

Abstract
The human papillomavirus (HPV) E7 oncoprotein binds cellular factors, preventing or retargeting their function and thereby making the infected cell conducive for viral replication. A key target of E7 is the product of the retinoblastoma susceptibility locus (pRb). This interaction results in the release of E2F transcription factors and drives the host cell into the S phase of the cell cycle. E7 binds pRb via a high-affinity binding site in conserved region 2 (CR2) and also targets a portion of cellular pRb for degradation via the proteasome. Evidence suggests that a secondary binding site exists in CR3, and that this interaction influences pRb deregulation. Additionally, evidence suggests that CR3 also participates in the degradation of pRb. We have systematically analyzed the molecular mechanisms by which CR3 contributes to deregulation of the pRb pathway by utilizing a comprehensive series of mutations in residues predicted to be exposed on the surface of HPV16 E7 CR3. Despite differences in the ability to interact with cullin 2, all CR3 mutants degrade pRb comparably to wild-type E7. We identified two specific patches of residues on the surface of CR3 that contribute to pRb binding independently of the high-affinity CR2 binding site. Mutants within CR3 that affect pRb binding are less effective than the wild-type E7 in overcoming pRb-induced cell cycle arrest. This demonstrates that the interaction between HPV16 E7 CR3 and pRb is functionally important for alteration of the cell cycle.
INTRODUCTION
Papillomaviruses are a group of DNA viruses that infect the skin and mucosal tissues of most vertebrates. More than 100 human papillomavirus (HPV) types have been identified, but far more are presumed to exist (21). A subset of HPVs is associated with lesions that frequently progress to cancer, and these HPVs are classified as high-risk types. Persistent infection by high-risk HPVs is related to 99.7% of all human cervical cancer cases (20), other genitourinary cancers, and a growing number of oral cancers (31). Although recently developed vaccines protect individuals from the two most frequently carcinogenic HPV types, HPV16 and HPV18, they offer limited protection against other cancer-causing HPVs and provide no benefit to individuals with a preexisting history of infection (13, 28, 48). The development of other therapies to treat HPV-induced malignancy may be facilitated by a greater understanding of the mechanisms of virally mediated cellular transformation.
The viral E6 and E7 oncoproteins are consistently expressed in HPV-induced cancers and are necessary to maintain malignant cell growth (2, 4, 37, 49, 51). Repression of their transcription by reexpression of the viral E2 regulatory protein induces rapid growth arrest and senescence of cervical cancer cells, suggesting that these cancer cells are addicted to E6 and E7 (33, 34). The HPV E7 protein is a multifunctional oncoprotein that interacts with a multitude of cellular factors (3, 6, 8, 11, 53). One of the main activities of E7 is to induce terminally differentiated cells to enter the cell cycle. This is accomplished in part by E7's association with the product of the retinoblastoma susceptibility locus (pRb) and the related pocket protein family members p107 and p130 (25). The interaction of E7 with pRb is essential for transformation and abrogation of the antiproliferative signals in cervical cancer (26, 47). One of the ways in which pRb has an essential role in proliferation control is through binding to and regulating its key effectors, the E2F family of transcription factors. E2Fs coordinate the transcription of genes that are necessary for cell cycle progression (10, 24, 43). During the cell cycle, as cells progress from the G1 to S phase, the sequential phosphorylation of pRb by cyclin/cyclin-dependent kinase complexes causes the release of E2F from pRb and activation of genes required for the entry into S phase (19, 41, 50).
The E7 protein contains three conserved regions (CR), termed CR1, CR2, and CR3. CR1 and CR2 are thought to be intrinsically disordered, while CR3 has a defined three-dimensional structure comprised of a unique β1β2α1β3α2 fold (40, 45). All three regions are required for abrogation of epithelial cell quiescence and contribute to cellular transformation (5, 26, 47). The role of the LxCxE motif within CR2 is well established and is necessary for the abrogation of antiproliferative signals and oncogenic transformation (26, 47). The LxCxE motif functions as a high-affinity binding site for pRb and related family members. However, other regions of E7 are also important for deregulating the pRb pathway, and E7 is known to exert its activity on pRb through multiple mechanisms. First, E7 binds the hypophosphorylated form of pRb in complex with E2F and blocks the association of E2F and pRb. Although the LxCxE motif in CR2 is necessary for interference with pRb-E2F binding (14, 44), a requirement for both the CR1 and CR3 regions has also been established (35). Precisely how these two regions contribute is not known. Second, E7 inactivates a fraction of the cellular pool of pRb by targeting it for proteasome-mediated degradation (9, 32, 36). Both the LxCxE motif and CR1 are necessary for efficient reduction of pRb steady-state levels (29, 38), while the role of CR3 has remained largely elusive. One study has shown that small changes within CR3 do not disrupt the pRb degradation capacity of E7 (35), but it has also been hypothesized that CR3 contributes to pRb degradation due to the observation that mutations in this region lead to reduced association with the cullin 2 E3 ubiquitin ligase complex (36, 38). Binding of the cullin 2 complex to E7 is mediated via its Zer1 subunit and is currently the only known mechanism by which HPV16 E7 targets pRb for destruction by the proteasome (36, 53). As the interaction of cullin 2 is exclusive to HPV16 E7, the mechanism by which other HPV E7 types degrade pRb remains unclear (36, 53). Lastly, strong evidence has been presented that E7 CR3 contributes to binding of pRb independently from the LxCxE motif, but the functional significance of this interaction is unknown (15, 40, 46).
Despite the large number of biological activities that are attributed to CR3, the role of this domain of E7 in deregulating the pRb pathway is still unclear. The study presented here was aimed at determining the role of E7 CR3 sequences in deregulating the pRb pathway. We examined the involvement of surface-exposed residues of HPV16 E7 CR3 in binding pRb, targeting it for degradation, and influencing pRb's ability to induce cell cycle arrest. Although CR3 is necessary for association with the cullin 2 complex and specific CR3 mutants affect cullin 2 interaction, none of these mutants were impaired for pRb degradation. However, we identified two specific patches on the surface of CR3 which contribute to pRb binding independently of the LxCxE motif in CR2. Additionally, we show for the first time that the CR3-pRb interaction is functionally important in overcoming pRb-induced cell cycle arrest.
MATERIALS AND METHODS
Plasmids.
The surface-exposed mutants of E7 were previously generated by site-directed mutagenesis, and their construction has been described (52). For pRb degradation assays, E7 mutations were subcloned into the BamHI and XhoI restriction sites of a modified pCMV-Neo-Bam mammalian expression vector. Constructs for del21-24 and C58G/C91G as well as C91G mutations were obtained from D. Galloway (Fred Hutchinson Cancer Research Center, University of Washington) and K. H. Vousden (National Cancer Institute at Frederick, Frederick, MD), respectively, and subcloned as necessary. For degradation assays carried out in H1299 cells, full-length E7 mutants were subcloned into the pCMV-Neo-Bam expression vector with a tandem N-terminal flag-hemagglutinin tag using BamHI and XhoI restriction sites. For expression in yeast, full-length E7 was cloned into a modified pJG4-5+ vector (Clontech) using EcoRI and SalI or XhoI sites (52) or, in the case of the HPV16 E7 CR3 region, using PvuII and XhoI. HPV6, HPV11, and HPV18 CR3 regions were PCR amplified using the following primers: HPV6-F, CGAATTCCGAAGTGGACGGACAAGATTCA; HPV6-R, GATCCTCGAGTTAGGTCTTCGGTGCGCAG; HPV11-F, CGAATTCAAGGTGGACAAACAAGACGCA; HPV11-R, TATGTCGACTTATGGTTTTGGTGCGCAGATGGG; HPV18-F, CGAATTCGATGGAGTTAATCATCAACAT; and HPV18-R, CCGCTCGAGTTACTGCTGGGATGC. PCR products were cloned into EcoRI and XhoI or SalI sites of the modified pJG4-5+. For expression of green fluorescent protein (GFP)-fused constructs, either full-length or PCR-amplified fragments of E7 were cloned into EcoRI and XbaI sites of pCAN-myc-EGFP. Either the wild-type or mutant CR3 region of HPV16 E7 (residues 39 to 98) was subcloned from their respective pJG4-5+ plasmid into pCAN-myc-EGFP. Alternatively, fragments of residues 1 to 39, 1 to 57, and 39 to 58 were first PCR amplified using the following primers: 1-39 F, GTCGAATTCATGCATGGAGATACACCTACATTGC; 1-39 R, ATTATCTAGAATCTCGAGTTAATCTATTTCATCCTCCTC; 1-57 F, CGAATTCGTAATCATGCATGGAGATAC; 1-57 R, CCTCTCGAGCTAAAAGGTTACAATATTGTAATG; 39-58 F, ATTAGAATTCGATGGTCCAGCTGGACAA; and 39-58 R, ATTATCTACAATCTCGAGTTAACAAAAGGTTACAATATT. They were subsequently cloned into the EcoRI and XbaI sites of pCAN-myc-EGFP. For recombinant protein production and purification, the CR3 region of each E7 construct was cloned into the pGEX4T1 (Invitrogen) vector that was modified to contain the tobacco etch virus (TEV) protease recognition sequence between the glutathione S-transferase (GST) tag and the protein of interest. The pCMV-pRb, pGEX4T1-pRbC, and pScodon-pRbABC constructs were previously described (12, 22, 23). Residues 379 to 792 of pRb were PCR amplified and cloned into the BamHI site of pGEX4T1. pBB14-Us9-EGFP has been described previously (39). pCMV-HA-cullin 2 was a gift from P. Branton (McGill University, Montreal, Canada); pcDNA3-HA-pRb was a gift from J. DeCaprio (Dana Farber Cancer Institute, Harvard Medical School, Boston, MA), and pcDNA3-His-βgal has been described previously (42). PSH1834 reporter plasmid for yeast two-hybrid analyses was recovered from the EGY48 yeast strain and contains eight operator sequences that respond to LexA. pCMV-HA-p21 plasmid has been described previously (16).
Cell culture, transfection, and pRb degradation assay.
Human Saos2, HT1080, and H1299 cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum and penicillin-streptomycin (100 U/ml). H1299 control and cullin 2 knockdown (KD) cells have been previously described (17, 18) and were maintained supplemented with 1 μg/ml of puromycin. For pRb degradation assays, Saos2 cells were seeded into 6-well plates at 3 × 105 cells per well and transfected 24 h later with 2 μg of pRb with or without 0.2 μg of E7 expression plasmid and with 0.1 μg of green fluorescent protein (GFP) or β-galactosidase (β-gal) expression plasmid as a control using FugeneHD (Roche) according to the instructions provided by the manufacturer, unless otherwise noted. The amount of plasmid DNA was balanced with empty pCMV vector where necessary. The amounts of pRb, GFP, and actin were assessed by Western blotting 48 h posttransfection. Assessment of pRb degradation in H1299 control or cullin 2 KD cells was carried out similarly, where 0.2 μg of HA-pRb expression plasmid was cotransfected with an increasing amount of wild-type or mutant Flag-HA-tagged E7-expressing plasmid (20, 200, or 2,000 ng) using X-tremeGENE HP (Roche). The amounts of pRb, E7, and actin were determined 24 h posttransfection by Western blotting. For coimmunoprecipitation experiments, HT1080 cells were seeded into 10-cm plates at 2 × 106 cells per plate and transfected 24 h later with 8 μg of total DNA with X-tremeGENE HP (Roche) according to the manufacturer's instructions. Twenty-four h posttransfection cells were collected for coimmunoprecipitation experiments. For cycloheximide treatment experiments, HT1080 cells were seeded into 6-well plates 24 h before transfection. For each mutant, the cells were transfected with 2 μg of E7 pCMV-Neo-Bam expression plasmid along with 0.1 μg of GFP expression plasmid using X-tremeGENE HP. At 24 h posttransfection, the cells were treated with 0.5 μg/μl cycloheximide for 0, 15, 30, and 60 min.
Antibodies.
The following antibodies were used: rat anti-hemagglutinin (anti-HA) (Roche) at 1:2,000 and rabbit anti-glucose-6-phosphate dehydrogenase (anti-G6PD), used as a loading control for yeast samples, at 1:80,000 (Sigma). For detection of pRb in the degradation assay, mouse anti-pRb (G3-245; BD Pharmingen) was used at 1:500 or mouse anti-pRb hybridoma lysate (clone C36) at 1:4. GFP was detected using rabbit anti-GFP (Living Colors; Clontech) at 1:2,000. Cullin 2 was detected using rabbit polyclonal anti-cul2 antibody at 1:10,000 (Bethyl Laboratories). Myc-tagged constructs were detected with mouse anti-myc hybridoma lysate (clone 9E10) used at 1:200. Mouse anti-β-gal (Promega) and rabbit anti-actin (Sigma) were used at 1:2,000. Mouse anti-E7 (8C9 clone; Invitrogen) was used at 1:200. Horseradish peroxidase (HRP)-conjugated goat anti-rabbit (Jackson Laboratories), goat anti-rat (Pierce), and rabbit anti-mouse (Jackson Laboratories) secondary antibodies were used.
Coimmunoprecipitation and Western blot analysis.
Cells were transfected in a 1:1 ratio of the myc-GFP fusion and hemagglutinin (HA)-tagged binding partner (cullin 2, pRb, or p21). Cells were harvested at 24 h posttransfection by scraping and washed once with 1× phosphate-buffered saline (PBS). Cells were lysed in NP-40 (50 mM Tris, pH 7.8, 150 mM NaCl, 0.1% NP-40) lysis buffer supplemented with 1× mammalian protease inhibitor cocktail (Sigma). Typically, 1 mg of cell lysate was mixed with 100 μl of anti-myc hybridoma (clone 9E10) or 1 μl of anti-GFP antibody and 100 μl of 10% slurry of protein A-Sepharose resin (Sigma) and incubated at 4°C for 1 to 2 h with nutating. Immunoprecipitates were washed three times with lysis buffer, resuspended in 2× lithium dodecyl sulfate (LDS) sample buffer, and boiled for 5 min. Samples were then separated by SDS-PAGE, transferred to a polyvinylidene difluoride membrane (GE), and blocked in 5% nonfat milk in Tris-buffered saline-Tween 20. Western blot analyses were carried out with mouse anti-myc hybridoma clone (9E10) or rat monoclonal anti-HA (clone 3F10; Roche).
Yeast two-hybrid assay.
Yeast two-hybrid analysis was performed in strain W303-1a (MATa leu2-3,112 his3-11,15 trp1-1 ura3-1 ade2-1, can1-100). Standard yeast culture medium was prepared using previously described methods (1). Yeast transformation was carried out using a modified lithium acetate procedure (30). Yeast cells were transformed with the reporter plasmid and pRb and E7 expression plasmids and then plated on selective medium plates and grown at 30°C for 2 to 4 days. The method for assaying β-galactosidase activity in yeast has been described previously (1).
GST pulldown assays.
Recombinant GST-tagged E7 CR3 (residues 39 to 98) and GST-tagged fragments of pRb (pRb-AB and pRb-C) were expressed in and purified from the BL21-RIL strain of Escherichia coli and pRbABC from the BL21-Gold strain per protocols provided by the affinity resin manufacturer (Amersham). For GST pulldown experiments with GST-CR3, Saos2 cells were transfected with pRb expression plasmid and lysates prepared 48 h posttransfection in NP-40 lysis buffer. Approximately 400 μg of Saos2 extract was used per reaction and mixed with an increasing amount of GST-CR3, 600 μl of NP-40 lysis buffer, and 20 μl of 50% glutathione Sepharose. Reaction mixtures were incubated at 4°C for 1 h and then washed two times with the lysis buffer. Samples were resuspended in 2× LDS sample buffer, boiled for 5 min, then separated by SDS-PAGE and examined by Western blotting for the amount of associated pRb. For GST pulldown assay with purified recombinant GST-pRbABC, -pRbAB, or -pRbC, HT1080 cells were transfected with myc-GFP-E7 39-98 expression plasmid. Lysates were prepared 24 h posttransfection in NP-40 lysis buffer. Approximately 1 mg of lysate was mixed with an increasing amount of GST-pRb fragment and 20 μl of 50% glutathione Sepharose. Reaction mixtures were incubated and samples processed as indicated above. Western blot analysis was used to assess the amount of associated myc-GFP-E7 39-98 by blotting with anti-myc.
In vitro pRb binding assay.
To obtain untagged E7 CR3 (residues 39 to 98), purified recombinant GST-tagged wild-type E7 CR3 and mutants were incubated with ∼0.2 μg/ml TEV protease overnight at 4°C. The purified protein concentration was determined using the Bio-Rad assay and was verified by running 0.5 μg of purified protein on a gel and silver staining (SilverQuest staining kit; Invitrogen) prior to using the samples in the assay. The ability of E7 CR3 to associate with GST-pRbABC was assessed at protein concentrations well below saturation levels (see Fig. 7A). Briefly, 0.6 μM E7 CR3 (residues 39 to 98) was incubated with 0.3 μM GST-pRbABC at room temperature for 1 h in a total volume of 300 μl of phosphate buffer (20 mM phosphate, pH 7.0, 200 mM NaCl, 1% Tween 20, 2 mM dithiothreitol) and 20 μl of 50% glutathione Sepharose. Samples were washed three times with phosphate buffer, boiled in 2× LDS sample buffer for 5 min, and separated by SDS-PAGE. Gels were silver stained to determine the amount of associated E7 CR3.
Fig 7.

Mapping the pRb binding surface on CR3 in vitro. (A) CR3 associates with the large pocket of pRb (pRbABC) in a dose-dependent manner. GST-pRbABC (0.3 μM) was incubated with increasing amounts of purified CR3 (residues 39 to 98), and the amount of associated CR3 was determined by silver staining. (B) Representative gel image of CR3-pRb binding as quantified for panel C. (C) Patch 1 residues most significantly contribute to pRb binding. Purified wild-type CR3 or the indicated mutant was assessed for the ability to bind pRb in a GST pulldown assay using 0.3 μM GST-pRbABC and 0.6 μM CR3. Data are presented relative to wild-type protein and are representative of a minimum of three independent assays. **, P ≤ 0.001; *, P = 0.032. (D) Based on the in vitro analysis, patch 1 and patch 2 residues significantly contributed to pRb binding. Colored in blue are patch 1 residues Y52, N53, V55, F57, C59, S63, T64, and T72, as well as R77, E80, and D81 within patch 2.
Cell cycle analysis.
A total of 6 × 105 or 1 × 106 Saos2 cells were seeded into 6- or 10-cm dishes, respectively. Cells were transfected with 1.5 μg of each Us9-EGFP expression plasmid (membrane-bound GFP) and pRb, as well as 3 μg of untagged, full-length E7 (pCMV-Neo-Bam plasmids), with FugeneHD (Roche). Twenty-four h posttransfection, cells were transferred to either 10- or 15-cm dishes (depending on the size of the original dish) using phosphate-buffered saline supplemented with 10 mM EDTA (PBS-EDTA). Forty-eight h after the transfer to a new dish, cells were collected with PBS-EDAT solution and washed once with PBS. Cell pellets were resuspended in 300 μl of 50% fetal bovine serum (FBS) solution (in PBS) and fixed by dropwise addition of 900 μl of ice-cold 70% ethanol. Samples were incubated on ice for at least 2 h and then washed two times with PBS. Cells were resuspended in and stained with 300 μl of propidium iodide staining solution (1% FBS, 10 μg/ml propidium iodide, 0.25 mg/ml RNase A in PBS). Cell cycle data were collected by flow cytometry on a FACSCalibur (BD Biosciences). DNA content was determined based on propidium iodide staining. Data were analyzed using FlowJo software, and the percentage of cells in G1 phase was determined using the Watson model.
RESULTS
CR3 mutations.
To examine the role that HPV16 E7 CR3 plays in pRb deregulation, we utilized the panel of putative surface-exposed mutants within CR3, here referred to as the surface-exposed mutants. These have been previously described by our group (52). Briefly, these mutations target residues whose side chains are predicted to be at least 25% surface exposed but not those that would participate in stabilizing the structural features of E7 (40, 45, 52). Hence, these mutations were constructed with the dual aims of preserving the structure of the CR3 region while disrupting the interaction surfaces available for cellular targets. In total we included in these studies 19 point mutants, one double mutant, and one mutant targeting the last three amino acids of CR3 (Fig. 1). As controls, we also utilized the deletion mutant, del21-24, which removes the LxCxE motif in CR2, and the C58G/C91G mutant, which targets two critical zinc-coordinating cysteines in CR3 and is thought to grossly perturb the folded structure of this region of E7.
Fig 1.

Schematic representation of HPV16 E7 and the position of mutations targeting residues within CR3. The conserved regions (CR1, CR2, and CR3) are depicted as boxes. The amino acid sequences of CR3 and the mutations used in this study are indicated. Also indicated are del21-24, which removes the LxCxE motif in CR2 of E7, and the C58G/C91G mutant, which targets zinc-coordinating cysteine residues within CR3.
Contribution of HPV16 E7 CR3 to pRb degradation.
The LxCxE motif and certain residues within CR1 of E7 were previously shown to be important for pRb degradation (32, 35). Other studies have also examined the role of CR3 and have found no contribution of any single residue to pRb degradation (7, 35). However, another study has shown that specific residues within CR3 contribute to cullin 2 complex binding, currently the exclusive mechanism known to be used by HPV16 E7 to degrade pRb (36). These findings led us to reexamine the role of CR3 in pRb degradation. Our first aim was to map the surface of CR3 which is required for pRb degradation utilizing the panel of surface-exposed mutants. Our approach was to test full-length wild-type E7 or E7 mutants for their ability to degrade pRb exogenously expressed in pRb-null Saos2 osteosarcoma cells. Cotransfection of pRb with increasing amounts of wild-type E7 led to a dose-dependent decrease in pRb levels (Fig. 2A). As expected, cotransfection of the del21-24 E7 mutant, which removes the high-affinity LxCxE site, did not reduce pRb levels. However, all 21 of the CR3 mutants that we tested retained the ability to degrade pRb comparably to wild-type E7 under nonsaturating conditions (Fig. 2B).
Fig 2.

All E7 CR3 mutants retain the ability to degrade pRb. (A) Dose-dependent degradation of pRb by wild-type E7 in Saos2 osteosarcoma cells. Saos2 cells were transfected with 1 μg of pRb expression plasmid, 0.1 μg GFP expression plasmid, and the indicated amount of wild-type E7 expression plasmid; 48 h posttransfection cells were collected and samples analyzed by Western blotting for pRb, GFP, and actin levels. (B) pRb degradation by E7 mutants. Full-length wild-type E7 or the indicated mutants were transfected with pRb and GFP expression plasmids at a 1:10 ratio of E7 to pRb, and at 48 h posttransfection levels of pRb were analyzed. The blot is a representative image; the bar graphs represent quantified pRb levels normalized to GFP and actin levels. The relative pRb levels are representative of five independent experiments.
Contribution of HPV16 E7 CR3 to cullin 2 binding and the role of cullin 2 in E7-mediated pRb degradation.
The findings described above led us to question whether the pRb degradation phenotypes observed could be due to the fact that each of these surface-exposed mutants maintains the ability to associate with the cullin 2 complex. This seemed unlikely, as others previously showed that mutations CVQ68-70AAA, del79-83, and C91S lose the ability to associate with the cullin 2 complex (36). Before testing the individual mutants for interaction with cullin 2, we first determined which portions of E7 mediated binding using GFP fused to fragments of E7 corresponding to residues 1 to 39 (CR1 and CR2), 39 to 58 (N terminus of CR3), and 39 to 98 (CR3). In coimmunoprecipitation experiments, neither residues 1 to 39 nor 39 to 58 of E7 could interact with the cullin 2 complex on their own. In contrast, residues 39 to 98 (CR3) of E7 were necessary and sufficient for this interaction (Fig. 3A). To test the properties of the surface-exposed mutants for cullin 2 binding, we expressed the entire panel in the context of the residue 39 to 98 fragment fused to GFP. In coimmunoprecipitation experiments with coexpressed HA-cullin 2, all mutants retained at least some ability to associate with this complex (Fig. 3B). However, mutants such as N53D, V55T, and S63D had a substantially reduced capacity to associate with the cullin 2 complex. Nonetheless, as described above, all of these mutations retained the ability to target pRb for degradation. These results suggest that either the relatively weak interaction with cullin 2 is sufficient for pRb degradation or that other, as-yet unidentified mechanisms are utilized for the degradation process. In addition to assessing the ability of our panel of surface-exposed mutants to bind the cullin 2 complex, we also included in these experiments the C58G/C91G mutant, which abrogates zinc coordination within CR3 and presumably disrupts proper folding of this domain (Fig. 3B). A similar mutation, C91S, in the context of full-length E7 was previously shown to disrupt cullin 2 binding (36). Unexpectedly, we found that C58G/C91G associated with cullin 2 at least as well as wild-type E7 residues 39 to 98 (Fig. 3B). When tested for the ability to target pRb for degradation in the Saos2 cell assay, the C58G/C91G mutant in the context of full-length E7 degraded pRb in a dose-dependent manner that was indistinguishable from wild-type E7 (Fig. 3C). The C91G mutation was similarly tested and behaved identically to C58G/C91G (data not shown). These findings suggest that both cullin 2 binding and pRb degradation function independently of correct folding of CR3.
Fig 3.

Contribution of cullin 2 complex to E7-induced pRb degradation. (A) CR3 of E7 is necessary and sufficient to bind cullin 2. HT1080 cells were transfected with an equal ratio of hemagglutinin (HA)-tagged cullin 2 expression plasmid to either myc-GFP or myc-GFP-fused E7 fragment (as indicated). Twenty-four h posttransfection, cell lysates were subjected to immunoprecipitation (IP) with anti-myc antibody, followed by Western blotting for cullin 2 with anti-HA antibody. (B) Cullin 2 binding capacity of CR3 mutants. HT1080 cells were cotransfected with HA-tagged cullin 2 and either myc-GFP or myc-GFP-fused E7 39-98 (wild type or indicated mutant). Coimmunoprecipitation was carried out as described above. (C) The C58G/C91G mutant retains the ability to degrade pRb. Saos2 cells were transfected with 1 μg pRb, 0.1 μg β-galactosidase, and the indicated amounts of full-length wild-type or C58G/C91G mutant E7. Samples were collected 48 h posttransfection, and the steady-state levels of pRb were analyzed by Western blotting. %pRb indicates the relative amount of remaining pRb in each lane. (D) Cullin 2 levels in H1299 control and H1299 cullin 2 knockdown (KD) cells. The levels of endogenous cullin 2 present in H1299 control and H1299 cullin 2 KD cells were analyzed using anti-cul2 antibody. (B) Dependence of E7 on cullin 2 for pRb degradation. pRb degradation assays were conducted in cullin 2 KD and control cells for indicated E7 mutants, as described in Materials and Methods.
To further study to role of cullin 2 in the pRb degradation process, we compared the ability of four E7 mutants identified to have reduced association with cullin 2 (N53D, V55T, R66E, and T72D) to degrade pRb in H1299 cells in which cullin 2 was stably knocked down (Fig. 3E). Each mutant degraded pRb in cullin 2 knockdown cells similarly to control cells, further suggesting that E7 is utilizing a cullin 2-independent mechanism to degrade pRb.
CR3 binds pRb independently of the LxCxE motif in vitro and in vivo.
Several reports have suggested that CR3 contributes to deregulation of pRb function by providing a secondary lower-affinity binding site. For example, full-length E7 binds pRb more strongly than mutants retaining the LxCxE motif but lacking CR3 (46). More recently, it was demonstrated in two independent studies that CR3 binds pRb in vitro (15, 45). We first confirmed that purified GST-CR3 of E7 was sufficient to independently pull down pRb from cell lysates in a dose-dependent manner (Fig. 4A). Utilizing the reciprocal approach, we also demonstrated that a recombinant large pocket of pRb (GST-pRbABC) could associate with GFP-CR3 from cell lysates obtained by overexpressing GFP-CR3 in HT1080 cells (Fig. 4B). Additionally, the small pocket of pRb (pRbAB) was sufficient for this interaction, consistent with a previous report (15). In contrast, the C terminus of pRb (pRbC) was not sufficient to interact with CR3 of E7. As CR3 has never been reported to interact with pRb in vivo, we examined the ability of GFP-CR3 to coimmunoprecipitate HA-pRb. As expected, an N-terminal fragment of E7 (residues 1 to 57, containing the high-affinity LxCxE binding site for pRb) was found to associate strongly with pRb by coimmunoprecipitation (Fig. 5A). Importantly, the CR3 region of E7 (residues 39 to 98) was also sufficient to coimmunoprecipitate pRb, albeit at a substantially lower level than the full-length E7 protein or the LxCxE-containing N-terminal fragment. Yeast two-hybrid analysis utilizing E7 fragments spanning residues 1 to 57 or 39 to 98 as prey and full-length pRb as bait showed that CR3 also interacted with pRb independently of the N terminus. In these assays, residues 39 to 98 bound pRb with approximately 30% of the activity observed with full-length E7 (Fig. 5B).
Fig 4.

E7 CR3 interacts with pRb independently of the LxCxE motif in vitro. (A) GST-CR3 associates with pRb from cell lysates. Increasing amounts of GST-CR3 (residues 39 to 98) were incubated with ∼400 μg of Saos2 cell lysate (previously transfected with pRb expression plasmid) and analyzed for the amount of associated pRb via Western blotting. (B) GFP-CR3 (residues 39 to 98) associates with the large and small pockets of pRb. HT1080 cells were transfected with myc-GFP or myc-GFP-fused E7 39-98. Twenty-four h posttransfection, cell lysates were prepared and ∼1 mg was used in each GST pulldown reaction. Five μg of GST was incubated with 1 mg of myc-GFP-E7 39-98 lysate as a control. Five μg of GST-pRbABC (large pocket), GST-pRbAB (small pocket), and GST-pRbC (C terminus of pRb) was incubated with 1 mg of myc-GFP lysate as a control. Increasing amounts of GST-pRb fragments (0.5, 1, or 5 μg) were incubated with 1 mg of myc-GFP-E7 39-98 lysate. Samples were washed and then analyzed by Western blotting for the amount of associated myc-GFP-E7 39-98. The bottom panel is the Ponceau stain of the membrane, illustrating the input levels of GST-pRb fragments. The left side of the Ponceau stain indicates the position and size of the ladder in kDa.
Fig 5.

E7 CR3 interacts with pRb independently of the LxCxE motif in vivo. (A) E7 CR3 interacts with pRb in vivo. HT1080 cells were cotransfected with equal amounts of HA-tagged pRb and myc-GFP or myc-GFP-E7 fragments. Twenty-four h posttransfection, cell lysates were prepared and subjected to immunoprecipitation with anti-myc antibody, followed by Western blotting for HA-pRb. (B) HPV16 E7 CR3 interacts with pRb in a yeast two-hybrid assay. Yeast cells were transformed with either empty vector, full-length E7, or the two indicated E7 fragments as prey proteins, along with pRb as bait and a LexA-responsive β-galactosidase reporter plasmid. Following transformation, yeast cells were analyzed for β-galactosidase activity. Data are presented as percent binding relative to that of full-length E7. (C) E7 proteins from other HPV types also associate with pRb. HT1080 cells were cotransfected with equal amounts of HA-tagged pRb and myc-GFP or myc-GFP-E7 from HPV16, HPV18, HPV6, or HPV11. Twenty-four h posttransfection, immunoprecipitation and Western blotting were carried out as described above. (D) CR3 regions from other HPV types also associate with pRb independently from the LxCxE motif. The C-terminal portions of HPV16, HPV18, HPV11, and HPV6 were tested for the ability to associate with pRb similarly to what was described for panel B. Data are represented as experimentally determined β-galactosidase activity.
CR3s from other HPV types also interact with pRb.
Utilizing coimmunoprecipitation experiments, we determined that full-length E7 from the high-risk HPV18 and low-risk HPV6 and HPV11 also associated with pRb in mammalian cells cotransfected with E7 and pRb (Fig. 5C) or utilizing endogenous pRb (data not shown). This was expected, as each of the E7 proteins contains a high-affinity LxCxE motif. We next tested whether the E7 CR3 regions from other HPV types could interact in vivo with pRb independently of the LxCxE binding site in CR2. In a yeast two-hybrid analysis, low-risk HPV6 and HPV11 E7 CR3 regions also interacted with pRb, whereas little or no activity was detected for the high-risk HPV18 CR3 (Fig. 5D). Similar to the coimmunoprecipitation experiments from mammalian cells, each of the full-length E7 proteins was also able to bind pRb in the yeast two-hybrid assay. These results indicate that while all of the E7 proteins tested could bind pRb, the ability to interact with pRb via CR3 is not a universal property of all E7 proteins.
Mapping the binding surface for pRb in CR3.
We initially used the yeast two-hybrid system as a rapid method to identify candidate residues from HPV16 E7 that are involved in binding pRb. We cloned our panel of mutants into a yeast two-hybrid prey vector as fragments expressing only residues 39 to 98 and assessed their capacity to bind pRb. With this approach, we identified a number of mutants with statistically significant differences in pRb binding. Specifically, V55T, F57A, R66E, M84S, and I93T were increased and Y52A, N53D, C59S, S63D, T64D, T72D, R77E, and G85A were decreased. The most significant increase in binding was observed for R66E, which displayed 10 times higher β-galactosidase activity than the wild-type CR3. In contrast, Y52A, N53D, C59S, and G85A all showed more than 50% reductions in activity (Fig. 6B). It should be noted that for the yeast two-hybrid analysis, CR3 mutants were previously assessed for any autoactivation potential (52). Although R66E had greatly increased pRb binding potential in the yeast two-hybrid analysis, this was not due to enhanced autoactivation. To rule out the possibility that differential expression played a role in the observed changes in binding capacities, we tested the majority of CR3 mutants which displayed increased or decreased binding (mutations with P ≤ 0.05) for expression in yeast cells. All mutants with decreased binding capacity were expressed comparably to the wild type except for S63D, which was expressed at a slightly lower level (Fig. 6C). Similarly, mutants with increased binding for pRb were expressed comparably to the wild type, with the possibility that the slight increase in binding seen with the D62K and I93T mutants could be the result of increased expression. It should be noted that full-length E7 wild type or mutants were also assessed for the capacity to associate with pRb in a yeast two-hybrid analysis (Fig. 6A). With this approach, we found that none of the mutants exhibited a substantially reduced ability to associate with pRb, suggesting that defects in binding via CR3 are masked by the high-affinity pRb binding site within CR2.
Fig 6.

Identification of CR3 residues involved in binding pRb using the yeast two-hybrid system. (A) Yeast two-hybrid analysis of the binding capabilities of full-length E7 mutants and pRb. Yeast cells were transformed with full-length pRb expression plasmid as bait and full-length wild-type or mutant E7 as prey, in addition to the LexA-responsive β-galactosidase reporter plasmid. Data are shown relative to wild-type E7 and are representative of four independent experiments. (B) Yeast two-hybrid analysis of the binding capabilities of E7 CR3 mutants. Yeast cells were transformed with full-length pRb expression plasmid as bait and wild-type or mutant fragment of E7 spanning residues 39 to 98, in addition to the LexA-responsive β-galactosidase reporter plasmid. Data are represented relative to wild-type E7 39-98. **, P ≤ 0.01; *, P ≤ 0.05. (C) Expression level of E7 CR3 mutants in yeast cells. The same yeast cultures used for the analysis of the pRb-CR3 interaction were also analyzed by Western blotting for E7 CR3 protein expression levels.
Confirmation of the pRb binding properties of E7 CR3 mutants in vitro.
Our initial analysis using the yeast two-hybrid system identified a number of residues that appeared to contribute to the interaction of CR3 with pRb. Interestingly, most of these residues map to two independent patches on the surface of CR3. Specifically, residues Y52, N53, V55, F57, C59, S63, T64, R66, and T72 map to one region, referred to as patch 1; residues R77, M84, and G85 map to a second region, referred to as patch 2. However, results from yeast two-hybrid assays may be influenced by the presence of additional factors in yeast. In order to conclusively determine if the identified residues contribute to pRb binding, we set up an in vitro GST pulldown assay utilizing the affinity-purified recombinant GST-fused large pocket of pRb and increasing molar amounts of purified wild-type CR3 (E7 residues 39 to 98). In this assay, CR3 associated with pRb in a dose-dependent manner (Fig. 7A). This analysis established appropriate protein concentrations for the assay, ensuring that the reactions were carried out at levels of CR3 below saturation. For all subsequent experiments, GST pulldown assays were set up with 0.3 μM pRbABC and a 2-fold molar excess of wild-type or mutant CR3 (0.6 μM).
To confirm the contribution of patch 1 to pRb binding, we focused on mutants of E7 with the most significantly altered binding (P ≤ 0.01) as determined by the yeast two-hybrid system (Fig. 6B). These included Y52A, N53D, V55T, F57A, C59S, S63D, T64D, R66E, and T72D. Of these mutants, all but R66E bound pRb at reduced levels in vitro (Fig. 7C and D). Taken together, the data from these two independent types of analyses clearly indicate that patch 1 contributes to pRb binding by CR3.
Patch 2 overlaps residues previously reported to contribute to pRb interaction, including R77, E80, and D81 (45). In agreement, E80K/D81K retained only ∼25% of the binding capacity for the large pocket of pRb in our in vitro tests. In contrast, R77E showed a substantial increase in pRb binding and G85A bound comparably to the wild type (Fig. 7C and D). These data suggest that negative charges present within patch 2 contribute to pRb interaction, as previously proposed (45).
The CR3-pRb interaction is important for E7 to overcome pRb-induced cell cycle arrest.
Although all surface-exposed mutants of E7 CR3 can target pRb for degradation, our data demonstrate that specific residues contribute to pRb binding. To determine the functional significance of this interaction, we assessed patch 1 and patch 2 mutants for their ability to overcome cell cycle arrest induced by reintroduction of pRb into pRb-null Saos2 cells. Reintroduction of pRb into Saos2 cells results in ∼80% of cells arresting in the G1 phase of the cell cycle (Fig. 8A). Coexpression of full-length wild-type E7 reverses this arrest, with ∼40% of the cells remaining in G1. The change in percentage of cells in G1 in the presence or absence of wild-type E7 was normalized to 100% (Fig. 8A). Although the LxCxE deletion mutant del21-24 was impaired for inducing G1 exit, it still retained 27% of wild-type activity. We tested all CR3 mutants that were assessed for pRb binding in vitro for their ability to overcome this pRb-induced cell cycle arrest. Of those mutants located within patch 1, we found that all except C59S, R66E, and T72D had a significantly reduced ability to overcome cell cycle arrest. Their activity ranged from 60 to 77% of that of wild-type E7. C59S and T72D showed a modest decrease in the ability to bind pRb in vitro but behaved similarly to wild-type E7 in their ability to overcome cell cycle arrest. R66E retained pRb binding in both the yeast two-hybrid and the in vitro studies and overcame cell cycle arrest, similar to wild-type E7. This finding is consistent with a previous analysis of R66A in similar functional assays (35). Of the patch 2 mutants, R77E behaved like wild-type E7, but G85A and E80K/D81K retained only about 60% of wild-type capacity to overcome the G1 arrest. The reduced ability of the mutants to overcome the pRb-induced cell cycle arrest was not related to a reduction in their stability (Fig. 8B). We also tested selected mutants for their ability to interact with p21Cip1/WAF1 by coimmunoprecipitation, as this regulator of G1 exit has previously been reported to interact with HPV16 E7 CR3 (27). However, no correlation was apparent between binding p21Cip1/WAF1 and ability to overcome a pRb-induced cell cycle arrest (Fig. 9). Taken together, CR3 mutants with reduced ability to bind pRb typically were less able to overcome a pRb-induced cell cycle arrest.
Fig 8.

CR3-pRb interaction is functionally important for overcoming cell cycle arrest. (A) Mutants with reduced capacity to associate with pRb via CR3 have defects in overcoming cell cycle arrest. Mutants analyzed for pRb binding in vitro were also assessed for their ability to overcome cell cycle arrest induced by reexpression of wild-type pRb in Saos2 cells. The percentage of cells in G1 phase of the cell cycle was determined by flow cytometry. Control cells represent the normally cycling Saos2 population. Saos2 cells expressing pRb only but not E7 are labeled as pRb only. For all other samples, cells express pRb and either the full-length wild-type or indicated mutant E7 protein. Data are representative of a minimum of three independent experiments. ***, P ≤ 0.001; **, P ≤ 0.01; and *, P ≤ 0.05 relative to the wild-type E7 sample. The relative activity of E7 mutants in overcoming cell cycle arrest was calculated and is indicated below the bar graph. (B) Analysis of E7 mutant stability. HT1080 cells were transfected with the expression plasmid for the indicated E7 mutants together with GFP, and 24 h posttransfection they were treated with cycloheximide for 15, 30, or 60 min. The levels of E7 were analyzed by Western blotting. DMSO, dimethylsulfoxide.
Fig 9.
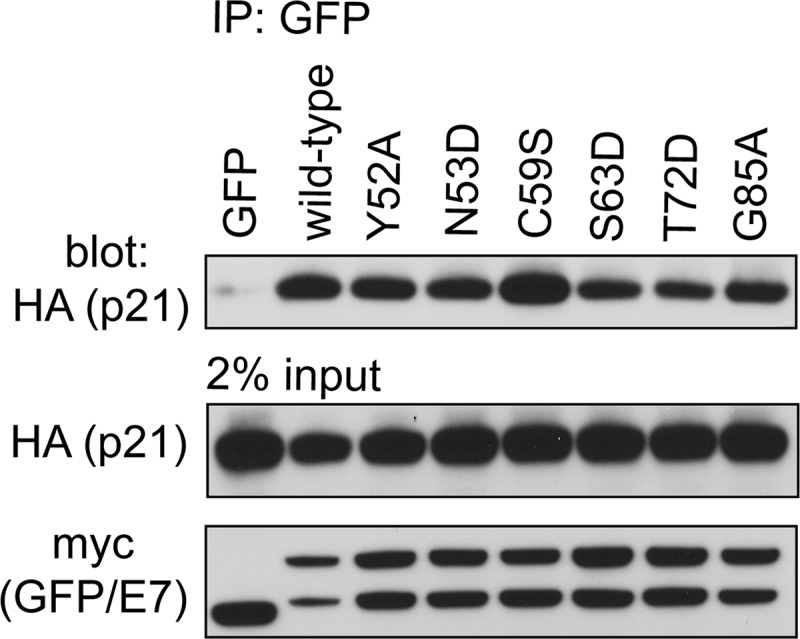
Association of E7 mutants with p21Cip1/WAF1. HT1080 cells were cotransfected with expression plasmids for HA epitope-tagged p21 and wild-type full-length E7 or the indicated mutant fused to a myc-GFP tag. Twenty-four h posttransfection, the cells were lysed and immunoprecipitation was performed with anti-GFP antibody, followed by Western blotting.
DISCUSSION
Although the role of HPV E7 in cellular transformation and cancer progression has been extensively studied, we still do not fully understand how E7 deregulates one of its most vital interacting partners, the retinoblastoma tumor suppressor (pRb). Precise assessment of E7-pRb protein interactions appears fundamental to understanding virally mediated subversion of cell cycle control and may allow novel shared features of viral and cellular pRb protein interaction partners to be uncovered. The objective of this study was to more precisely determine the role of E7 CR3 sequences in deregulating the pRb pathway.
For these studies, we utilized an extensive panel of surface-exposed mutants of HPV16 E7 CR3, which target residues that are accessible for interaction with cellular partners (Fig. 1). Unlike previous work, we avoided using deletion mutants or mutants which target highly conserved, structurally important hydrophobic residues of E7 CR3 (52). This approach allowed us to examine the role of each individual residue on the surface of CR3 in deregulating pRb function. Utilizing this panel of mutants, we first tested whether any of the introduced changes on the surface of E7 CR3 reduced its capacity to target pRb for degradation. Unexpectedly, the entire panel of 21 surface-exposed mutants was as effective as the wild-type HPV16 E7 protein in targeting pRb for degradation (Fig. 2B). The only mutant that was unable to induce pRb degradation was del21-24, which cannot associate with pRb via the LxCxE motif located in CR2. This is most consistent with a previous report that even mutants with substantial deletions in CR3 were still able to destabilize pRb (35).
The ability of all of the CR3 mutants to degrade pRb was unexpected, as this region is necessary for interaction with the cullin 2 E3 ubiquitin ligase which facilitates ubiquitination of pRb and subsequent proteasome-mediated degradation (36, 53). In agreement with other studies, we show that CR3 confers association with cullin 2 (Fig. 3A). Our finding that mutations in the zinc-coordinating cysteine residues of CR3 still bind cullin 2 suggests that this interaction occurs independently of the correct folding of CR3, perhaps via a linear interaction motif. Although some CR3 mutations result in reduced binding capacity for cullin 2, these mutants still degrade pRb as efficiently as wild-type E7 under nonsaturating conditions. We found this to be the case in the Saos2 degradation assay as well as in cells in which cullin 2 levels were diminished by stable expression of a short hairpin RNA targeting cullin 2. This suggests that a cullin 2-independent mechanism is utilized for pRb degradation. This is certainly possible, as the ability of HPV16 E7 to bind cullin 2 is not shared by the E7 proteins of any other HPV type so far tested (53).
Nearly 20 years ago, it was suggested that CR3 also functions as a secondary lower affinity binding site for pRb (46). However, this interaction was only recently confirmed by two independent reports (15, 45). Importantly, we have extended those in vitro studies to demonstrate that the interaction of CR3 with pRb occurs in mammalian cells in vivo by utilizing coimmunoprecipitation experiments (Fig. 5A). We also found that CR3 from other HPV types, specifically the low-risk HPV11 and HPV6, associate with pRb (Fig. 5D). Interestingly, CR3 from HPV18 E7 did not bind pRb, suggesting that this interaction is present in some, but not all, E7 proteins. To establish the functional consequences of the pRb-CR3 interaction, we first sought to identify the surface of CR3 that binds pRb. We assessed our panel of surface-exposed CR3 mutants for their pRb binding properties by utilizing the yeast two-hybrid assay. This identified two specific patches on the surface of CR3 that contribute to pRb binding. We confirmed these data in vitro using purified recombinant proteins and GST pulldown experiments with the large pocket of pRb (pRbABC) fused to GST and purified wild-type or mutant CR3. Although both yeast two-hybrid and in vitro interaction assays yield similar results for the majority of mutants, the two assays responded differently for a subset of the mutants. It is possible that additional factors present in yeast but not in the fully recombinant system influence the results. Nevertheless, these studies identified residues that comprise patch 1, which forms one continuous surface on CR3 and significantly contributes to pRb binding (Fig. 7D). These include residues Y52, N53, V55, F57, C59, S63, T64, and T72. Consistent with a previous report, we also identified residues within patch 2 that also contribute to pRb binding (40). Although we tested other mutants of E7 that are found within the patch 2 region, we found that only R77E and E80K/D81K had an impact on binding. This suggests that negative charges in this region contribute to pRb binding via electrostatic interactions, as previously proposed (45).
We have established that all CR3 mutants maintain the ability to degrade pRb, but that specific residues in this region of E7 have reduced binding to pRb. We therefore wanted to determine whether CR3 contributes functionally to deregulating the pRb pathway by targeting the remaining pools of undegraded pRb. Previous work has shown that CR3 of E7 is important for pRb-E2F complex interference, but that small alterations in CR3 do not lead to significant changes in the ability of E7 to interfere with pRb-E2F complex formation (35). Therefore, our approach was to assess the role of the CR3-pRb interaction more globally by determining the ability of CR3 mutants with decreased pRb binding to overcome cell cycle arrest induced by reintroduction of pRb into pRb-null Saos2 cells. We found that there is a high degree of correlation between the ability of E7 to bind pRb via the CR3 region and the ability of the corresponding full-length mutants to overcome cell cycle arrest. Most notably, mutations in residues within patch 1 that exhibited the most significant decrease in pRb binding also displayed a significant decrease in the ability to overcome pRb-induced cell cycle arrest. Additionally, mutants in patch 2 were also examined. E80K/D81K had a reduced ability to overcome cell cycle arrest, which correlated with its reduced ability to bind pRb in vitro. Importantly, these data establish that the ability of HPV16 E7 to induce pRb degradation is not sufficient to fully overcome a pRb-induced cell cycle arrest. The data suggest that the remaining pool of undegraded pRb is still partially able to block cell cycle entry, and that this is overcome by the CR3-pRb interaction.
The cell cycle analysis also identified the G85A mutant as being interesting. Although this mutant binds pRb similarly to wild-type E7 in vitro, it has a reduced ability to overcome cell cycle arrest. This mutant maintains the ability to interact with p21Cip1/WAF1 (Fig. 9), but it is possible that it has lost the capacity to interact with another important cellular target which is necessary for E7 to drive the cells into the S phase of the cell cycle.
Taken together, we have identified a new surface of HPV16 E7 that contributes to binding to pRb, which we have called patch 1. We also observed that negative charges located in patch 2 likely contribute to electrostatic interactions with pRb. Our data illustrate for the first time that the ability of E7 to bind pRb via CR3 is important in overcoming the cell cycle arrest functions of pRb. In light of these findings, we suggest that the CR3 domain of E7 is a candidate for targeted inactivation by small-molecule compounds. Specifically, development of small molecules that bind patch 1 of CR3 may disrupt the ability of E7 to perturb pRb function and have utility in the treatment of papillomas or HPV-induced cancers.
ACKNOWLEDGMENTS
This work was supported by a grant from the Canadian Institute of Health Research awarded to J.S.M. B.T. was supported by a Canadian Institute of Health Research doctoral award. G.S.S. was supported by the Canadian Cancer Society (018414).
H1299 cullin 2 KD and control cells were generously provided by P. Branton and P. Blanchette.
Footnotes
Published ahead of print 26 September 2012
REFERENCES
- 1. Adams A, Gottschling DE, Kaiser CA, Stearns T. 1997. Methods in yeast genetics. Cold Spring Harbor Press, Cold Spring Harbor, NY [Google Scholar]
- 2. Androphy EJ, Hubbert NL, Schiller JT, Lowy DR. 1987. Identification of the HPV-16 E6 protein from transformed mouse cells and human cervical carcinoma cell lines. EMBO J. 6:989–992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Avvakumov N, Torchia J, Mymryk JS. 2003. Interaction of the HPV E7 proteins with the pCAF acetyltransferase. Oncogene 22:3833–3841 [DOI] [PubMed] [Google Scholar]
- 4. Banks L, et al. 1987. Identification of human papillomavirus type 18 E6 polypeptide in cells derived from human cervical carcinomas. J. Gen. Virol. 68(Pt 5):1351–1359 [DOI] [PubMed] [Google Scholar]
- 5. Barbosa MS, et al. 1990. The region of the HPV E7 oncoprotein homologous to adenovirus E1a and Sv40 large T antigen contains separate domains for Rb binding and casein kinase II phosphorylation. EMBO J. 9:153–160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Berezutskaya E, Bagchi S. 1997. The human papillomavirus E7 oncoprotein functionally interacts with the S4 subunit of the 26 S proteasome. J. Biol. Chem. 272:30135–30140 [DOI] [PubMed] [Google Scholar]
- 7. Bodily JM, Mehta KP, Cruz L, Meyers C, Laimins LA. 2011. The E7 open reading frame acts in cis and in trans to mediate differentiation-dependent activities in the human papillomavirus type 16 life cycle. J. Virol. 85:8852–8862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bodily JM, Mehta KP, Laimins LA. 2011. Human papillomavirus E7 enhances hypoxia-inducible factor 1-mediated transcription by inhibiting binding of histone deacetylases. Cancer Res. 71:1187–1195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Boyer SN, Wazer DE, Band V. 1996. E7 protein of human papilloma virus-16 induces degradation of retinoblastoma protein through the ubiquitin-proteasome pathway. Cancer Res. 56:4620–4624 [PubMed] [Google Scholar]
- 10. Brehm A, et al. 1998. Retinoblastoma protein recruits histone deacetylase to repress transcription. Nature 391:597–601 [DOI] [PubMed] [Google Scholar]
- 11. Brehm A, et al. 1999. The E7 oncoprotein associates with Mi2 and histone deacetylase activity to promote cell growth. EMBO J. 18:2449–2458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cecchini MJ, Dick FA. 2011. The biochemical basis of CDK phosphorylation-independent regulation of E2F1 by the retinoblastoma protein. Biochem. J. 434:297–308 [DOI] [PubMed] [Google Scholar]
- 13. Centers for Disease Control and Prevention 2010. FDA licensure of bivalent human papillomavirus vaccine (HPV2, cervarix) for use in females and updated HPV vaccination recommendations from the Advisory Committee on Immunization Practices (ACIP). MMWR Morb. Mortal. Wkly. Rep. 59:626–629 [PubMed] [Google Scholar]
- 14. Chellappan S, et al. 1992. Adenovirus E1A, simian virus 40 tumor antigen, and human papillomavirus E7 protein share the capacity to disrupt the interaction between transcription factor E2F and the retinoblastoma gene product. Proc. Natl. Acad. Sci. U. S. A. 89:4549–4553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chemes LB, Sanchez IE, Smal C, de Prat-Gay G. 2010. Targeting mechanism of the retinoblastoma tumor suppressor by a prototypical viral oncoprotein. Structural modularity, intrinsic disorder and phosphorylation of human papillomavirus E7. FEBS J. 277:973–988 [DOI] [PubMed] [Google Scholar]
- 16. Chen J, Saha P, Kornbluth S, Dynlacht BD, Dutta A. 1996. Cyclin-binding motifs are essential for the function of p21CIP1. Mol. Cell. Biol. 16:4673–4682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cheng CY, Blanchette P, Branton PE. 2007. The adenovirus E4orf6 E3 ubiquitin ligase complex assembles in a novel fashion. Virology 364:36–44 [DOI] [PubMed] [Google Scholar]
- 18. Cheng CY, et al. 2011. The E4orf6/E1B55K E3 ubiquitin ligase complexes of human adenoviruses exhibit heterogeneity in composition and substrate specificity. J. Virol. 85:765–775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Classon M, Harlow E. 2002. The retinoblastoma tumour suppressor in development and cancer. Nat. Rev. Cancer 2:910–917 [DOI] [PubMed] [Google Scholar]
- 20. Dawar M, Deeks S, Dobson S. 2007. Human papillomavirus vaccines launch a new era in cervical cancer prevention. CMAJ 177:456–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. de Villiers EM, Fauquet C, Broker TR, Bernard HU, zur Hausen H. 2004. Classification of papillomaviruses. Virology 324:17–27 [DOI] [PubMed] [Google Scholar]
- 22. Dick FA, Dyson N. 2003. pRB contains an E2F1-specific binding domain that allows E2F1-induced apoptosis to be regulated separately from other E2F activities. Mol. Cell 12:639–649 [DOI] [PubMed] [Google Scholar]
- 23. Dick FA, Sailhamer E, Dyson NJ. 2000. Mutagenesis of the pRB pocket reveals that cell cycle arrest functions are separable from binding to viral oncoproteins. Mol. Cell. Biol. 20:3715–3727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dyson N. 1998. The regulation of E2F by pRB-family proteins. Genes Dev. 12:2245–2262 [DOI] [PubMed] [Google Scholar]
- 25. Dyson N, Howley PM, Munger K, Harlow E. 1989. The human papilloma virus-16 E7 oncoprotein is able to bind to the retinoblastoma gene product. Science 243:934–937 [DOI] [PubMed] [Google Scholar]
- 26. Edmonds C, Vousden KH. 1989. A point mutational analysis of human papillomavirus type 16 E7 protein. J. Virol. 63:2650–2656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Funk JO, et al. 1997. Inhibition of CDK activity and PCNA-dependent DNA replication by p21 is blocked by interaction with the HPV-16 E7 oncoprotein. Genes Dev. 11:2090–2100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Future II Study Group 2007. Quadrivalent vaccine against human papillomavirus to prevent high-grade cervical lesions. N. Engl. J. Med. 356:1915–1927 [DOI] [PubMed] [Google Scholar]
- 29. Giarre M, et al. 2001. Induction of pRb degradation by the human papillomavirus type 16 E7 protein is essential to efficiently overcome p16INK4a-imposed G1 cell cycle arrest. J. Virol. 75:4705–4712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gietz RD, Schiestl RH, Willems AR, Woods RA. 1995. Studies on the transformation of intact yeast cells by the LiAc/SS-DNA/PEG procedure. Yeast 11:355–360 [DOI] [PubMed] [Google Scholar]
- 31. Gillison ML, et al. 2000. Evidence for a causal association between human papillomavirus and a subset of head and neck cancers. J. Natl. Cancer Inst. 92:709–720 [DOI] [PubMed] [Google Scholar]
- 32. Gonzalez SL, Stremlau M, He X, Basile JR, Munger K. 2001. Degradation of the retinoblastoma tumor suppressor by the human papillomavirus type 16 E7 oncoprotein is important for functional inactivation and is separable from proteasomal degradation of E7. J. Virol. 75:7583–7591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Goodwin EC, DiMaio D. 2000. Repression of human papillomavirus oncogenes in HeLa cervical carcinoma cells causes the orderly reactivation of dormant tumor suppressor pathways. Proc. Natl. Acad. Sci. U. S. A. 97:12513–12518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Goodwin EC, et al. 2000. Rapid induction of senescence in human cervical carcinoma cells. Proc. Natl. Acad. Sci. U. S. A. 97:10978–10983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Helt AM, Galloway DA. 2001. Destabilization of the retinoblastoma tumor suppressor by human papillomavirus type 16 E7 is not sufficient to overcome cell cycle arrest in human keratinocytes. J. Virol. 75:6737–6747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Huh K, et al. 2007. Human papillomavirus type 16 E7 oncoprotein associates with the cullin 2 ubiquitin ligase complex, which contributes to degradation of the retinoblastoma tumor suppressor. J. Virol. 81:9737–9747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jeon S, Allen-Hoffmann BL, Lambert PF. 1995. Integration of human papillomavirus type 16 into the human genome correlates with a selective growth advantage of cells. J. Virol. 69:2989–2997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jones DL, Thompson DA, Munger K. 1997. Destabilization of the RB tumor suppressor protein and stabilization of p53 contribute to HPV type 16 E7-induced apoptosis. Virology 239:97–107 [DOI] [PubMed] [Google Scholar]
- 39. Kalejta RF, Brideau AD, Banfield BW, Beavis AJ. 1999. An integral membrane green fluorescent protein marker, Us9-GFP, is quantitatively retained in cells during propidium iodide-based cell cycle analysis by flow cytometry. Exp. Cell Res. 248:322–328 [DOI] [PubMed] [Google Scholar]
- 40. Liu X, Clements A, Zhao K, Marmorstein R. 2006. Structure of the human papillomavirus E7 oncoprotein and its mechanism for inactivation of the retinoblastoma tumor suppressor. J. Biol. Chem. 281:578–586 [DOI] [PubMed] [Google Scholar]
- 41. Longworth MS, Dyson NJ. 2010. pRb, a local chromatin organizer with global possibilities. Chromosoma 119:1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Massimi P, Shai A, Lambert P, Banks L. 2008. HPV E6 degradation of p53 and PDZ containing substrates in an E6AP null background. Oncogene 27:1800–1804 [DOI] [PubMed] [Google Scholar]
- 43. Morris EJ, Dyson NJ. 2001. Retinoblastoma protein partners. Adv. Cancer Res. 82:1–54 [DOI] [PubMed] [Google Scholar]
- 44. Nevins JR. 1992. E2F: a link between the Rb tumor suppressor protein and viral oncoproteins. Science 258:424–429 [DOI] [PubMed] [Google Scholar]
- 45. Ohlenschlager O, et al. 2006. Solution structure of the partially folded high-risk human papilloma virus 45 oncoprotein E7. Oncogene 25:5953–5959 [DOI] [PubMed] [Google Scholar]
- 46. Patrick DR, Oliff A, Heimbrook DC. 1994. Identification of a novel retinoblastoma gene product binding site on human papillomavirus type 16 E7 protein. J. Biol. Chem. 269:6842–6850 [PubMed] [Google Scholar]
- 47. Phelps WC, Munger K, Yee CL, Barnes JA, Howley PM. 1992. Structure-function analysis of the human papillomavirus type 16 E7 oncoprotein. J. Virol. 66:2418–2427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Romanowski B. 2011. Long term protection against cervical infection with the human papillomavirus: review of currently available vaccines. Hum. Vaccin. 7:161–169 [DOI] [PubMed] [Google Scholar]
- 49. Schneider-Gadicke A, Schwarz E. 1987. Transcription of human papillomavirus type-18 DNA in human cervical carcinoma cell lines. Haematol. Blood Transfus. 31:380–381 [DOI] [PubMed] [Google Scholar]
- 50. Sherr CJ. 2000. The Pezcoller lecture: cancer cell cycles revisited. Cancer Res. 60:3689–3695 [PubMed] [Google Scholar]
- 51. Smotkin D, Wettstein FO. 1987. The major human papillomavirus protein in cervical cancers is a cytoplasmic phosphoprotein. J. Virol. 61:1686–1689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Todorovic B, et al. 2011. Systematic analysis of the amino acid residues of human papillomavirus type 16 E7 conserved region 3 involved in dimerization and transformation. J. Virol. 85:10048–10057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. White EA, et al. 2012. Systematic identification of interactions between host cell proteins and E7 oncoproteins from diverse human papillomaviruses. Proc. Natl. Acad. Sci. U. S. A. 109:E260–E267 [DOI] [PMC free article] [PubMed] [Google Scholar]