitoylated and that this modification is also essential for IFITM3 binding to membranes (40). Our data thus suggest that Fyn phosphorylates IFITM3 at Y20 and thereby retains a subpopulation of IFITM3 at the plasma membrane.
One function of IFITM3 in endosomes appears to regulate compartment acidification and morphogenesis. The late endosomes and lysosomes are enlarged when IFITM3 is induced by alpha interferon or is overexpressed (12). IFITM3 may do so through interacting with Atp6v0b and thereby stabilizing the vacular ATPase proton pump (34). Supporting this, depletion of IFITM3 leads to a neutralized pH in endosomal compartments and the loss of clathrin from membrane compartments (34). This cellular function of IFITM3 in modulating the structure and property of endosomes may have allowed IFITM3 to inhibit virus entry at these membrane compartments (12).
The topology of IFITM3 on membranes has been described in two models. One model predicts that IFITM3 passes the membrane twice with its N and C termini present within the lumen of the endoplasmic reticulum (ER) or extracellular. In support of this prediction, the N or C terminus of IFITM3 has been detected on cell surfaces by immunostaining (4, 35). However, if this is the sole topology of IFITM3 on cell membranes, then Y20 would not have access to Fyn that is located at the cytoplasmic face of the cellular membranes and so Y20 could not be phosphorylated. This contradiction is reconciled by a second model proposing that IFITM3 is an intramembrane protein with its two termini projecting into the cytoplasm (39). The latter model is supported by the lack of glycosylation of IFITM3, which suggests that IFITM3 is not exposed to the ER lumen glycosylation machinery (39). Also, S-palmitoylation at the cysteine residues 71, 72, and 105 is essential for IFITM3 membrane association, which is a common feature of a number of cellular intramembrane proteins (39, 40). This intramembrane model places Y20 within the cytoplasm so Y20 can be phosphorylated. Also consistent with this model, the K24 amino acid was shown to be ubiquitinated, which can only occur if K24 is exposed to the cytoplasmic ubiquitin E3 ligases (39).
In summary, results of this study suggest that Y20 likely functions as a key residue of a putative sorting signal, 20-YEML-23, that resides within the N-terminal region of IFITM3 and likely plays an important role in regulating the distribution of IFITM3 at different cellular membrane compartments. Phosphorylation of Y20 may modulate the sorting activity of 20-YEML-23 and thereby affect IFITM3 subcellular location as well as antiviral function. Although Y20 is essential for IFITM3 to reside at the endosomes and thereby to restrict the entry of pH-dependent viruses, it is dispensable for inhibiting HIV-1, which enters cells in a pH-independent fashion.
ACKNOWLEDGMENTS
We thank David Levy and Warner Greene for providing valuable reagents. We also thank Richard Sloan for careful reading of the manuscript.
This study was supported by funding from the Canadian Institutes for Health Research, the Chinese Ministry of Health (2012ZX 10001–006), the 973 program (2010CB534907), and the CSC (China Scholarship Council) State Scholarship Fund.
Footnotes
Published ahead of print 10 October 2012
REFERENCES
- 1. Akiyama T, et al. 1987. Genistein, a specific inhibitor of tyrosine-specific protein kinases. J. Biol. Chem. 262:5592–5595 [PubMed] [Google Scholar]
- 2. Alland L, Peseckis SM, Atherton RE, Berthiaume L, Resh MD. 1994. Dual myristylation and palmitylation of Src family member p59fyn affects subcellular localization. J. Biol. Chem. 269:16701–16705 [PubMed] [Google Scholar]
- 3. Bonifacino JS, Traub LM. 2003. Signals for sorting of transmembrane proteins to endosomes and lysosomes. Annu. Rev. Biochem. 72:395–447 [DOI] [PubMed] [Google Scholar]
- 4. Brass AL, et al. 2009. The IFITM proteins mediate cellular resistance to influenza A H1N1 virus, West Nile virus, and dengue virus. Cell 139:1243–1254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cavrois M, De Noronha C, Greene WC. 2002. A sensitive and specific enzyme-based assay detecting HIV-1 virion fusion in primary T lymphocytes. Nat. Biotechnol. 20:1151–1154 [DOI] [PubMed] [Google Scholar]
- 6. Chan YK, Huang IC, Farzan M. 2012. IFITM proteins restrict antibody-dependent enhancement of dengue virus infection. PLoS One 7:e34508 doi:10.1371/journal.pone.0034508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chen BJ, Lamb RA. 2008. Mechanisms for enveloped virus budding: can some viruses do without an ESCRT? Virology 372:221–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chuang E, et al. 1997. Interaction of CTLA-4 with the clathrin-associated protein AP50 results in ligand-independent endocytosis that limits cell surface expression. J. Immunol. 159:144–151 [PubMed] [Google Scholar]
- 9. Côté M, Zheng YM, Albritton LM, Liu SL. 2008. Fusogenicity of Jaagsiekte sheep retrovirus envelope protein is dependent on low pH and is enhanced by cytoplasmic tail truncations. J. Virol. 82:2543–2554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Everitt AR, et al. 2012. IFITM3 restricts the morbidity and mortality associated with influenza. Nature 484:519–523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Favata MF, et al. 1998. Identification of a novel inhibitor of mitogen-activated protein kinase kinase. J. Biol. Chem. 273:18623–18632 [DOI] [PubMed] [Google Scholar]
- 12. Feeley EM, et al. 2011. IFITM3 inhibits influenza A virus infection by preventing cytosolic entry. PLoS Pathog. 7:e1002337 doi:10.1371/journal.ppat.1002337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Freed EO. 2002. Viral late domains. J. Virol. 76:4679–4687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hanke JH, et al. 1996. Discovery of a novel, potent, and Src family-selective tyrosine kinase inhibitor. Study of Lck- and FynT-dependent T cell activation. J. Biol. Chem. 271:695–701 [DOI] [PubMed] [Google Scholar]
- 15. Henne WM, Buchkovich NJ, Emr SD. 2011. The ESCRT pathway. Dev. Cell 21:77–91 [DOI] [PubMed] [Google Scholar]
- 16. Hickford DE, Frankenberg SR, Shaw G, Renfree MB. 2012. Evolution of vertebrate interferon inducible transmembrane proteins. BMC Genomics 13:155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Huang IC, et al. 2011. Distinct patterns of IFITM-mediated restriction of filoviruses, SARS coronavirus, and influenza A virus. PLoS Pathog. 7:e1001258 doi:10.1371/journal.ppat.1001258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jiang D, et al. 2010. Identification of five interferon-induced cellular proteins that inhibit West Nile virus and dengue virus infection. J. Virol. 84:8332–8341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lawe DC, et al. 2003. Essential role of Ca2+/calmodulin in early endosome antigen-1 localization. Mol. Biol. Cell 14:2935–2945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Levy DN, Aldrovandi GM, Kutsch O, Shaw GM. 2004. Dynamics of HIV-1 recombination in its natural target cells. Proc. Natl. Acad. Sci. U. S. A. 101:4204–4209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lewin AR, Reid LE, McMahon M, Stark GR, Kerr IM. 1991. Molecular analysis of a human interferon-inducible gene family. Eur. J. Biochem. 199:417–423 [DOI] [PubMed] [Google Scholar]
- 22. Lu J, et al. 2011. The IFITM proteins inhibit HIV-1 infection. J. Virol. 85:2126–2137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mercer J, Schelhaas M, Helenius A. 2010. Virus entry by endocytosis. Annu. Rev. Biochem. 79:803–833 [DOI] [PubMed] [Google Scholar]
- 24. Metzelaar MJ, et al. 1991. CD63 antigen. A novel lysosomal membrane glycoprotein, cloned by a screening procedure for intracellular antigens in eukaryotic cells. J. Biol. Chem. 266:3239–3245 [PubMed] [Google Scholar]
- 25. Miyauchi K, Kim Y, Latinovic O, Morozov V, Melikyan GB. 2009. HIV enters cells via endocytosis and dynamin-dependent fusion with endosomes. Cell 137:433–444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Moffatt P, et al. 2008. Bril: a novel bone-specific modulator of mineralization. J. Bone Miner. Res. 23:1497–1508 [DOI] [PubMed] [Google Scholar]
- 27. Sällman Almén M, Bringeland N, Fredriksson R, Schioth HB. 2012. The dispanins: a novel gene family of ancient origin that contains 14 human members. PLoS One 7:e31961 doi:10.1371/journal.pone.0031961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Schoggins JW, et al. 2011. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 472:481–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shiratori T, et al. 1997. Tyrosine phosphorylation controls internalization of CTLA-4 by regulating its interaction with clathrin-associated adaptor complex AP-2. Immunity 6:583–589 [DOI] [PubMed] [Google Scholar]
- 30. Siegrist F, Ebeling M, Certa U. 2011. The small interferon-induced transmembrane genes and proteins. J. Interferon Cytokine Res. 31:183–197 [DOI] [PubMed] [Google Scholar]
- 31. Stein BS, et al. 1987. pH-independent HIV entry into CD4-positive T cells via virus envelope fusion to the plasma membrane. Cell 49:659–668 [DOI] [PubMed] [Google Scholar]
- 32. Valsdottir R, et al. 2001. Identification of rabaptin-5, rabex-5, and GM130 as putative effectors of rab33b, a regulator of retrograde traffic between the Golgi apparatus and ER. FEBS Lett. 508:201–209 [DOI] [PubMed] [Google Scholar]
- 33. van't Hof W, Resh MD. 1997. Rapid plasma membrane anchoring of newly synthesized p59fyn: selective requirement for NH2-terminal myristoylation and palmitoylation at cysteine-3. J. Cell Biol. 136:1023–1035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wee YS, Roundy KM, Weis JJ, Weis JH. 30 March 2012. Interferon-inducible transmembrane proteins of the innate immune response act as membrane organizers by influencing clathrin and v-ATPase localization and function. Innate Immun. [Epub ahead of print.] doi:10.1177/1753425912443392 [DOI] [PubMed] [Google Scholar]
- 35. Weidner JM, et al. 2010. Interferon-induced cell membrane proteins, IFITM3 and tetherin, inhibit vesicular stomatitis virus infection via distinct mechanisms. J. Virol. 84:12646–12657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wolven A, Okamura H, Rosenblatt Y, Resh MD. 1997. Palmitoylation of p59fyn is reversible and sufficient for plasma membrane association. Mol. Biol. Cell 8:1159–1173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Xu W, Doshi A, Lei M, Eck MJ, Harrison SC. 1999. Crystal structures of c-Src reveal features of its autoinhibitory mechanism. Mol. Cell 3:629–638 [DOI] [PubMed] [Google Scholar]
- 38. Xu W, Harrison SC, Eck MJ. 1997. Three-dimensional structure of the tyrosine kinase c-Src. Nature 385:595–602 [DOI] [PubMed] [Google Scholar]
- 39. Yount JS, Karssemeijer RA, Hang HC. 2012. S-palmitoylation and ubiquitination differentially regulate interferon-induced transmembrane protein 3 (IFITM3)-mediated resistance to influenza virus. J. Biol. Chem. 287:19631–19641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yount JS, et al. 2010. Palmitoylome profiling reveals S-palmitoylation-dependent antiviral activity of IFITM3. Nat. Chem. Biol. 6:610–614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhang Y, Allison JP. 1997. Interaction of CTLA-4 with AP50, a clathrin-coated pit adaptor protein. Proc. Natl. Acad. Sci. U. S. A. 94:9273–9278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zucchi I, et al. 2004. Association of rat8 with Fyn protein kinase via lipid rafts is required for rat mammary cell differentiation in vitro. Proc. Natl. Acad. Sci. U. S. A. 101:1880–1885 [DOI] [PMC free article] [PubMed] [Google Scholar]