

Abstract
The baculovirus Autographa californica multicapsid nucleopolyhedrovirus (AcMNPV) initiates apoptosis in diverse insects through events triggered by virus DNA (vDNA) replication. To define the proapoptotic pathway and its role in antivirus defense, we investigated the link between the host's DNA damage response (DDR) and apoptosis. We report here that AcMNPV elicits a DDR in the model insect Drosophila melanogaster. Replication of vDNA activated DDR kinases, as evidenced by ATM-driven phosphorylation of the Drosophila histone H2AX homolog (H2Av), a critical regulator of the DDR. Ablation or inhibition of ATM repressed H2Av phosphorylation and blocked virus-induced apoptosis. The DDR kinase inhibitors caffeine and KU55933 also prevented virus-induced apoptosis in cells derived from the permissive AcMNPV host, Spodoptera frugiperda. This block occurred at a step upstream of virus-mediated depletion of the cellular inhibitor-of-apoptosis protein, an event that initiates apoptosis in Spodoptera and Drosophila. Thus, the DDR is a conserved, proapoptotic response to baculovirus infection. DDR inhibition also repressed vDNA replication and reduced virus yields 100,000-fold, demonstrating that the DDR contributes to virus production, despite its recognized antivirus role. In contrast to virus-induced phosphorylation of Drosophila H2Av, AcMNPV blocked phosphorylation of the Spodoptera H2AX homolog (SfH2AX). Remarkably, AcMNPV also suppressed SfH2AX phosphorylation following pharmacologically induced DNA damage. These findings indicate that AcMNPV alters canonical DDR signaling in permissive cells. We conclude that AcMNPV triggers a proapoptotic DDR that is subsequently modified, presumably to stimulate vDNA replication. Thus, manipulation of the DDR to facilitate multiplication is an evolutionarily conserved strategy among DNA viruses of insects and mammals.
INTRODUCTION
The molecular pathways by which infected cells sense viral pathogens and initiate an antiviral response serve as principal determinants of host immunity and virus pathogenicity. Beyond its canonical role in detecting damaged DNA and mediating DNA repair, the DNA damage response (DDR) functions as an intrinsic host defense that can limit virus multiplication and destroy infected cells by apoptosis (reviewed in reference 66). DNA viruses frequently degrade, mislocalize, modify, and even exploit components of the host DDR to counter these antiviral effects and to facilitate genome replication (66). DNA viruses also encode factors that prevent apoptosis to ensure cell survival and virus propagation (reviewed in reference 2). Collectively, these strategies not only enhance virus multiplication but also promote genomic instability and transformation by disrupting DNA repair and limiting apoptosis of damaged cells (66). These contributions to virus pathogenicity argue for a broader understanding of the mechanistic links between the virus-induced DDR and apoptosis, as well as the strategies whereby viruses evade and manipulate these host responses.
The baculoviruses are large DNA viruses that infect insects and can cause widespread apoptosis (reviewed in references 11 and 14). A critical role for the host DDR in baculovirus-induced apoptosis was suggested by the finding that the prototype baculovirus Autographa californica multicapsid nucleopolyhedrovirus (AcMNPV) triggers apoptosis through events associated with the replication of its 134-kb DNA genome (Fig. 1) (54, 64). The replication of mammalian virus DNA (vDNA) is a well-established initiator of the DDR, which is also a trigger for apoptosis in both vertebrates and invertebrates (reviewed in references 61 and 66). Indeed, AcMNPV DNA replication stimulates the DDR in cells derived from the permissive moth Spodoptera frugiperda (22). However, as is the case for many DNA viruses, the replication events that initiate the DDR are unknown. Baculovirus DNA replication intermediates generated by rolling-circle and recombination-mediated mechanisms (reviewed in reference 51) probably mimic damaged DNA that subsequently triggers the DDR. The DDR commences with the activation of one or more phosphatidylinositol 3-kinase-like kinases, including ATM (ataxia telangiectasia-mutated kinase) and ATR (ATM- and Rad3-related kinase) (reviewed in references 10, 29, and 66). Upon their recruitment to sites of damaged DNA, these kinases phosphorylate and activate multiple substrates, including the histone 2A variant H2AX (29). Used as a classical indicator of the DDR, phosphorylated H2AX (γ-H2AX) plays a central role in amplifying DDR signaling and recruiting additional DDR components, including DNA repair factors (reviewed in references 3 and 63). Ultimately, DDR signaling leads to cell cycle arrest for DNA repair or apoptosis for elimination of damaged cells (reviewed in references 40, 61, and 66).
Fig 1.
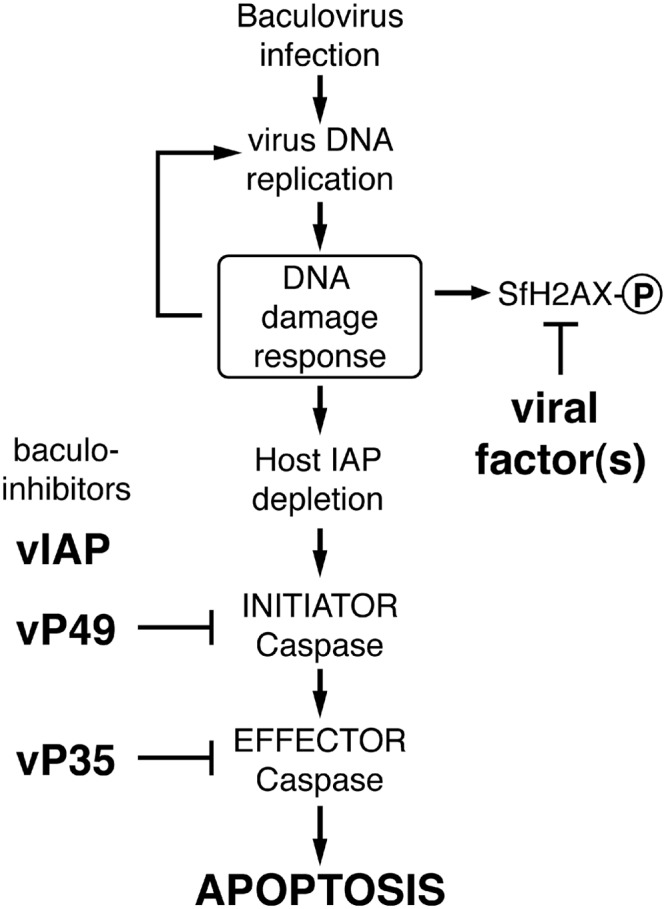
Model for baculovirus-induced DDR and apoptosis. Early in infection, unknown viral DNA replication events trigger the host's DNA damage response (DDR), which activates DDR kinases. Virus DNA replication is stimulated by the DDR, but cellular inhibitor-of-apoptosis (IAP) proteins are depleted. The loss of IAP activates initiator and effector caspases, which cause apoptotic death. To prevent apoptosis and thus virus elimination, baculoviruses encode apoptosis inhibitors, including viral IAP (vIAP) and caspase inhibitors (vP49 and vP35). This strategy allows viral exploitation of the otherwise proapoptotic DDR. As also reported here, one or more baculovirus factors repress phosphorylation of the Spodoptera histone variant SfH2AX, a key regulatory step in the DDR, and thus manipulate the host DDR presumably to benefit virus multiplication. SfH2AX-P denotes the phosphorylated form of the protein.
Baculoviruses efficiently prevent host apoptosis through the activity of virus-encoded apoptotic suppressors, including P35, P49, and viral inhibitor-of-apoptosis (vIAP) protein, which target the proteolytic caspases that execute apoptosis (Fig. 1) (11, 14). The suppression of apoptosis enables baculoviruses to potentially exploit the otherwise proapoptotic DDR to stimulate virus propagation. Caspase activation occurs upon virus-signaled destruction of the host's inhibitor-of-apoptosis (IAP) proteins, which normally repress caspase activity (reviewed in reference 17). Through unknown events associated with AcMNPV DNA replication, the principal IAP of Spodoptera is rapidly depleted as a result of instability motifs embedded within its N-terminal leader domain (9, 64). Likewise, AcMNPV DNA replication causes depletion of the Drosophila melanogaster IAP (DIAP1), which leads to widespread apoptosis in cultured cells derived from this distantly related insect species (54, 64). Thus, baculoviruses provide a unique opportunity to define conserved pathways by which vDNA replication engages the host apoptotic program.
Here we use AcMNPV and Drosophila with its well-defined DDR and cell death programs (reviewed in references 18 and 61) to investigate the link between vDNA replication, the DDR, and apoptosis. We report that AcMNPV triggers the Drosophila DDR, which in turn initiates caspase activation and apoptosis. AcMNPV DNA replication is required to activate the DDR, which involves ATM and includes phosphorylation of H2Av, the Drosophila H2AX homolog. Similarly, the DDR was required for AcMNPV DNA replication-induced apoptosis of permissive Spodoptera cells. This finding suggests that the DDR is a conserved, proapoptotic response to baculovirus infection. Importantly, the Spodoptera DDR conferred a dramatic stimulation of AcMNPV multiplication and vDNA replication. However, unlike Drosophila H2Av, virus-induced phosphorylation of the Spodoptera H2AX homolog, SfH2AX, was reduced or suppressed. Thus, in permissive cells AcMNPV alters canonical DDR signaling. Our findings suggest that baculoviruses use a novel strategy of manipulating the host DDR to promote vDNA replication and thus virus propagation.
MATERIALS AND METHODS
Cells and viruses.
Spodoptera frugiperda IPLB-SF21 (SF21) cells (65) and Drosophila melanogaster Schneider DL-1 cells (53) were maintained at 27°C in TC100 growth medium (Invitrogen) supplemented with 10% heat-inactivated fetal bovine serum (FBS) (HyClone Laboratories) or Schneider's growth medium (Invitrogen)–15% FBS, respectively.
Wild-type AcMNPV (L-1 strain) (30) and the AcMNPV recombinants wt/lacZ (p35+ iap− polyhedrin gene [polh−] lacZ+) (19), vΔ35K (p35− polh+) (19), and vOpIAP (pIE1prmOp-iap3HA/Δ35K/lacZ; Op-IAP3HA p35− polh− lacZ+) (34) were described previously. AcMNPV (HR3 strain) mutant ts8 was kindly provided by E. B. Carstens (Queen's University) (6, 16). For inoculation, extracellular budded virus (BV) in TC100–10% FBS was added to DL-1 and SF21 monolayers. After gentle rocking at room temperature for 1 h, the inoculum was replaced with FBS-supplemented medium and the cells were incubated at 27°C. Inoculations were conducted with the indicated multiplicity of infection (MOI) calculated as PFU per cell.
To measure BV production, virus inoculum (MOI, 0.5) was replaced with FBS-supplemented medium with vehicle (dimethyl sulfoxide [DMSO]) or the indicated drug. The concentration of drug was maintained by adding fresh drug-containing medium every 24 h. Growth medium was collected 72 h after infection, and the BV titers were determined by using SF21 cells and 50% tissue culture infective dose (TCID50) endpoint dilution (45).
Small-molecule inhibitors.
Topoisomerase II inhibitor etoposide, pancaspase inhibitor benzyloxycarbonyl-Val-Ala-Asp(OMe)-fluoromethylketone (zVAD-fmk), DNA polymerase inhibitor aphidicolin, and ATM inhibitor KU55933 were purchased from Calbiochem, dissolved in DMSO, and diluted in FBS-supplemented medium at the concentrations indicated. Caffeine (Alexis Biochemicals) was dissolved directly in FBS medium. Where indicated, cells were overlaid with drug-containing FBS medium and incubated at 27°C.
Plasmids. (i) dsRNA production.
pBluescript K/S+ (Invitrogen) plasmids containing portions of the enhanced green fluorescence protein (GFP) gene (egfp) or the AcMNPV genes ie-1, p143, lef-1, or p47 were described previously (54, 55). Portions of Drosophila atm (GenBank accession number NM_001043247) and atr (GenBank accession number NM_078645) open reading frames (ORFs) were PCR amplified using DL-1 genomic DNA and the following primer sets: ATM #1, 5′-CGAGCTCCGGCAATGCGAAAAATCTCCG-3′ and 5′-GGGTACCCGATCTCAAAGTTAAGGCCTCGC-3′ (ORF nucleotides 1305 to 2005); ATM #2, 5′-GGAGCTCAGCGTTCTGCTGGAAGATGC-3′ and 5′-AGGTACCGGTCAATTGGTTGGCGGC-3′ (ORF nucleotides 6097 to 6834); and ATR, 5′-CGAGCTCGAGAGCTGCCTAAGTGAACCG-3′ and 5′-AGGTACCGATGGCTCCCAGTTCTCCG-3′ (ORF nucleotides 2911 to 3639). PCR products were digested with SacI and KpnI (restriction sites underlined) and inserted into pBluescript K/S+.
(ii) GFPHA and GFPHA-P143.
To generate plasmid pGFPHA, which encodes GFP with a C-terminal hemagglutinin (HA) epitope, the GFP-encoding sequence of pAcIE1hr/EGFP-AcIE1/PA (43) was PCR amplified and inserted into the ie-1 promoter-based expression vector pIE1prm/PA. Expression plasmid pGFPHA-P143, which encodes the AcMNPV DNA helicase P143 fused to the C terminus of GFPHA, was constructed by insertion of the appropriate AatII fragment from pHSEpiHis/P143 (48) (kindly provided by Lorena Passarelli, Kansas State University) into AatII-digested pGFPHA. P35 expression plasmid pIE1prm/P35/PA was described previously (62).
(iii) GFPHA-SfH2AXwt and -SfH2AXS135A.
The predicted ORF of Spodoptera frugiperda h2ax (sfh2ax) was identified via a BLAST search of the Spodoptera frugiperda cDNA database, SpodoBase (39), by using the lepidopteran species Bombyx mori h2ax sequence (GenBank accession number NM_001160195) as the query. The putative sfh2ax ORF was PCR amplified from a λ phage cDNA library of SF21 poly(A)+ RNA (26) by using primers 5′-AGCTAGCCCGACCTCCACGTTTAGAAGTG-3′ and 5′-GCAAGCTTTTAATACTCTTGTGATGAGCTATTAGTGTTATGTTGAC-3′. The PCR product was digested with NheI and HindIII (restriction sites underlined above) and inserted into pBluescript K/S+. This pBS/SfH2AX vector was digested with PstI and HindIII, and the SfH2AX-containing fragment was inserted into the hr5 enhancer-containing expression vector pIE1prm/hr5/PA (8). The SfH2AX amino acid substitution S135A was generated via site-directed mutagenesis of pIE1prm/hr5/SfH2AX/PA by using standard PCR methods. The GFPHA-encoding sequence of pGFPHA was PCR amplified and inserted into NheI-digested pIE1prm/hr5/SfH2AX/PA vectors to generate pIE1prm/hr5/GFPHA-SfH2AX/PA expression constructs. pIE1prm/GFPHA-SfH2AX/PA lacking the hr5 enhancer was constructed via AatII/SacI digestion of pIE1prm/hr5/GFPHA-SfH2AX/PA and insertion of the SfH2AX-containing fragment into pGFPHA.
Transfection with dsRNA and plasmid DNA.
RNA silencing (RNAi) was conducted by using gene-specific double-stranded RNA (dsRNA) as described previously (55). After transfection with dsRNA mixed with cationic liposomes consisting of N-[1-(2,3 dioleoyloxy)propyl]-N,N,N-trimethylammonium methyl sulfate-l-α-phophatidylethanolamine, dioleoyl (C18:1, [cis]-9) (DOTAP-DOPE), DL-1 cells were maintained as monolayers for 2 days at 27°C, after which they were harvested, counted, and treated as indicated. Visual inspection revealed that all dsRNA preparations used here had no obvious effect on cell viability. For plasmid transfections, SF21 monolayers (106 cells per 35-mm plate) were overlaid with TC100 medium containing cesium chloride (CsCl)-purified plasmid previously mixed with DOTAP-DOPE as described previously (26).
GFPHA-SfH2AX stably transfected cells.
SF21 cells were transfected with pIE1prm/GFPHA-SfH2AX/PA and neomycin resistance plasmid pIE1/neo/PA and selected with Geneticin (G418 sulfate; Gibco-BRL) as described previously (8). Following selection, a cloned cell line demonstrating GFP fluorescence was isolated via serial dilution of cells into 96-well culture plates.
Immunoblot assays and antisera.
Cells were collected, lysed in 1% sodium dodecyl sulfate (SDS)–1% β-mercaptoethanol, and subjected to SDS-polyacrylamide gel electrophoresis. Proteins were transferred to Immobilon-P polyvinylidene difluoride (Millipore) or nitrocellulose (Osmonics, Inc.) membranes that were then incubated with the following antisera, diluted as indicated: polyclonal anti-H2AvD pS137 (1:1,000) (Rockland Immunochemicals), polyclonal anti-IE1 (1:10,000) (44), polyclonal anti-DrICE (1:5,000) (27), affinity-purified polyclonal anti-SfIAP (1,1:000) (9), polyclonal anti-Sf-caspase-1 (1:1,000) (26), monoclonal anti-P35 (1:5,000) (kindly provided by Yuri Lazebnik, Cold Spring Harbor Laboratory), monoclonal anti-HA (1:1,000) (Covance), polyclonal anti-gamma H2A.X pS139 (1:1,000) (ab11174; Abcam), and monoclonal anti-actin (1:4,000) (BD Biosciences). Signal development was conducted by using alkaline phosphatase-conjugated goat anti-rabbit or goat anti-mouse immunoglobulin G (Jackson ImmunoResearch Laboratories, Inc.) with the CDP-Star Chemiluminescent detection system (Roche). Films were scanned at 300 dpi by using an Epson TWAIN Pro scanner and prepared using Adobe Photoshop CS2 and Adobe Illustrator CS2.
Cell death assays.
Viable, nonapoptotic DL-1 and SF21 cells were counted at 2 h and 24 h after infection in the presence or absence of the indicated inhibitors by using a phase-contrast microscope (Axiovert 135TV; Zeiss) equipped with a Microfire camera (Optronics) and Picture frame software (Optronics) as described previously (21, 56). The numbers of surviving cells were determined from six nonoverlapping fields of view from triplicate infections. Percent cell death is reported as the average reduction ± standard deviation in the number of viable cells for each condition relative to 2 h after infection in the absence of inhibitors.
Quantitation of AcMNPV DNA.
SF21 cells were infected with AcMNPV wt/lacZ and treated with or without the indicated drugs as described above. At 12 h after infection, infected cells were collected and total cellular DNA was extracted. AcMNPV DNA was quantified by using quantitative real-time PCR as described previously (54). Samples were normalized for cell equivalents by using the Spodoptera frugiperda sfiap gene. Linearized plasmids containing sfiap or the AcMNPV HindIII-T genome fragment were used to generate standard curves. Values are reported as the average levels of viral DNA ± standard deviations from triplicate infections relative to that of infected cells treated without drug.
Confocal microscopy.
SF21 cells (2 × 106 cells) were seeded onto glass coverslips in 35-mm plates and transfected with plasmids encoding the indicated proteins. After 24 h, the cells were fixed with 4% formaldehyde in TC100 for 30 min, washed in PHEM buffer (60 mM PIPES [piperazine-N,N′-bis(2-ethanesulfonic acid)], pH 6.9, 25 mM HEPES, 10 mM EGTA, 2 mM MgCl2), overlaid with 0.5 μg/ml 4′, 6-diamidine-2-phenylindole dihydrochloride (DAPI) (Roche) in PHEM buffer for 10 min, and then washed with PHEM buffer. For infections, SF21 cells on coverslips were transfected with plasmid, inoculated 24 h later with AcMNPV wt/lacZ, and incubated in growth medium with or without drug. After 24 h, infected cells were fixed as described above. DAPI and GFP fluorescence were monitored by excitation at wavelengths (λ) of 408 and 488, respectively. Fluorescence images were collected with a Nikon Eclipse TE2000-U confocal microscope (kindly provided by Tom Martin, University of Wisconsin—Madison) and prepared by using Adobe Photoshop CS2.
Nucleotide sequence accession number.
The nucleotide sequence of the Spodoptera h2ax (sfh2ax) ORF identified in this study was submitted to the GenBank database under the accession number JX154397.
RESULTS
AcMNPV triggers a DNA damage response in Drosophila.
Baculovirus AcMNPV causes widespread and uniform apoptosis of Drosophila DL-1 cells as a result of virus DNA (vDNA) replication (54). Because apoptosis and the DNA damage response are well characterized in Drosophila (18, 61), we have used this advantageous system to define the linkage between these two pathways during infection. In AcMNPV-inoculated DL-1 cells, vDNA replicates with kinetics comparable to that in cells from permissive hosts (54). Thus, to determine whether vDNA replication triggers a DNA damage response (DDR), we assessed DDR activation during the course of infection. To this end, we used phosphorylated H2Av (γ-H2Av) as a sensitive indicator of the Drosophila DDR. This histone 2A variant is rapidly phosphorylated at the nuclear sites of damage by Drosophila kinases ATM and ATR, which function at the apex of DDR signaling (33, 49). Robust phosphorylation was confirmed in DL-1 cells (Fig. 2A, lanes 1 and 2) as γ-H2Av was readily detected by phospho-specific H2Av antiserum after treatment with etoposide, a potent DNA damage-inducing topoisomerase inhibitor (7). Upon inoculation with AcMNPV, γ-H2Av levels also increased, beginning with vDNA replication at 6 h (Fig. 2A, lanes 3 to 6). Virus-induced γ-H2Av peaked by 12 h and remained through 24 h after inoculation (lanes 7 to 11). The AcMNPV immediate early transactivator IE1 was detected before the increase in γ-H2Av (lanes 4 and 5). These findings suggested that AcMNPV induces the DDR in Drosophila.
Fig 2.

Baculovirus-induced phosphorylation of Drosophila H2Av. (A) Time course of infection. Drosophila DL-1 monolayers were mock infected (mi) or inoculated (+ virus) with P35+ AcMNPV recombinant wt/lacZ (MOI, 10). Uninfected monolayers were also treated with DMSO vehicle (veh) or 100 μM etoposide (etop). Whole-cell lysates prepared at the specified times (hours) thereafter were subjected to immunoblot analysis using phospho-specific anti-H2Av serum (top), anti-IE1 serum (middle), or anti-actin (bottom) to verify protein loading. H2Av-P denotes phosphorylated Drosophila H2Av. Protein molecular size standards (in kilodaltons) are designated on the left. (B) ATM- and ATR-specific RNAi of infected cells. DL-1 cells were transfected with no dsRNA (none) or with egfp-, atm-1, atm-2, or atr-specific dsRNAs and 48 h later mock infected (mi) or inoculated (+virus) with AcMNPV wt/lacZ (MOI, 10). Whole-cell lysates prepared 12 h after infection were subjected to immunoblot analysis as described for panel A. (C) ATM- and ATR-specific RNAi of etoposide-treated cells. DL-1 cells were transfected with the indicated dsRNAs and treated 48 h later with DMSO (vehicle) or etoposide (100 μM). Whole-cell lysates prepared 2 h later were subjected to immunoblot analysis as described for panel A.
To confirm that the host DDR was responsible for virus-induced H2Av phosphorylation, we used RNA silencing (RNAi) to ablate the DDR kinases ATM and ATR. Treatment of DL-1 cells with either of two nonoverlapping, atm-specific dsRNAs before infection reduced virus-induced γ-H2Av to background levels (Fig. 2B, lanes 8 and 9). Conversely, atr-specific dsRNA (lane 10) had no effect on γ-H2Av levels, which were comparable to those of control egfp dsRNA-treated (lane 7) or -untreated (lane 6) cells. None of the dsRNAs induced γ-H2Av in mock-infected cells (lanes 2 to 5), nor did they affect virus entry or early gene expression as judged by comparable levels of intracellular viral IE1 (Fig. 2B). To verify the efficacy of RNA silencing, we monitored γ-H2Av in DL-1 cells treated with etoposide to pharmacologically induce DNA damage. As expected, atm-specific but not egfp-specific dsRNAs reduced etoposide-induced γ-H2Av (Fig. 2C, lanes 7 to 9). Collectively, these findings indicated that AcMNPV elicits an ATM-driven DDR in Drosophila. Because we were unable to confirm ATR ablation by RNAi (lane 10), it remains possible that ATR also contributes to this DDR.
Baculovirus DNA replication activates the Drosophila DDR.
On the basis that H2Av phosphorylation coincided with the initiation of vDNA synthesis (Fig. 2A), we predicted that vDNA replication triggers the DDR. We tested this possibility by silencing essential AcMNPV replicative genes and thereby inhibiting vDNA synthesis. AcMNPV IE1 is required for replicative gene expression and contributes directly to vDNA synthesis (54, 60, 62). Upon treatment with ie-1-specific dsRNA, IE1 was ablated and virus-induced γ-H2Av was reduced to background levels (Fig. 3A, lane 9). Silencing DNA helicase P143 or DNA primase LEF-1, both of which are essential for AcMNPV DNA replication, caused comparable reductions in γ-H2Av (lanes 10 and 11). In contrast, silencing the late viral RNA polymerase P47, which is not required for vDNA synthesis (46, 54), had no effect on virus-induced γ-H2Av (lane 12). None of the dsRNAs induced γ-H2Av in the absence of virus (lanes 1 to 6). We concluded that baculovirus DNA replication and not late gene expression is required for activation of the Drosophila DDR.
Fig 3.

Role of vDNA replication in triggering H2Av phosphorylation. (A) RNAi of baculovirus replicative genes. DL-1 cells transfected with no dsRNA (none), control egfp dsRNA, or virus-specific ie-1, p143, lef-1, or p47 dsRNAs were mock infected (mi) or inoculated (+virus) 48 h later with AcMNPV wt/lacZ (MOI, 10). Whole-cell lysates prepared 12 h after infection were subjected to immunoblot analysis using the indicated antisera. (B) Caspase inhibition. DL-1 cells were mock infected (mi) or inoculated (+virus) with AcMNPV wt/lacZ (MOI, 10) in the presence (+) or absence (−) of zVAD-fmk (200 μM), lysed 24 h later, and subjected to immunoblot analysis using the indicated antisera. The DrICE proform (pro-DrICE) and active large subunit (active DrICE) are indicated. H2Av-P denotes phosphorylated H2Av.
The end stages of caspase-dependent apoptosis include fragmentation of host chromosomal DNA (50). To rule out the possibility that the DDR was triggered by caspase-mediated DNA damage, we monitored γ-H2Av under conditions of complete caspase inhibition. Treatment with zVAD-fmk, a potent cell-permeable inhibitor of Drosophila caspases, had no effect on virus-induced levels of γ-H2Av (Fig. 3B, lanes 3 and 4). Confirming the efficacy of zVAD-fmk, virus-induced processing of the Drosophila effector caspase DrICE to its active large and small subunits (27, 64) was fully blocked (compare lanes 3 and 4). We concluded that the DDR was caspase independent and thus triggered by vDNA replication-dependent events upstream of apoptosis.
ATM/ATR inhibitors block baculovirus-induced apoptosis in Drosophila.
To assess the contribution of the DDR to apoptosis triggered by infection, we determined the effects of known inhibitors of DDR kinases on AcMNPV-induced apoptosis. Caffeine is a broad but effective inhibitor of ATM, ATR, and cell cycle checkpoint kinases (52). Caffeine suppressed AcMNPV-induced apoptosis in a dose-dependent manner. DL-1 cells remained intact and exhibited little or no apoptotic debris after inoculation with a p35-deficient AcMNPV mutant in the presence of 10 mM caffeine (Fig. 4A, panel iii), the minimum concentration for inhibition. Conversely, in the absence of drug, cell death and debris were abundant (panel ii), as expected (27, 71, 73). By 24 h after inoculation, caffeine reduced virus-induced apoptosis ∼6-fold compared to that of untreated controls (Fig. 4B). This level was comparable to that of cells treated with aphidicolin, an inhibitor of AcMNPV DNA synthesis that prevents virus-induced apoptosis (12). Confirming caffeine-mediated inhibition of apoptosis, proteolytic processing of the Drosophila effector caspase DrICE to its active large and small subunits was blocked in a dose-dependent manner (Fig. 4C). Active DrICE is a principal mediator of baculovirus-induced apoptosis in DL-1 cells (27, 64). Ruling out the possibility that caffeine blocked virus entry or early gene expression, levels of intracellular IE1 were comparable up through 10 mM caffeine (Fig. 4C). Importantly, caffeine prevented both virus- and etoposide-induced phosphorylation of H2Av in a dose-dependent manner (Fig. 4C and D). Thus, caffeine is a potent inhibitor of the Drosophila DDR. In contrast, the drug KU55933, which is an inhibitor of human ATM (20), failed to block H2Av phosphorylation in infected DL-1 cells (data not shown). As predicted, therefore, KU55933 had no effect on AcMNPV-induced apoptosis of these cells. Collectively, our findings indicated that the DDR contributes to baculovirus-induced apoptosis.
Fig 4.

Inhibition of virus-induced apoptosis and γ-H2Av in Drosophila. (A) Apoptotic cytolysis. DL-1 cells were photographed (magnification, ×200) at 2 h and 24 h after inoculation (+virus) with p35-deficient AcMNPV in the presence (+ caffeine [10 mM]) or absence of caffeine. (B) Cell death. Intact, nonapoptotic DL-1 cells inoculated either 2 h or 24 h earlier with p35-deficient AcMNPV in the absence (−) or presence of 10 mM caffeine (caff), DMSO (veh), or 5 μg/ml aphidicolin (aphid) were counted by using computer-aided microscopy. Values shown represent the percent (± standard deviation) cell death calculated from surviving cells averaged for three independent plates and compared to that of untreated cells at 2 h after infection. A representative experiment is shown. (C) Inhibition of γ-H2Av and caspase activation. Whole-cell DL-1 lysates prepared 24 h after mock infection (mi) or inoculation (+virus) with AcMNPV wt/lacZ (MOI, 10) in the absence (−) or presence of the indicated concentrations of caffeine were subjected to immunoblot analysis as indicated. (D) Inhibition of etoposide-induced γ-H2Av. DL-1 monolayers were treated for 1 h in the absence (−) or presence of the indicated concentrations of caffeine followed by DMSO vehicle (veh) or etoposide (100 μM) for 2 h. Whole-cell lysates were subjected to immunoblot analysis.
ATM/ATR inhibitors block baculovirus-induced apoptosis in Spodoptera.
AcMNPV also triggers a DDR in cultured cells of the natural lepidopteran host Spodoptera frugiperda (22). To further examine the connection between virus-induced apoptosis and the DDR, we compared the responses in Drosophila and Spodoptera, two evolutionarily distinct insect lineages that diverged ∼350 million years ago (15). As in DL-1 cells, 10 mM caffeine suppressed p35-deficient AcMNPV-induced apoptosis of SF21 cells (Fig. 5A). Compared to vehicle-treated or untreated cells, caffeine reduced cell death at 24 h more than 2-fold (Fig. 5B). Treatment of SF21 cells with the ATM-specific inhibitor KU55933 (20) also suppressed virus-induced apoptosis (Fig. 5A and B). The level of inhibition by the ATM/ATR inhibitors was comparable to that of aphidicolin, a potent inhibitor of AcMNPV DNA synthesis. These findings suggested that one or more Spodoptera ATM/ATR homologs contribute to baculovirus-induced apoptosis.
Fig 5.

Effects of ATM/ATR inhibition on AcMNPV-induced apoptosis in Spodoptera. (A) Apoptotic cytolysis. SF21 cells were photographed (magnification, ×200) at 2 h and 24 h after inoculation (+virus) with p35-deficient AcMNPV in the absence (−) or presence of ATM/ATR inhibitors caffeine (10 mM), KU55933 (50 μM), or DMSO (vehicle). (B) Cell death. Intact, nonapoptotic SF21 cells inoculated 2 h or 24 h earlier with p35-deficient AcMNPV in the absence (−) or presence of 10 mM caffeine (caff), 50 μM KU55933 (KU), DMSO (veh), or 5 μg/ml aphidicolin (aphid) were counted by using computer-aided microscopy. Values shown represent the percent (± standard deviation) cell death calculated from surviving cells averaged for three independent plates and compared to that of untreated cells at 2 h after infection. A representative experiment is shown. (C) SfIAP depletion and caspase activation. SF21 cells were mock infected (mi) or inoculated (+virus) with AcMNPV wt/lacZ (MOI, 10) in the absence (−) or presence of DMSO (veh), 50 μM KU55933 (KU), or 10 mM caffeine (caff). Whole-cell lysates were prepared at the indicated times (hours) after infection and subjected to immunoblot analysis by using anti-SfIAP (α-SfIAP; top), anti-Sf-caspase-1 (α-Sfcasp-1; middle), or anti-actin (α-Actin; bottom). The Sfcaspase-1 proform (pro-Sfcasp-1) and active large subunit (active Sfcasp-1) are indicated. The asterisk denotes a nonspecific protein recognized by anti-SfIAP.
The inhibitor-of-apoptosis (IAP) proteins regulate caspase-dependent apoptosis in vertebrates and invertebrates (11, 14, 17). Upon baculovirus infection, the principal cellular IAP in Spodoptera, SfIAP, is rapidly destroyed by a mechanism triggered by vDNA replication (9, 64). Loss of SfIAP causes caspase activation and apoptotic cell death. Treatment of SF21 cells with either caffeine or KU55933 delayed virus-induced depletion of SfIAP compared to vehicle-treated cells (Fig. 5C). Consistent with the delay in SfIAP loss, these inhibitors reduced the accumulation of active subunits of Sf-caspase-1 (Fig. 5C, lanes 5 to 8), which is the principal effector caspase of Spodoptera (1, 26, 73). Caffeine or KU55933 had no effect on SfIAP or Sf-caspase-1 levels in mock-infected cells over the course of the experiment (data not shown). Thus, caffeine and KU55933 suppressed apoptosis at a step upstream of virus-induced depletion of SfIAP in Spodoptera.
The Spodoptera DDR stimulates AcMNPV DNA replication.
Despite the detrimental effects of virus-induced apoptosis, some viruses hijack components of the host's DNA damage response, including ATM and ATR, to promote virus multiplication (66). Caffeine's inhibitory effect on AcMNPV has suggested that baculoviruses do the same (22). To further explore this possibility, we also quantified the effects of ATM and ATR inhibition on AcMNPV replication. In preliminary experiments, we discovered that the potency of caffeine and KU55933 declined significantly through 24 h at 27°C (data not shown); consequently, each drug was replenished every 24 h during infection. By 72 h, caffeine and KU55933 suppressed budded virus yields ∼100,000-fold compared to untreated or vehicle-treated controls (Fig. 6A). Neither drug interfered with early synthesis of AcMNPV proteins IE1, IE0, or P35 (data not shown), indicating that virus entry and early gene expression and function were unaffected. These findings suggested that Spodoptera ATM/ATR homologs are required for replication events occurring downstream of early virus gene expression. Thus, to determine the effect of ATM/ATR inhibition on baculovirus DNA synthesis, we used quantitative real-time PCR to measure AcMNPV DNA accumulation. During the exponential increase in vDNA synthesis in SF21 cells (12 h), caffeine and KU55933 reduced vDNA accumulation ∼18- and ∼8-fold, respectively, compared to controls (Fig. 6B). The DNA synthesis inhibitor aphidicolin reduced vDNA accumulation similarly. These findings confirmed that ATM/ATR inhibitors suppressed one or more events required for AcMNPV DNA replication.
Fig 6.

Effect of ATM/ATR inhibitors on baculovirus multiplication. (A) Budded virus (BV). Extracellular BV was collected from SF21 cells 72 h after inoculation with AcMNPV wt/lacZ (MOI, 0.5) in the absence (−) or presence of 10 mM caffeine (caff), DMSO (veh), or 50 μM KU55933 (KU); drugs were replenished every 24 h. BV yields were quantified as TCID50 and are reported as the average virus concentration (PFU/ml) ± standard deviation of triplicate samples; a representative experiment is shown. (B) vDNA replication. Total DNA was extracted from SF21 cells 12 h after infection with AcMNPV wt/lacZ (MOI, 10) in the absence (−) or presence of 10 mM caffeine (caff), DMSO (veh), 50 μM KU55933 (KU), or 5 μg/ml aphidicolin (aphid). AcMNPV DNA levels were quantified by real-time PCR using the Spodoptera sfiap gene to normalize for cell equivalents of genomic DNA. Values are reported as the average quantities of virus DNA ± standard deviations from triplicate infections relative to that of infected cells in the absence of drug. (C) AcMNPV DNA replication centers. SF21 cells were transfected with GFPHA-P143-encoding plasmid as well as P35-encoding plasmid to prevent potential caspase activation and cell death associated with expression of the replicative viral helicase. Twenty-four hours later, cells were mock infected (mi) or infected (+virus) with AcMNPV wt/lacZ (MOI, 10) in the presence (+caff) or absence (−) of 10 mM caffeine. After 24 h, the cells were fixed, stained with DAPI, and viewed by fluorescence microscopy. Representative fields are shown. White arrows denote marginalized host chromatin at the nuclear periphery. Asterisks mark viral DNA replication centers. Bar, 5 μm.
To investigate replicative events affected by ATM/ATR inhibition, we monitored formation of vDNA replication centers by using confocal microscopy. Comprised of AcMNPV DNA and replicative factors, including vDNA helicase P143, these replication centers expand dramatically within the infected nucleus, forcing host chromatin to the nuclear periphery (38, 41). Viral DNA replication centers and marginalized host DNA are readily detected by DAPI staining of AcMNPV-infected cells (Fig. 6C, panel iv). The replication center is depicted by the presence of helicase P143, which can be visualized by coexpression of a GFP-P143 fusion protein in infected cells (panels v and vi). In uninfected cells, GFP-P143 localizes exclusively to the cytoplasm (panels ii and iii) because this helicase must interact with virus DNA-binding protein LEF-3 for nuclear import (72). Upon caffeine treatment of infected cells, viral replication centers failed to enlarge. Rather, DAPI-stained DNA was retained in small punctate foci and GFP-P143 was evenly distributed through the nucleus (panels vii to ix). The nuclear presence of P143 indicated that caffeine had no effect on LEF-3-mediated import. These findings confirmed that caffeine blocks one or more early events required for vDNA replication and are consistent with host ATM/ATR playing a critical role in the replication process.
DNA damage triggers phosphorylation of Spodoptera histone H2AX.
To better define the biochemical activities of ATM and ATR during infection, we obtained histone H2AX from Spodoptera for use as a substrate and a sensitive marker for Spodoptera DDR activation. Using nucleotide sequence similarity with the Bombyx mori h2ax homolog, we identified and cloned the Spodoptera h2ax (sfh2ax) gene from an SF21 cDNA library (26). Sequence analysis revealed that SfH2AX is a 138-residue protein with conserved DNA-binding residues within the histone 2A core and a C-terminal consensus site at Ser135 for phosphorylation by ATM and ATR (Fig. 7). Upon fusion with GFP (Fig. 8A), GFPHA-SfH2AX localized preferentially to the nuclei of plasmid-transfected SF21 cells (Fig. 8B, panels v and vi), consistent with previous reports for nuclear import and chromatin incorporation of GFP-histone fusion proteins (24, 59). In contrast, GFP alone was evenly distributed within the cell (panels ii and iii). Following infection with AcMNPV, GFPHA-SfH2AX was excluded from vDNA replication centers and redistributed to the nuclear periphery, colocalizing with DAPI-stained host chromatin (panels x to xii). Altogether, these findings suggested that GFPHA-SfH2AX functions as expected for a typical histone within both uninfected and baculovirus-infected cells (38).
Fig 7.

H2AX homologs. The predicted amino acid sequence of the Spodoptera frugiperda H2AX homolog (GenBank accession no. JX154397) is compared to that of Bombyx mori (NP_001153667.1), Drosophila melanogaster (NP_524519.1), and Homo sapiens (NP_002096.1). The conserved C-terminal ATM/ATR phosphorylation site (SQ) is highlighted. Conserved DNA-binding residues (Arg and Lys) (solid boxes) and a conserved ubiquitination site (dashed box) within the H2A core (gray) are marked. Alignments were created using the ClustalW2 multiple sequence alignment program (28) (http://www.ebi.ac.uk/Tools/msa/clustalw2/).
Fig 8.

Localization of Spodoptera SfH2AX. (A) Fusion proteins. GFPHA-SfH2AX (top) contains Spodoptera SfH2AX (138 residues) fused to the C terminus of GFPHA (bottom). The required Ser residue (Ser135) comprising the consensus ATM/ATR phosphorylation site (SQ) within SfH2AX was replaced by Ala in GFPHA-SfH2AXS135A (middle). HA epitope tags are indicated. (B) Fluorescence microscopy. SF21 cells were fixed 24 h after transfection with plasmids expressing the indicated proteins (panels i to ix). Cells were also inoculated (+ virus) with AcMNPV wt/lacZ (MOI, 10) 24 h after transfection and fixed 24 h later (panels x to xii). All cells were stained with DAPI and viewed by fluorescence microscopy. Representative fields at the same magnification are shown. White arrows denote marginalized host chromatin at the nuclear periphery of infected cells. Asterisks mark viral DNA replication centers. Bar, 5 μm.
To demonstrate that SfH2AX is an ATM/ATR substrate, we compared phosphorylation of GFPHA-SfH2AX with that of S135A-mutated GFPHA-SfH2AX, in which the consensus ATM/ATR phosphorylation residue Ser135 was replaced by Ala (Fig. 8A). Both fusion proteins localized to the nucleus of plasmid-transfected cells (Fig. 8B, panels iv to ix). Moreover, the DNA damage-inducing agent etoposide triggered phosphorylation of a protein with a size expected for γ-GFPHA-SfH2AX (45 kDa) as detected by phospho-specific H2AX antiserum (Fig. 9, lane 2). Caffeine and KU55933 blocked this phosphorylation (lanes 3 and 4). In contrast, etoposide failed to induce phosphorylation of S135A-mutated GFPHA-SfH2AX (lane 8), which was present at levels comparable to those of the wild-type fusion protein (compare lanes 2 and 8). Neither protein was phosphorylated when treated with vehicle alone (lanes 1 and 7). We concluded that GFPHA-SfH2AX is phosphorylated by Spodoptera ATM/ATR homologs at its consensus residue in response to DNA damage and therefore recapitulates the function of endogenous H2AX proteins (29, 61). Because the phosphorylation signature of GFPHA-SfH2AX is specific and robust, it represents a highly sensitive indicator of the Spodoptera DDR. As such, we used it to further characterize the interplay between baculoviruses and the host DDR.
Fig 9.

Etoposide-induced phosphorylation of GFPHA-SfH2AX. SF21 cells transfected 24 h earlier with plasmids encoding wild-type GFPHA-SfH2AX or GFPHA-SfH2AXS135A were treated with DMSO vehicle (veh) (lanes 1 and 7) or 100 μM etoposide (etop) (lanes 2, 3, 4, and 8). Simultaneously, the etoposide-dosed cells were left untreated (−) (lane 2) or treated with 10 mM caffeine (caff) (lane 3) or 50 μM KU55933 (KU) (lane 4). Transfected cells were also mock infected (mi) (lanes 5 and 9) or inoculated (virus) (lanes 6 and 10) with AcMNPV wt/lacZ (MOI, 10). Whole-cell lysates prepared 1 h after drug treatment or 12 h after infection were subjected to immunoblot analysis by using phospho-specific anti-H2AX (top), anti-HA (middle top), anti-IE1 (middle bottom), or anti-actin (bottom). GFP-H2AX-P denotes the phosphorylated form of the protein.
AcMNPV represses SfH2AX phosphorylation.
The demonstrated role of Spodoptera ATM/ATR in baculovirus DNA replication suggested that the host DDR contributes to optimum virus multiplication (Fig. 6; see also reference 22). Due to the critical nature of γ-H2AX in DDR signaling, we predicted that AcMNPV infection of Spodoptera SF21 cells would trigger phosphorylation of SfH2AX. To our surprise, however, GFPHA-SfH2AX-producing SF21 cells exhibited only background levels of phosphorylation after infection (Fig. 9, compare lanes 5 and 6). This low level of γ-GFPHA-SfH2AX was comparable to that of infected cells producing S135A-mutated GFP-SfH2AX (compare lanes 6 and 10). Ruling out the possibility that GFPHA-SfH2AX was destabilized, AcMNPV had no effect on total levels of either the wild-type or S135A-mutated forms of the protein (Fig. 9, compare lanes 5 to 6 and 9 to 10). The presence of intracellular IE1 and IE0 confirmed infection (lanes 6 and 10).
This finding raised the interesting possibility that AcMNPV modulates host phosphorylation of SfH2AX and thus manipulates aspects of the DDR to its own advantage. To investigate, we first tested AcMNPV's ability to affect SfH2AX phosphorylation in response to an independent DNA-damaging agent, etoposide. To this end, we treated cells with increasing doses of etoposide at various times after AcMNPV infection and monitored levels of γ-GFPHA-SfH2AX. At 3 h after infection, etoposide-induced levels of γ-GFPHA-SfH2AX were indistinguishable in infected versus uninfected cells (Fig. 10A, compare lanes 1 to 3 with 4 to 6). However, by 6 h, γ-GFPHA-SfH2AX was reduced within infected cells (lanes 10 to 12). By 12 h, only background levels of γ-GFPHA-SfH2AX were detected following etoposide treatment of infected cells (lanes 16 to 18). Similarly, virus infection suppressed γ-GFPHA-SfH2AX induced by a second DNA-damaging agent, bleocin (data not shown). In each case, the total level of intracellular GFPHA-SfH2AX was unchanged (Fig. 10A, lanes 1 to 18) and IE1 and IE0 were readily detected in infected cells. These findings suggested that AcMNPV mediates a time-dependent impairment of GFPHA-SfH2AX phosphorylation.
Fig 10.

AcMNPV inhibition of SfH2AX phosphorylation. (A) Time course of infection. SF21 cells transfected 24 h earlier with plasmid encoding GFPHA-SfH2AX were mock infected (mi) or inoculated (+virus) with AcMNPV wt/lacZ (MOI, 10). At the specified times (hours) after infection, cells were treated for 30 min with DMSO vehicle (−) or the indicated concentration of etoposide, lysed, and subjected to immunoblot analysis as indicated. (B) Effect of MOI. SF21 cells transfected 24 h earlier with plasmid encoding GFPHA-SfH2AX were mock infected (mi) or inoculated with AcMNPV wt/lacZ at the indicated MOIs. After a 30-min treatment with 100 μM etoposide at 12 h after infection, the cells were lysed and subjected to immunoblot analysis as indicated. (C) AcMNPV mutant ts8. The temperature-sensitive substitution V934M within DNA helicase p143 (gene Ac95) prohibits vDNA replication and late viral gene expression at 33°C but not 24°C (32). (D) Comparison of γ-GFPHA-SfH2AX at permissive and nonpermissive temperatures. SF21 cells stably expressing GFPHA-SfH2AX were mock infected (mi) or inoculated with wild-type AcMNPV (wt) or temperature-sensitive ts8 and maintained at either the permissive (24°C) or the nonpermissive (33°C) temperature. After a 1-h treatment with DMSO (veh) or 100 mM etoposide at 12 h after infection, the cells were lysed and subjected to immunoblot analysis as indicated.
To verify that inhibition of SfH2AX phosphorylation is a direct result of infection, we monitored this phenomenon at different MOIs. If AcMNPV actively alters SfH2AX phosphorylation, then increased virus gene dosage that is conferred by high MOIs would enhance this effect. Indeed, as the MOI was increased, etoposide-induced GFPHA-SfH2AX phosphorylation was reduced (Fig. 10B); at the highest MOIs, only background levels of γ-GFPHA-SfH2AX were detected (lanes 4 and 5). As expected, infection had no effect on the steady-state levels of GFPHA-SfH2AX (lanes 1 to 5). Indicative of increased gene dosage, IE1/IE0 levels accumulated with MOI (Fig. 10B). We concluded that alteration of SfH2AX phosphorylation was directly proportional to the level of input virus and thus gene dosage. This finding is consistent with the presence of a virus-encoded activity that affects γ-SfH2AX during vDNA replication.
AcMNPV repression of SfH2AX phosphorylation is independent of vDNA replication.
Impairment of SfH2AX phosphorylation coincided with the start of vDNA replication by ∼6 h after infection (Fig. 10A). Thus, as a means to investigate the virus-specific genes or events responsible, we determined whether AcMNPV DNA replication or late gene expression was required for this inhibition. To this end, we used AcMNPV mutant ts8 (Fig. 10C), a virus that carries a temperature-sensitive defect within the DNA helicase gene p143 (32). At the nonpermissive temperature, ts8 expresses early viral genes but fails to synthesize viral DNA, express late genes, or produce infectious virus (16). Thus, we monitored the capacity of ts8 to affect etoposide-induced phosphorylation of GFPHA-SfH2AX at permissive and nonpermissive temperatures. At the permissive temperature (24°C), ts8 blocked etoposide-induced GFPHA-SfH2AX phosphorylation as well as wild-type AcMNPV (Fig. 10D, lanes 2 to 4). Similarly, at the nonpermissive temperature (33°C), inhibition of SfH2AX phosphorylation by ts8 was comparable to that by wild-type virus (lanes 6 to 8). Neither virus had any effect on the total intracellular levels of GFPHA-SfH2AX (lanes 1 to 8). We confirmed the temperature sensitivity of ts8 (data not shown) as indicated by the absence of late viral protein synthesis and the lack of occluded virus (OV) production at nonpermissive temperature (16, 25, 32). Collectively, these findings demonstrated that P143-mediated vDNA synthesis and virus late gene expression are not required for virus-mediated reduction of SfH2AX phosphorylation. These data suggested that one or more early AcMNPV factors produced prior to vDNA synthesis alter SfH2AX phosphorylation and that this activity modifies the host DDR for optimum virus multiplication.
DISCUSSION
In this study, we report that the prototype baculovirus AcMNPV elicits a DNA damage response in cells derived from evolutionarily divergent insects, including Drosophila (order Diptera) and Spodoptera (order Lepidoptera). Events associated with vDNA replication activated host DDR kinases, including ATM, which were required for depletion of cellular IAPs and subsequent caspase activation. Despite this proapoptotic antiviral role, the host DDR stimulated AcMNPV DNA replication and contributed significantly to virus yields. Thus, by virtue of baculovirus apoptotic suppressors that block cell death, these DNA viruses exploit the host DDR to promote multiplication (Fig. 1). Indeed, we discovered here that AcMNPV alters the DDR through virus-encoded activities that affect phosphorylation of host histone variant H2AX, a central regulator of the DDR. Thus, viral manipulation of the host DDR (66) is an efficient multiplication strategy conserved among DNA viruses of both insects and mammals.
Baculovirus activation of host DDR-mediated apoptosis.
The best-studied invertebrate DDR is that of Drosophila. Many key DDR factors of mammals (e.g., ATM, ATR, H2AX, Mre11, Rad50, Nbs1, Chk2, p53, etc.) have Drosophila orthologs (61). We demonstrated here that AcMNPV DNA replication activates the Drosophila DDR, including the DDR kinase ATM, at a step upstream from caspase activation and cellular DNA fragmentation. In virus-infected DL-1 cells, ATM activity was blocked by RNAi ablation of ATM and AcMNPV replicative factors but not by caspase inhibition, as demonstrated by effects on ATM-mediated phosphorylation of the Drosophila histone variant H2Av (Fig. 2 and 3). The ATM inhibitor caffeine also blocked or reduced H2Av phosphorylation following etoposide-induced DNA damage, verifying the role of ATM in the Drosophila DDR. Importantly, caffeine also suppressed H2Av phosphorylation and apoptosis in virus-infected Drosophila cells (Fig. 4). Consistent with the established role of the DDR as an inducer of Drosophila apoptosis (61), our findings suggest that the DDR activated by vDNA replication triggers apoptosis. Upon activation by the DDR, the Drosophila P53 ortholog (DmP53) can upregulate proapoptotic factors that destabilize cellular IAPs to promote caspase activation and apoptosis (5, 36). However, DmP53-independent pathways for apoptosis also exist (35, 68). Thus, whether DmP53 or other proapoptotic factors contribute to baculovirus-induced apoptosis requires further study.
Cellular IAP is rapidly depleted as an apical step during apoptosis of virus-infected cells of Drosophila and Spodoptera, two insect lineages that diverged ∼350 million years ago (15, 54, 56, 64). We demonstrated here that blocking the DDR with kinase inhibitors caffeine and KU55933 delayed virus-induced depletion of Spodoptera SfIAP and thus suppressed caspase activation and apoptosis (Fig. 5). We confirmed that both drugs inhibited the DDR of Drosophila and Spodoptera (Fig. 4 and 9). Thus, our findings indicate that loss of cellular IAP function is a consequence of DDR activation. Because the Spodoptera DDR stimulates AcMNPV DNA replication (see below and reference 22), it is also possible that the DDR accelerates apoptosis by contributing directly to vDNA synthetic events in a type of feedback loop (Fig. 1). Although the exact mechanism(s) by which the DDR promotes IAP destruction and thus apoptosis remains to be defined, it is clear that the DDR plays an evolutionarily conserved role as a proapoptotic antiviral defense.
Host DDR-mediated enhancement of baculovirus multiplication.
Despite its role in promoting virus-induced apoptosis, we observed that the DDR also stimulated viral DNA synthesis and contributed significantly to virus production. Caffeine- and KU55933-mediated inhibition of DDR kinases restricted accumulation of AcMNPV DNA as potently as the DNA synthesis inhibitor aphidicolin (Fig. 6). Yields of budded virus were reduced ∼100,000-fold. Thus, our findings agree with those of Huang et al. (22), who found that the AcMNPV-induced DDR contributes to virus replication. Notably, the baculoviruses can usurp host DDR activities to promote vDNA replication by virtue of their ability to efficiently block the proapoptotic outcome at downstream steps with caspase inhibitors (Fig. 1).
How the DDR facilitates replication of DNA viruses is poorly understood (66). For human cytomegalovirus (HCMV) and herpes simplex virus 1 (HSV-1), the host DDR contributes to vDNA replication (13, 31, 58, 69). These viruses recruit DDR and DNA repair factors to the replication compartments, suggesting these factors participate in recombination-dependent replication (reviewed in references 67 and 70). We observed that the Spodoptera DDR is required for formation of nuclear AcMNPV replication centers (Fig. 6). Given the high level of recombination during baculovirus DNA replication (reviewed in references 42 and 51), it is likely that host DDR components are recruited to these virus centers for a similar purpose. DDR kinases such as ATM may also phosphorylate baculovirus replicative factors to activate them for vDNA synthesis. This mechanism has been proposed for ATM-driven phosphorylation of simian virus 40 (SV40) large T-antigen, which is required for optimal SV40 replication (57). Indeed, we recently demonstrated that phosphorylation of the essential replicative factor IE1 of AcMNPV coincides with vDNA replication (62). IE1 phosphorylation was mediated by a caffeine-sensitive kinase and was required for IE1 replicative activity. Thus, IE1 may be regulated by the DDR. The DDR may also contribute to baculovirus-induced cell cycle arrest and thereby establish a cellular environment more favorable for vDNA replication (4, 23, 47). Further studies should define the mechanisms whereby the host DDR facilitates baculovirus replication.
AcMNPV manipulation of the host DDR.
ATM-mediated phosphorylation of histone variant H2AX is a key step in the regulation and amplification of the DDR (3, 63). γ-H2AX recruits DDR components and repair factors to the site of DNA damage and is a sensitive marker for DDR activation in diverse organisms. AcMNPV induced phosphorylation of H2Av during the peak of vDNA synthesis in Drosophila (Fig. 2). In Spodoptera, etoposide-induced DNA damage also caused ATM-mediated phosphorylation of SfH2AX (Fig. 9). Thus, we were surprised that γ-SfH2AX was reduced to background levels upon AcMNPV infection. To increase sensitivity, we used GFPHA-SfH2AX, which localized to the nucleus (Fig. 8) and behaved as a bona fide ATM substrate in a residue (Ser135)-specific manner (Fig. 9). We confirmed that AcMNPV repressed etoposide-induced γ-SfH2AX in an MOI-dependent manner (Fig. 10). Moreover, this repression coincided with the period of peak vDNA synthesis. Additional experiments (data not shown) indicated that this repressive activity declines as vDNA synthesis subsides late in infection. Hence, our findings suggest an important role for repression of histone SfH2AX phosphorylation and thus modulation of the host DDR for baculovirus DNA replication (see below). Moreover, these preliminary findings explain discrepancies between our studies and those of Huang et al. (22), wherein SfH2AX was shown to be phosphorylated late in infection.
By using a conditional lethal mutant (ts8) for DNA replication, we determined that late gene expression had no effect on repression of γ-SfH2AX. These findings suggest the involvement of one or more early AcMNPV genes in Spodoptera (Fig. 1). Additional studies are required to determine why AcMNPV is less efficient for repressing γ-H2Av during infection of Drosophila cells, which support expression of some early AcMNPV genes (37, 54). Preliminary studies suggest that AcMNPV represses γ-H2Av that is triggered by etoposide-induced DNA damage in Drosophila (data not shown). Thus, in this context, AcMNPV may function in part to repress DNA damage-induced γ-H2Av. Efforts to identify the AcMNPV factor(s) responsible are under way.
DNA viruses regularly disable antiviral facets of the DDR to exploit other beneficial aspects for DNA replication (66). Our study suggests that virus-mediated repression of γ-H2AX is another such tactic. We hypothesize that this repressive activity is a baculovirus strategy for manipulating the host DDR to render it more favorable for vDNA replication. Relevant to this hypothesis, we observed that GFPHA-SfH2AX was marginalized to the nuclear periphery away from AcMNPV replication centers (Fig. 8C). This finding and those of another study (38) suggest that SfH2AX associates exclusively with host chromatin, not baculovirus DNA, during infection. Because γ-H2AX recruits DDR and DNA repair components, it may be advantageous to prevent retention of DDR factors on host chromatin, thereby ensuring their availability for vDNA replication by suppressing SfH2AX phosphorylation. This process may be especially critical at the onset of vDNA replication, prior to marginalization of host H2AX/chromatin and the establishment of discrete vDNA centers. Further studies should provide insight into the mechanism of γ-H2AX repression and its benefits for baculovirus replication.
ACKNOWLEDGMENTS
We thank David J. Taggart for pGFPHA and pGFPHA-P143 and Nathaniel Byers for affinity purification of SfIAP antiserum. We also thank Tom Martin (U.W.—Madison) for providing access to the confocal microscope used in this study.
This work was supported in part by Public Health Service grants AI25557 and AI40482 from the National Institute of Allergy and Infectious Diseases (P.D.F.) and NIH predoctoral traineeship T32 GM07125 (J.K.M.).
Footnotes
Published ahead of print 3 October 2012
REFERENCES
- 1. Ahmad M, et al. 1997. Spodoptera frugiperda caspase-1, a novel insect death protease that cleaves the nuclear immunophilin FKBP46, is the target of the baculovirus antiapoptotic protein P35. J. Biol. Chem. 272:1421–1424 [DOI] [PubMed] [Google Scholar]
- 2. Best SM. 2008. Viral subversion of apoptotic enzymes: escape from death row. Annu. Rev. Microbiol. 62:171–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bonner WM, et al. 2008. GammaH2AX and cancer. Nat. Rev. Cancer 8:957–967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Braunagel SC, Parr R, Belyavskyi M, Summers MD. 1998. Autographa californica nucleopolyhedrovirus infection results in Sf9 cell cycle arrest at G2/M phase. Virology 244:195–211 [DOI] [PubMed] [Google Scholar]
- 5. Brodsky MH, et al. 2004. Drosophila melanogaster MNK/Chk2 and p53 regulate multiple DNA repair and apoptotic pathways following DNA damage. Mol. Cell. Biol. 24:1219–1231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Brown M, Crawford AM, Faulkner PF. 1979. Genetic analysis of a baculovirus Autographa californica nuclear polyhedrosis virus 1. Isolation of temperature sensitive mutants and assortment into complementation groups. J. Virol. 31:190–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Caldecott K, Banks G, Jeggo P. 1990. DNA double-strand break repair pathways and cellular tolerance to inhibitors of topoisomerase II. Cancer Res. 50:5778–5783 [PubMed] [Google Scholar]
- 8. Cartier JL, Hershberger PA, Friesen PD. 1994. Suppression of apoptosis in insect cells stably transfected with baculovirus p35: dominant interference by N-terminal sequences p35(1-76). J. Virol. 68:7728–7737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cerio RJ, Vandergaast R, Friesen PD. 2010. Host insect inhibitor-of-apoptosis SfIAP functionally replaces baculovirus IAP but is differentially regulated by its N-terminal leader. J. Virol. 84:11448–11460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cimprich KA, Cortez D. 2008. ATR: an essential regulator of genome integrity. Nat. Rev. Mol. Cell Biol. 9:616–627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Clem RJ. 2007. Baculoviruses and apoptosis: a diversity of genes and responses. Curr. Drug Targets 8:1069–1074 [DOI] [PubMed] [Google Scholar]
- 12. Clem RJ, Miller LK. 1994. Control of programmed cell death by the baculovirus genes p35 and iap. Mol. Cell. Biol. 14:5212–5222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. E X, et al. 2011. An E2F1-mediated DNA damage response contributes to the replication of human cytomegalovirus. PLoS Pathog. 7:e1001342 doi:10.1371/journal.ppat.1001342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Friesen PD. 2007. Insect viruses, p 707–736 In Knipe DM, et al. (ed), Fields virology, 5th ed, vol 1 Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 15. Gaunt MW, Miles MA. 2002. An insect molecular clock dates the origin of the insects and accords with palaeontological and biogeographic landmarks. Mol. Biol. Evol. 19:748–761 [DOI] [PubMed] [Google Scholar]
- 16. Gordon JD, Carstens EB. 1984. Phenotypic characterization and physical mapping of a temperature-sensitive mutant of Autographa californica nuclear polyhedrosis virus defective in DNA synthesis. Virology 138:69–81 [DOI] [PubMed] [Google Scholar]
- 17. Gyrd-Hansen M, Meier P. 2010. IAPs: from caspase inhibitors to modulators of NF-kappaB, inflammation and cancer. Nat. Rev. Cancer 10:561–574 [DOI] [PubMed] [Google Scholar]
- 18. Hay BA, Guo M. 2006. Caspase-dependent cell death in Drosophila. Annu. Rev. Cell Dev. Biol. 22:623–650 [DOI] [PubMed] [Google Scholar]
- 19. Hershberger PA, Dickson JA, Friesen PD. 1992. Site-specific mutagenesis of the 35-kilodalton protein gene encoded by Autographa californica nuclear polyhedrosis virus: cell line-specific effects on virus replication. J. Virol. 66:5525–5533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hickson I, et al. 2004. Identification and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM. Cancer Res. 64:9152–9159 [DOI] [PubMed] [Google Scholar]
- 21. Hozak RR, Manji GA, Friesen PD. 2000. The BIR motifs mediate dominant interference and oligomerization of inhibitor of apoptosis Op-IAP. Mol. Cell. Biol. 20:1877–1885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Huang N, et al. 2011. Baculovirus infection induces a DNA damage response that is required for efficient viral replication. J. Virol. 85:12547–12556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ikeda M, Kobayashi M. 1999. Cell-cycle perturbation in Sf9 cells infected with Autographa californica nucleopolyhedrovirus. Virology 258:176–188 [DOI] [PubMed] [Google Scholar]
- 24. Kanda T, Sullivan KF, Wahl GM. 1998. Histone-GFP fusion protein enables sensitive analysis of chromosome dynamics in living mammalian cells. Curr. Biol. 8:377–385 [DOI] [PubMed] [Google Scholar]
- 25. LaCount DJ, Friesen PD. 1997. Role of early and late replication events in induction of apoptosis by baculoviruses. J. Virol. 71:1530–1537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. LaCount DJ, Hanson SF, Schneider CL, Friesen PD. 2000. Caspase inhibitor P35 and inhibitor of apoptosis Op-IAP block in vivo proteolytic activation of an effector caspase at different steps. J. Biol. Chem. 275:15657–15664 [DOI] [PubMed] [Google Scholar]
- 27. Lannan E, Vandergaast R, Friesen PD. 2007. Baculovirus caspase inhibitors P49 and P35 block virus-induced apoptosis downstream of effector caspase DrICE activation in Drosophila melanogaster cells. J. Virol. 81:9319–9330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Larkin MA, et al. 2007. Clustal W and Clustal X version 2.0. Bioinformatics 23:2947–2948 [DOI] [PubMed] [Google Scholar]
- 29. Lavin MF. 2008. Ataxia-telangiectasia: from a rare disorder to a paradigm for cell signalling and cancer. Nat. Rev. Mol. Cell Biol. 9:759–769 [DOI] [PubMed] [Google Scholar]
- 30. Lee HH, Miller LK. 1978. Isolation of genotypic variants of Autographa californica nuclear polyhedrosis virus. J. Virol. 27:754–767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lilley CE, Carson CT, Muotri AR, Gage FH, Weitzman MD. 2005. DNA repair proteins affect the lifecycle of herpes simplex virus 1. Proc. Natl. Acad. Sci. U. S. A. 102:5844–5849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lu A, Carstens EB. 1991. Nucleotide sequence of a gene essential for viral DNA replication in the baculovirus Autographa californica nuclear polyhedrosis virus. Virology 181:336–347 [DOI] [PubMed] [Google Scholar]
- 33. Madigan JP, Chotkowski HL, Glaser RL. 2002. DNA double-strand break-induced phosphorylation of Drosophila histone variant H2Av helps prevent radiation-induced apoptosis. Nucleic Acids Res. 30:3698–3705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Manji GA, Hozak RR, LaCount DJ, Friesen PD. 1997. Baculovirus inhibitor of apoptosis functions at or upstream of the apoptotic suppressor P35 to prevent programmed cell death. J. Virol. 71:4509–4516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. McNamee LM, Brodsky MH. 2009. p53-independent apoptosis limits DNA damage-induced aneuploidy. Genetics 182:423–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Moon N-S, et al. 2008. E2F and p53 induce apoptosis independently during Drosophila development but intersect in the context of DNA damage. PLoS Genet. 4:e1000153 doi:10.1371/journal.pgen.1000153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Morris TD, Miller LK. 1992. Promoter influence on baculovirus-mediated gene expression in permissive and nonpermissive insect cell lines. J. Virol. 66:7397–7405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Nagamine T, Kawasaki Y, Abe A, Matsumoto S. 2008. Nuclear marginalization of host cell chromatin associated with expansion of two discrete virus-induced subnuclear compartments during baculovirus infection. J. Virol. 82:6409–6418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Negre V, et al. 2006. SPODOBASE: an EST database for the lepidopteran crop pest Spodoptera. BMC Bioinformatics 7:322 doi:10.1186/1471-2105-7-322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Norbury CJ, Zhivotovsky B. 2004. DNA damage-induced apoptosis. Oncogene 23:2797–2808 [DOI] [PubMed] [Google Scholar]
- 41. Okano K, Mikhailov VS, Maeda S. 1999. Colocalization of baculovirus IE-1 and two DNA-binding proteins, DBP and LEF-3, to viral replication factories. J. Virol. 73:110–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Okano K, Vanarsdall AL, Mikhailov VS, Rohrmann GF. 2006. Conserved molecular systems of the Baculoviridae. Virology 344:77–87 [DOI] [PubMed] [Google Scholar]
- 43. Olson VA, Wetter JA, Friesen PD. 2002. Baculovirus transregulator IE1 requires a dimeric nuclear localization element for nuclear import and promoter activation. J. Virol. 76:9505–9515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Olson VA, Wetter JA, Friesen PD. 2001. Oligomerization mediated by a helix-loop-helix-like domain of baculovirus IE1 is required for early promoter transactivation. J. Virol. 75:6042–6051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. O'Reilly D, Miller LK, Luckow A. (ed). 1994. Baculovirus expression vectors: a laboratory manual, p 132–134 Oxford University Press, New York, NY [Google Scholar]
- 46. Passarelli AL, Guarino LA. 2007. Baculovirus late and very late gene regulation. Curr. Drug Targets 8:1103–1115 [DOI] [PubMed] [Google Scholar]
- 47. Prikhod'ko EA, Miller LK. 1998. Role of baculovirus IE2 and its RING finger in cell cycle arrest. J. Virol. 72:684–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rapp JC, Wilson JA, Miller LK. 1998. Nineteen baculovirus open reading frames, including LEF-12, support late gene expression. J. Virol. 72:10197–10206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Rogakou EP, Boon C, Redon C, Bonner WM. 1999. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J. Cell Biol. 146:905–916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Rogakou EP, Nieves-Neira W, Boon C, Pommier Y, Bonner WM. 2000. Initiation of DNA fragmentation during apoptosis induces phosphorylation of H2AX histone at serine 139. J. Biol. Chem. 275:9390–9395 [DOI] [PubMed] [Google Scholar]
- 51. Rohrmann GF. 2011. Baculovirus molecular biology. National Center for Biotechnology Information, US National Library of Medicine, Bethesda, MD [Google Scholar]
- 52. Sarkaria JN, et al. 1999. Inhibition of ATM and ATR kinase activities by the radiosensitizing agent, caffeine. Cancer Res. 59:4375–4382 [PubMed] [Google Scholar]
- 53. Schneider I. 1972. Cell lines derived from late embryonic stages of Drosophila melanogaster. J. Embryol. Exp. Morphol. 27:353–365 [PubMed] [Google Scholar]
- 54. Schultz KLW, Friesen PD. 2009. Baculovirus DNA replication-specific expression factors trigger apoptosis and shutoff of host protein synthesis during infection. J. Virol. 83:11123–11132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Schultz KLW, Wetter JA, Fiore DC, Friesen PD. 2009. Transactivator IE1 is required for baculovirus early replication events that trigger apoptosis in permissive and nonpermissive cells. J. Virol. 83:262–272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Settles EW, Friesen PD. 2008. Flock house virus induces apoptosis by depletion of Drosophila inhibitor-of-apoptosis protein DIAP1. J. Virol. 82:1378–1388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Shi Y, Dodson GE, Shaikh S, Rundell K, Tibbetts RS. 2005. Ataxia-telangiectasia-mutated (ATM) is a T-antigen kinase that controls SV40 viral replication in vivo. J. Biol. Chem. 280:40195–40200 [DOI] [PubMed] [Google Scholar]
- 58. Shirata N, et al. 2005. Activation of ataxia telangiectasia-mutated DNA damage checkpoint signal transduction elicited by herpes simplex virus infection. J. Biol. Chem. 280:30336–30341 [DOI] [PubMed] [Google Scholar]
- 59. Siino JS, et al. 2002. Photobleaching of GFP-labeled H2AX in chromatin: H2AX has low diffusional mobility in the nucleus. Biochem. Biophys. Res. Commun. 297:1318–1323 [DOI] [PubMed] [Google Scholar]
- 60. Stewart TM, Huijskens I, Willis LG, Theilmann DA. 2005. The Autographa californica multiple nucleopolyhedrovirus ie0-ie1 gene complex is essential for wild-type virus replication, but either IE0 or IE1 can support virus growth. J. Virol. 79:4619–4629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Su TT. 2006. Cellular responses to DNA damage: one signal, multiple choices. Annu. Rev. Genet. 40:187–208 [DOI] [PubMed] [Google Scholar]
- 62. Taggart DJ, Mitchell JK, Friesen PD. 2012. A conserved N-terminal domain mediates required DNA replication activities and phosphorylation of the transcriptional activator IE1 of Autographa californica multicapsid nucleopolyhedrovirus. J. Virol. 86:6575–6585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. van Attikum H, Gasser SM. 2009. Crosstalk between histone modifications during the DNA damage response. Trends Cell Biol. 19:207–217 [DOI] [PubMed] [Google Scholar]
- 64. Vandergaast R, Schultz KLW, Cerio RJ, Friesen PD. 2011. Active depletion of host cell inhibitor-of-apoptosis proteins triggers apoptosis upon baculovirus DNA replication. J. Virol. 85:8348–8358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Vaughn JL, Goodwin RH, Tompkins GJ, McCawley P. 1977. The establishment of two cell lines from the insect Spodoptera frugiperda (Lepidoptera; Noctuidae). In Vitro 13:213–217 [DOI] [PubMed] [Google Scholar]
- 66. Weitzman MD, Lilley CE, Chaurushiya MS. 2010. Genomes in conflict: maintaining genome integrity during virus infection. Annu. Rev. Microbiol. 64:61–81 [DOI] [PubMed] [Google Scholar]
- 67. Weller SK. 2010. Herpes simplex virus reorganizes the cellular DNA repair and protein quality control machinery. PLoS Pathog. 6:e1001105 doi:10.1371/journal.ppat.1001105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Wichmann A, Jaklevic B, Su TT. 2006. Ionizing radiation induces caspase-dependent but Chk2- and p53-independent cell death in Drosophila melanogaster. Proc. Natl. Acad. Sci. U. S. A. 103:9952–9957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Wilkinson DE, Weller SK. 2004. Recruitment of cellular recombination and repair proteins to sites of herpes simplex virus type 1 DNA replication is dependent on the composition of viral proteins within prereplicative sites and correlates with the induction of the DNA damage response. J. Virol. 78:4783–4796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Wilkinson DE, Weller SK. 2003. The role of DNA recombination in herpes simplex virus DNA replication. IUBMB Life 55:451–458 [DOI] [PubMed] [Google Scholar]
- 71. Wright CW, Means JC, Penabaz T, Clem RJ. 2005. The baculovirus anti-apoptotic protein Op-IAP does not inhibit Drosophila caspases or apoptosis in Drosophila S2 cells and instead sensitizes S2 cells to virus-induced apoptosis. Virology 335:61–71 [DOI] [PubMed] [Google Scholar]
- 72. Wu Y, Carstens EB. 1998. A baculovirus single-stranded DNA binding protein, LEF-3, mediates the nuclear localization of the putative helicase P143. Virology 247:32–40 [DOI] [PubMed] [Google Scholar]
- 73. Zoog SJ, Schiller JJ, Wetter JA, Chejanovsky N, Friesen PD. 2002. Baculovirus apoptotic suppressor P49 is a substrate inhibitor of initiator caspases resistant to P35 in vivo. EMBO J. 21:5130–5140 [DOI] [PMC free article] [PubMed] [Google Scholar]