

Abstract
The Epstein-Barr virus (EBV) latent-to-lytic switch is mediated by the viral proteins BZLF1 (Z), BRLF1 (R), and BRRF1 (Na). Since we previously showed that DNA-damaging agents (including chemotherapy and irradiation) can induce EBV lytic reactivation and recently demonstrated that wild-type p53 contributes to lytic reactivation, we investigated the role of the ATM kinase during EBV reactivation. ATM phosphorylates and activates p53, as well as numerous other substrates involved in the cellular DNA damage response. Using an ATM inhibitor (KU55933), we found that ATM activity is required for efficient induction of EBV lytic gene expression by a variety of different stimuli, including a histone deacetylase (HDAC) inhibitor, the transforming growth factor β (TGF-β) cytokine, a demethylating agent (5-azacytidine), B cell receptor engagement with anti-IgG antibody, hydrogen peroxide, and the proteosome inhibitor bortezomib. In EBV-infected AGS (gastric) cells, knockdown of ATM, or p53, expression inhibits EBV reactivation. Conversely, treatment of these cells with nutlin-3 (which activates p53 and ATM) robustly induces lytic reactivation in a p53- and ATM-dependent manner. The ability of the EBV R and Na proteins to induce lytic reactivation in EBV-infected AGS cells is ATM dependent. However, overexpression of Z induces lytic gene expression in the presence or absence of ATM activity. Our results suggest that ATM enhances Z promoter activity in the context of the intact EBV genome and that p53 contributes to the ATM effect. Nevertheless, since we found that ATM inhibitors also reduce lytic reactivation in Burkitt lymphoma cells that have no p53, additional ATM substrates must also contribute to the ATM effect.
INTRODUCTION
Epstein-Barr virus (EBV) is a gammaherpesvirus that is the cause of infectious mononucleosis and is associated with a variety of epithelial and B cell cancers, including nasopharyngeal carcinoma (NPC), a subset of gastric carcinomas, Burkitt lymphoma (BL), and Hodgkin's disease (77, 102). Like all herpesviruses, EBV can infect cells in either latent or lytic forms. Following infection of humans, EBV establishes long-term viral latency in the memory B cell compartment but can be reactivated to undergo the lytic form of infection following plasma cell differentiation (51). EBV infection of normal epithelial cells generally results in lytic infection (32, 93, 94), although EBV+ epithelial cell tumors such as NPCs and gastric carcinomas are primarily composed of cells with latent forms of infection (48, 77). Both the latent and lytic forms of EBV infection are essential for the long-term success of the virus, and the latent-lytic switch is tightly controlled by both cellular and viral factors.
The two EBV immediate-early (IE) proteins, BZLF1 (Z, also known as Zta, ZEBRA, or EB1) and BRLF1 (R), are transcription factors that activate expression of lytic viral promoters, and overexpression of either the Z or R IE protein is sufficient to reactivate the lytic form of EBV infection in many latently infected cell lines (2, 16, 18–20, 23, 27, 34, 39, 47, 48, 57, 69, 76, 80, 89, 99). In addition, expression of the BRRF1-encoded early gene product, Na, is sufficient to reactivate the lytic form of EBV infection in some latently infected epithelial cell lines, due to the ability of Na to activate the Z promoter indirectly through cellular factors (38, 42). Therefore, the cellular and viral proteins that regulate Z, R, and Na gene expression play a key role in determining if EBV infection is latent or lytic.
Cells containing the latent forms of EBV infection can be switched to the lytic form of infection by using a variety of different lytic reactivation-inducing stimuli, including histone deacetylase (HDAC) inhibitors (HDACi) (29, 58), B cell receptor (BCR) engagement with anti-IgG antibody (65, 88), transforming growth factor β (TGF-β) (25), the proteosome inhibitor bortezomib (85), the demethylating agent 5-azacytidine (4), radiation (95), and chemotherapy agents (28). These stimuli are thought to promote lytic EBV reactivation primarily through their ability to regulate cellular factors that control the activity of the Z and R promoters. In addition, cellular factors that regulate Z and R transcriptional function likewise contribute to the balance between latent and lytic infection; for example, we recently reported that the B cell-specific Oct-2 protein promotes viral latency by directly interacting with Z and inhibiting its transcriptional function, while the cellular Oct-1 protein promotes viral reactivation by interacting directly with R and enhancing its transcriptional function (78, 79).
At least two of the various different lytic reactivation-inducing stimuli (chemotherapy and radiation) are well-known DNA-damaging agents, although the mechanism(s) by which these two particular agents induce lytic reactivation has not been well studied. Since we recently showed that the tumor suppressor protein p53 greatly facilitates lytic reactivation induced by overexpression of the EBV Na (BRRF1) and R proteins (38), and DNA damage potently activates p53 transcriptional function (84, 86), DNA-damaging agents could potentially induce EBV reactivation (at least partially) via activation of p53. Recently, p53 was also shown to be required for HDACi-mediated lytic reactivation in both EBV-positive nasopharyngeal carcinoma cell lines and lymphoblastoid B cell lines (13, 17, 38), further confirming a critical role of p53 function in controlling EBV reactivation in a variety of different settings.
The ataxia telangiectasia-mutated (ATM) protein is a cellular serine/threonine kinase that plays a key role in mediating the cellular response to DNA damage (5, 41, 45). In response to DNA damage, ATM becomes activated via phosphorylation at Ser-1981 and phosphorylates multiple substrates, including p53, γH2AX, KAP1, and Chk2 (87). ATM-mediated phosphorylation of p53 on serine 15 plays a critical role in activating p53 function in response to DNA damage (101), suggesting that the ability of DNA-damaging agents to reactivate EBV might be mediated through ATM. The ATM kinase is also activated in response to reactive oxygen species (ROS) in the absence of DNA damage (36, 37). ROS activates ATM by inducing a conformational alteration in the protein that is not associated with Ser-1981 phosphorylation (37), and thus, measuring the amount of ATM phosphorylation at this site (although commonly used to monitor ATM activation in cells) does not detect ROS-mediated ATM activation.
Interestingly, ATM inhibits the ability of EBV to transform primary B cells (72), and ATM kinase expression is reduced in EBV-associated nasopharyngeal carcinomas (9). Of note, several EBV latent proteins (EBNA1, EBNA3C, and LMP1) have been shown to inhibit the DNA damage response (10, 35), and LMP1 in particular has been reported to inhibit ATM expression (35). In regard to lytic EBV viral DNA replication, one report has suggested that ATM promotes lytic viral DNA replication (55), while another paper has stated that ATM is not required for lytic viral DNA replication (50). However, whether ATM regulates the initial stage of the latent-to-lytic switch in EBV-infected cells is unknown.
In this study, we have used an ATM kinase inhibitor, as well as ATM knockdown, to explore the role of ATM during EBV reactivation in response to multiple different types of lytic reactivation-inducing stimuli. We unexpectedly find that ATM not only plays a crucial role in facilitating viral reactivation in response to classic DNA-damaging agents such as chemotherapy but also is required for efficient lytic reactivation in response to HDACi, TGF-β, B cell receptor engagement with anti-IgG, the demethylating agent 5-azacytidine, the proteosome inhibitor bortezomib, and H2O2. ATM is also important for Na- and R-, but not Z-, induced lytic gene expression, suggesting that ATM enhances activation of the Z promoter by cellular and viral proteins. In contrast, we find that ATM is not required for EBV lytic DNA replication per se. Finally, although we show that p53 is important for efficient EBV reactivation in cell lines that express wild-type (WT) p53, we find that ATM activity is also required for efficient EBV reactivation in cell lines that do not express wild-type p53. These results suggest that ATM plays an unexpectedly important role in promoting lytic EBV reactivation via both p53-dependent and p53-independent mechanisms.
MATERIALS AND METHODS
Cell lines.
AGS-Akata/BX1 cells (wild-type p53) were a gift from Lindsey Hutt-Fletcher and are gastric carcinoma cells superinfected with the Akata strain of EBV, which contains the green fluorescent protein (GFP) open reading frame under the control of the BXLF1 promoter (8, 63). AGS-Akata cells were maintained in F-12 medium supplemented with 10% fetal bovine serum (FBS), 1% penicillin-streptomycin (Pen-Strep), and G418 (400 μg/ml). EBV-negative AGS gastric carcinoma cells (obtained from the American Type Culture Collection [ATCC]) were maintained in F-12 medium supplemented with 10% FBS and 1% Pen-Strep. Mutu I and Kem I cells (a gift from Alan Rickinson and Jeff Sample) are EBV-positive Burkitt lymphoma type I latency cells with mutant p53 and were maintained in RPMI 1640 supplemented with 10% FBS and 1% Pen-Strep (33, 41, 73, 81, 96). Kem III cells (a gift from Alan Rickinson and Jeff Sample) are EBV-positive Burkitt lymphoma type III cells with wild-type p53 (41, 81). Raji cells (EBV-positive Burkitt lymphoma, from the ATCC) were maintained in RPMI 1640 supplemented with 10% FBS and Pen-Strep. Akata-GFP BL cells (a gift from Kenzo Takada [received from Bill Sugden]) are derived from a type I latency Burkitt lymphoma line (Akata BL) that lost the endogenous EBV genome and then was superinfected with an Akata EBV containing inserted GFP and G418 resistance genes as previously described (46). Akata (and Akata-GFP) BLs do not express p53 (26) and were maintained in RPMI 1640 supplemented with 10% FBS, 1% Pen-Strep, and 500 μg/ml G418. EBV-infected 293 cells (a gift from H.-J. Delecluse) have been previously described (21, 27) and were maintained in Dulbecco modified Eagle medium (DMEM) supplemented with hygromycin (100 μg/ml), 10% FBS, and Pen-Strep. EBV-negative Daudi cells (a gift from Kenzo Takada via Bill Sugden) are Burkitt lymphoma cells that lost the endogenous EBV genome and were maintained in RPMI 1640 supplemented with 10% FBS and 1% Pen-Strep (70). Lymphoblastoid cell line (LCL) donor 4 cells are early-passage lymphoblastoid B cells transformed with wild-type B95.8 EBV as previously described and were maintained in RPMI 1640 supplemented with 10% FBS and 1% Pen-Strep (42). LCL donor 4 cells are thought to have wild-type p53 based on the literature (96).
To generate AGS-WT (B95.8) and AGS-PKmut cell lines, previously described 293 cells latently infected with wild-type (WT) B95.8 or PK (BGLF4) mutant EBV (66) were transfected with Z and R and gp110 expression plasmids to induce virus production. Three days posttransfection, virus was harvested and filtered through an 0.8-μm filter. EBV-negative Daudi cells were then incubated with the WT or PKmut virus for 1 h and then cocultured with EBV-negative AGS cells for several days. The Daudi cells were then removed, and infected AGS cells were selected with hygromycin (100 μg/ml) to establish stable cell lines.
Plasmids.
Plasmid DNA was purified using Qiagen maxiprep columns according to the manufacturer's protocol. pSG5 was obtained from Stratagene. pSG5-Z, containing the genomic Z sequence transcribed from a simian virus 40 (SV40) promoter, and pSG5-R, containing the genomic sequence encoding R beginning from the second exon but lacking the Na-encoding first exon, were a gift from Diane Hayward (39). pSG5-FLAGNa contains a FLAG tag on the amino terminus of the Na open reading frame (38). pRK-BALF4 codes for the EBV glycoprotein 110 and was a gift from H.-J. Delecluse (71).
DNA transfection.
DNA was transfected into 293 cells using Lipofectamine 2000 reagent (Invitrogen) according to the manufacturer's protocol. AGS-Akata cells were transfected with DNA using Fugene HD reagent (Promega) according to the manufacturer's protocol.
Chemical reagents.
The following chemical reagents were used to induce EBV lytic infection, and cells were treated for 48 h unless otherwise stated: phorbol 12-myristate 13-acetate (TPA; 20 ng/ml; Sigma), paclitaxel (5 μg/ml; Calbiochem), sodium butyrate (3 mM; Sigma), cis-diammineplatinum(II) dichloride (cisplatin; 10 μM; Sigma), suberoylanilide hydroxamic acid (SAHA; 5 μM; Cayman Chemical), hydrogen peroxide (H2O2; 300 μM; Mallinckrodt Chemical), recombinant human TGF-β (5 μg/ml; R&D Systems), anti-human IgG (10 μg/ml; Sigma), 5-aza-2′-deoxycytidine (5′-azacytidine; 1 μM; Sigma), nutlin-3 (10 μM; Calbiochem), and bortezomib (20 nM; Selleck Chemical). For ATM inhibitor experiments, cells were pretreated for 1 h with the ATM inhibitor KU55933 (10 μM; Calbiochem).
Virus titration assay.
Virus titration assays were performed as previously described (42). EBV-infected 293 cells were transfected with BZLF1 and BALF4 (gp110) expression vectors and treated with or without the ATM inhibitor KU55933. Experiments with each condition were performed in triplicate. Supernatant was harvested 72 h posttransfection and filtered through an 0.8-μm-pore-size filter. Raji cells (2 × 105 cells/infection) were infected with various amounts of virus and incubated at 37°C. Phorbol 12-myristate 13-acetate (TPA; 20 ng/ml) and sodium butyrate (3 mM final concentration) were added 24 h after infection. GFP-positive Raji cells were counted 48 h postinfection to determine viral titer.
Western blot analysis.
Western blot analysis was performed as previously described (1, 6). Cell lysates were harvested in SUMO buffer containing protease inhibitor cocktail (Roche) and quantified using the DC protein assay kit, also known as the SUMO protein assay, according to the manufacturer's protocol (Bio-Rad). Equivalent amounts of protein were separated in sodium dodecyl sulfate-10% polyacrylamide gels and transferred to membranes. Membranes were blocked in phosphate-buffered saline (PBS) containing 5% milk and 0.1% Tween 20 solution and incubated with primary antibody. Immunoblots were probed with the following antibodies: anti-FLAG (Sigma; 1:2,000), anti-R (Argene; 1:250), anti-Z (Santa Cruz BZ-1; 1:250), anti-EAD (BMRF1; Vector; 1:250), anti-β-actin (Sigma; 1:5,000), anti-p53 (Santa Cruz; 1:250), anti-LMP1 (Dako; 1:250), anti-p53pSer15 (Cell Signaling Technology; 1:1,000), and anti-ATM (Cell Signaling Technology; 1:1,000).
Infection and packaging of lentivirus.
Lentivirus vectors expressing short hairpin RNAs (shRNAs) against a control sequence (Sigma catalog no. SHC002 or Addgene plasmid 1864 [82]), ATM (Open Biosystems catalog no. RHS4533-NM_000051), or p53 (Open Biosystems catalog no. RHS4533-NM_000546) were used to produce lentivirus as previously described according to the Open Biosystems protocol (79). 293T cells were transfected with the lentivirus expression plasmids and the packaging plasmids psPAX2 (Addgene plasmid 12260, a gift of Didier Trono) and VSVG (Addgene plasmid 12259, a gift of Didier Trono) using Lipofectamine 2000 reagent (Invitrogen). Supernatant was collected 3 days posttransfection and filtered through an 0.8-μm filter. The indicated EBV-positive cell lines were then transduced by incubation with filtered medium. After 3 days of incubation, stable cell lines were selected with 1 μg/ml puromycin. AGS-Akata cell lines were continuously passaged in puromycin (1 μg/ml) and G418 (400 μg/ml). Mutu I cells were selected for 18 days in puromycin (1 μg/ml) prior to induction with TGF-β.
Reverse transcription-PCR (RT-PCR).
RNA was isolated from Mutu I cells treated with or without ATM inhibitor (10 μM) and/or TGF-β (5 μg/ml) 18 h postinduction using a Qiagen RNeasy minikit. RNA was quantitated, DNase treated, and used to make cDNA with the ImProm-II reverse transcription system kit (Promega) according to the manufacturer's protocol. PCR was then performed using the cDNA as a template to detect Z and beta-2 microglobulin (B2M) transcripts. The following primers were used for PCR: Z-For (5′-CACGGTAGTGCTGCAGCAGTTGC-3′), Z-Rev (5′-CCCAGAATCAACAGACTAACCAAGC-3′), B2M-For (5′-TTCTGGCCTGGAGGGCATCC-3′), and B2M-Rev (5′-ATCTTCAAACCTGCATGATG-3′).
RESULTS
ATM kinase activity is required for activation of efficient EBV early lytic gene expression by various lytic reactivation-inducing stimuli in several different Burkitt lymphoma lines.
To examine if ATM activity mediates the ability of the DNA damage-inducing chemotherapy agent paclitaxel to induce EBV early lytic gene expression, we treated Akata BL cells with or without paclitaxel in the presence or absence of the ATM inhibitor KU55933. KU55933 is an ATP competitor highly specific to the ATM kinase (40). As anticipated, we found that paclitaxel was unable to activate expression of the EBV lytic viral proteins, BMRF1 and Z, in the presence of the ATM inhibitor (Fig. 1A). Unexpectedly, however, in the same experiment we found that the ATM inhibitor also prevented lytic reactivation in response to the HDAC inhibitor sodium butyrate. Since the Akata BL line does not express p53 (26), these results suggest that ATM promotes lytic EBV reactivation by both HDACi and paclitaxel in a p53-independent manner.
Fig 1.

ATM kinase activity is required for activation of early lytic EBV protein expression by various lytic reactivation-inducing stimuli in EBV+ B cells. Akata EBV+ (A), Kem I (B), or Mutu I (C to E) cells were pretreated with the ATM inhibitor KU55933 (10 μM) for 1 h and induced with various lytic reactivation-inducing stimuli, including sodium butyrate (butyrate, 3 mM), paclitaxel (5 μg/ml), TGF-β (5 μg/ml), anti-human IgG (αIgG, 10 μg/ml), bortezomib (20 nM), or 5′-azacytidine (5′-Aza, 1 μM). At 2 days postinduction, cells were harvested and Western blot analysis was performed to examine lytic EBV protein expression using antibodies against BMRF1, Z, or R. Antibody against LMP1 was used to demonstrate type III latency following 5′-azacytidine treatment in Mutu I cells (E). β-Actin was used as a loading control. DMSO, dimethyl sulfoxide.
To determine if ATM activity is required for EBV early lytic gene expression in multiple different cell lines and to determine whether this requirement is dependent upon the type of lytic reactivation-inducing agent used, we treated two additional EBV+ BL lines (Mutu I and Kem I) with different types of lytic reactivation-inducing agents in the presence and absence of the ATM inhibitor. The results of these studies indicated that ATM is also required for efficient lytic reactivation in response to the cytokine TGF-β (Fig. 1B and C) in both Kem I cells and Mutu I cells. Similarly, the amount of lytic protein expression induced following activation of the B cell receptor signaling pathway (using anti-IgG) in Mutu I cells was likewise attenuated in the presence of the ATM inhibitor (Fig. 1C). The ability of the proteosome inhibitor bortezomib to induce lytic protein expression in Mutu 1 cells was also reduced by ATM inhibition (Fig. 1D).
Finally, we also compared the abilities of the DNA-demethylating agent 5′-azacytidine to induce lytic protein expression, versus the switch between type I and type III latency, in the presence and absence of the ATM inhibitor in Mutu I cells. 5-Azacytidine has been shown to convert BL cells with a restricted form of viral latency (type I) to a less restricted form (type III) (62) and also to induce lytic protein expression (4). Although ATM inhibitor treatment had no effect on the ability of 5-azacytidine to induce expression of the LMP1 protein (expressed in type III but not type I latency), it blocked 5-azacytidine-mediated activation of lytic proteins (Fig. 1E). Together, these results demonstrate that activity of the ATM kinase is important for activation of EBV early lytic gene expression in response to a variety of different types of lytic reactivation-inducing stimuli in EBV-positive B cells. Additionally, since each of the BL lines used in the above studies does not express wild-type p53 (26, 41, 96), these results suggest that the ATM effect is independent of wild-type p53 function.
ATM activity is required for activation of EBV early lytic gene expression by various lytic reactivation-inducing stimuli in epithelial lines.
We next asked if ATM kinase activity is also important for EBV reactivation in epithelial cells. For these studies, an EBV-positive gastric carcinoma line, AGS-Akata, was treated with several different lytic reactivation-inducing stimuli, including sodium butyrate, SAHA (an HDACi), and cisplatin, in the presence or absence of the ATM inhibitor. As shown in Fig. 2A, the ATM inhibitor reduced the amount of lytic Z and BMRF1 protein expression induced by all three agents tested. Since ROS have recently been shown to activate ATM kinase activity (36, 37), we next asked if hydrogen peroxide treatment of AGS-Akata cells reactivates EBV and, if so, whether this effect requires ATM. We found that hydrogen peroxide efficiently induces lytic protein expression in AGS-Akata cells and that this effect is indeed ATM dependent (Fig. 2B). These results suggest that ATM kinase activity is also important for EBV reactivation in EBV-positive epithelial cells.
Fig 2.

ATM kinase activity is required for activation of early lytic EBV protein expression by various inducing stimuli in EBV+ epithelial cells. (A) AGS-Akata cells were pretreated with the ATM kinase inhibitor KU55933 (10 μM) for 1 h and induced with various inducing stimuli, including sodium butyrate (butyrate, 3 mM), SAHA (5 μM), and cisplatin (10 μM). Cells were harvested 2 days posttreatment, and Western blot analysis was performed to examine lytic BMRF1 and Z protein expression. (B) AGS-Akata cells were pretreated with the ATM kinase inhibitor KU55933 (10 μM) for 1 h and induced with hydrogen peroxide (H2O2, 300 μM). After 2 days, the cells were harvested and Western blot analysis was performed to examine lytic EBV infection using antibodies against BMRF1, Z, and R. β-Actin was used as a loading control. DMSO, dimethyl sulfoxide.
Knockdown of ATM inhibits EBV early lytic gene expression.
Although the ATM inhibitor KU55933 is reported to be highly specific to ATM activity, we next used knockdown technology to confirm whether ATM expression is required for efficient viral reactivation in AGS-Akata cells. AGS-Akata cells were infected with two different lentiviruses expressing different shRNAs directed against ATM, or two different control shRNA vectors, and stably infected cell lines were selected with puromycin. The various AGS-Akata lines were then treated with or without hydrogen peroxide, and immunoblot analysis was performed to examine the amount of lytic EBV protein expression. As shown in Fig. 3A, both of the ATM shRNA-expressing lines had decreased ATM expression compared to the lines infected with either of the two control shRNA vectors. Furthermore, neither of the two cell lines knocked down for ATM expression could be induced into the lytic form of viral protein expression by hydrogen peroxide (Fig. 3A). These results confirm that the ATM kinase is essential for efficient EBV lytic reactivation by ROS.
Fig 3.

Knockdown of ATM inhibits early lytic EBV protein expression. (A) AGS-Akata cells were transduced with two different lentiviruses expressing either a control shRNA (clones 1 and 2) or two different ATM-targeting shRNAs (clones 1 and 2). Stable cell lines were selected with puromycin and induced with hydrogen peroxide (300 μM). Two days later, cell lysates were harvested and Western blot analysis was performed to examine BMRF1 and Z lytic protein expression. Antibody against ATM was used to confirm that ATM was knocked down. β-Actin was used as a loading control. (B) Mutu I cells were transduced with lentiviruses expressing either two different control (clones 1 and 2) or two different ATM-targeting (clones 1 and 2) shRNAs. Cells were selected with puromycin for 18 days and induced with TGF-β (5 μg/ml). One day postinduction, cell lysates were harvested and Western blot analysis was performed to examine BMRF1 and Z lytic protein expression. Antibody against ATM was used to confirm knockdown of ATM expression. β-Actin was used as a loading control. DMSO, dimethyl sulfoxide.
We next used shRNA against ATM to determine if knocking down ATM inhibited TGF-β-induced lytic reactivation in Mutu I BL cells. Mutu I cells were transduced with lentiviruses expressing either two different control or two different ATM-targeting shRNAs. Cells were selected with puromycin for 18 days and induced with or without TGF-β for 2 days. As shown in Fig. 3B, TGF-β did not induce lytic protein expression of BMRF1 or Z as efficiently when ATM expression levels were decreased. These results suggest that ATM is also important for TGF-β-induced EBV early lytic protein expression.
ATM is required for Na- and R-, but not Z-, induced EBV lytic protein expression in AGS-Akata cells.
Since we previously showed that lytic EBV reactivation can be induced in AGS-Akata cells following overexpression of the viral Z, R, or Na protein (38), we next examined the effect of the ATM kinase inhibitor on the ability of each of these viral proteins to induce lytic protein expression. AGS-Akata cells were transfected with control, Na, Z, or R expression vectors in the presence or absence of the ATM inhibitor, and lytic protein expression was examined by Western blot analysis. The ATM inhibitor reduced the amount of endogenous viral BZLF1 and early BMRF1 expression induced by either Na or R transfection (Fig. 4A and B). However, the ATM inhibitor did not affect the ability of transfected Z protein to activate BMRF1 expression (Fig. 4B). These results suggest that ATM activity may be primarily required for activation of the Z promoter, since bypassing the need to activate this promoter by supplying a Z expression vector abrogated the need for ATM.
Fig 4.

ATM kinase activity is required for Na- and R-, but not Z-, induced early lytic EBV protein expression. (A and B) AGS-Akata cells were transfected with control, FLAG-Na (A), Z, or R (B) expression plasmids and treated with or without the ATM kinase inhibitor KU55933 (10 μM). At 48 h after transfection, the cells were harvested and Western blot analysis was performed to examine BMRF1, R, and Z lytic protein expression. The asterisk indicates endogenous Z expression (Akata strain), which runs at a higher molecular weight than does transfected Z (B95.8 strain). A long exposure of Z is included to demonstrate R-induced endogenous Z expression. FLAG antibody was used to detect Na expression. β-Actin was used as a loading control. (C) Mutu I cells were treated with ATM inhibitor (10 μM) for 1 h followed by treatment with dimethyl sulfoxide or TGF-β (5 μg/ml). At 18 h postinduction, RNA was isolated from the cells and treated with DNase and RT-PCR was performed using primers to detect BZLF1 or beta-2 microglobulin (B2M) transcripts.
To determine if ATM kinase activity is important for activation of BZLF1 gene transcription, RNA was isolated from Mutu I cells treated with or without the ATM inhibitor, in the presence or absence of the TGF-β cytokine. RT-PCR was performed on the RNA samples using primers to amplify the BZLF1 transcript or the control beta-2 microglobulin (B2M) cellular gene transcript. As shown in Fig. 4C, BZLF1 transcription was induced in the presence of TGF-β, and the ATM inhibitor blocked this effect. These results further support the hypothesis that ATM activity is important for activation of the BZLF1 promoter.
The EBV-PK is not required for lytic induction by hydrogen peroxide.
The EBV protein kinase (EBV-PK; encoded by the BGLF4 gene) has been shown to activate ATM via phosphorylation of Tip60, and it has been suggested that EBV-PK-mediated activation of ATM is important for viral replication (55). Therefore, we determined if EBV-PK is required for hydrogen peroxide-mediated EBV lytic reactivation. AGS cell lines stably infected with wild-type (WT) B95.8 virus, or a previously described EBV-PK mutant virus (66), were generated and shown to have the expected phenotype in regard to expressing the hyperphosphorylated form of the BMRF1 protein (an easily observed EBV-PK-dependent form of BMRF1) (15, 31) (Fig. 5). To determine if EBV-PK is required for induction of Z expression by H2O2, AGS-PKmut cells were treated with or without H2O2 in the presence or absence of the ATM inhibitor (KU55933) and examined by Western blot analysis for Z expression 24 h later (Fig. 5). H2O2 treatment activated Z expression in cells lacking the EBV-encoded protein kinase, and this effect required ATM. Therefore, these results suggest that EBV-PK-mediated activation of ATM is not required for induction of lytic viral reactivation by chemicals such as hydrogen peroxide, which can efficiently activate ATM in EBV-negative cells (37).
Fig 5.
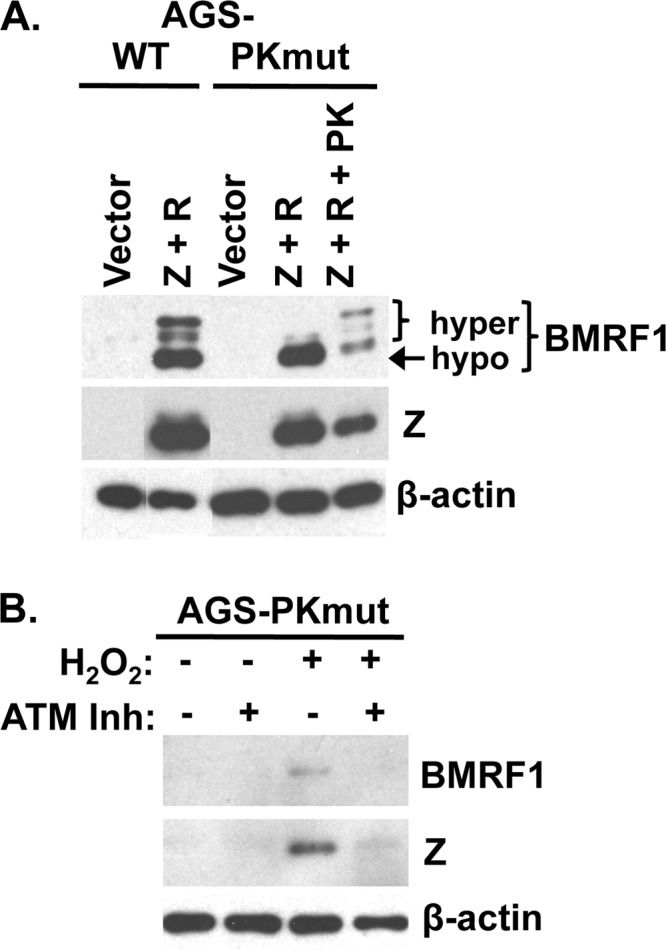
EBV-PK is not required for early lytic protein expression induced by hydrogen peroxide. (A) AGS-WT and PKmut cells were generated and transfected with the indicated combinations of Z, R, and EBV-PK expression vectors, and Western blot analysis was performed to detect hyper- and hypophosphorylated BMRF1. Antibodies against Z and β-actin were also included as controls. (B) AGS-PKmut cells were treated with or without H2O2 (300 μM) in the presence or absence of the ATM inhibitor KU55933 (10 μM), and Western blot analysis using antibody against BMRF1 or Z was used to detect early lytic protein expression. β-Actin was included as a loading control.
p53 is required for hydrogen peroxide-induced EBV lytic reactivation.
Since reactive oxygen species are known to activate p53 (14) and we previously demonstrated that wild-type p53 is important for Na- and R-mediated EBV lytic reactivation in AGS-Akata cells (38), we next asked whether the ability of hydrogen peroxide treatment to induce lytic reactivation in this cell type requires p53. AGS-Akata cells were knocked down for ATM or p53 expression using lentiviruses expressing shRNAs against ATM or p53 and then treated with or without hydrogen peroxide. In cells knocked down for either ATM or p53 expression, the lytic reactivation-inducing effect of hydrogen peroxide was highly attenuated (Fig. 6). Furthermore, we found that hydrogen peroxide treatment of AGS-Akata cells (which have wild-type p53) induces p53 phosphorylation at serine 15 and that this phosphorylation was reduced by both the ATM and p53 shRNAs. These results suggest that the ability of hydrogen peroxide to induce lytic viral reactivation in AGS-Akata cells is both ATM and p53 dependent and that ATM is required for efficient p53-Ser15 phosphorylation following hydrogen peroxide treatment of AGS-Akata cells.
Fig 6.
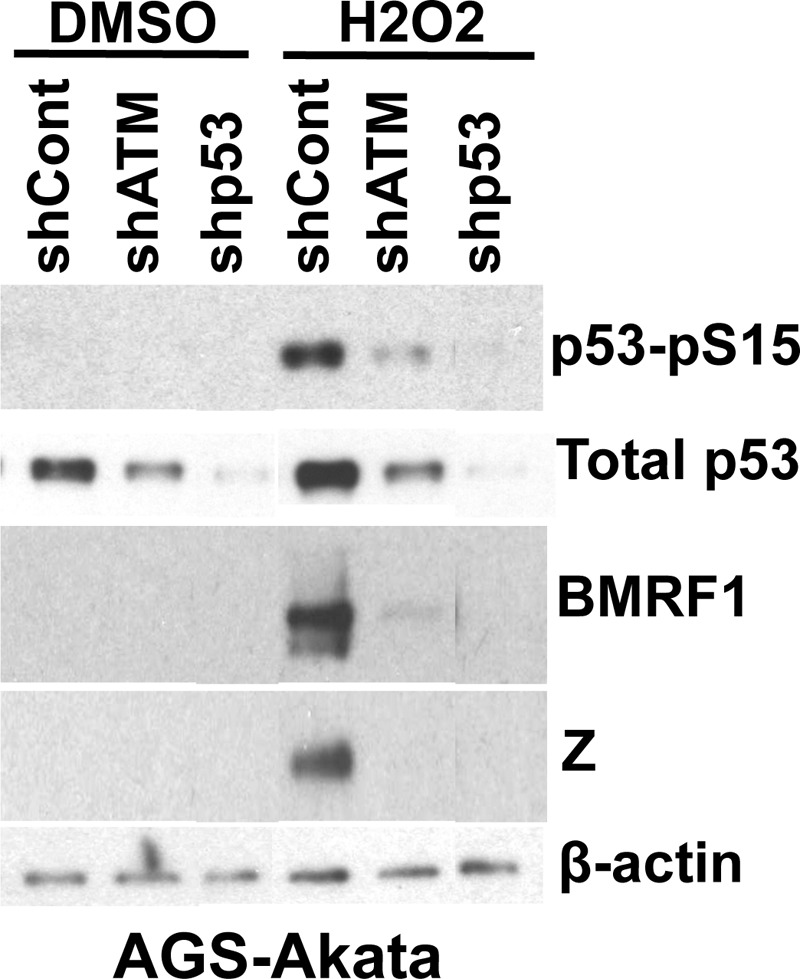
p53 and ATM are required for hydrogen peroxide-induced early lytic protein expression. AGS-Akata cells were knocked down for ATM or p53 expression using shRNA-expressing lentiviruses. Stable cell lines were treated with or without hydrogen peroxide (H2O2, 300 μM), and Western blot analysis was performed to detect BMRF1 or Z lytic protein expression. Western blots were also probed for total p53 and p53 phosphorylated on Ser15. β-Actin was used as a loading control. DMSO, dimethyl sulfoxide.
ATM is not required for lytic EBV DNA replication when Zp activation is bypassed by transfection of Z protein.
Whether ATM is required for EBV lytic DNA replication per se (downstream of early lytic protein activation) is unclear, since two previous papers have yielded conflicting results (50, 55). Therefore, to further investigate the requirement for ATM in EBV DNA replication, we overexpressed the Z protein in 293 cells latently infected with wild-type EBV in the presence or absence of the ATM inhibitor. Virus was harvested 3 days posttransfection, and virus titers were determined using a green Raji cell assay. As seen in AGS-Akata cells, we found that ATM is not required for Z-induced lytic protein expression in 293-EBV cells (Fig. 7B). Furthermore, we found that the ATM inhibitor certainly did not inhibit (and in fact somewhat increased) Z-induced virus production (Fig. 7A). Similar results were obtained in a second experiment. These results suggest that ATM is not required for lytic EBV DNA replication per se, at least when the Z protein is overexpressed and the need to activate the Zp in the endogenous viral genome is bypassed.
Fig 7.
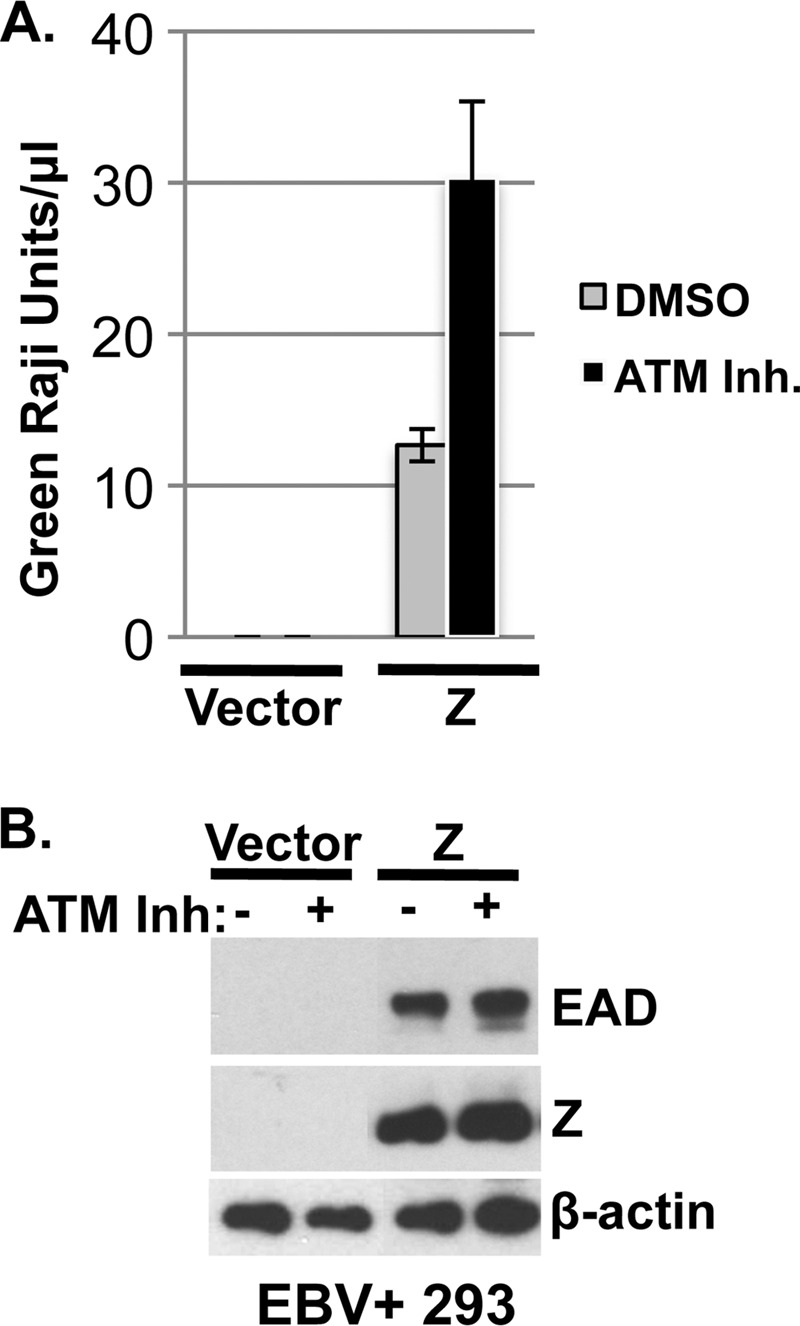
ATM kinase activity is not required for Z-induced EBV virus production. EBV+ 293 cells were transfected with or without Z and treated with either dimethyl sulfoxide (DMSO) or the ATM inhibitor KU55933 (10 μM). Each condition was performed in triplicate. (A) Three days posttransfection, supernatant was collected and filtered and virus titers were determined using a green Raji unit titration assay. The number of green Raji units per μl of virus was counted and graphed for each condition. (B) Western blot analysis of the transfected EBV+ 293 cell lysates was performed to examine Z, BMRF1, and β-actin expression levels.
Nutlin-3 activates EBV lytic protein expression in B cell lines that have wild-type p53 (Kem III and LCLs).
Since wild-type p53 activation is important for EBV lytic reactivation in at least a subset of EBV-positive cell lines, we next asked if nutlin-3, a drug which inhibits the interaction between MDM2 and p53 and thus stabilizes p53 expression (92), can reactivate EBV lytic infection in B cell lines that have wild-type p53. The Kem III BL line and an early-passage lymphoblastoid cell line (LCL) (LCL donor 4), which both express wild-type p53 (41, 96), were treated with nutlin-3 or vehicle control for 2 days, and immunoblot analysis was performed to examine EBV lytic protein expression, total p53 protein expression, and p53-Ser15 phosphorylation (Fig. 8). In both cell lines, BMRF1 and Z expression was upregulated in response to nutlin-3. Additionally, total p53 and p53-Ser15 phosphorylation was enhanced, as expected, by nutlin-3 treatment. In contrast, nutlin-3 treatment did not induce lytic reactivation in EBV+ BL lines (including Kem I and Mutu I) that do not express wild-type p53 (data not shown). Together, these results demonstrate that nutlin-3 can induce EBV lytic reactivation in EBV-infected B cells that express wild-type p53.
Fig 8.

Nutlin-3 induces EBV lytic protein expression in a p53- and ATM-dependent manner. (A and B) Kem III (A) or LCL donor 4 (B) cells were treated with dimethyl sulfoxide (DMSO) or nutlin-3 (10 μM) for 2 days, and cells were harvested for Western blot analysis. Antibodies against BMRF1 and Z were used to examine EBV lytic protein expression, and antibodies against total p53 or p53-phospho-Ser15 were used to confirm activation of p53. β-Actin was used as a loading control. (C) AGS-Akata cells transduced with lentivirus expressing either control or shRNA against p53 were treated with DMSO or nutlin-3 (10 μM) for 2 days, and Western blot analysis was performed on cell lysates using antibodies against BMRF1, Z, p53-phospho-Ser15, and β-actin. (D) LCL donor 4 cells were pretreated with DMSO or the ATM inhibitor KU55933 for 1 h and induced with or without nutlin-3 for 48 h. Western blot analysis was performed to examine BMRF1, p53-phospho-Ser15, and β-actin expression.
Both p53 and ATM kinase activity are required for nutlin-3-induced EBV lytic reactivation.
We next examined whether p53 activity is required for the ability of nutlin-3 to induce EBV lytic protein expression. AGS-Akata cells stably infected with control or p53 shRNA lentiviruses were treated with or without nutlin-3 and examined for BMRF1, Z, and p53-pSer15 expression by Western blot analysis (Fig. 8C). Nutlin-3 treatment efficiently induced lytic viral protein expression in AGS-Akata cells, and this effect required p53. These results confirm that p53 is required for the ability of nutlin-3 to induce EBV lytic reactivation. Of note, nutlin-3 was also recently shown to upregulate lytic gene transcription in Kaposi's sarcoma-associated herpesvirus (KSHV)-infected cells (97).
Since p53 can be activated by multiple different signaling pathways, and phosphorylation of p53 serine 15 can be mediated not only through ATM but also by the DNA-PK and ATR kinases (53, 91), we also asked if ATM kinase activity is specifically required for nutlin-3-induced EBV lytic reactivation. LCL donor 4 cells were treated with or without nutlin-3 in the presence or absence of the ATM inhibitor, and Western blot analysis was performed. As shown in Fig. 8D, the ATM inhibitor prevented nutlin-3-induced activation of BMRF1 expression, while it only partially blocked p53 serine 15 phosphorylation. These results suggest that ATM kinase activity is required for nutlin-3-induced EBV lytic reactivation.
DISCUSSION
We previously demonstrated that treatment of EBV-positive tumor cells with DNA-damaging agents such as chemotherapy or irradiation can induce lytic reactivation (28, 95), and we and others have recently shown that p53 is required for lytic reactivation by HDACi (13, 38). Since ATM is induced by DNA damage and plays an important role in activating p53 function, in this paper we have explored the role of ATM in the ability of chemotherapy, versus other lytic reactivation-inducing agents (with multiple different proposed mechanisms of action), to reactivate EBV. Using an ATM-specific inhibitor (KU55933), as well as an ATM knockdown approach, we found that ATM activity is important for EBV early lytic gene expression in both EBV-positive B cells and EBV-positive epithelial cells regardless of the lytic reactivation-inducing agent used. These studies reveal an unexpected and critical role of ATM activation in mediating the latent-to-lytic EBV switch.
In EBV-positive Burkitt lines, we found that ATM activity is required for lytic EBV gene expression induced by agents with multiple different proposed mechanisms of action, including HDACi, chemotherapy, TGF-β, B cell receptor engagement with anti-IgG, the proteosome inhibitor bortezomib, and the demethylating agent 5-azacytidine (Fig. 1). Additionally, our results show that ATM kinase activity is important for reactivation in the EBV-positive epithelial cell line AGS-Akata when cells are induced with HDACi (sodium butyrate or SAHA), the chemotherapy reagent cisplatin, or hydrogen peroxide (Fig. 2). It is important to note that most of these lytic induction agents have been reported to induce ATM activation, including HDACi (45), chemotherapy (22), hydrogen peroxide (36, 37), 5-azacytidine (41), bortezomib (43), and TGF-β (100). In addition, since it is now known that ROS induce ATM activation and BCR signaling results in ROS (60, 61), it is likely that BCR signaling also results in ATM activation. Although our results suggest that ATM kinase activity is important for EBV lytic reactivation by a variety of inducing stimuli, it is possible that under certain conditions, lytic reactivation may occur in the absence of ATM activity. Of note, we found that treatment of the Akata BL cell line (which is uniquely responsive to lytic induction in response to anti-IgG) with a high concentration, but not a lower concentration, of anti-IgG antibody appears to bypass the need for ATM activity (data not shown). We speculate that ATM-related kinases such as ATR may substitute for ATM activity in allowing EBV reactivation under some circumstances.
ATM activation occurs in response to both DNA damage and oxidative stress. While DNA damage induces phosphorylation of ATM at Ser-1981, hydrogen peroxide was recently shown to induce activation of ATM via a conformational alteration in the protein that does not result in Ser-1981 phosphorylation (37). Furthermore, oxidative stress has been shown to activate ATM without necessarily leading to activation of DNA damage-responsive genes (36). Recent studies demonstrated that hydrogen peroxide induces reactivation of another gammaherpesvirus, Kaposi's sarcoma-associated herpesvirus (KSHV) (56, 98). Additionally, hydrogen peroxide has been shown to induce Z transcription (52). Here we demonstrate that induction of lytic EBV infection by inducing oxidative stress with hydrogen peroxide requires ATM kinase activation (Fig. 2). It remains unclear if EBV lytic proteins, including Z, R, or Na, are able to induce ROS. However, the KSHV R homologue (KSHV RTA) has been shown to activate ROS, and this effect is important for RTA-mediated viral reactivation (56, 98). Therefore, in the future it will be interesting to determine if one or more of the EBV lytic proteins likewise induces ROS and promotes EBV lytic infection at least partially through this mechanism.
Additionally, we found that ATM activity is required for lytic reactivation induced by overexpression of the EBV Na or R protein, but not Z, suggesting that ATM is required for the ability of Na, R, and presumably certain cellular transcription factors to activate the Z promoter in the context of the endogenous viral genome (Fig. 4A and B). Furthermore, we demonstrated that ATM activity is required for Z transcription in EBV+ cells induced with TGF-β, further suggesting that ATM is required for activation of the Z promoter (Fig. 4C). We also found that the EBV-encoded protein kinase, EBV-PK, which has been shown to activate ATM through phosphorylation and activation of Tip60 (55), is not required for lytic reactivation induced by hydrogen peroxide (Fig. 5). Thus, activation of the Z promoter by this agent is independent of EBV-PK activation of ATM.
Our group and others have shown that p53 is important for EBV lytic reactivation in many cell lines (13, 17, 38), and p53 has been reported to form a complex with SP1 that binds to, and activates, the Z promoter (17). In addition, p53 was recently shown to inhibit expression of the ZEB-1 and ZEB-2 proteins (49), which are potent repressors of the Z promoter (24), by inducing expression of microRNAs that degrade ZEB-1/-2. Furthermore, similar to our findings with ATM, we previously showed that Na and R, but not Z, require p53 to induce lytic gene expression (38).
Thus, we initially hypothesized that the major role of ATM during lytic EBV reactivation might be to enhance p53 transcriptional function. Although our data here confirm that p53 plays an important role during early stages of EBV lytic reactivation in cells that contain wild-type p53, we find that ATM must also promote EBV reactivation via p53-independent mechanisms, since ATM inhibition also blocked EBV reactivation in p53-negative and p53 mutant cells. Thus, additional cellular ATM substrates must also function to regulate viral gene expression. Since ATM phosphorylates over 700 cellular proteins (64), we are unable to examine the role of each known ATM substrate on viral reactivation. In addition to p53, we have examined the effect of the cellular KAP1 protein on lytic EBV gene expression, since the unphosphorylated form of KAP1 promotes heterochromatin and KSHV viral latency (12, 83), whereas the ATM-phosphorylated form (Ser824) relaxes heterochromatin (11). We did not find that either phosphomimetic p53 or KAP1 mutants could compensate for the effect of ATM knockdown on EBV reactivation (data not shown). Thus, we speculate that multiple different ATM targets are important for EBV reactivation. One such ATM target may be the histone variant H2AX, since the ATM-phosphorylated form of H2AX (which is phosphorylated at the same residue by the murid herpesvirus 68 [MHV68] viral kinase) has been reported to promote viral reactivation of the MHV68 gammaherpesvirus in certain cell types (11, 90).
Interestingly, inhibition of ATM enhances the ability of EBV to transform primary B cells in vitro (72), and at least three different EBV latency proteins (EBNA1, EBNA3C, and LMP1) inhibit the DNA damage response (10, 72). LMP1 decreases expression of both ATM and p53 (35, 44). Furthermore, both LMP1 and LMP2A inhibit p53-mediated apoptosis (7, 30, 75). LMP1 and LMP2A have both been reported to inhibit lytic viral reactivation in vitro (3, 67, 68, 74), and we recently reported that LMP1- and LMP2A-expressing B cells do not express early lytic viral proteins in EBV-infected humanized mice, even when mice are infected with a “superlytic” EBV mutant that results in high-level Z/BMRF1 expression (59). Our results here suggest that LMP1 and/or LMP2A might inhibit lytic EBV reactivation at least partially through inhibition of ATM and/or p53 function.
Previous studies examining if ATM is required for EBV DNA replication downstream of early lytic protein activation have yielded conflicting results. Here, we demonstrate that ATM is not required for lytic EBV DNA replication when Zp activation is bypassed by transfection of the Z protein (Fig. 7). Interestingly, the group that found ATM to be dispensable for lytic viral replication induced replication by transfecting cells with Z (50), while the group that found ATM to be required for viral DNA replication induced replication by treating cells with anti-IgG and did not examine the effect of the inhibitor on early lytic protein expression (55). Therefore, our data suggest that ATM is important for EBV lytic infection primarily at the level of Zp activation and not for viral DNA replication per se.
Based on these results and the work of others, we propose a model in which ATM contributes to the initial steps of EBV lytic reactivation (Fig. 9). In this model, cellular stresses, including DNA damage or production of ROS, lead to ATM activation. ATM activation then leads to phosphorylation of ATM target substrates that activate the EBV Zp through effects on viral chromatin structure (for example, H2AX phosphorylation) and/or by increasing p53 transcriptional function. Expression of Z then leads to activation of R expression, and together Z and R induce expression of all the lytic EBV proteins, including the EBV-encoded protein kinase (EBV-PK). Since the EBV protein kinase (encoded by the BGLF4 gene) has been shown to enhance ATM activity through phosphorylation of Tip60 (54, 55), this leads to further amplification of ATM kinase activity. Viral DNA replication, which occurs downstream of Z and R activation, may also further activate ATM, although ATM activity is not required for viral DNA replication.
Fig 9.

Hypothesized model by which ATM contributes to lytic EBV reactivation. Cellular stresses such as DNA damage and reactive oxygen species (ROS) induce ATM activation. ATM then activates the EBV immediate-early promoter, Zp, through effects on viral chromatin structure (for example, by enhancing KAP1 and/or H2AX phosphorylation) and/or by increasing p53 transcriptional function. Induction of Zp activity leads to expression of the Z IE protein, which results in R expression. Z and R induce transcription of the early lytic viral proteins, including the EBV-encoded protein kinase (PK), which then further activates ATM activity through phosphorylation of Tip60. Viral DNA replication, which occurs downstream of Z and R activation, may further activate ATM (data not shown).
Finally, our results here suggest that drugs which activate ATM and/or p53 might be useful for “lytic induction therapy.” Lytic induction therapy, in which the lytic form of EBV infection is purposely activated in latently infected tumor cells, has been proposed as a novel way to promote EBV-dependent killing of tumor cells. We demonstrate that nutlin-3, a drug which stabilizes p53 expression by antagonizing its interaction with the E3 ligase MDM2, is able to induce lytic reactivation in an ATM- and p53-dependent manner in EBV-infected cells expressing wild-type p53.
ACKNOWLEDGMENTS
We thank Janet Mertz for reviewing the manuscript.
This work was supported by grants T32 AI078985, R01-CA58853, R01-CA66519, and P01-CA022443 from the National Institutes of Health.
Footnotes
Published ahead of print 26 September 2012
REFERENCES
- 1. Adamson AL, Kenney S. 2001. Epstein-Barr virus immediate-early protein BZLF1 is SUMO-1 modified and disrupts promyelocytic leukemia bodies. J. Virol. 75:2388–2399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Adamson AL, Kenney SC. 1998. Rescue of the Epstein-Barr virus BZLF1 mutant, Z(S186A), early gene activation defect by the BRLF1 gene product. Virology 251:187–197 [DOI] [PubMed] [Google Scholar]
- 3. Adler B, et al. 2002. Control of Epstein-Barr virus reactivation by activated CD40 and viral latent membrane protein 1. Proc. Natl. Acad. Sci. U. S. A. 99:437–442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ben-Sasson SA, Klein G. 1981. Activation of the Epstein-Barr virus genome by 5-aza-cytidine in latently infected human lymphoid lines. Int. J. Cancer 28:131–135 [DOI] [PubMed] [Google Scholar]
- 5. Bhatti S, et al. 2011. ATM protein kinase: the linchpin of cellular defenses to stress. Cell. Mol. Life Sci. 68:2977–3006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bhende PM, Seaman WT, Delecluse HJ, Kenney SC. 2005. BZLF1 activation of the methylated form of the BRLF1 immediate-early promoter is regulated by BZLF1 residue 186. J. Virol. 79:7338–7348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bieging KT, Amick AC, Longnecker R. 2009. Epstein-Barr virus LMP2A bypasses p53 inactivation in a MYC model of lymphomagenesis. Proc. Natl. Acad. Sci. U. S. A. 106:17945–17950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Borza CM, Hutt-Fletcher LM. 2002. Alternate replication in B cells and epithelial cells switches tropism of Epstein-Barr virus. Nat. Med. 8:594–599 [DOI] [PubMed] [Google Scholar]
- 9. Bose S, et al. 2009. The ATM tumour suppressor gene is down-regulated in EBV-associated nasopharyngeal carcinoma. J. Pathol. 217:345–352 [DOI] [PubMed] [Google Scholar]
- 10. Cai Q, et al. 2011. Epstein-Barr virus nuclear antigen 3C stabilizes Gemin3 to block p53-mediated apoptosis. PLoS Pathog. 7:e1002418 doi:10.1371/journal.ppat.1002418 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 11. Cann KL, Dellaire G. 2011. Heterochromatin and the DNA damage response: the need to relax. Biochem. Cell Biol. 89:45–60 [DOI] [PubMed] [Google Scholar]
- 12. Chang PC, et al. 2009. Kruppel-associated box domain-associated protein-1 as a latency regulator for Kaposi's sarcoma-associated herpesvirus and its modulation by the viral protein kinase. Cancer Res. 69:5681–5689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chang SS, et al. 2008. Critical role of p53 in histone deacetylase inhibitor-induced Epstein-Barr virus Zta expression. J. Virol. 82:7745–7751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chen K, Albano A, Ho A, Keaney JF., Jr 2003. Activation of p53 by oxidative stress involves platelet-derived growth factor-beta receptor-mediated ataxia telangiectasia mutated (ATM) kinase activation. J. Biol. Chem. 278:39527–39533 [DOI] [PubMed] [Google Scholar]
- 15. Chen MR, Chang SJ, Huang H, Chen JY. 2000. A protein kinase activity associated with Epstein-Barr virus BGLF4 phosphorylates the viral early antigen EA-D in vitro. J. Virol. 74:3093–3104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chevallier-Greco A, et al. 1986. Both Epstein-Barr virus (EBV)-encoded trans-acting factors, EB1 and EB2, are required to activate transcription from an EBV early promoter. EMBO J. 5:3243–3249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chua HH, et al. 2012. p53 and Sp1 cooperate to regulate the expression of Epstein-Barr viral Zta protein. J. Med. Virol. 84:1279–1288 [DOI] [PubMed] [Google Scholar]
- 18. Countryman J, Jenson H, Seibl R, Wolf H, Miller G. 1987. Polymorphic proteins encoded within BZLF1 of defective and standard Epstein-Barr viruses disrupt latency. J. Virol. 61:3672–3679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Countryman J, Miller G. 1985. Activation of expression of latent Epstein-Barr herpesvirus after gene transfer with a small cloned subfragment of heterogeneous viral DNA. Proc. Natl. Acad. Sci. U. S. A. 82:4085–4089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Darr CD, Mauser A, Kenney S. 2001. Epstein-Barr virus immediate-early protein BRLF1 induces the lytic form of viral replication through a mechanism involving phosphatidylinositol-3 kinase activation. J. Virol. 75:6135–6142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Delecluse HJ, Hilsendegen T, Pich D, Zeidler R, Hammerschmidt W. 1998. Propagation and recovery of intact, infectious Epstein-Barr virus from prokaryotic to human cells. Proc. Natl. Acad. Sci. U. S. A. 95:8245–8250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Derheimer FA, Kastan MB. 2010. Multiple roles of ATM in monitoring and maintaining DNA integrity. FEBS Lett. 584:3675–3681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dickerson SJ, et al. 2009. Methylation-dependent binding of the Epstein-Barr virus BZLF1 protein to viral promoters. PLoS Pathog. 5:e1000356 doi:10.1371/journal.ppat.1000356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ellis AL, Wang Z, Yu X, Mertz JE. 2010. Either ZEB1 or ZEB2/SIP1 can play a central role in regulating the Epstein-Barr virus latent-lytic switch in a cell-type-specific manner. J. Virol. 84:6139–6152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fahmi H, Cochet C, Hmama Z, Opolon P, Joab I. 2000. Transforming growth factor beta 1 stimulates expression of the Epstein-Barr virus BZLF1 immediate-early gene product ZEBRA by an indirect mechanism which requires the MAPK kinase pathway. J. Virol. 74:5810–5818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Farrell PJ, Allan GJ, Shanahan F, Vousden KH, Crook T. 1991. p53 is frequently mutated in Burkitt's lymphoma cell lines. EMBO J. 10:2879–2887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Feederle R, et al. 2000. The Epstein-Barr virus lytic program is controlled by the co-operative functions of two transactivators. EMBO J. 19:3080–3089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Feng WH, Israel B, Raab-Traub N, Busson P, Kenney SC. 2002. Chemotherapy induces lytic EBV replication and confers ganciclovir susceptibility to EBV-positive epithelial cell tumors. Cancer Res. 62:1920–1926 [PubMed] [Google Scholar]
- 29. Feng WH, Kenney SC. 2006. Valproic acid enhances the efficacy of chemotherapy in EBV-positive tumors by increasing lytic viral gene expression. Cancer Res. 66:8762–8769 [DOI] [PubMed] [Google Scholar]
- 30. Forte E, Luftig MA. 2009. MDM2-dependent inhibition of p53 is required for Epstein-Barr virus B-cell growth transformation and infected-cell survival. J. Virol. 83:2491–2499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gershburg E, Pagano JS. 2002. Phosphorylation of the Epstein-Barr virus (EBV) DNA polymerase processivity factor EA-D by the EBV-encoded protein kinase and effects of the L-riboside benzimidazole 1263W94. J. Virol. 76:998–1003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Greenspan JS, et al. 1985. Replication of Epstein-Barr virus within the epithelial cells of oral “hairy” leukoplakia, an AIDS-associated lesion. N. Engl. J. Med. 313:1564–1571 [DOI] [PubMed] [Google Scholar]
- 33. Gregory CD, Rowe M, Rickinson AB. 1990. Different Epstein-Barr virus-B cell interactions in phenotypically distinct clones of a Burkitt's lymphoma cell line. J. Gen. Virol. 71:1481–1495 [DOI] [PubMed] [Google Scholar]
- 34. Gruffat H, Manet E, Rigolet A, Sergeant A. 1990. The enhancer factor R of Epstein-Barr virus (EBV) is a sequence-specific DNA binding protein. Nucleic Acids Res. 18:6835–6843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gruhne B, Sompallae R, Masucci MG. 2009. Three Epstein-Barr virus latency proteins independently promote genomic instability by inducing DNA damage, inhibiting DNA repair and inactivating cell cycle checkpoints. Oncogene 28:3997–4008 [DOI] [PubMed] [Google Scholar]
- 36. Guo Z, Deshpande R, Paull TT. 2010. ATM activation in the presence of oxidative stress. Cell Cycle 9:4805–4811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Guo Z, Kozlov S, Lavin MF, Person MD, Paull TT. 2010. ATM activation by oxidative stress. Science 330:517–521 [DOI] [PubMed] [Google Scholar]
- 38. Hagemeier SR, Barlow EA, Kleman AA, Kenney SC. 2011. The Epstein-Barr virus BRRF1 protein, Na, induces lytic infection in a TRAF2- and p53-dependent manner. J. Virol. 85:4318–4329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hardwick JM, Lieberman PM, Hayward SD. 1988. A new Epstein-Barr virus transactivator, R, induces expression of a cytoplasmic early antigen. J. Virol. 62:2274–2284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hickson I, et al. 2004. Identification and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM. Cancer Res. 64:9152–9159 [DOI] [PubMed] [Google Scholar]
- 41. Hoglund A, Nilsson LM, Forshell LP, Maclean KH, Nilsson JA. 2009. Myc sensitizes p53-deficient cancer cells to the DNA-damaging effects of the DNA methyltransferase inhibitor decitabine. Blood 113:4281–4288 [DOI] [PubMed] [Google Scholar]
- 42. Hong GK, et al. 2004. The BRRF1 early gene of Epstein-Barr virus encodes a transcription factor that enhances induction of lytic infection by BRLF1. J. Virol. 78:4983–4992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hong YS, et al. 2012. Bortezomib induces G2-M arrest in human colon cancer cells through ROS-inducible phosphorylation of ATM-CHK1. Int. J. Oncol. 41:76–82 [DOI] [PubMed] [Google Scholar]
- 44. Husaini R, Ahmad M, Soo-Beng Khoo A. 2011. Epstein-Barr virus latent membrane protein LMP1 reduces p53 protein levels independent of the PI3K-Akt pathway. BMC Res. Notes 4:551 doi:10.1186/1756-0500-4-551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ju R, Muller MT. 2003. Histone deacetylase inhibitors activate p21(WAF1) expression via ATM. Cancer Res. 63:2891–2897 [PubMed] [Google Scholar]
- 46. Kanda T, Yajima M, Ahsan N, Tanaka M, Takada K. 2004. Production of high-titer Epstein-Barr virus recombinants derived from Akata cells by using a bacterial artificial chromosome system. J. Virol. 78:7004–7015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kenney S, et al. 1989. The Epstein-Barr virus (EBV) BZLF1 immediate-early gene product differentially affects latent versus productive EBV promoters. J. Virol. 63:1729–1736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kieff E, Rickinson AB. 2007. Epstein-Barr virus and its replication, p 2603–2654 In Knipe DM, et al. (ed), Fields virology, 5th ed Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 49. Kim T, et al. 2011. p53 regulates epithelial-mesenchymal transition through microRNAs targeting ZEB1 and ZEB2. J. Exp. Med. 208:875–883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kudoh A, et al. 2005. Epstein-Barr virus lytic replication elicits ATM checkpoint signal transduction while providing an S-phase-like cellular environment. J. Biol. Chem. 280:8156–8163 [DOI] [PubMed] [Google Scholar]
- 51. Laichalk LL, Thorley-Lawson DA. 2005. Terminal differentiation into plasma cells initiates the replicative cycle of Epstein-Barr virus in vivo. J. Virol. 79:1296–1307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lassoued S, Gargouri B, El Feki Ael F, Attia H, Van Pelt J. 2010. Transcription of the Epstein-Barr virus lytic cycle activator BZLF-1 during oxidative stress induction. Biol. Trace Elem. Res. 137:13–22 [DOI] [PubMed] [Google Scholar]
- 53. Lees-Miller SP, Sakaguchi K, Ullrich SJ, Appella E, Anderson CW. 1992. Human DNA-activated protein kinase phosphorylates serines 15 and 37 in the amino-terminal transactivation domain of human p53. Mol. Cell. Biol. 12:5041–5049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Li R, et al. 2012. SUMO binding by the Epstein-Barr virus protein kinase BGLF4 is crucial for BGLF4 function. J. Virol. 86:5412–5421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Li R, et al. 2011. Conserved herpesvirus kinases target the DNA damage response pathway and TIP60 histone acetyltransferase to promote virus replication. Cell Host Microbe 10:390–400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Li X, Feng J, Sun R. 2011. Oxidative stress induces reactivation of Kaposi's sarcoma-associated herpesvirus and death of primary effusion lymphoma cells. J. Virol. 85:715–724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lieberman PM, Hardwick JM, Sample J, Hayward GS, Hayward SD. 1990. The zta transactivator involved in induction of lytic cycle gene expression in Epstein-Barr virus-infected lymphocytes binds to both AP-1 and ZRE sites in target promoter and enhancer regions. J. Virol. 64:1143–1155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Luka J, Kallin B, Klein G. 1979. Induction of the Epstein-Barr virus (EBV) cycle in latently infected cells by n-butyrate. Virology 94:228–231 [DOI] [PubMed] [Google Scholar]
- 59. Ma SD, et al. 2012. An Epstein-Barr virus (EBV) mutant with enhanced BZLF1 expression causes lymphomas with abortive lytic EBV infection in a humanized mouse model. J. Virol. 86:7976–7987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Maly FE, et al. 1988. The superoxide generating system of B cell lines. Structural homology with the phagocytic oxidase and triggering via surface Ig. J. Immunol. 140:2334–2339 [PubMed] [Google Scholar]
- 61. Maly FE, et al. 1989. Superoxide-dependent nitroblue tetrazolium reduction and expression of cytochrome b-245 components by human tonsillar B lymphocytes and B cell lines. J. Immunol. 142:1260–1267 [PubMed] [Google Scholar]
- 62. Masucci MG, et al. 1989. 5-Azacytidine up regulates the expression of Epstein-Barr virus nuclear antigen 2 (EBNA-2) through EBNA-6 and latent membrane protein in the Burkitt's lymphoma line Rael. J. Virol. 63:3135–3141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Matozaki T, et al. 1992. Missense mutations and a deletion of the p53 gene in human gastric cancer. Biochem. Biophys. Res. Commun. 182:215–223 [DOI] [PubMed] [Google Scholar]
- 64. Matsuoka S, et al. 2007. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 316:1160–1166 [DOI] [PubMed] [Google Scholar]
- 65. Mellinghoff I, et al. 1991. Early events in Epstein-Barr virus genome expression after activation: regulation by second messengers of B cell activation. Virology 185:922–928 [DOI] [PubMed] [Google Scholar]
- 66. Meng Q, Hagemeier SR, Kuny CV, Kalejta RF, Kenney SC. 2010. Simian virus 40 T/t antigens and lamin A/C small interfering RNA rescue the phenotype of an Epstein-Barr virus protein kinase (BGLF4) mutant. J. Virol. 84:4524–4533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Miller CL, Lee JH, Kieff E, Longnecker R. 1994. An integral membrane protein (LMP2) blocks reactivation of Epstein-Barr virus from latency following surface immunoglobulin crosslinking. Proc. Natl. Acad. Sci. U. S. A. 91:772–776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Miller CL, Longnecker R, Kieff E. 1993. Epstein-Barr virus latent membrane protein 2A blocks calcium mobilization in B lymphocytes. J. Virol. 67:3087–3094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Murata T, et al. 2009. TORC2, a coactivator of cAMP-response element-binding protein, promotes Epstein-Barr virus reactivation from latency through interaction with viral BZLF1 protein. J. Biol. Chem. 284:8033–8041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Nanbo A, Inoue K, Adachi-Takasawa K, Takada K. 2002. Epstein-Barr virus RNA confers resistance to interferon-alpha-induced apoptosis in Burkitt's lymphoma. EMBO J. 21:954–965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Neuhierl B, Feederle R, Hammerschmidt W, Delecluse HJ. 2002. Glycoprotein gp110 of Epstein-Barr virus determines viral tropism and efficiency of infection. Proc. Natl. Acad. Sci. U. S. A. 99:15036–15041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Nikitin PA, et al. 2010. An ATM/Chk2-mediated DNA damage-responsive signaling pathway suppresses Epstein-Barr virus transformation of primary human B cells. Cell Host Microbe 8:510–522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Nonkwelo C, Skinner J, Bell A, Rickinson A, Sample J. 1996. Transcription start sites downstream of the Epstein-Barr virus (EBV) Fp promoter in early-passage Burkitt lymphoma cells define a fourth promoter for expression of the EBV EBNA-1 protein. J. Virol. 70:623–627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Prince S, et al. 2003. Latent membrane protein 1 inhibits Epstein-Barr virus lytic cycle induction and progress via different mechanisms. J. Virol. 77:5000–5007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Pujals A, et al. 2011. Treatment with a BH3 mimetic overcomes the resistance of latency III EBV (+) cells to p53-mediated apoptosis. Cell Death Dis. 2:e184 doi:10.1038/cddis.2011.67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Ragoczy T, Heston L, Miller G. 1998. The Epstein-Barr virus Rta protein activates lytic cycle genes and can disrupt latency in B lymphocytes. J. Virol. 72:7978–7984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Rickenson AB, Kieff E. 2007. Epstein-Barr virus, p 2655–2700 In Knipe DM, et al. (ed), Fields virology, 5th ed Lippincott Williams & Williams, Philadelphia, PA [Google Scholar]
- 78. Robinson AR, Kwek SS, Hagemeier SR, Wille CK, Kenney SC. 2011. Cellular transcription factor Oct-1 interacts with the Epstein-Barr virus BRLF1 protein to promote disruption of viral latency. J. Virol. 85:8940–8953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Robinson AR, Kwek SS, Kenney SC. 2012. The B-cell specific transcription factor, Oct-2, promotes Epstein-Barr virus latency by inhibiting the viral immediate-early protein, BZLF1. PLoS Pathog. 8:e1002516 doi:10.1371/journal.ppat.1002516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Rooney CM, Rowe DT, Ragot T, Farrell PJ. 1989. The spliced BZLF1 gene of Epstein-Barr virus (EBV) transactivates an early EBV promoter and induces the virus productive cycle. J. Virol. 63:3109–3116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Rowe M, et al. 1995. Restoration of endogenous antigen processing in Burkitt's lymphoma cells by Epstein-Barr virus latent membrane protein-1: coordinate up-regulation of peptide transporters and HLA-class I antigen expression. Eur. J. Immunol. 25:1374–1384 [DOI] [PubMed] [Google Scholar]
- 82. Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. 2005. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307:1098–1101 [DOI] [PubMed] [Google Scholar]
- 83. Schultz DC, Friedman JR, Rauscher FJ., III 2001. Targeting histone deacetylase complexes via KRAB-zinc finger proteins: the PHD and bromodomains of KAP-1 form a cooperative unit that recruits a novel isoform of the Mi-2alpha subunit of NuRD. Genes Dev. 15:428–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Shieh SY, Ikeda M, Taya Y, Prives C. 1997. DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell 91:325–334 [DOI] [PubMed] [Google Scholar]
- 85. Shirley CM, et al. 2011. Bortezomib induction of C/EBPbeta mediates Epstein-Barr virus lytic activation in Burkitt lymphoma. Blood 117:6297–6303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Siliciano JD, et al. 1997. DNA damage induces phosphorylation of the amino terminus of p53. Genes Dev. 11:3471–3481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Smith J, Tho LM, Xu N, Gillespie DA. 2010. The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Adv. Cancer Res. 108:73–112 [DOI] [PubMed] [Google Scholar]
- 88. Takada K, Ono Y. 1989. Synchronous and sequential activation of latently infected Epstein-Barr virus genomes. J. Virol. 63:445–449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Takada K, Shimizu N, Sakuma S, Ono Y. 1986. trans activation of the latent Epstein-Barr virus (EBV) genome after transfection of the EBV DNA fragment. J. Virol. 57:1016–1022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Tarakanova VL, et al. 2007. Gamma-herpesvirus kinase actively initiates a DNA damage response by inducing phosphorylation of H2AX to foster viral replication. Cell Host Microbe 1:275–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Tibbetts RS, et al. 1999. A role for ATR in the DNA damage-induced phosphorylation of p53. Genes Dev. 13:152–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Vassilev LT, et al. 2004. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 303:844–848 [DOI] [PubMed] [Google Scholar]
- 93. Walling DM, Flaitz CM, Nichols CM, Hudnall SD, Adler-Storthz K. 2001. Persistent productive Epstein-Barr virus replication in normal epithelial cells in vivo. J. Infect. Dis. 184:1499–1507 [DOI] [PubMed] [Google Scholar]
- 94. Webster-Cyriaque J, Middeldorp J, Raab-Traub N. 2000. Hairy leukoplakia: an unusual combination of transforming and permissive Epstein-Barr virus infections. J. Virol. 74:7610–7618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Westphal EM, Blackstock W, Feng W, Israel B, Kenney SC. 2000. Activation of lytic Epstein-Barr virus (EBV) infection by radiation and sodium butyrate in vitro and in vivo: a potential method for treating EBV-positive malignancies. Cancer Res. 60:5781–5788 [PubMed] [Google Scholar]
- 96. Wiman KG, Magnusson KP, Ramqvist T, Klein G. 1991. Mutant p53 detected in a majority of Burkitt lymphoma cell lines by monoclonal antibody PAb240. Oncogene 6:1633–1639 [PubMed] [Google Scholar]
- 97. Ye F, Lattif AA, Xie J, Weinberg A, Gao S. 2012. Nutlin-3 induces apoptosis, disrupts viral latency and inhibits expression of angiopoietin-2 in Kaposi sarcoma tumor cells. Cell Cycle 11:1393–1399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Ye F, et al. 2011. Reactive oxygen species hydrogen peroxide mediates Kaposi's sarcoma-associated herpesvirus reactivation from latency. PLoS Pathog. 7:e1002054 doi:10.1371/journal.ppat.1002054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Zalani S, Holley-Guthrie E, Kenney S. 1996. Epstein-Barr viral latency is disrupted by the immediate-early BRLF1 protein through a cell-specific mechanism. Proc. Natl. Acad. Sci. U. S. A. 93:9194–9199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Zhang S, et al. 2006. TGFbeta1-induced activation of ATM and p53 mediates apoptosis in a Smad7-dependent manner. Cell Cycle 5:2787–2795 [DOI] [PubMed] [Google Scholar]
- 101. Zhang XP, Liu F, Wang W. 2011. Two-phase dynamics of p53 in the DNA damage response. Proc. Natl. Acad. Sci. U. S. A. 108:8990–8995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. zur Hausen H, et al. 1970. EBV DNA in biopsies of Burkitt tumours and anaplastic carcinomas of the nasopharynx. Nature 228:1056–1058 [DOI] [PubMed] [Google Scholar]