

Abstract
Here, we investigated thiol-redox-mediated phospholipase D (PLD) signaling as a mechanism of mercury cytotoxicity in mouse aortic endothelial cell (MAEC) in vitro model utilizing the novel lipid-soluble thiol-redox antioxidant and heavy metal chelator, N,N′-bis(2-mercaptoethyl)isophthalamide (NBMI) and the novel PLD-specific inhibitor, 5-fluoro-2-indolyl des-chlorohalopemide (FIPI). Our results demonstrated (i) mercury in the form of mercury(II) chloride, methylmercury, and thimerosal induced PLD activation in a dose- and time-dependent manner; (ii) NBMI and FIPI completely attenuated mercury- and oxidant-induced PLD activation; (iii) mercury induced upstream phosphorylation of extracellular-regulated kinase 1/2 (ERK1/2) leading to downstream threonine phosphorylation of PLD1 which was attenuated by NBMI; (iv) mercury caused loss of intracellular glutathione which was restored by NBMI; and (v) NBMI and FIPI attenuated mercury- and oxidant-induced cytotoxicity in MAECs. For the first time, this study demonstrated that redox-dependent and PLD-mediated bioactive lipid signaling was involved in mercury-induced vascular EC cytotoxicity which was protected by NBMI and FIPI.
Keywords: mercury, vasculotoxicity, PLD, endothelial cell, NBMI, thiol redox, antioxidant, FIPI, mercaptoethylisophthalamide, bioactive lipid signaling
Introduction
Mercury (Hg), a transition element, is a serious environmental heavy metal pollutant with established toxicity in humans. Mercury has been reported to cause cytotoxicity, neurotoxicity, and immunotoxicity with no known nutritional actions and physiological role.1-3 Mercury enters the air, water, and soil environments from the industrial processing, medical uses, and anthropogenic activities. Inorganic Hg undergoes biomethylation leading to the formation of methylmercury which is a highly toxic form of the element.4 Methylmercury bioaccumulates in the aquatic food chain which results in Hg toxicity in fish and mammalian systems. Methylmercury has been shown to cause hypertension in rats, and mental retardation and cerebral palsy in infants.5 Methylmercury is known to cause oxidative stress in biological systems through the generation of reactive oxidant species (ROS).6,7 Human exposure to the significantly more toxic methylmercury through environmental contamination is well documented.8 Mercury vapor released from the elemental Hg (Hg0) in dental amalgams is the chief source of Hg exposure among dental patients.9 In the human mouth, elemental Hg is biotransformed into methylmercury, an organic toxic form of the metal. Thimerosal, an ethylmercury compound used as a preservative in influenza and other vaccines, poses controversies and concerns as an elicitor of autism and neurological defects in infants and children.10-14 In addition to dental amalgams and herbal medicines, cosmetics (beauty products) have been recently reported to cause nonoccupational exposure of humans to Hg.15
Exposure to Hg is emerging as a risk factor associated with the cardiovascular diseases (CVDs) in humans.16-20 Levels of Hg in toenail and urine have been directly correlated with the elevated risk of myocardial infarction and coronary heart disease.5,18,19 Mercury exposure has also been linked to asthma and hypertension.21 Regardless of the reported association of exposure to Hg with CVD in humans, the role of vascular endothelial cells (ECs) in the Hg-mediated CVD is not known. Vascular endothelium is crucial for the structure and function of the blood vessels toward maintaining the homeostasis of the circulatory system.22 The dysfunction of EC has been shown to cause vascular damage and leak and the breakdown of the cardiovascular system.23,24 Hence, Hg-induced vasculotoxicity at the EC level appears to be involved in the Hg-induced CVDs.
Phospholipase D (PLD) is a crucial signaling enzyme ubiquitous in mammalian cells, including vascular ECs, which generates the bioactive lipid signal mediators, phosphatidic acid (PA), diacylglycerol (DAG), and lysophosphatidic acid (LPA) from the hydrolysis of membrane phospholipids.25-27 These bioactive lipids regulate important cellular functions such as cell proliferation and differentiation.25,26 Earlier, we have reported that Hg induces the activation of PLD in bovine pulmonary artery ECs (BPAECs) which is associated with the loss of thiols, ROS generation, and intracellular calcium.7,22 Earlier, we have also demonstrated that oxidant-induced PLD signaling is regulated by extracellular-regulated kinase 1/2 (ERK1/2) in vascular ECs.25 However, the thiol-redox-mediated upstream PLD-signaling mechanism and downstream Hg-induced cytotoxicity in vascular ECs are not known. Therefore, here we hypothesized that Hg would induce PLD activation through thiol-redox-regulated upstream ERK1/2 activation, which would further lead to the formation of the bioactive lipid signal mediator, PA, mediating the cytotoxicity of Hg in vascular ECs.
Plasma membranes of animal cells rich in phospholipids are the prime targets for toxicants such as Hg. Both the inorganic and organic forms of Hg have been shown to bind to the phospholipids, in addition to the –SH-containing proteins in the animal cell membranes28-30 and thus cause alterations in the structure and function of cell membrane. Therefore, it can be hypothesized that the hydrophobic cell membrane phospholipid domain is a prime target for the adverse actions of Hg. This is further supported by our earlier report that Hg induces membrane phospholipid hydrolysis through the activation of PLD and PLA2.7,22,31 Mercury is also known to alter/deplete the thiol-redox status, induce oxidative stress, and complex with protein ligands in biological systems.32 In order to counteract this at cellular level, a multifunctional pharmacological compound with antioxidant properties, thiol-redox protection, and heavy metal chelating ability are needed. Unlike the commonly used water-soluble thiol protectants and heavy metal chelators such as N-acetyl-l-cysteine (NAC), 2,3-dimercaptopropane-1-sulfonate (DMPS), and meso-2,3-dimercaptosuccinic acid (DMSA),33 a hydrophobic and lipid-soluble heavy metal chelator would be ideal to sequester Hg in the phospholipid membrane bilayer microenvironment of the cell (Figure 1A, B). A lipid-soluble hydrophobic compound, N,N′-bis(2-mercaptoethyl)isophthalamide (NBMI), has been synthesized for use in remediation of Hg-polluted soil and water environs (Figure 1C).34 N,N′-bis(2-Mercaptoethyl)isophthalamide appears to possess unique features of lipid solubility and lodging in the membrane phospholipid bilayer, Hg chelation, and thiol-redox stabilization at the cell membrane level.
Figure 1.
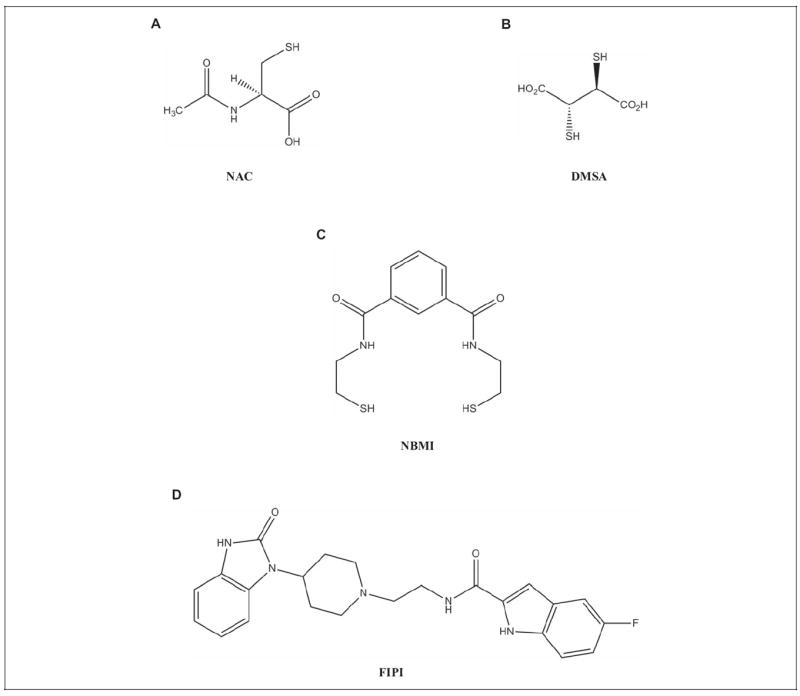
Chemical structure of pharmacological compounds. A, N-Acetyl-l-cysteine (NAC). B, meso-2,3-Dimercaptosuccinic acid (DMSA). C, N,N′-bis(2-Mercaptoethyl)isopthalamide (NBMI). D, 5-Fluoro-2-indolyl des-chlorohalopemide (FIPI).
Therefore, here, we utilized NBMI to dissect out the thiol-redox-dependent activation of PLD signaling and its mediation of Hg cytotoxicity in vascular ECs. The currently available novel PLD-specific inhibitor, 5-fluoro-2-indolyl des-chlorohalopemide (FIPI), allowed us to establish the role of PLD and PLD-derived bioactive lipid mediators in the Hg-induced lipid signaling and cytotoxicity in vascular ECs in the present study (Figure 1D). Water-soluble thiol antioxidants and heavy metal chelators (NAC and DMSA) at higher concentrations are required for in vivo treatment of heavy metal toxicity as they are cleared from the body in urine more readily as their lipid-soluble counterparts. In addition, small-molecule thiol protectants, such as NAC, are known to act as pro-oxidants and exacerbate the oxidative stress. However, lipid-soluble antioxidants such as NBMI and the lipid-soluble PLD inhibitor (FIPI) are more likely to accumulate in lipid microdomains of the cells (membranes) and tissues (fat deposits) and can be mobilized to the sites of action. Cumulatively, it is anticipated that the benefit of using the lipid-soluble thiol protectant such as NBMI and the PLD-specific inhibitor, FIPI, for in vivo treatment of heavy metal-mediated oxidative stress and toxicity, would offer the requirement of lower doses of those compounds in addition to their longer duration of action relative to water-soluble thiol antioxidants and heavy metal chelators (NAC and DMSA). For the first time, our current study demonstrated that the Hg-induced PLD activation in mouse aortic ECs (MAECs) was thiol-redox dependent and ERK regulated. Also, the current study revealed that both NBMI and FIPI offered protection against the Hg-induced cytotoxicity, establishing the role of thiol redox and PLD-generated bioactive lipid signal mediator (PA) therein.
Materials and Methods
Materials
The MAECs used in this study were provided by Dr Robert Auerbach at the University of Wisconsin, Madison, Wisconsin. Phosphate-buffered saline (PBS) was obtained from Biofluids Inc (Rockville, Maryland). Minimal essential medium (MEM), nonessential amino acids, trypsin, fetal bovine serum (FBS), penicillin/streptomycin, phosphate-free modified Dulbecco-modified Eagle medium (DMEM), DMEM high glucose, tissue culture reagents, 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide reduction kit (MTT assay kit), lactate dehydrogenase (LDH) cytotoxicity assay kit (LDH release assay kit), t-octylphenoxypolyethoxyethanol (Triton X-100), bovine serum albumin (BSA), diamide, glutathione (GSH), mercury(II) chloride, methylmercury chloride, thimerosal, NAC, dimethylsulfoxide (DMSO), DMSA, and analytical reagents of highest purity were purchased from Sigma Chemical Co (St Louis, Missouri). 4-Hydroxy-2-nonenal (4-HNE) was obtained from Biomol International L.P. (Plymouth Meeting, Pennsylvania). PD98059 were obtained from Calbiochem (San Diego, California). Primary antibodies for PLD1, PLD2, phosphotheonine-PLD1, ERK-1, ERK-2, and phospho-ERK-1/2 raised in rabbit were obtained from Cell Signaling Technology, Inc (Danvers, Massachusetts). Phosphatidylbutanol (PBt) was obtained from Avanti Polar Lipids (Alabaster, Alabama). [32P]Orthophosphate (carrier-free) was obtained from New England Nuclear (Wilmington, Delaware). Anti-rabbit AlexaFluor 488-conjugated antibody and 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI) were purchased from Molecular Probes Invitrogen Co (Carlsbad, California). Glutathione chemiluminescence assay kit (GSH-Glo) was obtained from Promega Corporation (Madison, Wisconsin). Paraformaldehyde was purchased from Electron Microscopy Sciences (Fort Washington, Pennsylvania). Polyoxyethylene sorbitan monolaurate (Tween-20) was purchased from Bio-Rad Laboratories (Hercules, California). Horseradish peroxidase (HRP)-conjugated anti-rabbit secondary antibody and the enhanced chemiluminescence (ECL) kit for the detection of proteins by Western blotting were obtained from Amersham (Arlington Heights, Illinois). Diperoxovanadate (DPV) was a generous gift from Dr Ramasarma of the Indian Institute of Sciences, Bangalore, India.
N,N′-bis(2-Mercaptoethyl)Isophthalamide Synthesis
N,N′-bis(2-Mercaptoethyl)isophthalamide was synthesized using a modification of the method of Matlock et al.34,35 Three grams of 2-aminoethylthiol hydrochloride was dissolved in 25 mL of chloroform and 3.7 mL of triethylamine and placed in an ice bath with stirring; 2.68 g of isophthaloyl chloride was dissolved in 25 mL of chloroform and slowly added to the solution containing 2-aminoethylthiol and allowed to stir for 2 hours on ice. Precipitation of the NBMI was induced by adding about 100 mL of 0.1 N hydrochloric acid (HCl) slowly to the stirring mixture. The resulting precipitate was collected by filtration and washed 2 times with a water–chloroform (50/50) mixture and then 2 times with 0.1 N HCl and 3 times with distilled water. The resulting white powder was dried under vacuum and the yielded the product NBMI, showing a 70% yield. Gram amounts of this powder were dissolved in pure ethanol and recrystallized twice resulting in the final product. The purity of NBMI was determined by liquid chromatography–mass spectrometry (LC-MS)/MS. Column was a Waters X-Bridge C-18 (150 mm × 3.0 mm, 5 mm particle size; Waters Corporation, Milford, MA). The mobile phase consisted of (A) aqueous with 0.1% formic acid and (B) methanol with 0.1% formic acid. A gradient system was used and the total run time was 30 minutes. The elution conditions expressed as percentage of B is as follows: 0 to 30 seconds, 10% B; 30 seconds to 10 minutes, 10% to 90% B; 10 to 19 minutes, 90% B; 19 to 20 minutes, 90% to 10% B; and 20 to 30 minutes, 10% B. Injection volume was 10 μL. Flow rate was 250 μL/min and the retention time for NBMI was 9.81 minutes. Analysis was done on a Varian LC 1200 L Triple Quadruple MS using a positive electrospray ionization (ESI) source. Chemicals used in the LC-MS/MS analysis were purchased from Sigma-Aldrich, St Louis, Missouri. N,N′-bis(2-Mercaptoethyl)isophthalamide used in the current study was of >99.8% purity.
5-Fluoro-2-Indolyl des-Chlorohalopemide Synthesis
5-Fluoro-2-indolyl des-chlorohalopemide was prepared as described earlier.36-39
Cell Culture
The MAECs were cultured in DMEM (high glucose) supplemented with 5% (volume/volume [vol/vol]) FBS, 100 U/mL penicillin and streptomycin, and 1% (vol/vol) nonessential amino acids at 37°C under a humidified 95% air–5% carbon dioxide (CO2) atmosphere, according to our previously published method.40 Cells in culture were maintained at 37°C in a humidified environment of 95% air–5% CO2 and grown to contact-inhibited monolayers with typical cobblestone morphology. When confluence was reached, cells were trypsinized and subcultured in T 75-cm2 flasks or 12.5 or 35 or 100 mm sterile tissue culture dishes or 96-well sterile tissue culture plates. Confluent cells showed cobblestone morphology under light microscope. All experiments were conducted between 8 and 20 passages (80%-90% confluence). All media and treatments were carefully adjusted to pH 7.4 before use.
Assay of PLD Enzyme Activity
Phospholipase D activity was quantified using our established method of measuring the formation of [32P]-radiolabeled PBt.27,39 Cellular lipids were extracted and PBt was isolated using our published methods of lipid extraction and thin layer chromatographic separation, respectively. Radioactivity was measured using liquid scintillation counting and quantified as DPM normalized to 106 counts in the total cellular lipid extract or as percentage of control (vehicle-treated cells).
Lactate Dehydrogenase Release Assay for Cytotoxicity
Cytotoxicity in MAECs was determined by assaying the extent of release of LDH from cells according to our previously published method.39 At the end of treatment, LDH released into the medium was determined spectrophotometrically using the commercial LDH assay kit according to the manufacturer’s recommendations (Sigma Chemical Co).
MTT Reduction Assay for Cytotoxicity
Cytotoxicity in MAECs was determined by assaying the extent of reduction in MTT in intact cells using the commercial MTT reduction assay kit as previously reported.24 At the end of the experimental treatments, MTT solution (10% vol/vol in MEM) was added and the cells were incubated for 3 hours, following which MTT solvent was added in an amount equal to the original culture volume. Absorbance of the reduced MTT was determined spectrophotometrically, according to the manufacturer’s recommendations (Sigma Chemical Co).
Sodium Dodecyl Sulfate–Polyacrylamide Gel Electrophoresis and Western Blotting
Preparation of cell lysates and Western blotting were carried out according to our previously published methods.25 Protein content in the cell lysates was determined by the BCA protein assay (Thermo Scientific, Rockford, IL). Cell lysates containing 40 μg of protein were loaded onto a 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) gel and proteins were separated at 90 V for 2.5 hours. Proteins resolved on gels were electrotransferred onto the polyvinylidene difluoride (PVDF) membranes at 200 mA for 2 hours at 4°C. The PVDF membranes with proteins were then washed with 0.05% Tween-20 in Tris-buffered saline (TBST) following which they were blocked with TBST containing 5% milk for 1 hour and then incubated with the primary antibody (1:1000 dilution) at 4°C overnight in TBST containing 5% BSA. Membranes were then treated with HRP-conjugated anti-rabbit immunoglobulin G ([IgG] 1:2000 dilution) in TBST containing 5% milk at room temperature for 1 hour followed by washing for 3 times with TBST. The immunoblots were then developed with the ECL reagents, according to the manufacturer’s recommendations. Images on films were scanned and quantified using ImageJ software.
Confocal Immunofluorescence Microscopy
Translocation of PLD1 and PLD2 were analyzed by confocal laser scanning microscopy according to our earlier published method.24 The MAECs grown on sterile glass coverslips (75% confluence) were subjected to desired treatments and then were rinsed 3 times with PBS containing 0.01% Tween-20 (PBST) and fixed with 3.4% paraformaldehyde in PBS for 5 minutes at room temperature. The cells were then rinsed 3 times with PBST and permeabilized with 0.25% Triton-X-100 prepared in PBST for 5 minutes. The cells were then washed 3 times with PBST 0.01% and treated with 1% BSA blocking buffer for 30 minutes at room temperature. The cells were incubated overnight at room temperature with the primary antibody (anti-PLD1 and anti-PLD2 antibodies at 1:200 dilution) in 1% BSA solution in PBST. After rinsing 3 times with PBST, the cells were treated with AlexaFluor 488-conjugated antibody (1:100 dilution) in 1% BSA in PBST for 1 hour. The cells were washed 3 times with PBST 0.01%, mounted, and examined under Zeiss LSM 510 Confocal/Multi-photon Microscope at 490-nm excitation and 530-nm emission under ×63 magnification. The images were captured digitally.
Glutathione Determination
Intracellular soluble thiol (GSH) levels were determined using the GSH-Glo assay kit as reported earlier.39 The MAECs grown up to 90% confluence in 96-well sterile plates were treated with MEM alone or MEM containing desired concentrations of treatments under a humidified 95% air–5% CO2 atmosphere. Following incubation, intracellular GSH levels were determined according to the manufacturer’s recommendations (Promega Corp, Madison, Wisconsin).
Preparation of Solutions Containing Pharmacological Agents
Stock solutions of the hydrophobic pharmacological agents including NBMI, FIPI, and PD98059 were freshly prepared in DMSO and diluted in MEM for treatment of cells. The final DMSO concentration in the cell treatment medium was 0.1% (vol/vol). All other solutions of water-soluble pharmacological compounds were freshly prepared in MEM for treatment of cells.
Statistical Analysis
All experiments were done in triplicate. Data were expressed as mean ± standard deviation (SD). Statistical analysis was carried out by analysis of variance (ANOVA) using SigmaStat (Version 2.0, Jandel). The level of statistical significance was taken as P <.05.
Results
N,N′-bis(2-Mercaptoethyl)Isophthalamide Attenuates Hg-Induced PLD Activation in MAECs
Earlier, we have reported that Hg in 3 different forms (inorganic mercury(II) chloride, organic methylmercury, and pharmaceutical thimerosal) induces PLD activation in BPAECs.7 However, in the current study, we used the MAEC model of aortic origin which is different from the earlier used BPAEC model of arterial origin. Therefore, here, we investigated whether Hg in 3 different forms (mercury(II) chloride, methylmercury, and thimerosal) would cause activation of PLD in MAECs similar to that observed in BPAECs as reported by us earlier.7 In the current study, we further investigated whether the novel thiol-redox antioxidant and heavy metal chelator, NBMI, would attenuate the Hg-induced PLD activation in MAECs. One hour following treatment of cells, mercury(II) chloride, methylmercury, and thimerosal induced significant activation of PLD ([32P]PBt formation) in a dose-dependent manner in comparison to untreated control cells. Mercury(II) chloride induced 2-, 4-, and 10-fold, methylmercury induced 0.5-, 2-, and 3-fold, and thimerosal induced 5-, 10-, and 12-fold increase in PLD activation in a dose-dependent fashion at 5, 10, and 25 μmol/L dose, respectively, as compared to untreated control cells (Figure 2A). N,N′-bis(2-Mercaptoethyl)isophthalamide (10, 25, and 50 μmol/L) significantly attenuated PLD activation induced by mercury(II) chloride (25 μmol/L), methylmercury (10 μmol/L), and thimerosal (25 μmol/L) in MAECs at 1 hour of exposure (Figure 2B-D). N,N′-bis(2-Mercaptoethyl)isophthalamide was effective in attenuating the mercury(II) chloride-induced PLD activation (48%, 65%, and 86% inhibition at 10, 25, and 50 μmol/L, respectively) as compared to cells treated with mercury(II) chloride alone. The methylmercury-induced [32P]PBt formation was also attenuated by NBMI (67%, 53%, and 72% inhibition at 10, 25, and 50 μmol/L, respectively) as compared to cells treated with methylmercury alone. N,N′-bis(2-Mercaptoethyl)isophthalamide also attenuated the thimerosal-induced PLD activation (58%, 76%, and 80% inhibition at 10, 25, and 50 μmol/L, respectively) as compared to cells treated with thimerosal alone. These results showed that organic, inorganic, and pharmaceutical Hg significantly induced PLD activation in MAECs in a dose-dependent manner. Also, the results revealed that the novel thiol-redox antioxidant and heavy metal chelator, NBMI, significantly attenuated the Hg-induced PLD activation in a dose-dependent manner in MAECs.
Figure 2.
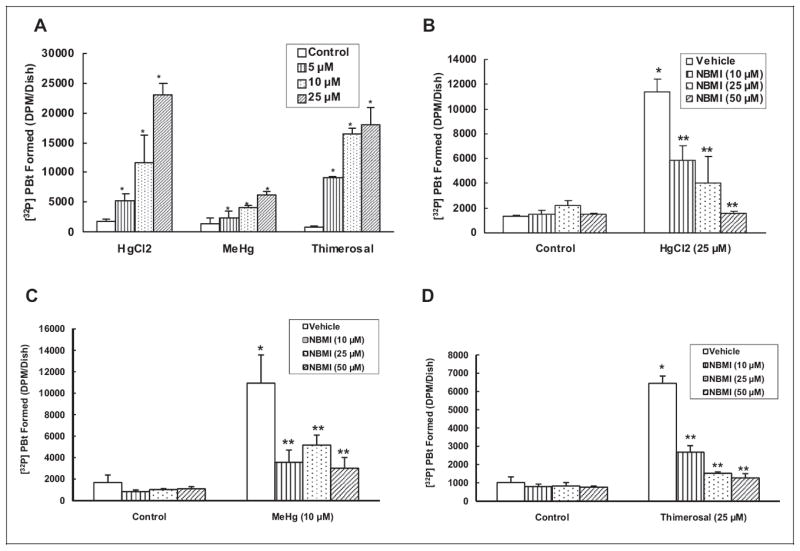
N,N′-bis(2-Mercaptoethyl)isopthalamide (NBMI) attenuates the mercury-induced phospholipase D (PLD) activation in a dose-dependent fashion in mouse aortic endothelial cells (MAECs). The MAECs (5 × 105 cells/35 mm dish) were labeled with [32P]orthophosphate in phosphate-free Dulbecco-modified Eagle medium (DMEM) for 12 hours. Following [32P]orthophosphate labeling, MAECs were treated with minimal essential medium (MEM) alone or MEM containing different concentrations (5, 10, and 25 μmol/L) of mercury(II) chloride, methylmercury, and thimerosal for 1 hour (A) in the presence of 0.05% (volume/volume [vol/vol]) 1-butanol. The MAECs were also labeled with [32P]orthophosphate, then pretreated with MEM or different concentrations of MEM containing NBMI (10, 25, and 50 μmol/L) for 1 hour, then treated with mercury(II) chloride (25 μmol/L) (B); methylmercury (10 μmol/L) (C); and thimerosal (25 μmol/L); (D), for 1 hour in the presence of 0.05% (vol/vol) 1-butanol. At the end of the incubation period, [32P] PBt formed was determined. Data represent mean ± standard deviation (SD) calculated from 3 independent experiments. *Significantly different at P < .05 as compared to cells treated with MEM alone. **Significantly different at P < .05 as compared to cells treated with MEM containing mercury alone.
N,N′-bis(2-Mercaptoethyl)Isophthalamide Attenuates Hg-Induced PLD Activation With Efficacy Similar to the Established Thiol Protectants
Earlier, we reported that the Hg-induced PLD activation in BPAECs is thiol-redox dependent.7 We have also shown that the widely used thiol protectants NAC and DMSA attenuate the Hg-induced PLD activation in BPAECs.7 Therefore, here, we investigated the efficacy of NBMI, the novel thiol-redox antioxidant, by comparison with 2 well-established thiol protectants, NAC and DMSA, in attenuating the Hg-induced activation of PLD in MAECs. The NBMI treatment offered significant attenuation of the Hg-induced PLD activation comparable to equal concentrations of NAC treatment (66%-69% of the methylmercury-induced activation and 73%-81% of the thimerosal-induced activation at 50 μmol/L, respectively) in comparison to MAECs treated with methylmercury (10 μmol/L) and thimerosal (25 μmol/L) alone for 1 hour (Figure 3A, B). The NBMI showed marginally greater attenuation of Hg-induced PLD activation than equal concentrations of DMSA (74%-72% of the methylmercury-induced activation and 66%-43% of the thimerosal-induced activation at 50 μmol/L, respectively) as compared to MAECs treated with methylmercury (10 μmol/L) and thimerosal (25 μmol/L) alone for 1 hour (Figure 3C, D). These results demonstrated that the novel thiol-redox antioxidant, NBMI, was equally effective in attenuating the Hg-induced PLD activation in MAECs as the 2 well-established thiol protectants, NAC and DMSA, tested here under identical conditions.
Figure 3.
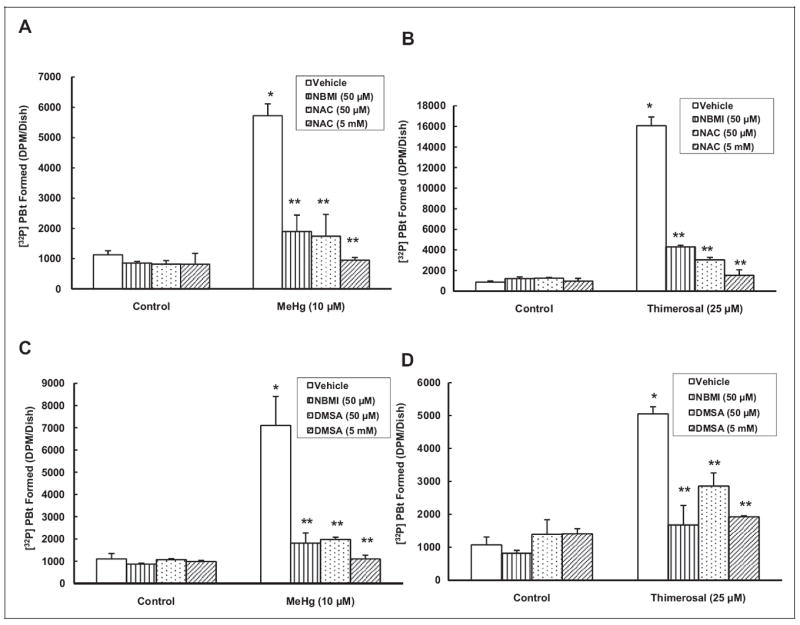
N,N′-bis(2-Mercaptoethyl)isopthalamide (NBMI) attenuates the mercury-induced phospholipase D (PLD) activation in similar fashion to N-acetyl-l-cysteine (NAC) and meso-2,3-dimercaptosuccinic acid (DMSA) in mouse aortic endothelial cells (MAECs). The MAECs (5 × 105 cells/35 mm dish) were labeled with [32P]orthophosphate in phosphate-free Dulbecco-modified Eagle medium (DMEM) for 12 hours. Following [32P]orthophosphate labeling, cells were pretreated with minimal essential medium (MEM) alone or MEM containing NBMI (50 μmol/L) and MEM containing NAC (50 μmol/L, A and 5 mmol/L, B) or DMSA (50 (C) μmol/L and 5 mmol/L (D)) for 1 hour. After pretreatment, cells were treated with MEM alone or MEM containing methylmercury (10 μmol/L) or thimerosal (25 μmol/L) for 1 hour in the presence of 0.05% (volume/volume [vol/vol]) 1-butanol. At the end of the incubation period, [32P]phosphatidylbutanol ([32P]PBt) formed was determined. Data represent mean ± standard deviation (SD) calculated from 3 independent experiments. *Significantly different at P < .05 as compared to cells treated with MEM alone. **Significantly different at P < .05 as compared to cells treated with MEM containing mercury alone.
N,N′-bis(2-Mercaptoethyl)Isophthalamide Attenuates Oxidant-Induced PLD Activation
Oxidants have been shown to activate EC PLD through thiol-redox-mediated regulation.26 Thiol protectants have been shown to attenuate the oxidant-induced PLD activation in ECs.26 Earlier, we reported that the Hg-induced PLD activation in BPAECs is mediated by ROS and oxidative stress.7 Consequently, here, we investigated whether the novel thiol-redox antioxidant, NBMI, would attenuate the oxidant-induced PLD activation in ECs. Treatment of MAECs with the well-established oxidants 4-HNE (100 μmol/L) and DPV (10 μmol/L) for 1 hour caused significant (7- and 5-fold, respectively) activation of PLD as compared to untreated control cells (Figure 4A, B). The NBMI treatment showed significant dose-dependent attenuation of PLD activation induced by 4-HNE (39%, 68%, and 74% inhibition at 10, 25, and 50 μmol/L, respectively) and DPV (59%, 77%, and 75% inhibition at 10, 25, and 50 μmol/L, respectively) in MAECs as compared to cells treated with the same oxidants alone. These results demonstrated that oxidants such as 4-HNE and DPV induced PLD activation and the novel thiol-redox antioxidant, NBMI, effectively attenuated the oxidant-induced PLD activation in MAECs in a dose-dependent fashion.
Figure 4.
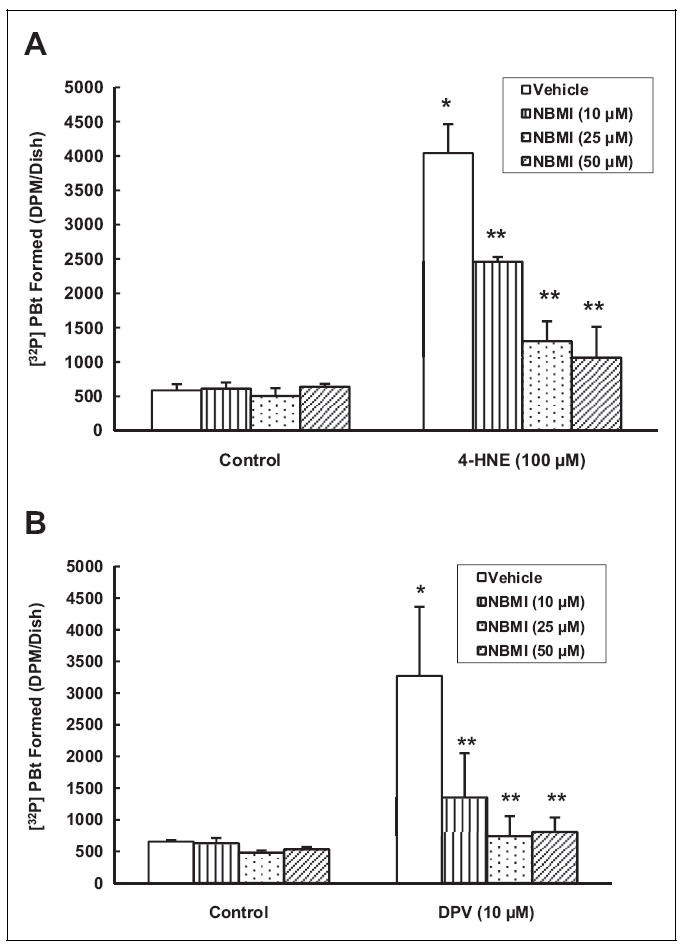
N,N′-bis(2-Mercaptoethyl)isopthalamide (NBMI) attenuates the oxidant-induced phospholipase D (PLD) activation in mouse aortic endothelial cells (MAECs). The MAECs (5 × 105 cells/35 mm dish) were labeled with [32P]orthophosphate in phosphate-free Dulbecco-modified Eagle medium (DMEM) for 12 hours. Following [32P]orthophosphate labeling, cells were pretreated with minimal essential medium (MEM) alone or MEM containing different concentrations (10, 25, and 50 μmol/L) NBMI for 1 hour. Following NBMI pretreatment, cells were treated with 4-hydroxy-2-nonenal ([4-HNE] 100 μmol/L; A) or diperoxovanadate ([DPV] 10 μmol/L; B) for 1 hour in the presence of 0.05% (volume/volume [vol/vol]) 1-butanol. At the end of the incubation period, [32P]phosphatidylbutanol ([32P]PBt) formed was determined. Data represent mean ± standard deviation (SD) calculated from 3 independent experiments. *Significantly different at P < .05 as compared to cells treated with MEM alone. **Significantly different at P < .05 as compared to cells treated with MEM containing oxidant alone.
Phospholipase D-Specific Inhibitor, FIPI, Attenuates Hg-Induced PLD Activation
Earlier, we have demonstrated that the novel PLD-specific inhibitor, FIPI, attenuates the bleomycin-induced PLD activation in lung microvascular ECs.39 However, the effectiveness of FIPI in attenuating the Hg-induced PLD activation in ECs has not been reported so far. Therefore, here, we conducted studies to establish the efficacy of the newly available PLD-specific inhibitor, FIPI, in attenuating the Hg-induced PLD activation in MAECs as determined by [32P]PBt formation. 5-Fluoro-2-indolyl des-chlorohalopemide (250, 500 nmol/L, and 1 μmol/L) exhibited significant, dose-dependent attenuation of the mercury(II) chloride-induced PLD activation (77%, 80%, and 88% at 250, 500 nmol/L, and 1 μmol/L, respectively) as compared to MAECs treated with mercury(II) chloride (25 μmol/L) alone for 1 hour (Figure 5A). Methylmercury-induced PLD activation was also attenuated by FIPI (43%, 52%, and 59% at 250 nmol/L, 500 nmol/L, and 1 μmol/L, respectively) as compared to MAECs treated with methylmercury (10 μmol/L) alone for 1 hour (Figure 5B). Furthermore, FIPI attenuated the thimerosal-induced PLD activation (62%, 59%, and 65% at 250, 500 nmol/L, and 1 μmol/L, respectively) as compared to cells treated with thimerosal (25 μmol/L) alone for 1 hour (Figure 5C). These results revealed that the novel PLD-specific inhibitor, FIPI, effectively attenuated the Hg-induced PLD activation in MAECs in a dose-dependent manner.
Figure 5.
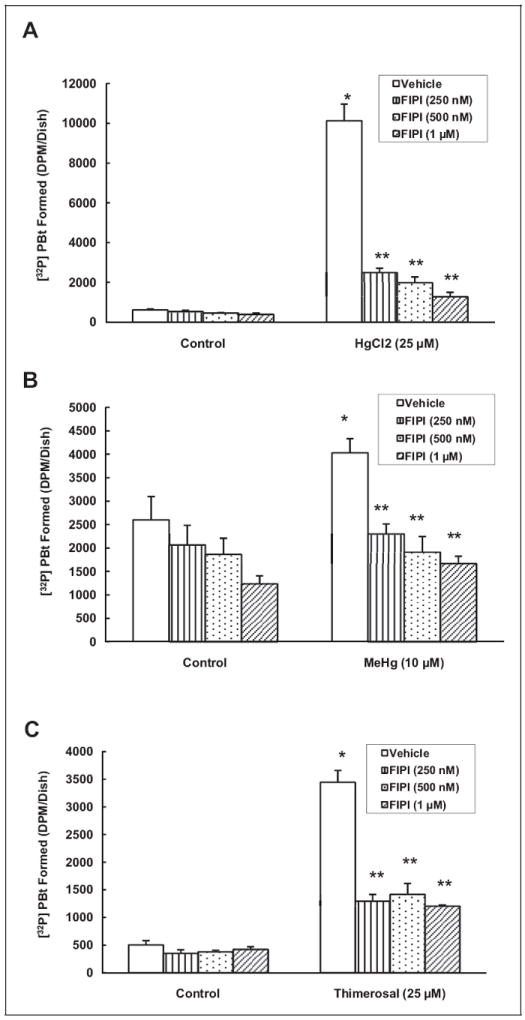
Phospholipase D (PLD)-specific inhibitor, 5-fluoro-2-indolyl des-chlorohalopemide (FIPI), attenuates the mercury-induced PLD activation in mouse aortic endothelial cells (MAECs). The MAECs (5 × 105 cells/35 mm dish) were labeled with [32P]orthophosphate in phosphate-free Dulbecco-modified Eagle medium (DMEM) alone or phosphate-free DMEM containing different concentrations (250, 500 nmol/L, and 1 μmol/L) of FIPI for 12 hours. Following [32P]orthophosphate labeling, cells were treated with minimal essential medium (MEM) alone or MEM containing mercury(II) chloride (25 μmol/L; A), methylmercury (10 μmol/L; B) and thimerosal (25 μmol/L; C) for 1 hour in the presence of 0.05% (volume/volume [vol/vol]) 1-butanol. At the end of the incubation period, [32P]phosphatidylbutanol ([32P]PBt) formed was determined. Data represent mean ± standard deviation (SD) calculated from 3 independent experiments. *Significantly different at P < .05 as compared to cells treated with MEM alone. **Significantly different at P < .05 as compared to cells treated with MEM containing mercury alone.
Phospholipase D-Specific Inhibitor, FIPI, Attenuates Oxidant-Induced PLD Activation
It has been shown that oxidants and Hg induce PLD activation and FIPI, the PLD-specific inhibitor, attenuates the bleomycin-induced PLD activation in vascular ECs.26,39 Our earlier experiments in the current study also revealed that FIPI significantly attenuated the Hg-induced PLD activation in MAECs. Accordingly, here, we conducted studies to establish the specificity of FIPI to inhibit the oxidant- and agonist 12-O-tetradecanoylphorbol-13-acetate (TPA)-induced PLD activation in MAECs. Following pretreatment with FIPI for 12 hours, MAECs were treated with oxidants (hydrogen peroxide [H2O2], 100 μmol/L, diamide, 100 μmol/L, or 4-HNE, 100 μmol/L) for 1 hour. TPA was used as an agonist to induce PLD activation through protein kinase C (PKC)–signaling pathway.41 TPA-induced PLD activation was attenuated by FIPI in a dose-dependent manner (89%, 91%, and 87% by 250, 500 nmol/L, and 1 μmol/L FIPI, respectively) as compared to cells treated with TPA alone (Figure 6A). The FIPI, at the same doses, significantly attenuated PLD activation induced by H2O2 (53% and 45% attenuation at 250 and 500 nmol/L, respectively), diamide (48%, 56%, and 44% attenuation at 250, 500 nmol/L and 1 μmol/L, respectively), and 4-HNE (37%, 42%, and 50% attenuation at 250, 500 nmol/L, and 1 μmol/L, respectively) as compared to cells treated with the same oxidants alone for 1 hour (Figure 6B-D). These results revealed that the novel PLD-specific inhibitor, FIPI, attenuated the agonist- and oxidant-induced PLD activation in a dose-dependent manner in MAECs.
Figure 6.
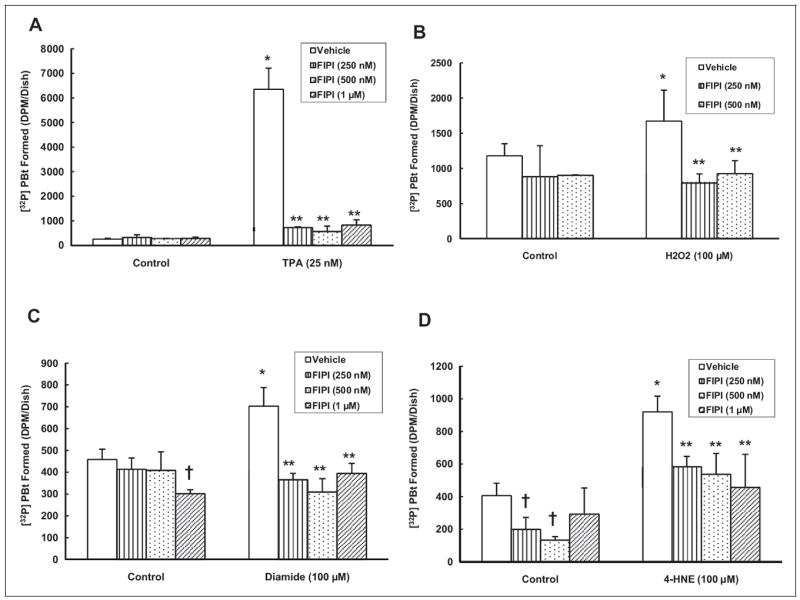
5-Fluoro-2-indolyl des-chlorohalopemide (FIPI) attenuates the oxidant-induced phospholipase D (PLD) activation in mouse aortic endothelial cells (MAECs). The MAECs (5 × 105 cells/35 mm dish) were labeled with [32P]orthophosphate in phosphate-free Dulbecco-modified Eagle medium (DMEM) alone or phosphate-free DMEM containing different concentrations (250, 500 nmol/L, and 1 μmol/L) of FIPI for 12 hours. Following [32P]orthophosphate labeling, cells were treated with minimal essential medium (MEM) or MEM containing TPA (25 nmol/L; A), H2O2 (100 μmol/L; B), diamide (100 μmol/L; C), and 4-hydroxy-2-nonenal ([4-HNE] 100 μmol/L; D) for 1 hour in the presence of 0.05% (volume/ volume [vol/vol]) 1-butanol. At the end of the incubation period, [32P]phosphatidylbutanol ([32P]PBt) formed was determined. Data represent mean ± standard deviation (SD) calculated from 3 independent experiments. *Significantly different at P < .05 as compared to cells treated with MEM alone. **Significantly different at P < .05 as compared to cells treated with MEM containing agonist or oxidant alone.
N,N′-bis(2-Mercaptoethyl)Isophthalamide Attenuates Hg-Induced PLD1 and PLD2 In Situ Translocation
Our earlier study revealed that pro-oxidants like vitamin C induce translocation of PLD1 and PLD2 isoforms in lung microvascular ECs.25 Also, we have reported that Hg induces PLD activation in BPAECs through ROS formation, oxidative stress, and thiol alteration.7 Hence, here, we investigated whether Hg would induce in situ translocation of PLD1 and PLD2 isoforms and whether NBMI would attenuate the process in MAECs. In untreated control MAECs, little to no PLD staining was visible as revealed by confocal immunofluorescence microscopy imaging. However, both methylmercury and thimerosal caused enhanced translocation of both PLD1 and PLD2 isoforms in situ in MAECS. Also, NBMI treatment showed significant attenuation of the Hg-induced PLD1 and PLD2 translocation in cells. These results demonstrated that Hg caused PLD translocation in situ in MAECs which was effectively attenuated by the novel thiol-redox antioxidant, NBMI (Figure 7).
Figure 7.
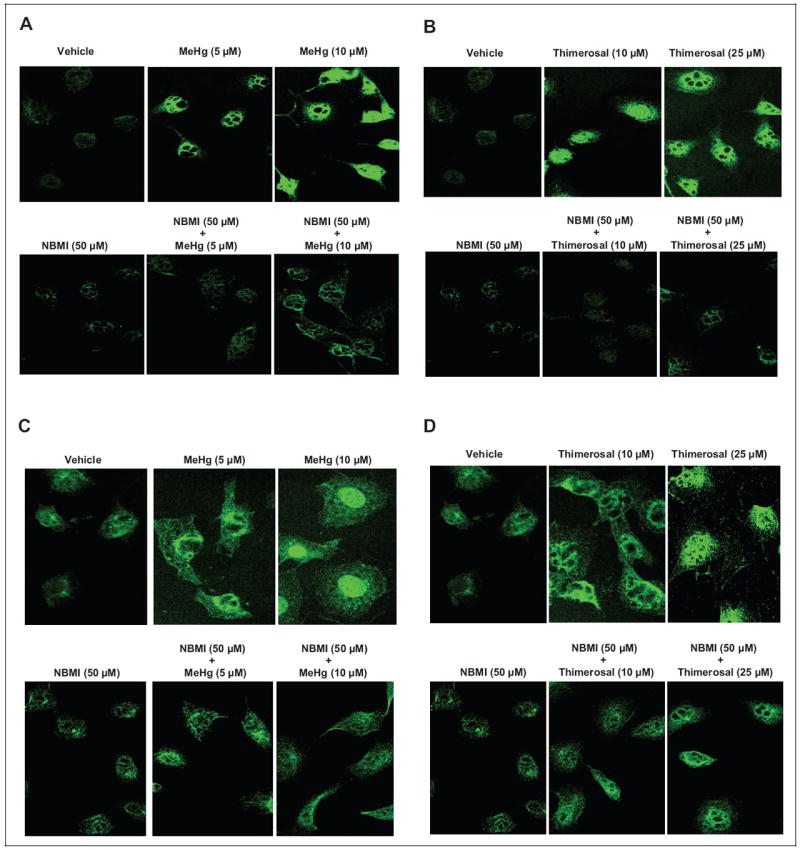
N,N′-bis(2-Mercaptoethyl)isopthalamide (NBMI) attenuates the mercury-induced phospholipase D1 (PLD1) and PLD2 in situ translocation. Mouse aortic endothelial cells (MAECs) cultured on coverslips (5 × 105 cells/35 mm dish) were pretreated with minimal essential medium (MEM) alone or MEM containing NBMI (50 μmol/L) for 1 hour then treated with MEM alone or MEM containing methylmercury (5 and 10 μmol/L) and thimerosal (10 and 25 μmol/L) for 1 hour. At the end of the incubation period, the cells were fixed, stained for PLD1 (A and B) and PLD2 (C and D), and visualized using confocal fluorescent microscopy. Each digitally captured image is a representative picture obtained from 3 independent experiments conducted under identical conditions.
Mercury Induces Phosphorylation of ERK1/2 and PLD1
Oxidants and metals have been shown to induce phosphorylation and activation of ERK1/2 in different cellular systems.42,43 Also, it has been reported that oxidants activate PLD in ECs through phosphorylation of PLD1 mediated by upstream activation and phosphorylation of ERK1/2.25 Therefore, here, we investigated whether Hg would induce phosphorylation of ERK1/2 which in turn would mediate the phosphorylation of PLD1 in MAECs. The MAECs were treated with Hg (methylmercury, 10 μmol/L or thimerosal, 25 μmol/L) for 0 to 60 minutes and then examined for phosphorylation of ERK1/2 and threonine phosphorylation of PLD1 by SDS-PAGE and Western blotting. Methylmercury induced a 1.56-fold increase in ERK1/2 phosphorylation at 5 minutes and 1.44-and 1.23-fold increase at 15 and 30 minutes, respectively (Figure 8A). Methylmercury also induced 2.17-, 2.71-, and 5.02-fold PLD1 threonine phosphorylation at 5, 15, and 30 minutes of methylmercury treatment, respectively, in MAECs relative to untreated control cells (Figure 8B). Thimerosal induced 1.21- and 1.59-fold increase in ERK1/2 phosphorylation at 5 and 60 minutes, respectively, relative to untreated control cells (Figure 8C). Also, thimerosal induced 4.80- and 10.00-fold increase in PLD1 phosphorylation at 5 and 60 minutes of treatment, respectively, relative to untreated control cells (Figure 8D). These results demonstrated that mercury induced robust phosphorylation of ERK1/2 at the initial time of treatment, whereas the induction of intense threonine phosphorylation of PLD1 occurred at later times of treatment, suggesting activation of ERK1/2 upstream of PLD1 threonine phosphorylation.
Figure 8.
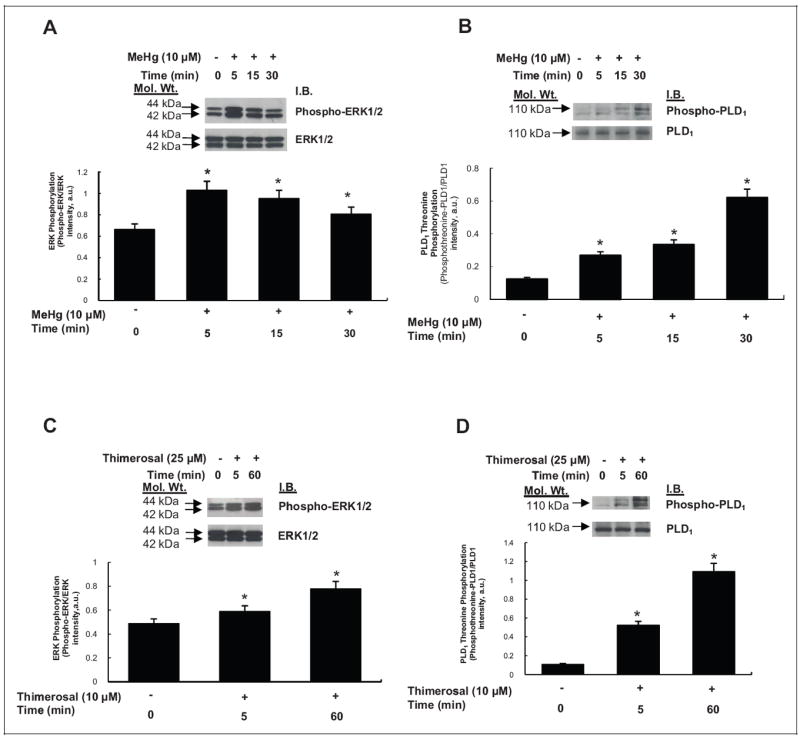
Mercury induces phosphorylation of extracellular-regulated kinase 1/2 (ERK1/2) and phospholipase D1 (PLD1) in mouse aortic endothelial cells (MAECs). The MAECs (5 × 105 cells/35 mm dish) were treated with minimal essential medium (MEM) alone or MEM containing methylmercury (10 μmol/L) for 5, 15, and 30 minutes (A and B) and MEM containing thimerosal (25 μmol/L) for 5 and 60 minutes (C and D). Following treatment, proteins in cell lysates were subjected to sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and Western blotting with phospho-ERK1/2-specific, ERK1/2-specific, phosphotheonine-PLD1-specific, and PLD1-specific monoclonal antibodies. The intensity of the protein bands were digitally determined as described in Materials and Methods. *Significantly different at P < .05 as compared to cells treated with MEM alone.
Extracellular-Regulated Kinase 1/2 Upstream Inhibitor, PD98059, Attenuates Hg-Induced PLD Activation
Several studies have shown ERK1/2 to act as an upstream activator of PLD in intracellular signaling pathways.25,42 Therefore, here, we investigated the modulatory effect of ERK1/2-signaling inhibition on the Hg-induced PLD activation using the ERK1/2 upstream inhibitor, PD98059, and measuring (i) [32P]PBt formation, (ii) ERK1/2 phosphorylation, and (iii) PLD1 threonine phosphorylation. The MAECs were pretreated with PD98059 (0 or 10 μmol/L) for 1 hour then treated with methylmercury (0 or 10 μmol/L) for 1 hour. PD98059 significantly attenuated the methylmercury-induced [32P]PBt formation to near the level in untreated control cells (Figure 9A). Methylmercury-induced ERK1/2 phosphorylation and PLD1 threonine phosphorylation as observed by SDS-PAGE and Western blotting also decreased in PD98059-treated MAECs almost to the levels in untreated control cells (Figure 9B, C). These results revealed that the ERK1/2 upstream inhibitor attenuated the Hg-induced activation of PLD1, ERK1/2 phosphorylation, and PLD1 threonine phosphorylation in MAECs, suggesting that ERK1/2 regulated the Hg-induced activation of PLD1 upstream.
Figure 9.
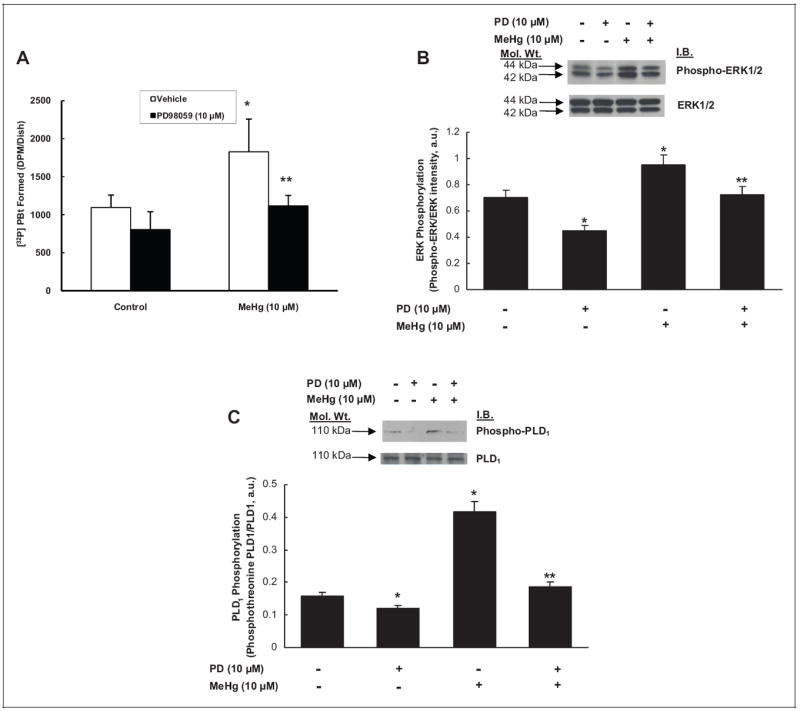
Extracellular-regulated kinase 1/2 (ERK1/2) upstream inhibitor, PD98059, attenuates the mercury-induced phospholipase D (PLD) activation, ERK1/2 phosphorylation, and PLD1 phosphorylation in mouse aortic endothelial cells (MAECs). The MAECs (5 × 105 cells/ 35 mm dish) were labeled with [32P]orthophosphate in phosphate-free Dulbecco-modified Eagle medium (DMEM) for 12 hours. Following [32P]orthophosphate labeling, cells were pretreated with minimal essential medium (MEM) alone or MEM containing ERK1/2 upstream inhibitor, PD98059 (10 μmol/L), for 1 hour. Following pretreatment, cells were treated with MEM alone or MEM containing methylmercury (10 μmol/L) for 1 hour. At the end of the incubation period, [32P]phosphatidylbutanol ([32P]PBt) formed was determined (A). B, MAECs (5 × 105 cells/35 mm dish) pretreated with MEM alone or MEM containing PD98059 (10 μmol/L) for 1 hour were treated with MEM alone or methylmercury (10 μmol/L) for 1 hour. Following treatment, proteins in cell lysates were subjected to sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and Western blotting with phospho-ERK1/2-specific, ERK1/2-specific, (C) phosphotheo-nine-PLD1-specific, and PLD1-specific monoclonal antibodies. The intensity of the protein bands were digitally determined as described in Materials and Methods. Data represent mean ± standard deviation (SD) calculated from 3 independent experiments. *Significantly different at P < .05 as compared to cells treated with MEM alone. **Significantly different at P < .05 as compared to cells treated with MEM containing mercury alone.
N,N′-bis(2-Mercaptoethyl)Isophthalamide Attenuates Hg-Induced Phosphorylation of ERK1/2 and PLD1
Having shown in this study that Hg induced [32P]PBt formation (PLD activation), upstream phosphorylation of ERK1/2, PLD1 threonine phosphorylation downstream of ERK1/2 phosphorylation, and NBMI attenuated the Hg-induced [32P]PBt formation, next we investigated whether NBMI would modulate the Hg-induced phosphorylation of ERK1/2 and PLD1 threonine phosphorylation in MAECs. Cells were pretreated with NBMI (0 or 25 μmol/L) for 1 hour prior to being treated with methylmercury (0 or 10 μmol/L) for 1 hour. Treatment with NBMI caused a significant attenuation of the Hg-induced phosphorylation of ERK1/2 and PLD1 threonine phosphorylation, almost to control levels, as compared to cells treated with Hg alone (Figure 10A, B). These results revealed that the novel thiol stabilizer, NBMI, attenuated the Hg-induced phosphorylation of ERK1/2 and PLD1 threonine phosphorylation in MAECs, suggesting the role of redox-regulation therein.
Figure 10.
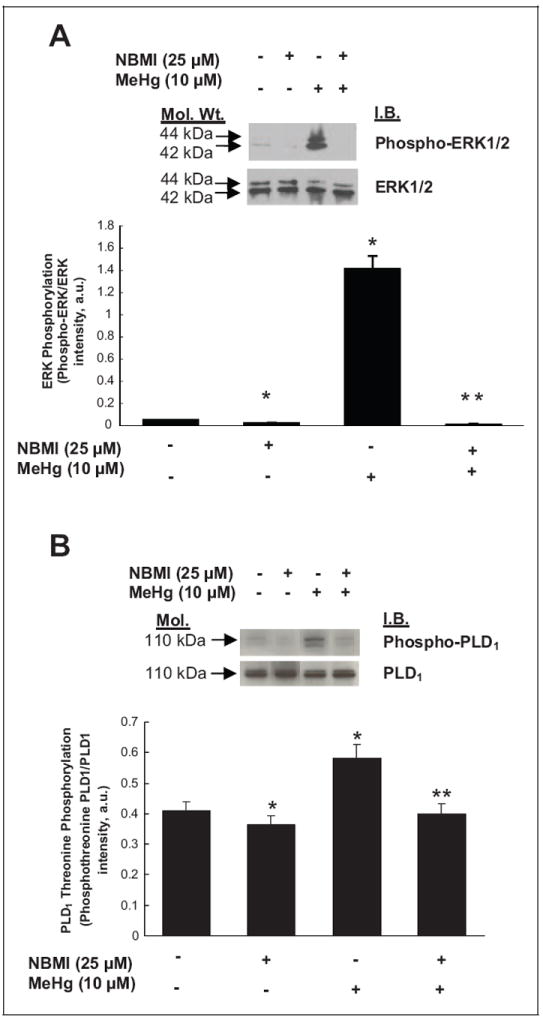
N,N′-bis(2-Mercaptoethyl)isopthalamide (NBMI) attenuates the mercury-induced phosphorylation of extracellular-regulated kinase 1/2 (ERK1/2) and phospholipase D1 (PLD1) in mouse aortic endothelialcells (MAECs). The MAECs (5 × 105 cells/35 mm dish) pretreated with minimal essential medium (MEM) alone or MEM containing NBMI (25 μmol/L) for 1 hour were treated with MEM alone or MEM containing methylmercury (10 μmol/L) for 1 hour. Following treatment, proteins in cell lysates were subjected to sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and Western blotting with (A) phospho-ERK1/2-specific, ERK1/2-specific, (B) phosphotheonine-PLD1-specific and PLD1-specific monoclonal antibodies. The intensity of the protein bands were digitally determined as described in Materials and Methods section. Data represent mean ± standard deviation (SD) calculated from 3 independent experiments. *Significantly different at P < .05 as compared to cells treated with MEM alone. **Significantly different at P < .05 as compared to cells treated with MEM containing mercury alone.
N,N′-bis(2-Mercaptoethyl)Isophthalamide Protects Against Hg- and Oxidant-Induced GSH Loss
Our earlier studies have demonstrated that the oxidant- and Hg-induced PLD activation in ECs is thiol redox regulated and is associated with the loss of thiols including GSH.7,26 Therefore, here we hypothesized that Hg and oxidants would cause the depletion of intracellular soluble thiols concomitant with the PLD activation in MAECs. In addition, we hypothesized that the loss of intracellular soluble thiols could be protected by the novel thiol-redox antioxidant, NBMI. To determine this relationship, MAECs were pretreated with NBMI (0 or 50 μmol/L) for 1 hour prior to treatment with Hg (methylmercury, 10 μmol/L or thimerosal, 25 μmol/L) or oxidant (H2O2, 100 μmol/L or diamide, 250 μmol/L) for 1 hour. Both Hg and oxidant treatment demonstrated significant loss of GSH in MAECs. N,N′-bis(2-Mercaptoethyl)isophthalamide offered significant attenuation of the Hg- and oxidant-induced loss of GSH in cells, restoring the levels of GSH in treated cells to the levels measured in untreated control cells. These results demonstrated that the novel thiol-redox antioxidant, NBMI, protected the Hg- and oxidant-induced loss of the intracellular soluble thiol, GSH (Figure 11).
Figure 11.
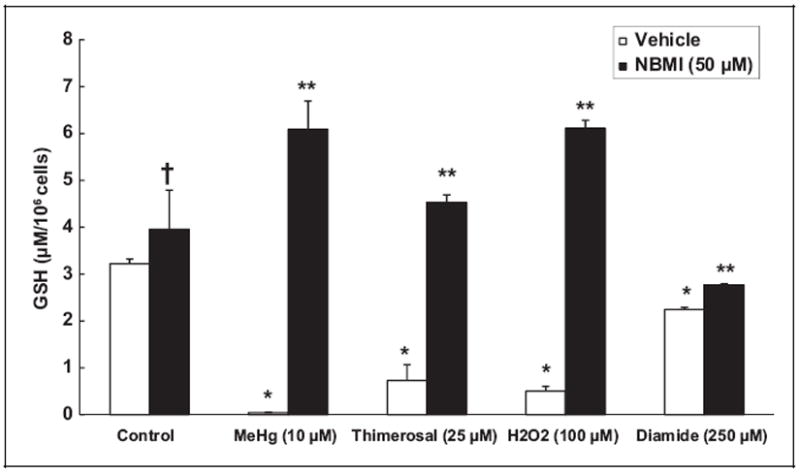
N,N′-bis(2-Mercaptoethyl)isopthalamide (NBMI) protects against the mercury- and oxidant-induced glutathione (GSH) loss. Mouse aortic endothelial cells ([MAECs] 5 × 105 cells/35 mm dish) pretreated with minimal essential medium (MEM) alone or MEM containing NBMI (50 μmol/L) for 1 hour were treated with MEM alone or MEM containing methylmercury (10 μmol/L) or thimerosal (25 μmol/L) or H2O2 (100 μmol/L) or diamide (250 μmol/L). At the end of the incubation period, the intracellular soluble thiol (glutathione [GSH]) concentrations were determined. Data represent mean ± standard deviation (SD) calculated from 3 independent experiments. *Significantly different at P < .05 as compared to cells treated with MEM alone. **Significantly different at P < .05 as compared to cells treated with MEM containing mercury or oxidant alone.
Both FIPI and NBMI Protect Against Hg- and Oxidant-Induced Cytotoxicity
Phospholipase D-mediated PA signaling has been implicated in several signaling pathways and the role of PA has been previously reported in the oxidant-induced cytotoxicity in ECs.39 Hence, we hypothesized that Hg and oxidants which induced significant activation of PLD would also cause cytotoxicity in MAECs. Furthermore, we hypothesized that NBMI and FIPI, which attenuated the Hg- and oxidant-induced PLD activation, would offer protection against the Hg- and oxidant-induced cytotoxicity in MAECs. In order to investigate this hypothesis, MAECs were pretreated with FIPI (0, 250 nmol/L, and 1 μmol/L for 12 hours) or NBMI (0 or 50 μmol/L for 1 hour) prior to treatment with Hg (methylmercury, 10 μmol/L, or thimerosal, 25 μmol/L) or oxidant (diamide, 250 μmol/L) for 1 hour, and cytotoxicity (LDH release and MTT reduction) was measured at the end of treatment. Both Hg and diamide caused significant cytotoxicity in MAECs as compared with untreated control cells. N,N′-bis(2-Mercaptoethyl)isophthalamide offered significant attenuation of the Hg- and diamide-induced LDH release by MAECs (Figure 12A, B). Also, FIPI significantly attenuated Hg- and diamide-induced inhibition of MTT reduction in MAECs (Figure 12C, D). Thus, these results revealed that the novel thiol-redox antioxidant, NBMI, and the novel PLD-specific inhibitor, FIPI, effectively protected against the Hg- and oxidant-induced cytotoxicity in MAECs.
Figure 12.
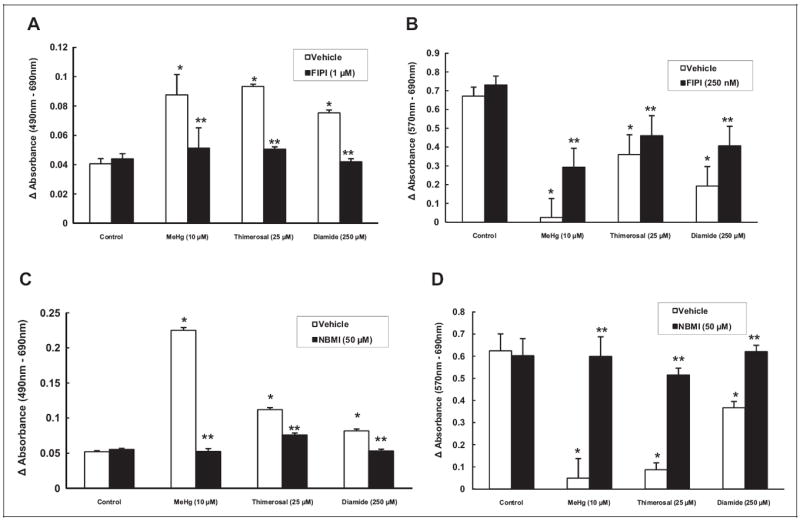
5-Fluoro-2-indolyl des-chlorohalopemide (FIPI) and N,N′-bis(2-mercaptoethyl)isopthalamide (NBMI) protect against the mercury- and oxidant-induced cytotoxicity in mouse aortic endothelial cells (MAECs). The MAECs (2.5 × 105 cells/17.5 mm dish) were pretreated with minimal essential medium (MEM) alone, MEM containing FIPI (10 μmol/L) for 12 hours, or MEM containing NBMI (50 μmol/L) for 1 hour. Pretreated cells were then treated with MEM alone or MEM containing methylmercury (10 μmol/L) or thimerosal (25 μmol/L) or diamide (250 μmol/L) for 1 hour. At the end of the incubation period, release of lactate dehydrogenase (LDH) into the medium (A and C) and decrease in 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide (MTT) reduction (B and D; as an index of cytotoxicity) were determined spectro-photometrically. Data represent mean ± standard deviation (SD) calculated from 3 independent experiments. *Significantly different at P < .05 as compared to cells treated with MEM alone. **Significantly different at P < .05 as compared to cells treated with MEM containing mercury or oxidant alone.
Discussion
In the current study, we investigated the thiol-redox-mediated and ERK-regulated PLD signaling as a mechanism of Hg cytotoxicity in MAECs utilizing the novel lipid-soluble, thiol-redox antioxidant and heavy metal chelator, NBMI, and the novel PLD-specific inhibitor, FIPI. The results revealed that (i) all the 3 chosen forms of Hg including the inorganic form of Hg (mercury(II) chloride), environmental organic form of Hg (methylmercury), and the pharmaceutical organic form (thi-merosal) induced PLD activation in a dose- and time-dependent manner which was completely attenuated by NBMI and FIPI; (ii) NBMI and FIPI also attenuated the oxidant-induced PLD activation; (iii) Hg induced in situ translocation of PLD1 and PLD2 that was attenuated by NBMI; (iv) Hg induced upstream phosphorylation of ERK1/2 which caused the downstream threonine phosphorylation of PLD1 which was attenuated by NBMI; (v) Hg depleted the intracellular GSH levels which were restored by NBMI; and (vi) NBMI and FIPI offered protection against the Hg- and oxidant-induced cytotoxicity in MAECs. This is the first observation which demonstrated that thiol-redox-dependent and ERK1/2-regulated PLD activation led to the Hg-induced vascular EC cytotoxicity through bioactive lipid signal mediators which was protected by the novel thiol-redox antioxidant and heavy metal chelator, NBMI, and novel PLD-specific inhibitor, FIPI (Figure 13).
Figure 13.
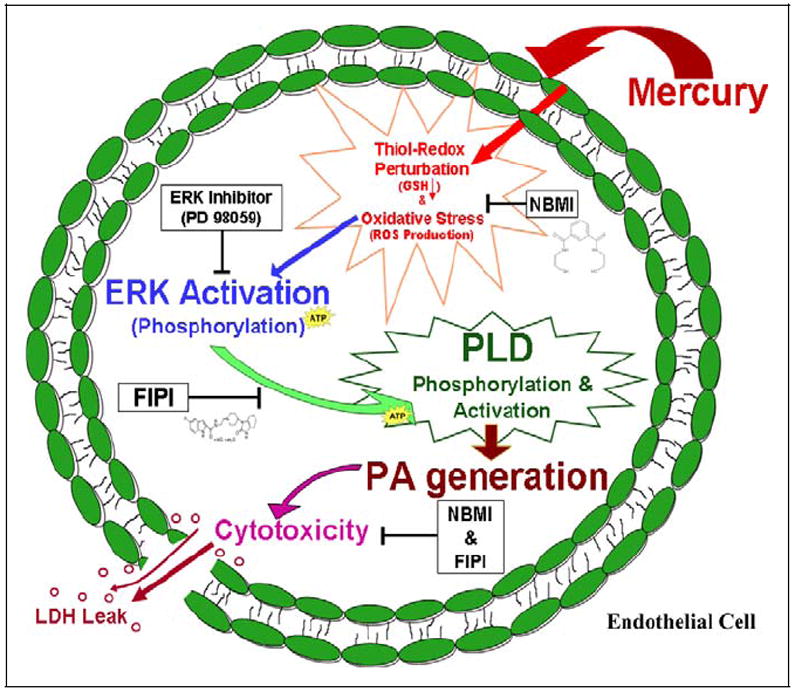
Proposed mechanism of protection by N,N′-bis(2-mercaptoethyl)isopthalamide (NBMI) and 5-fluoro-2-indolyl des-chlorohalopemide (FIPI) against mercury-induced cytotoxicity in aortic endothelial cells (ECs) through the attenuation of thiol-redox–regulated and extracellular-regulated kinase 1/2 (ERK1/2)–mediated activation of phospholipase D (PLD) and bioactive lipid signal mediator formation.
Phospholipases catalyze the hydrolysis of membrane phospholipids, leading to the generation of bioactive lipid second messengers which play crucial roles in cellular signaling cascades.25,27 Phospholipase D, one such phospholipid hydrolase and a lipid signaling enzyme, is ubiquitously present in all mammalian cells that preferentially acts on phosphatidylcholine (PC) generating PA and choline. Phosphatidic acid is further metabolized into either 1,2-DAG by phosphatidate phosphohydrolase or to LPA by phospholipase A1/A2.25,27 Agonist-mediated activation of PLD regulates signal transduction in mammalian cells.25 Phospholipase D-generated lipid mediators including PA and LPA are involved in critical cellular functions such as mitogenesis in fibroblasts, oxidative burst in neutrophils, elevation of intracellular calcium levels, and activation of protein kinases.25 The 2 major isoenzymes of PLD, PLD1, and PLD2, have been well characterized and cloned in mammalian cells and are selectively activated by different cofactors such as Arf, Rho, and Cdc42.41 Phosphatidylinositol 4,5-bisphosphate (PIP2), a lipid mediator, has been shown to activate both PLD1 and PLD2 in vitro.41
The current study revealed that all the 3 chosen forms of Hg activated PLD and induced the formation of PBt (an index of PA formation) in MAECs in a dose- and time-dependent fashion. In addition, the study also demonstrated that the Hg-induced PLD activation was regulated upstream by the thiol redox in MAECs as supported by our finding that Hg caused the loss of GSH which was restored by the novel thiol-redox agent, NBMI, leading to the attenuation of PLD activation in the cells. Our current finding is also supported by our earlier report that Hg as mercury (II) chloride, methylmercury, and thimerosal activates PLD in the BPAECs which is regulated by thiol redox and ROS.7 However, in the current study, we utilized the MAECs as our in vitro vascular EC model which represents the aortic endothelium and is different from our previously utilized BPAEC model that originates from the pulmonary arteries. The rationale for using the MAECs in the current study as the in vitro model was to justify the systemic vascular endothelium as the target for Hg adverse effects, especially the activation of the lipid signaling cascades such as PLD. Our study also revealed that in addition to NBMI, the classic thiol-redox antioxidant, NAC, and the thiol-chelating drug, DMSA, also attenuated the Hg-induced PLD activation in MAECs. This is also supported by our earlier reported finding that the Hg-induced PLD activation in BPAECs is attenuated by both NAC and DMSA, which are both water-soluble drugs.7 However, the novel thiol-redox antioxidant, NBMI, used in this study is hydrophobic and lipid-soluble which might have rendered the compound to exhibit its actions more effectively in the hydrophobic sites of the phospholipid bilayer of the cell membrane such as attenuating the Hg-induced PLD activation in MAECs. This study, thus clearly established that the Hg-induced PLD activation and the generation of the lipid signal mediator, PA, were thiol-redox regulated. This was further confirmed in the current study by the observations that the PLD activation in MAECs induced by oxidants such as 4-HNE (a lipid peroxidation-derived reactive carbonyl) and DPV (an oxidant and thiol-redox perturbing agent) was also attenuated by NBMI. Oxidant-induced PLD activation in ECs through ROS generation and thiol-redox dysregulation and attenuation of the enzyme activation by thiol-redox enhancer/stabilizer, NAC, has been documented.25,27,39,41 Diamide, a thiol-depleting agent, has been reported to cause PLD activation in vascular ECs that is attenuated by the thiol-protecting agents such as NAC and mercaptopropionylglycine.26 Translocation and relocalization of PLD isoenzymes in situ have been shown to be associated with the oxidant-induced PLD activation in vascular ECs.25 Along those lines, the current study also demonstrated that the Hg-induced in situ translocation of PLD1 and PLD2 isoenzymes in MAECs was also attenuated by the thiolredox stabilizer, NBMI. Our findings clearly revealed that NBMI was not only effective in attenuating the Hg-induced PLD activation but was also effective in attenuating the oxidant-induced enzyme activation in MAECs, suggesting its effectiveness in restoring thiol-redox balance as Hg, 4-HNE, and DPV are known to cause PLD activation in ECs through thiol-redox alteration.7,26,44
Others and we have reported that oxidants induce PLD activation in vascular ECs, vascular smooth muscle cells, and fibroblasts,25,41 which is attenuated by antioxidants. It is important to mention here that the oxidant-induced PLD activation is also regulated by the upstream activation of certain key protein kinases, leading to the phosphorylation of PLD in vascular ECs. Hydrogen peroxide-induced PLD activation in ECs has been reported to be regulated upstream by the activation of protein tyrosine kinases.45 Diperoxovanadate has been observed to stimulate PLD activity in vascular ECs through the upstream activation and regulation of Src kinase and p38 mitogen-activated protein kinase (p38 MAPK).41,42 Vitamin C, a pro-oxidant, has been reported to activate PLD in lung microvascular ECs through the upstream thiol-redox regulation and activation of ERK1/2 and p38 MAPK.25 These studies have clearly established that the phosphorylation of PLD isoenzymes is an essential requirement upstream of activation of PLD by oxidants in vascular ECs. The MAPKs, the stress-activated protein kinases, are broadly divided into 3 classes, namely (i) ERK, (ii) c-Jun N-terminal kinase (JNK), and (iii) p38 MAPK, all of which take part in diverse cellular signaling cascades, upon activation through phosphorylation of both threonine and tyrosine residues by dual-specific serine–threonine MAPK kinases.25 Mercury has been shown to activate MAPKs in model systems such as the H4IIE cells, renal cortical slices, and macrophages.43,46,47 As the mechanism of the Hg-induced PLD activation in vascular ECs has not been established so far, here, we investigated whether ERK1/2 would be involved in the upstream activation of PLD in MAECs through threonine phosphorylation of the enzyme. Our current results are in accord with the reports made by others by revealing that ERK1/2 upstream-specific inhibitor (PD98059) not only attenuated the Hg-induced PLD activation but also attenuated the Hg-induced upstream ERK1/2 phosphorylation and PLD1 threonine phosphorylation in MAECs, suggesting that ERK1/2 was the upstream regulator of Hg-induced PLD activation. In this context, we were able to utilize the only commercially available antiphosphothreonine PLD1 antibody to determine the ERK1/2-mediated threonine phosphorylation of PLD1 in the Hg-treated MAECs by SDS-PAGE and Western blotting. Due to the nonavailability of antiphosphothreonine PLD2 antibody, we were unable to determine the ERK1/2-mediated threonine phosphorylation of PLD2 in the Hg-treated MAECs. However, threonine phosphorylation of PLD2 induced by Hg in MAECs could be possible and was not ruled out in our current study. More noticeably, the upstream phosphorylation of ERK1/2 and the downstream threonine phosphorylation of PLD1 and activation of PLD induced by Hg in MAECs were effectively attenuated by the novel thiol-redox stabilizer, NBMI, further strengthening the notion that thiol redox played a critical role in regulating the Hg-induced PLD activation through upstream ERK1/2-mediated threonine phosphorylation of the enzyme. Nevertheless, the exact mechanism of the Hg-induced ERK1/2 activation and the resultant site-specific threonine phosphorylation in PLD1 in ECs warrant further investigation.
The regulation of the agonist-induced PLD activation is apparently complex involving PKC, heterotrimeric G proteins, small molecular weight G proteins, and protein tyrosine kinases/protein tyrosine phosphatases.25 Hormones, growth factors, neurotransmitters, cytokines, and ROS induce PLD activation in a wide variety of mammalian cell systems and tissues.25,41 Agonist-specific and cell-specific mechanisms, through signaling modulation by PKC, intracellular calcium, heterotrimeric G proteins, protein tyrosine kinases/phosphatases, small molecular weight G proteins or Rho GTPase class, and MAPKs, regulate the agonist-induced PLD activation in cellular systems. Oxidant-specific signaling mechanisms regulate the oxidant-induced PLD activation in cells through PKC or protein tyrosine kinases or MAPKs involving thiol redox.25,39 Protein tyrosine kinases and PKC have been reported to regulate the H2O2-induced PLD activation in Swiss 3T3 fibroblasts.25,41 Earlier, we have also shown the involvement of intracellular calcium in the Hg-induced PLD activation in vascular ECs.22 Although the current study did not examine the involvement of PKC, protein tyrosine kinases, and heterotrimeric and small molecular weight G proteins, their role in the Hg-induced PLD activation in MAECs was not ruled out, considering the fact that Hg has been shown to perturb the thiol-redox status and induced ROS in vascular ECs.7,48
In the current study, we took advantage of the availability of the novel PLD-specific inhibitor, FIPI, to not only confirm the PLD activation induced by Hg but also to establish for the first time its specificity to cause inhibition of the agonist- and oxidant-induced PLD activation in ECs. Our study demonstrated that FIPI effectively attenuated the Hg-, oxidant-, and TPA-induced PLD activation in MAECs. This finding was further supported by our earlier report that bleomycin-induced and thiol-redox-regulated PLD activation in lung microvascular ECs is attenuated by FIPI.39 Mercury is reported to cause PLD activation through thiol-redox dysregulation and ROS.7 Oxidants are shown to activate PLD through thiol-redox alteration, protein tyrosine kinase activation, and MAPK signaling in ECs.25,26,41,42 TPA is an established agonist to cause activation of PLD through PKC signaling in ECs.49 Therefore, our current study established that FIPI effectively inhibited PLD activation in MAECs, irrespective of agonist-mediated induction and upstream signaling regulation involving thiol redox, ROS, protein tyrosine kinases, MAPK, and PKC and further suggested that FIPI can be used as a PLD-specific inhibitor in studies aimed at PLD-mediated bioactive cellular lipid signaling cascades. Also, the current study clearly revealed that FIPI protected against the Hg-induced cytotoxicity in MAECs suggesting the role of PLD therein. This finding was supported by our earlier report that the bleomycin-induced cytotoxicity through bioactive lipid (PA) formation in lung microvascular ECs is protected by the PLD-specific inhibitor, FIPI. From our current study, it could be surmised that the bioactive lipids generated by PLD including PA, LPA, and DAG would have caused cytotoxicity of Hg in MAECs.
More striking observation of the current study was the protection of the Hg-induced cytotoxicity by the novel thiol-redox antioxidant, NBMI, in MAECs through restoring the loss of GSH and attenuating the ERK1/2-mediated PLD signaling. It has been established that Hg preferentially reacts with cellular thiols, and GSH being the most critical intracellular soluble thiol antioxidant is depleted during Hg toxicity.50 Association of GSH depletion during thimerosal neurotoxicity and its protection by GSH precursors (GSH ethyl ester and NAC) have been documented.50 Low-molecular-weight thiols such as l-cysteine, dl-homocysteine, l-methionine, and NAC have been shown to protect against the thimerosal-induced neurotoxicity in cerebellar granule cells.51 Thiol chelators including NAC and dithiothreitol have been shown to protect against the dimethylmercury-induced toxicity in rats.52 Common metal chelators such as calcium disodium ethylenediaminetetraacetate (CaNa2EDTA), d-penicillamine, 2,3-dimercaptopropane-1-sulfonate (DMPS), and DMSA have been reported to offer protection against the Hg-induced neurotoxicity in cortical cells.53 These reports have established that the thiol-protective compounds offer protection against Hg-induced toxicity and further support our current findings that NBMI offered protection against Hg cytotoxicity in MAECs through stabilization of thiol-redox-mediated ERK1/2 and PLD signaling. The results of the current study also demonstrated that DMSA, the water-soluble thiol chelator, was equally effective as NBMI, the lipid-soluble thiol antioxidant and chelator in attenuating the Hg-induced PLD activation in MAECs. However, it should be emphasized that DMSA is only a heavy metal chelator, whereas NBMI is both a heavy metal chelator and a thiol antioxidant. Moreover, the present study was only aimed at investigating the efficacy of the novel lipid-soluble thiol antioxidant and heavy metal chelator, NBMI. Although NAC and GSH have been used for treating Hg toxicity, caution has to be exercised while using these thiols for treatment of Hg toxicity, as the low-molecular-weight thiols containing cysteine are identified to be critical in the transport and body disposition of the metal regulated by molecular mimicry.33 In addition, the low-molecular-weight thiol-protective compounds such as NAC and GSH undergo autoxidation/oxidative attack and also act as pro-oxidants further contributing to oxidative stress, which should be also taken into serious consideration while using them for treatment of Hg toxicity.
Structurally, NBMI possesses dicarboxybenzoate moiety as that occurs naturally in fruits that is bound to 2 cysteamines. Therefore, it is reasonable to expect that NBMI could act as both a Hg chelator and free radical scavenger. The ability of NBMI to complex with trace heavy metals including Hg has been reported.54 Hence, the cytoprotective action of NBMI against the Hg-induced cytotoxicity through thiol-redox regulation, ERK1/2 activation, and PLD activation as observed in the current investigation could have been operated by the ability of NBMI in stabilizing/protecting the cellular thiol redox, attenuating oxidative stress, and sequestering Hg. In addition, the lipophilic nature of NBMI would have offered additional advantage to the molecule to specifically lodge in the phospholipid-rich hydrophobic domains of the cell membrane where the redox-regulated signaling cascades would take place, leading to the generation of PLD-catalyzed bioactive lipids which were responsible for the Hg-induced cytotoxicity in vascular ECs. The emergence of novel bifunctional chelating drugs with thiol-redox antioxidant action such as NBMI will have tremendous impact on the evolving therapies effective for the treatment of Hg toxicity. Current treatments for Hg toxicity mostly include the water-soluble chelators and low-molecular-weight thiols. However, the lipid-soluble drugs such as NBMI and FIPI will be more efficacious in protecting against the Hg- and oxidant-induced cytotoxicity by acting at the membrane hydrophobic lipid interface where the redox reactions, phospholipid hydrolysis, and oxidant signaling in cells take place.
Acknowledgments
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: the Dorothy M. Davis Heart and Lung Research Institute and the Division of Pulmonary, Allergy, Critical Care, and Sleep Medicine of the Ohio State University College of Medicine, the National Institute of Health (HL093463), the International Academy of Oral Medicine and Toxicology (IAOMT), and the Alan D. Clark, MD, Memorial Research Fund.
Footnotes
Declaration of Conflicting Interests
Boyd E. Haley, a coauthor of this communication, is Professor Emeritus in the Department of Chemistry at the University of Kentucky, Lexington, KY, USA with a 20% research appointment and is on an active NSF grant in that institution. Dr Haley is also the President of CTI Science which is a possible conflict of interest since they hold the license to the patents concerning the compound, NBMI. However, this research project had not been funded or had not been supported or carried out in part or full by CTI Science in any way. Neither Dr Haley nor CTI Science had any influence or control on this research project. This research project was entirely initiated and supervised by Narasimham L. Parinandi at the Ohio State University, Columbus, OH, USA, with the compound, NBMI synthesized by Niladri Gupta (a graduate student of Boyd E. Haley) in the Department of Chemistry at the University of Kentucky, Lexington, KY, USA, under the supervision of Dr Haley. Boyd E. Haley is Chair of the Scientific Advisory Committee of the International Academy of Oral Medicine and Toxicology (IAOMT), who advises the IAOMT Board regarding scientific matters, but he does not have any control over IAOMT in their grant funding decisions. The IAOMT award has been given exclusively to Narasimham L. Parinandi to conduct research without any influence or control of Boyd E. Haley on this research project. Overall, there are no conflicts of interest.
References
- 1.Clarkson TW, Magos L, Myers GJ. The toxicology of mercury-current exposures and clinical manifestations. N Engl J Med. 2003;349(18):1731–1737. doi: 10.1056/NEJMra022471. [DOI] [PubMed] [Google Scholar]
- 2.Bolger PM, Schwetz BA. Mercury and health. N Engl J Med. 2002;347(22):1735–1736. doi: 10.1056/NEJMp020139. [DOI] [PubMed] [Google Scholar]
- 3.Mutter J, Naumann J, Schneider R, Walach H, Haley B. Mercury and autism: accelerating evidence? Neuro Endocrinol Lett. 2005;26(5):439–446. [PubMed] [Google Scholar]
- 4.Renneberg AJ, Dudas MJ. Transformations of elemental mercury to inorganic and organic forms in mercury and hydrocarbon co-contaminated soils. Chemosphere. 2001;45(6-7):1103–1109. doi: 10.1016/s0045-6535(01)00122-9. [DOI] [PubMed] [Google Scholar]
- 5.Guallar E, Sanz-Gallardo MI, van’t Veer P, et al. Mercury, fish oils, and the risk of myocardial infarction. N Engl J Med. 2002;347(22):1747–1754. doi: 10.1056/NEJMoa020157. [DOI] [PubMed] [Google Scholar]
- 6.Wolf MB, Baynes JW. Cadmium and mercury cause an oxidative stress-induced endothelial dysfunction. Biometals. 2007;20(1):73–81. doi: 10.1007/s10534-006-9016-0. [DOI] [PubMed] [Google Scholar]
- 7.Hagele TJ, Mazerik JN, Gregory A, et al. Mercury activates vascular endothelial cell phospholipase D through thiols and oxidative stress. Int J Toxicol. 2007;26(1):57–69. doi: 10.1080/10915810601120509. [DOI] [PubMed] [Google Scholar]
- 8.Dopp E, Hartmann LM, Florea AM, Rettenmeier AW, Hirner AV. Environmental distribution, analysis, and toxicity of organometal(loid) compounds. Crit Rev Toxicol. 2004;34(3):301–333. doi: 10.1080/10408440490270160. [DOI] [PubMed] [Google Scholar]
- 9.Wilhelm M, Muller F, Idel H. Biological monitoring of mercury vapour exposure by scalp hair analysis in comparison to blood and urine. Toxicol Lett. 1996;88(1-3):221–226. doi: 10.1016/0378-4274(96)03741-1. [DOI] [PubMed] [Google Scholar]
- 10.Doja A, Roberts W. Immunizations and autism: a review of the literature. Can J Neurol Sci. 2006;33(4):341–346. doi: 10.1017/s031716710000528x. [DOI] [PubMed] [Google Scholar]
- 11.Counter SA, Buchanan LH. Mercury exposure in children: a review. Toxicol Appl Pharmacol. 2004;198(2):209–230. doi: 10.1016/j.taap.2003.11.032. [DOI] [PubMed] [Google Scholar]
- 12.Cave SF. The history of vaccinations in the light of the autism epidemic. Altern Ther Health Med. 2008;14(6):54–57. [PubMed] [Google Scholar]
- 13.Barregard L, Rekic D, Horvat M, Elmberg L, Lundh T, Zachrisson O. Toxicokinetics of mercury after long-term repeated exposure to thimerosal-containing vaccine. Toxicol Sci. 2011;120(2):499–506. doi: 10.1093/toxsci/kfr009. [DOI] [PubMed] [Google Scholar]
- 14.Bose-O’Reilly S, McCarty KM, Steckling N, Lettmeier B. Mercury exposure and children’s health. Curr Probl Pediatr Adolesc Health Care. 2010;40(8):186–215. doi: 10.1016/j.cppeds.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Washam C. Beastly beauty products: exposure to inorganic mercury in skin-lightening creams. Environ Health Perspect. 2011;119(2):A80. doi: 10.1289/ehp.119-a80b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chan HM, Egeland GM. Fish consumption, mercury exposure, and heart diseases. Nutr Rev. 2004;62(2):68–72. doi: 10.1111/j.1753-4887.2004.tb00027.x. [DOI] [PubMed] [Google Scholar]
- 17.Kannan K, Smith RG, Jr, Lee RF, et al. Distribution of total mercury and methyl mercury in water, sediment, and fish from south Florida estuaries. Arch Environ Contam Toxicol. 1998;34(2):109–118. doi: 10.1007/s002449900294. [DOI] [PubMed] [Google Scholar]
- 18.Kim DS, Lee EH, Yu SD, Cha JH, Ahn SC. Heavy metal as risk factor of cardiovascular disease—an analysis of blood lead and urinary mercury. J Prev Med Public Health. 2005;38(4):401–407. [PubMed] [Google Scholar]
- 19.Yoshizawa K, Rimm EB, Morris JS, et al. Mercury and the risk of coronary heart disease in men. N Engl J Med. 2002;347(22):1755–1760. doi: 10.1056/NEJMoa021437. [DOI] [PubMed] [Google Scholar]
- 20.Boffetta P, Sallsten G, Garcia-Gomez M, et al. Mortality from cardiovascular diseases and exposure to inorganic mercury. Occup Environ Med. 2001;58(7):461–466. doi: 10.1136/oem.58.7.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wakita Y. Hypertension induced by methyl mercury in rats. Toxicol Appl Pharmacol. 1987;89(1):144–147. doi: 10.1016/0041-008x(87)90185-2. [DOI] [PubMed] [Google Scholar]
- 22.Peltz A, Sherwani SI, Kotha SR, et al. Calcium and calmodulin regulate mercury-induced phospholipase D activation in vascular endothelial cells. Int J Toxicol. 2009;28(3):190–206. doi: 10.1177/1091581809338077. [DOI] [PubMed] [Google Scholar]
- 23.Dudek SM, Garcia JG. Cytoskeletal regulation of pulmonary vascular permeability. J Appl Physiol. 2001;91(4):1487–1500. doi: 10.1152/jappl.2001.91.4.1487. [DOI] [PubMed] [Google Scholar]
- 24.Sliman SM, Eubank TD, Kotha SR, et al. Hyperglycemic oxoaldehyde, glyoxal, causes barrier dysfunction, cytoskeletal alterations, and inhibition of angiogenesis in vascular endothelial cells: amino-guanidine protection. Mol Cell Biochem. 2010;333(1-2):9–26. doi: 10.1007/s11010-009-0199-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Varadharaj S, Steinhour E, Hunter MG, et al. Vitamin C-induced activation of phospholipase D in lung microvascular endothelial cells: regulation by MAP kinases. Cell Signal. 2006;18(9):1396–1407. doi: 10.1016/j.cellsig.2005.10.019. [DOI] [PubMed] [Google Scholar]
- 26.Parinandi NL, Scribner WM, Vepa S, Shi S, Natarajan V. Phospholipase D activation in endothelial cells is redox sensitive. Antioxid Redox Signal. 1999;1(2):193–210. doi: 10.1089/ars.1999.1.2-193. [DOI] [PubMed] [Google Scholar]
- 27.Steinhour E, Sherwani SI, Mazerik JN, et al. Redox-active antioxidant modulation of lipid signaling in vascular endothelial cells: vitamin C induces activation of phospholipase D through phospholipase A2, lipoxygenase, and cyclooxygenase. Mol Cell Biochem. 2008;315(1-2):97–112. doi: 10.1007/s11010-008-9793-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nakada S, Inoue K, Nojima S, Imura N. Change in permeability of liposomes caused by methylmercury and inorganic mercury. Chem Biol Interact. 1978;22(1):15–23. doi: 10.1016/0009-2797(78)90146-1. [DOI] [PubMed] [Google Scholar]
- 29.Le MT, Gailer J, Prenner EJ. Hg2+ and Cd2+ interact differently with biomimetic erythrocyte membranes. Biometals. 2009;22(2):261–274. doi: 10.1007/s10534-008-9162-7. [DOI] [PubMed] [Google Scholar]
- 30.Broniatowski M, Flasinski M, Dynarowicz-Latka P, Majewski J. Grazing incidence diffraction and X-ray reflectivity studies of the interactions of inorganic mercury salts with membrane lipids in Langmuir monolayers at the air/water interface. J Phys Chem B. 2010;114(29):9474–9484. doi: 10.1021/jp101668n. [DOI] [PubMed] [Google Scholar]
- 31.Mazerik JN, Mikkilineni H, Kuppusamy VA, et al. Mercury activates phospholipase a(2) and induces formation of arachidonic acid metabolites in vascular endothelial cells. Toxicol Mech Methods. 2007;17(9):541–557. doi: 10.1080/15376510701380505. [DOI] [PubMed] [Google Scholar]
- 32.Ercal N, Gurer-Orhan H, Aykin-Burns N. Toxic metals and oxidative stress part I: mechanisms involved in metal-induced oxidative damage. Curr Top Med Chem. 2001;1(6):529–539. doi: 10.2174/1568026013394831. [DOI] [PubMed] [Google Scholar]
- 33.Rooney JP. The role of thiols, dithiols, nutritional factors and interacting ligands in the toxicology of mercury. Toxicology. 2007;234(3):145–156. doi: 10.1016/j.tox.2007.02.016. [DOI] [PubMed] [Google Scholar]
- 34.Matlock MM, Howerton BS, Atwood DA. Irreversible precipitation of mercury and lead. J Hazard Mater. 2001;84(1):73–82. doi: 10.1016/s0304-3894(01)00190-x. [DOI] [PubMed] [Google Scholar]
- 35.Matlock MM, Howerton BS, Atwood DA. 6,586,600. Lexington, KY: University of Kentucky; Novel Multidentate Sulfur Containing Ligands. U S Patent No. 2003 Jul 1;
- 36.Su W, Chen Q, Frohman MA. Targeting phospholipase D with small-molecule inhibitors as a potential therapeutic approach for cancer metastasis. Future Oncol. 2009;5(9):1477–1486. doi: 10.2217/fon.09.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Monovich L, Mugrage B, Quadros E, et al. Optimization of halopemide for phospholipase D2 inhibition. Bioorg Med Chem Lett. 2007;17(8):2310–2311. doi: 10.1016/j.bmcl.2007.01.059. [DOI] [PubMed] [Google Scholar]
- 38.Su W, Yeku O, Olepu S, et al. 5-Fluoro-2-indolyl des-chlorohalopemide (FIPI), a phospholipase D pharmacological inhibitor that alters cell spreading and inhibits chemotaxis. Mol Pharmacol. 2009;75(3):437–446. doi: 10.1124/mol.108.053298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Patel RB, Kotha SR, Sherwani SI, et al. Pulmonary fibrosis inducer, bleomycin, causes redox-sensitive activation of phospholipase D and cytotoxicity through formation of bioactive lipid signal mediator, phosphatidic acid, in lung microvascular endothelial cells. Int J Toxicol. 2011;30(1):69–90. doi: 10.1177/1091581810388850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pandian RP, Kutala VK, Liaugminas A, Parinandi NL, Kuppusamy P. Lipopolysaccharide-induced alterations in oxygen consumption and radical generation in endothelial cells. Mol Cell Biochem. 2005;278(1-2):119–127. doi: 10.1007/s11010-005-6936-x. [DOI] [PubMed] [Google Scholar]
- 41.Parinandi NL, Roy S, Shi S, et al. Role of Src kinase in diperoxovanadate-mediated activation of phospholipase D in endothelial cells. Arch Biochem Biophys. 2001;396(2):231–243. doi: 10.1006/abbi.2001.2609. [DOI] [PubMed] [Google Scholar]
- 42.Natarajan V, Scribner WM, Morris AJ, et al. Role of p38 MAP kinase in diperoxovanadate-induced phospholipase D activation in endothelial cells. Am J Physiol Lung Cell Mol Physiol. 2001;281(2):L435–L449. doi: 10.1152/ajplung.2001.281.2.L435. [DOI] [PubMed] [Google Scholar]
- 43.Son MH, Kang KW, Lee CH, Kim SG. Potentiation of cadmium-induced cytotoxicity by sulfur amino acid deprivation through activation of extracellular signal-regulated kinase1/2 (ERK1/2) in conjunction with p38 kinase or c-jun N-terminal kinase (JNK). Complete inhibition of the potentiated toxicity by U0126 an ERK1/2 and p38 kinase inhibitor. Biochem Pharmacol. 2001;62(10):1379–1390. doi: 10.1016/s0006-2952(01)00780-8. [DOI] [PubMed] [Google Scholar]
- 44.Natarajan V, Scribner WM, Vepa S. Phosphatase inhibitors potentiate 4-hydroxynonenal-induced phospholipase D activation in vascular endothelial cells. Am J Respir Cell Mol Biol. 1997;17(2):251–259. doi: 10.1165/ajrcmb.17.2.2623. [DOI] [PubMed] [Google Scholar]
- 45.Natarajan V, Vepa S, Verma RS, Scribner WM. Role of protein tyrosine phosphorylation in H2O2-induced activation of endothelial cell phospholipase D. Am J Physiol. 1996;271(3 pt 1):L400–L408. doi: 10.1152/ajplung.1996.271.3.L400. [DOI] [PubMed] [Google Scholar]
- 46.Turney KD, Parrish AR, Orozco J, Gandolfi AJ. Selective activation in the MAPK pathway by Hg(II) in precision-cut rabbit renal cortical slices. Toxicol Appl Pharmacol. 1999;160(3):262–270. doi: 10.1006/taap.1999.8772. [DOI] [PubMed] [Google Scholar]
- 47.Kim SH, Sharma RP. Mercury-induced apoptosis and necrosis in murine macrophages: role of calcium-induced reactive oxygen species and p38 mitogen-activated protein kinase signaling. Toxicol Appl Pharmacol. 2004;196(1):47–57. doi: 10.1016/j.taap.2003.11.020. [DOI] [PubMed] [Google Scholar]
- 48.Mazerik JN, Hagele T, Sherwani S, et al. Phospholipase A2 activation regulates cytotoxicity of methylmercury in vascular endothelial cells. Int J Toxicol. 2007;26(6):553–569. doi: 10.1080/10915810701707759. [DOI] [PubMed] [Google Scholar]
- 49.Roy S, Parinandi N, Zeigelstein R, et al. Hyperoxia alters phorbol ester-induced phospholipase D activation in bovine lung microvascular endothelial cells. Antioxid Redox Signal. 2003;5(2):217–228. doi: 10.1089/152308603764816578. [DOI] [PubMed] [Google Scholar]
- 50.James SJ, Slikker W, 3rd, Melnyk S, New E, Pogribna M, Jernigan S. Thimerosal neurotoxicity is associated with glutathione depletion: protection with glutathione precursors. Neurotoxicology. 2005;26(1):1–8. doi: 10.1016/j.neuro.2004.07.012. [DOI] [PubMed] [Google Scholar]
- 51.Zieminska E, Toczylowska B, Stafiej A, Lazarewicz JW. Low molecular weight thiols reduce thimerosal neurotoxicity in vitro: modulation by proteins. Toxicology. 2010;276(3):154–163. doi: 10.1016/j.tox.2010.07.023. [DOI] [PubMed] [Google Scholar]
- 52.Joshi D, Mittal DK, Shrivastava S, Shukla S. Protective role of thiol chelators against dimethylmercury induced toxicity in male rats. Bull Environ Contam Toxicol. 2010;84(5):613–617. doi: 10.1007/s00128-010-9982-3. [DOI] [PubMed] [Google Scholar]
- 53.Rush T, Hjelmhaug J, Lobner D. Effects of chelators on mercury, iron, and lead neurotoxicity in cortical culture. Neurotoxicology. 2009;30(1):47–51. doi: 10.1016/j.neuro.2008.10.009. [DOI] [PubMed] [Google Scholar]
- 54.Zaman KM, Blue LY, Huggins FE, Atwood DA. Cd, Hg, and Pb compounds of benzene-1, 3-diamidoethanethiol (BDETH(2)) Inorg Chem. 2007;46(6):1975–1980. doi: 10.1021/ic0607639. [DOI] [PubMed] [Google Scholar]