

Abstract
The mechanisms of lung microvascular complications and pulmonary hypertension known to be associated with idiopathic pulmonary fibrosis (IPF), a debilitating lung disease, are not known. Therefore, we investigated whether bleomycin, the widely used experimental IPF inducer, would be capable of activating phospholipase D (PLD) and generating the bioactive lipid signal-mediator phosphatidic acid (PA) in our established bovine lung microvascular endothelial cell (BLMVEC) model. Our results revealed that bleomycin induced the activation of PLD and generation of PA in a dose-dependent (5, 10, and 100 μg) and time-dependent (2-12 hours) fashion that were significantly attenuated by the PLD-specific inhibitor, 5-fluoro-2-indolyl des-chlorohalopemide (FIPI). PLD activation and PA generation induced by bleomycin (5 μg) were significantly attenuated by the thiol protectant (N-acetyl-L-cysteine), antioxidants, and iron chelators suggesting the role of reactive oxygen species (ROS), lipid peroxidation, and iron therein. Furthermore, our study demonstrated the formation of ROS and loss of glutathione (GSH) in cells following bleomycin treatment, confirming oxidative stress as a key player in the bleomycin-induced PLD activation and PA generation in ECs. More noticeably, PLD activation and PA generation were observed to happen upstream of bleomycin-induced cytotoxicity in BLMVECs, which was protected by FIPI. This was also supported by our current findings that exposure of cells to exogenous PA led to internalization of PA and cytotoxicity in BLMVECs. For the first time, this study revealed novel mechanism of the bleomycin-induced redox-sensitive activation of PLD that led to the generation of PA, which was capable of inducing lung EC cytotoxicity, thus suggesting possible bioactive lipid-signaling mechanism/mechanisms of microvascular disorders encountered in IPF.
Keywords: bioactive lipid signaling, interstitial pulmonary fibrosis, lung microvascular endothelial cell, oxidative stress, phosphatidic acid, phospholipase D, thiol redox
Introduction
Cellular membranes consist of phospholipids, proteins, and carbohydrate moieties. The membrane phospholipids are critical for the cellular homeostasis and survival at the levels of both the cell structure and function. Cellular signaling is one of the crucial cell functions required for homeostasis. Phospholipase D (PLD) is an important membrane phospholipid hydrolase, which generates a potent cellular bioactive lipid signal mediator, phosphatidic acid (PA) that is further metabolized into potent bioactive lipid signal mediators such as diacylglycerol (DAG) and lysophosphatidic acid (LPA).1-3 PLD has been shown to be involved in several pathophysiological conditions such as cardiovascular diseases and cancer.4,5 Thus, the PLD-generated bioactive lipid mediators (PA, DAG, and LPA) regulate the cellular cytoskeleton, especially the actin cytoskeleton, which leads to the regulation and/or alteration of the cell shape and movement.
Phospholipase D activation in mammalian cells is under the control of several regulators including hormones, growth factors, G protein-coupled receptors, and other bioactive signal mediators through the regulation of protein kinases such as protein kinase C (PKC), tyrosine kinase, and mitogen-activated protein kinases.1 Oxidative stress and oxidant exposure (reactive oxygen species [ROS]) are known to activate PLD in mammalian cells.6 Vascular endothelial cells (ECs), the inner monolayer lining of blood vessels which form the barrier, are susceptible to oxidative stress that leads to vascular EC disruption and vascular leak. It is emerging that oxidative stress-induced activation of PLD and associated lipid-signaling enzymes are among key responsible players in EC dysfunction, leading to pathophysiological conditions in lung vasculature during oxidant exposure such as infection, pulmonary hypertension (PH), lung fibrosis, and hyperoxic lung damage. Oxidative stress also leads to redox perturbation in vascular ECs such as alterations in the soluble thiols (glutathione [GSH]) and protein thiols.2 However, the thiol redox modulation of oxidative stress-mediated activation of PLD in lung ECs is not completely understood. Therefore, here, it is envisioned that oxidative stress causes an imbalance in the thiol redox of the lung vascular ECs so that the redox-sensitive PLD is activated, resulting in the generation of the bioactive lipid mediator, PA, that ultimately signals the vascular EC dysfunction and damage.
Idiopathic pulmonary fibrosis (IPF) is a degenerative, chronic, and progressive fibrosing lung disorder of the tissue that lines and separates the alveoli with unknown etiology for which the mechanisms of the onset and progression of the disease are not thoroughly understood.7 The prevalence of IPF in the United States is estimated in the range of 35 000 to 55 000 cases as recorded in 2005.8 Extended exposure to environmental and occupational agents including metal, wood, and stone dusts has been associated with the tissue damage that leads to IPF among the idiopathic lung diseases.9,10 In IPF, the tissue that lines and separates the alveoli becomes scarred due to lung damage. This scarring causes the tissue to become inelastic and hard. The buildup of scar tissue causes difficulties in breathing and results in respiratory failure. Interstitial lung diseases (ILDs) including the sarcoidosis, IPF, and pulmonary Langerhans cell histiocytosis have been shown to be associated with PH.11,12 Lung parenchymal and vascular remodeling indicates the high prevalence (30%-40%) of PH among the ILD patients.13 Although both the lung epithelium and endothelium have been shown to be the critical cellular players in IPF, microvascular injury has been shown as an initial event in lungs during IPF.14 In addition, microvascular injury has been emphasized as an important event in the evolution of IPF.15 Therefore, the lung microvasculature, specifically the lung microvascular endothelium is apparently an important target in the lung fibrotic events. This offers a premise to hypothesize that lung microvasculature is a key player in the mechanism of onset/progression of IPF.
Several cellular signaling mechanisms involving caveolae, caveolins, protein kinases, focal adhesion kinase, protein kinase B, and mitogen-activated protein kinases (MAPKs) have been shown to operate in the IPF.16-18 Studies with experimental models have revealed the role of oxidative stress and antioxidant imbalance in the initiation and progression of IPF, and accordingly redox modulatory therapy has been proposed for the treatment of the disease.7,19 The lung microvasculature and vascular endothelium are critical regions in IPF, oxidative stress and redox alterations appear to drive the initiation and progression of IPF. Oxidants are known to activate the lung vascular endothelial PLD, leading to the generation of bioactive lipid mediators. Therefore, it is compelling to rationalize that oxidant-mediated lung microvascular endothelial PLD signaling is likely to play a crucial role in the initiation and propagation of IPF. To date, no reports have been made on the activation of PLD by the experimental lung fibrosis inducer, bleomycin, in lung cells including the vascular endothelium. Therefore, here, we have hypothesized that bleomycin induces oxidant-mediated and redox-dependent PLD activation upstream that leads to the downstream cytotoxicity through the PLD-generated bioactive lipid mediator, PA in the lung microvascular ECs in culture. In order to test this hypothesis, we used the well-established bovine lung microvascular ECs (BLMVECs) and exposed them to the experimental lung fibrosis inducer, bleomycin, to establish the thiol redox regulation of PLD activation in lung ECs and define the connection between the upstream PLD activation and downstream cytotoxicity that could be responsible for the oxidant-mediated lung vascular dysfunction similar to that encountered in IPF conditions. In order to investigate the comparative responses to bleomycin treatment exhibited by another lung microvascular EC model, we also included the well-established human lung microvascular ECs (HLMVECs) in the current study. For the first time, the results of the current study revealed that the experimental lung fibrosis inducer, bleomycin, caused the upstream activation of PLD and generated the bioactive lipid signal mediator, PA, in oxidant-mediated and redox-sensitive mechanism that led to the cytotoxicity in the lung microvascular ECs in culture, suggesting the possible role of PLD in IPF.
Materials and Methods
Materials
Bovine lung microvascular ECs (passage 4) were purchased from VEC Technologies (New York). Phosphate-buffered saline (PBS) was obtained from Biofluids Inc (Rockville, Maryland). Human lung microvascular ECs and endothelial growth medium-2-microvascular ([EGM-2-MV] containing 5% fetal bovine serum [FBS], hydrocortisone, human recombinant vascular endothelial growth factor, recombinant human fibroblast growth factor-B, recombinant insulin-like growth factor 1, human recombinant epidermal growth factor, ascorbic acid, gentamycin, and amphotericin-B) were purchased from ProCell GmbH (Heidelberg, Germany). Minimal essential medium (MEM), nonessential amino acids, trypsin, FBS, penicillin/streptomycin, Dulbecco modified Eagle medium (DMEM) phosphate-free modified medium, tissue culture reagents, 3-[4,5-dimethylthiazol-2-yl]-2, 5-diphenyl tetrazolium bromide reduction kit (MTT assay kit), lactate dehydrogenase cytotoxicity assay kit (LDH release assay kit), and analytical reagents of highest purity were all purchased from Sigma Chemical Co (St Louis, Missouri). Phosphatidylbutanol (PBt), fluorescent C6:0-18:1 NBD-PA, 1-oleoyl-2-{6-[(7-nitro-2-1, 3-benzoxadiazol-4-yl) amino]hexanoyl}-sn-glycero-3-phosphate (C18:2 PA), and 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphate (C16:0-18:1 PA) were obtained from Avanti Polar Lipids (Alabaster, Alabama). [32P]orthophosphate (carrier-free) was obtained from New England Nuclear (Wilmington, Delaware). Bleomycin was obtained from Teva Parenteral Medicines (Irvine, California). Desferal was obtained from Calbiochem (San Diego, California). Polyclonal antibody raised against 4-hydroxy-2-nonenal (anti-HNE) was obtained from Enzo Life Sciences (Farmingdale, New York). Endothelial cell growth factor was obtained from Upstate Biotechnology (Lake Placid, New York). Anti-rabbit AlexaFluor 488-conjugated antibody and 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI) were purchased from Molecular Probes Invitrogen Co (Carlsbad, California). 5-Fluoro- 2-indolyl des-chlorohalopemide hydrochloride hydrate (FIPI) was prepared as described earlier.5,20,21 Glutathione assay kit (GSH-Glo) was obtained from Promega Corporation (Madison, Wisconsin).
Cell Culture
Bovine lung microvascular ECs were grown to confluence in MEM supplemented with 10% (vol/vol [v/v]) FBS, 100 units/mL penicillin and streptomycin, 5 μg/mL EC growth factor, and 1% (v/v) nonessential amino acids at 37°C under a humidified 95% air and 5% CO2 atmosphere, as described earlier.1 Bovine lung microvascular ECs, from passages 7 to 15, were used in the experiments. Human lung microvascular ECs were cultured in the EGM-2-MV medium under a humidified 95% air and 5% CO2 atmosphere. Human lung microvascular ECs, from passages 2 to 6, were used in the experiments. Endothelial cells cultured in 35-mm or 60-mm sterile dishes or T-75 cm sterile flasks to ~95% confluence under a humidified 95% air and 5% CO2 atmosphere were used for treatments with bleomycin and desired pharmacological agents. Minimal essential medium containing bleomycin and other pharmacological agents were carefully adjusted to pH 7.4 for cellular treatments.
Phospholipase D Activation in Intact ECs
Phospholipase D activity in ECs was determined according to our previously published procedure.1,2,6 Endothelial cells in 35-mm dishes (5 × 105 cells/dish) were labeled with [32P]orthophosphate (5 μCi/mL) in DMEM phosphate-free medium containing 2% (v/v) FBS for 6 to 14 hours.6 Following experimental treatments, [32P]-labeled phosphatidylbutanol ([32P]PBt) formed from PLD activation and transphosphatidylation reaction in cellular lipid extracts, as an index of PLD activity in intact cells, was separated by thin-layer chromatography (TLC).1,6 Radioactivity associated with the [32P]PBt was quantified by liquid scintillation counting, and data were expressed as Disintegrations per minute (DPM) normalized to 106 counts in the total cellular lipid extract or as percentage of control (vehicle-treated cells).
Phosphatidic Acid Formation in Intact ECs
At the end of the experiments, treatments of cells were terminated by the addition of 1 mL methanol:HCl (100:1, v/v). Lipids were extracted essentially according to the method of Bligh and Dyer, as described previously.1 [32P]-labeled PA formed from PLD activation, as an index of PLD activity in intact cells, was separated by TLC.1,6 Autoradiogram was developed from the TLC plate with [32P]PA separated from other phospholipids. Intensity of the radioactivity associated with the [32P]PA was quantified using the Scion Image program and data were expressed as percentage of control (untreated cells).
Reactive Oxygen Species Measurement by 2′-7′-Dichlorofluorescin Diacetate Fluorescence
Formation of ROS in BLMVECs in 35-mm dishes (5 × 105 cells/dish) was determined by 2′-7′-dichlorofluorescin diacetate (DCFDA) fluorescence in cells preloaded with 10 μmol/L DCFDA for 30 minutes in complete MEM at 37°C in a 95% air and 5% CO2 environment prior to experimental treatments, according to our previously published procedure.22,23 2′-7′-Dichlorofluorescin diacetate fluorescence as an index of intracellular ROS generation was visualized under Olympus fluorescence microscope at ×50 magnification with excitation and emission set at 490 and 530 nm, respectively. Intracellular ROS formation was quantified by measuring DCFDA fluorescence in lysates of cells on a Bio-Tex ELx808 fluorescent plate reader set at 490 nm excitation and 530 nm emission, using appropriate blanks.22,23 The extent of intracellular ROS formation was expressed as the arbitrary fluorescence units.
Glutathione Determination
Intracellular soluble thiol (GSH) levels were determined using the GSH-Glo GSH assay kit. Bovine lung microvascular ECs grown up to 90% confluence in 96 well plates were treated with MEM alone or MEM containing desired concentrations of bleomycin for 12 hours under a humidified 95% air and 5% CO2 atmosphere. Following incubation, intracellular GSH levels were determined according to the manufacturer’s recommendations (Promega Corp, Madison, Wisconsin).
Immunofluorescence Microscopy
Bovine lung microvascular ECs cultured on sterile coverslips (Harvard Apparatus, 22 mm2) in 35-mm sterile dishes at a density of 104 cells/dish were treated with MEM alone and MEM containing bleomycin for 12 hours under a humidified atmosphere of 95% air and 5% CO2 at 37°C. Following the experimental treatments, cells were incubated for 12 hours at room temperature with rabbit primary anti-4HNE antibody (1:200 dilution, v/v) to detect lipid peroxidation. After the primary antibody treatment, cells were incubated with the secondary anti-rabbit AlexaFluor 488-conjugated antibody (1:100 dilution, vol/vol), for 1 hour at room temperature. The coverslips with cells were then mounted on a glass slide with the antifade mounting medium, Fluoromount-G, viewed with Ziess Confocal microscope at a magnification of ×60, pictures were captured digitally, and the extent of 4-HNE formation was determined from the fluorescence intensity using the Scion Image software.6
Cytotoxicity Assay
In the current study, we utilized 3 different cytotoxicity assay viz (i) cell morphology examination, which demonstrates the loss of cell shape, necrosis, and cytoskeletal alterations (attachment) due to toxic effects; (ii) LDH leak assay, which reveals the plasma membrane damage and leak of cellular macromolecules in response to the toxic insult; and (iii) MTT reduction assay, which demonstrates the mitochondrial dysfunction due to their inability to reduce MTT during toxic insult.
Morphology Assay of Cytotoxicity
Morphological alterations in BLMVECs cultured in 35-mm dishes up to 70% confluence, following experimental treatments, were examined as an index of cytotoxicity according to our previously published methods.24 Images of cell morphology were digitally captured with the Olympus microscope at ×20 magnification.
Lactate Dehydrogenase Release Assay of Cytotoxicity
Cytotoxicity in ECs was determined by assaying the extent of release of LDH from cells according to our previously published method.24 At the end of treatment, the medium was collected and the LDH released into the medium was determined spectrophotometrically using the commercial LDH assay kit according to the manufacturer’s recommendations (Sigma Chemical Co).
3-[4,5-Dimethylthiazol-2-yl]-2, 5-Diphenyl Tetrazolium Bromide Reduction Assay of Cytotoxicity
Cytotoxicity in ECs was determined by assaying the extent of reduction of MTT in intact cells using the commercial MTT reduction assay kit. At the end of the experimental treatments, MTT solution (10% by vol in MEM) was added and the cells were incubated for 3 hours, following which MTT solvent was added in an amount equal to the original culture volume. Absorbance of the reduced MTT was determined spectrophotometrically according to the manufacturer’s recommendations (Sigma Chemical Co).
Uptake and Distribution of PA
Bovine lung microvascular ECs cultured on sterile coverslips (Harvard Apparatus, 22 mm2) in 35-mm sterile dishes, at a density of 104 cells/dish, were treated with MEM alone and MEM containing fluorescent C6:0-18:1 NBD-PA (5 μmol/L) for 24 hours under a humidified atmosphere of 95% air and 5% CO2 at 37°C. At the end of the incubation period, cells attached to coverslips were washed with 1× PBS, fixed with 3.7% of paraformaldehyde for 10 minutes, permeabilized with 0.25% Triton X-100 in Tris buffer saline Tween-20 (TBST) containing 0.01% Tween-20 for 5 minutes, then blocked for 30 minutes with 1% BSA in 0.01% TBST, and incubated for 5 minutes at room temperature with 1% (v/v) DAPI in PBS to visualize the nuclei. The coverslips with cells were then mounted on a glass slide with the antifade mounting medium, Fluoromount-G, viewed under Ziess Confocal fluorescence microscope at a magnification of ×60, and pictures were captured digitally.
Protein Determination
Protein was determined by the bicinchoninic acid (BCA) protein assay (Thermo Scientific, Rockford, Illinois).
Statistical Analysis
All experiments were done in triplicate. Data were expressed as mean ± standard deviation (SD). Statistical analysis was carried out by analysis of variance (ANOVA) using SigmaStat (Jandel Scientific, San Rafael, California). The level of statistical significance was taken as P < .05.
Results
Bleomycin Induces PLD Activation in Dose- and Time-dependent Fashion in ECs
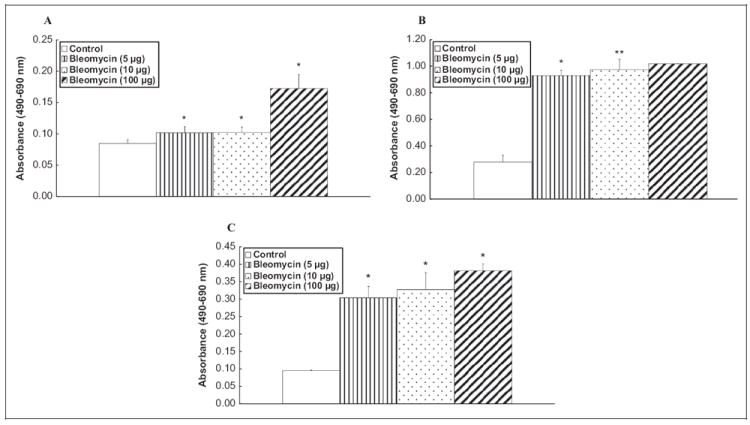
Here, we conducted studies to (i) determine whether bleomycin would increase PLD activation in lung ECs and (ii) compare the bleomycin-induced PLD activation in 2 different established lung EC models such as BLMVECs (of bovine origin) and HLMVECs (of human origin). Bleomycin, in a dose-dependent manner (5, 10, and 100 μg), at 4 and 12 hours of incubation, induced significant activation of PLD ([32P]PBt formation) in BLMVECs as compared with the vehicle-treated control cells (2-, 2-, and 3-fold for 4 hours and 5-, 6-, and 12-fold for 12 hours at 5, 10, and 100 μg, respectively; Figure 1A). Treatment of ECs with bleomycin (5 μg) for 4, 8, 12, and 24 h induced significant increases (2-, 3-, 4-, and 3-fold) in the activation of PLD as compared with that in the cells exposed to the MEM alone (Figure 1B). Bleomycin, in a dose-dependent manner (5, 10, and 100 μg), at 8 and 12 hours of incubation, induced significant activation of PLD in HLMVECs as compared with that in the vehicle-treated control cells (2-, 2-, and 3-fold for 8 hours and 2-, 3-, and 3-fold for 12 hours at 5, 10, and 100 μg, respectively; Figure 1C). These results revealed that bleomycin induced PLD activation in dose- and time-dependent manner in both BLMVECs and HLMVECs. These results demonstrated that bleomycin induced PLD activation in chosen EC systems including those of bovine (BLMVECs) and human (HLMVECs) origins. However, subsequent experiments in the current study were conducted using the ECs of bovine origin (BLMVECs).
Figure 1.

Bleomycin induces PLD activation in dose- and time-dependant fashion in ECs. Bovine lung microvascular ECs (BLMVECs) and HLMVECs (5 × 105 cells/35-mm dish) were labeled with [32P]orthophosphate in DMEM phosphate-free medium for 6 to 12 hours. Following [32P]orthophosphate labeling, BLMVECs were treated with MEM alone or MEM containing different concentrations (5, 10, and 100 μg) of bleomycin (A), or for different time periods (4-24 hours; B), and HLMVECs were treated with MEM alone or MEM containing different concentrations of bleomycin (5, 10, and 100 μg) for 8 and 12 hours (C) in presence of 0.05% (vol/vol) 1-butanol. At the end of incubation period, [32P]PBt formed was determined. Data represent mean ± SD calculated from 3 independent experiments. *Significantly different at P < .05 as compared to cells treated with MEM alone. HLMVECs indicates human lung microvascular endothelial cells; PLD, phospholipase D; DMEM, Dulbecco modified Eagle medium; MEM, minimal essential medium; [32P]PBt, [32P]-labeled phosphatidylbutanol; ECs, endothelial cells.
Thiol Protectants Attenuate Bleomycin-Induced PLD Activation in ECs
Altered thiol redox has been shown to activate PLD in ECs. Therefore, to establish the role of thiols in bleomycin-induced PLD activation in ECs, here, the effects of well-established thiol protective agents (N-acetyl-L-cysteine [NAC] and Dithiothreitol [DTT]) were investigated on PLD ([32P]PBt formation) activation induced by bleomycin. Cells were pretreated for 2 hours with MEM alone or MEM containing the chosen thiol protectant (0.5, 1, and 5 mmol/L) and then co-treated for 12 hours with bleomycin (5 μg) and the chosen thiol protectant. NAC, a widely used thiol protectant and antioxidant, caused effective and significant attenuation of bleomycin-induced PLD activation in ECs (22%, 70%, and 97% of inhibition for concentrations of 0.5, 1, and 5 mmol/L, respectively; Figure 2A). DTT, a sulfhydryl protective agent, also offered effective and significant attenuation of the bleomycin-induced PLD activation in ECs (43%, 67%, and 65% of inhibition at concentrations of 0.5, 1, and 5 mmol/L, respectively; Figure 2B). These results revealed that thiol protectants effectively attenuated the bleomycin-induced PLD activation in ECs, further suggesting the involvement of cellular thiol redox status in the bleomycin-induced activation of PLD in ECs.
Figure 2.

Thiol protectants attenuate bleomycin-induced PLD activation in ECs. Bovine lung microvascular ECs ([BLMVECs] 5 × 105 cells/ 35-mm dish) were labeled with [32P]orthophosphate in DMEM phosphate-free medium for 12 hours. Following [32P]orthophosphate labeling, cells were pretreated for 2 hours with MEM alone or MEM containing N-acetyl-l-cysteine [NAC] (0.5, 1, and 5 mmol/L; A) or Dithiothreitol [DTT] (0.5, 1, and 5 mmol/L; B) and then subjected to treatment with MEM alone or MEM containing the thiol protectant or MEM containing bleomycin (5 μg) or MEM containing the selected thiol protectant and bleomycin (5 μg) for 12 hours in the presence of 0.05% (vol/vol) 1-butanol. At the end of incubation period, [32P]PBt formed was determined as. Data represent mean ± SD calculated from 3 independent experiments. *Significantly different at P < .05 as compared to cells treated with MEM alone. **Significantly different at P < .05 as compared to cells treated with MEM containing bleomycin alone. DMEM indicates Dulbecco modified Eagle medium; MEM, minimal essential medium; [32P]PBt, [32P]-labeled phosphatidylbutanol; PLD, phospholipase D; ECs, endothelial cells.
Heavy Metal Chelators Attenuate Bleomycin-Induced PLD Activation in ECs
Oxidative stress and ROS production have been shown to activate PLD in ECs. Among heavy metals, particularly Fe2+ has been shown to be involved in the generation of ROS especially through Fenton-type reactions.25 Bleomycin complexes with iron to become an active drug.26 Therefore, to establish the role of iron-mediated oxidative stress in bleomycin-induced PLD activation in ECs, here, the effects of well-established heavy metal chelators (EDTA, desferal, and Dimercaptosuccinic Acid [DMSA]) were investigated on PLD activation ([32P]PBt formation) induced by bleomycin in BLMVECs. Cells were pretreated for 2 hours with MEM alone or MEM containing the chosen heavy metal chelator (2 mmol/L of EDTA and desferal, and 0.5, 1, 5 mmol/L of DMSA) and then co-treated for 12 hours with bleomycin (5 μg) and the chosen metal chelator. EDTA and desferal, widely used Fe2+ chelators, caused effective and significant attenuation of the bleomycin-induced PLD activation in ECs (32% and 27% of inhibition, respectively; Figure 3A and B). DMSA, a heavy metal chelator and a thiol redox stabilizer, also offered effective and significant attenuation of the bleomycin-induced PLD activation in ECs (39%, 64%, and 64% of inhibition at concentrations of 0.5, 1, and 5 mmol/L, respectively; Figure 3C). These results revealed that heavy metal chelators, especially Fe2+-specific chelators, effectively attenuated the bleomycin-induced PLD activation in ECs, further suggesting the involvement of ROS production in the bleomycin-induced enzyme activation in ECs.
Figure 3.
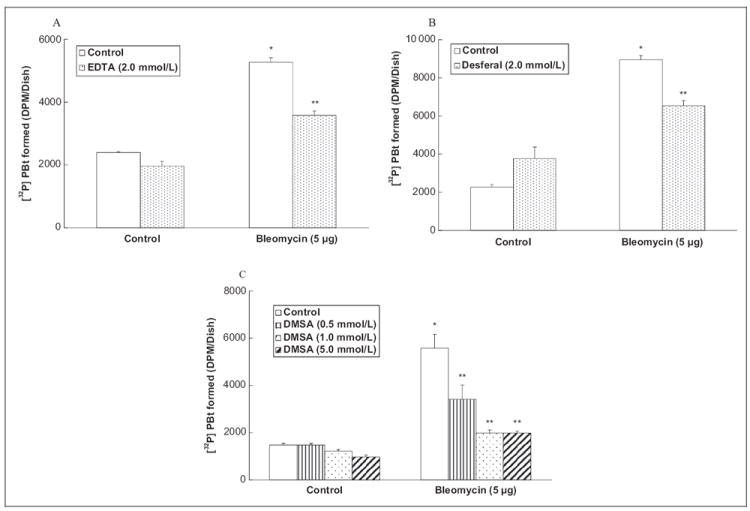
Heavy metal chelators attenuate bleomycin-induced PLD activation in ECs. Bovine lung microvascular ECs ([BLMVECs] 5 × 105 cells/35-mm dish) were labeled with [32P]orthophosphate in DMEM phosphate-free medium for 12 hours. Following [32P]orthophosphate labeling, cells were pretreated for 2 hours with MEM alone or MEM containing EDTA (2 mmol/L; A) or desferal (2 mmol/L; B) or DMSA (0.5, 1, 5 mmol/L; C) and then subjected to treatment with MEM alone or MEM containing the selected chelating agent or MEM containing bleomycin (5 μg) or MEM containing the selected chelating agent and bleomycin (5 μg) for 12 hours in the presence of 0.05% (vol/vol) 1-butanol. At the end of the incubation period, [32P]PBt formed was determined. Data represent mean ± SD calculated from 3 independent experiments. *Significantly different at P < .05 as compared to cells treated with MEM alone. **Significantly different at P < .05 as compared to cells treated with MEM containing bleomycin alone DMEM indicates Dulbecco modified Eagle medium; MEM, minimal essential medium; [32P]PBt, [32P]-labeled phosphatidylbutanol; PLD, phospholipase D; ECs, endothelial cells; DMSA, Dimercaptosuccinic Acid.
Antioxidants Attenuate Bleomycin-Induced PLD Activation in ECs
Oxidative stress, ROS production, and altered thiol redox has been shown to activate PLD in ECs.1,2 Moreover, bleomycin is established as an oxidant drug. We also found earlier in this study that heavy metal chelators and thiol protectants effectively and significantly attenuated the bleomycin-induced PLD activation in ECs. Therefore, in order to establish the role of oxidative stress in the bleomycin-induced PLD activation in ECs, here, the effects of established antioxidants (Pyrrolidine Dithiocarbamate [PDTC], propyl gallate, vitamin C, and Manganese Tetrakis (4-Benzoic Acid) Porphyrin [MnTBAP]) were investigated on the PLD activation induced by bleomycin. Cells were pretreated for 2 hours with MEM alone or MEM containing the chosen antioxidant (0.5, 1, and 5 mmol/L of PDTC, 500 μmol/L of propyl gallate and vitamin C, and 20 μmol/L of MnTBAP) and then co-treated for 12 hours with bleomycin (5 μg) and the chosen antioxidant. PDTC caused effective and significant attenuation of the bleomycin-induced PLD activation in ECs (25%, 49%, and 72% of inhibition at 0.5, 1, and 5 mmol/L, respectively; Figure 4A). Propyl gallate, a phenolic antioxidant and vitamin C, also offered effective and significant attenuation of the bleomycin-induced PLD activation in ECs (49% and 46%, respectively; Figure 4B and C). MnTBAP, a superoxide scavenger, also offered effective and significant attenuation of the bleomycin-induced PLD activation in ECs (21%; Figure 4D). These results revealed that antioxidants effectively attenuated the bleomycin-induced PLD activation in ECs, further suggesting the involvement of ROS in the bleomycin-induced enzyme activation in ECs.
Figure 4.
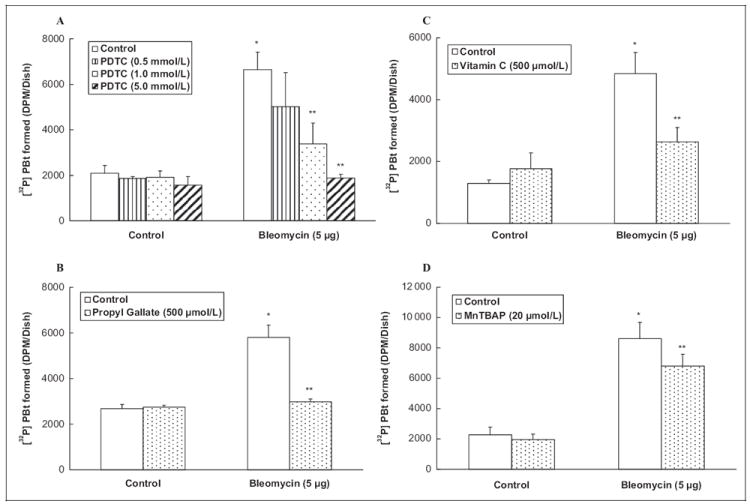
Antioxidants attenuate bleomycin-induced PLD activation in ECs. Bovine lung microvascular ECs ([BLMVECs] 5 × 105 cells/35-mm dish) were labeled with [32P]orthophosphate in DMEM phosphate-free medium for 12 hours. Following [32P]orthophosphate labeling, cells were pretreated for 2 hours with MEM alone or MEM containing Pyrrolidine Dithiocarbamate (PDTC) (0.5, 1, 5 mmol/L; A) or propyl gallate (500 μmol/ L; B) or vitamin C (500 μmol/L; C) or Manganese Tetrakis (4-Benzoic Acid) Porphyrin (MnTBAP) (20 μmol/L; D) and then subjected to treatment with MEM alone or MEM containing the selected antioxidant or MEM containing bleomycin (5 μg) or MEM containing the selected antioxidant and bleomycin (5 μg) for 12 hours in the presence of 0.05% (vol/vol) 1-butanol. At the end of incubation period, [32P]PBt formed was determined. Data represent mean ± SD calculated from 3 independent experiments. *Significantly different at P < .05 as compared to cells treated with MEM alone. **Significantly different at P < .05 as compared to cells treated with MEM containing bleomycin alone. PLD indicates phospholipase D; DMEM, Dulbecco modified Eagle medium; MEM, minimal essential medium; [32P]PBt, [32P]-labeled phosphatidylbutanol; ECs, endothelial cells.
Lipid Peroxidation Inhibitors Attenuate Bleomycin-Induced PLD Activation in ECs
Oxidative stress and ROS production have been shown to induce lipid peroxidation in ECs.1 Therefore, in order to establish the role of lipid peroxidation in the bleomycin-induced PLD activation in ECs, here, the effects of well-established lipid peroxidation-inhibiting antioxidants (nordihydroguaiaretic acid [NDGA] and trolox) were investigated on PLD activation ([32P]PBt formation) induced by bleomycin. Cells were pretreated for 2 hours with MEM alone or MEM containing the chosen lipid peroxidation-inhibiting antioxidant (10, 20, and 50 μmol/L of NDGA, and 100, 200, and 500 μmol/L of trolox) and then co-treated for 12 hours with bleomycin (5 μg) and the chosen antioxidant. Nordihydroguaiaretic acid, a plant-derived polyphenol, caused effective and significant attenuation of the bleomycin-induced PLD activation in ECs (45%, 72%, and 67% of inhibition at 10, 20, and 50 μmol/L NDGA, respectively; Figure 5A). Trolox, a water-soluble form of vitamin E, also offered effective and significant attenuation of the bleomycin-induced PLD activation in ECs (61%, 69%, and 71% of inhibition at concentrations of 100, 200, and 500 μmol/L trolox, respectively; Figure 5B). These results revealed that lipid peroxidation-inhibiting antioxidants effectively attenuated the bleomycin-induced PLD activation in ECs, further suggesting the involvement of lipid peroxidation in the bleomycin-induced activation of PLD in ECs.
Figure 5.
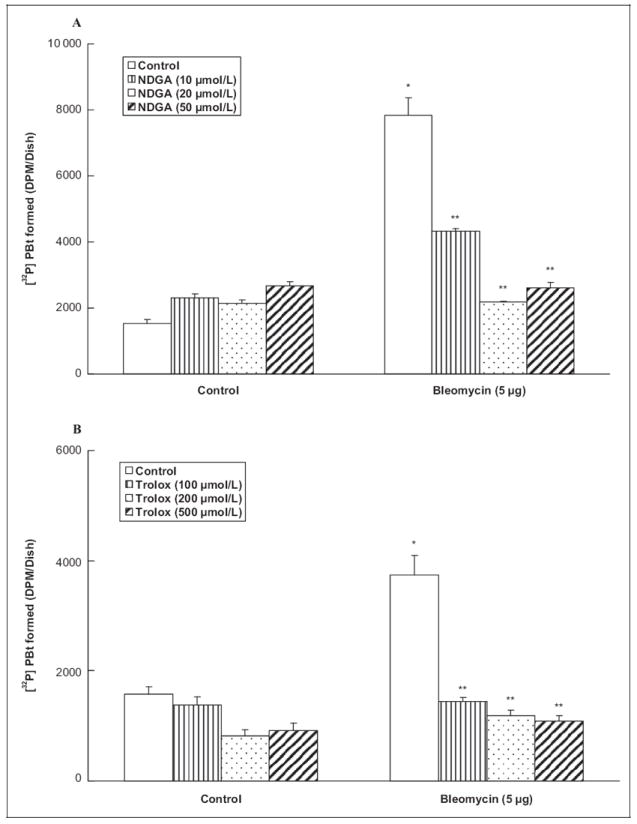
Lipid peroxidation inhibitors attenuate bleomycin-induced PLD activation in ECs. Bovine lung microvascular ECs ([ECs] 5 × 105 cells/35-mm dish) were labeled with [32P]orthophosphate in DMEM phosphate-free medium for 12 hours. Following [32P]orthophosphate labeling, cells were pretreated for 2 hours with MEM alone or MEM containing NDGA (10, 20, 50 μmol/L; A) or trolox (100, 200, 500 μmol/L; B) and then subjected to treatment with MEM alone or MEM containing the selected lipid peroxidation inhibitor, or MEM containing bleomycin (5 μg) or MEM containing the selected lipid peroxidation inhibitor and bleomycin (5 μg) for 12 hours in the presence of 0.05% (vol/vol) 1-butanol. At the end of the incubation period, [32P]PBt formed was determined. Data represent mean ± SD calculated from 3 independent experiments. *Significantly different at P < .05 as compared to cells treated with MEM alone. **Significantly different at P < .05 as compared to cells treated with MEM containing bleomycin alone. PLD indicates phospholipase D; DMEM, Dulbecco modified Eagle medium; MEM, minimal essential medium; [32P]PBt, [32P]-labeled phosphatidylbutanol; ECs, endothelial cells; NDGA, nordihydroguaiaretic acid.
Bleomycin Induces ROS Generation and Depletes GSH in ECs
The pharmacological action of bleomycin has been attributed to its ability to induce ROS formation.26 Earlier in this study, we showed that the bleomycin-induced PLD activation was attenuated by antioxidants, and therefore here we hypothesized that bleomycin would induce the intracellular ROS formation in ECs. Therefore, here we investigated the ability of bleomycin to induce the intracellular ROS formation in BLMVECs by determining the DCFDA fluorescence. Bleomycin, in a dose-dependent manner (5, 10, and 100 μg), at 1 hour of treatment, induced significant ROS formation in BLMVECs as compared with that in the vehicle-treated control cells (Figure 6A). Bleomycin, at 0.5 hour of treatment, effectively induced the ROS formation in BLMVECs as compared with that in the control untreated cells (1.5-, 1.8-, and 2-fold at 5, 10, and 100 μg, respectively; Figure 6B). Hence, these results demonstrated that bleomycin induced the intracellular ROS formation in ECs. Earlier in this study, we showed that thiol protectants attenuated the bleomycin-induced PLD activation in ECs. Therefore, here we hypothesized that bleomycin would cause the depletion of intracellular GSH. To test this postulate, intra-cellular soluble thiols (GSH) were measured. Bleomycin in a dose-dependent manner induced significant depletion of cellular GSH in BLMVECs at 12 hours of exposure as compared with the same in the vehicle-treated control cells (60%, 57%, and 45% of decrease at 5, 10, and 100 μg, respectively; Figure 6C). These results demonstrated that bleomycin caused the loss of intracellular GSH leading to the altered thiol redox status in BLMVECs.
Figure 6.
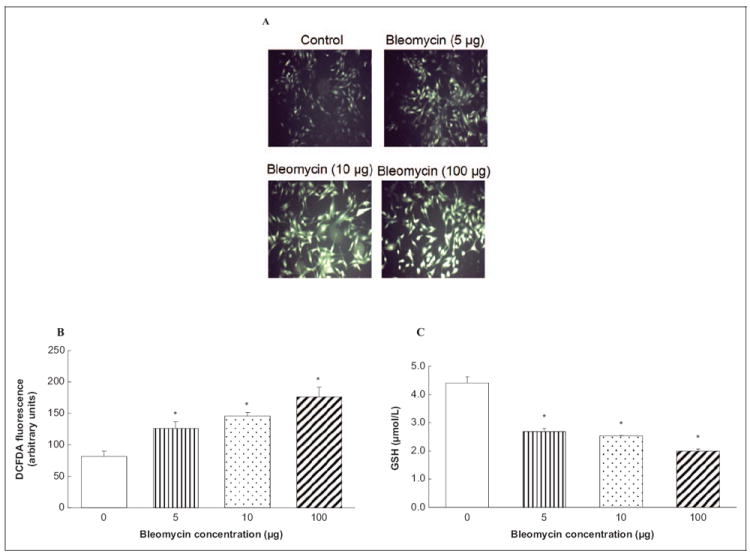
Bleomycin induces ROS generation and depletes GSH in ECs. Bovine lung microvascular ECs ([BLMVECs 5 × 105 cells/35-mm dish) were preloaded with 10 μmol/L DCFDA for 30 minutes in complete MEM to determine ROS generation. Following the DCFDA loading, cells were subjected to treatment with MEM alone or MEM containing bleomycin (5, 10, and 100 μg) for 1 hour (A) and 0.5 hour (B). At the end of the incubation period, the DCFDA fluorescence (as an index of ROS formation) was determined as described under Materials and Methods section. Each micrograph is a representative picture obtained from 3 independent experiments conducted under identical conditions. Bovine lung microvascular ECs (1 × 105 cells/96 well plates) were subjected to treatment with MEM alone or MEM containing bleomycin (5, 10, and 100 μg) for 12 hours to determine intracellular GSH levels (C). At the end of the incubation period, the intracellular soluble thiol (GSH) concentrations were determined. Data represent mean ± SD calculated from 3 independent experiments. *Significantly different at P < .05 as compared to cells treated with MEM alone. DCFDA indicates 2′-7′-dichlorofluorescin diacetate; MEM, minimal essential medium; ROS, reactive oxygen species; GSH, glutathione; ECs, endothelial cells.
Bleomycin Induces Lipid Peroxidation in ECs
Earlier in this study, we showed that bleomycin caused the production of ROS in ECs. Reactive oxygen species is known to cause lipid peroxidation of polyunsaturated fatty acids and produce highly reactive carbonyls such as 4-HNE.27 To observe whether bleomycin would induce lipid peroxidation in ECs, cells were treated with bleomycin for 12 hours, fixed, and stained with the anti-4-HNE antibody. Using confocal microscopy, bleomycin was found to induce the formation of 4-HNE (as an index of lipid peroxidation) in a dose-dependant manner (6-, 6-, and 10-fold increase at 5, 10, and 100 μg of bleomycin, respectively; Figure 7A and B).
Figure 7.

Bleomycin induces lipid peroxidation in ECs. Bovine lung microvascular ECs [BLMVECs] on coverslips (5 × 105 cells/35-mm dish) were subjected to treatment with MEM alone or MEM containing bleomycin (5, 10, and 100 μg) for 12 hours to assay the extent of lipid peroxidation. At the end of the incubation period, the cells were fixed, stained for 4-HNE formation, and visualized using confocal fluorescence microscopy. The fluorescence intensities were measured. Each micrograph is a representative picture obtained from 3 independent experiments conducted under identical conditions. Data represent mean ± SD calculated from 3 independent experiments. *Significantly different at P < .05 as compared to cells treated with vehicle alone. 4-HNE indicates 4-hydroxy-2-nonenal; MEM, minimal essential medium; ECs, endothelial cells.
Bleomycin Induces Morphological Alterations Which are Protected by PLD-Specific Inhibitor (FIPI) in ECs
As PLD is a lipid-signaling enzyme and plays an important role in cellular functions through the generation of bioactive lipid signal mediators, here we investigated the effects of bleomycin on the cell morphology as an index of disruption of cellular structure and cytotoxicity. Bovine lung microvascular ECs were treated with bleomycin (5, 10, and 100 μg) for 4, 8, 12, and 24 hours and then subjected to cell morphology examination by light microscopy. Bleomycin, in a time- and dose-dependant manner, caused morphological alterations (Figure 8A). The results revealed that bleomycin at a dose of 5 μg caused initiation of morphological alterations at 4 hours of treatment, which were prominent at 12 hours of treatment in BLMVECs. In order to determine that PLD activation would be involved in the bleomycin-induced cell morphology alterations (cytotoxicity), we treated the BLMVECs with the PLD-specific inhibitor, FIPI, to inhibit PLD activity and investigated the bleomycin-induced cell morphological alterations. Cells were pretreated with FIPI (1 μmol/L) for 12 hours to ensure the optimal uptake of the inhibitor and co-treated with bleomycin (5 μg) for 4, 8, 12 and 24 hours, following which their morphology was examined microscopically. 5-Fluoro-2-indolyl des-chlorohalopemide significantly attenuated the bleomycin-induced cell morphological alterations at all time points, suggesting PLD activation in the bleomycin-induced cytotoxic morphological alterations in BLMVECs (Figure 8B).
Figure 8.
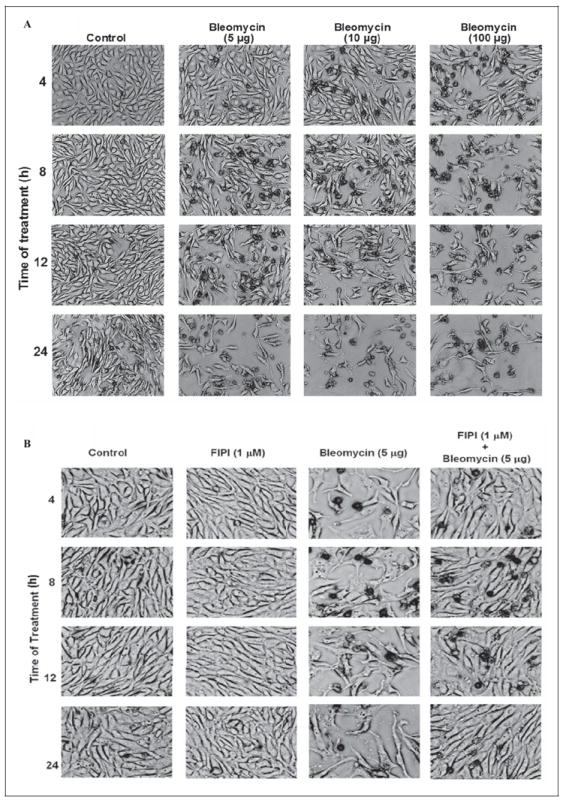
Bleomycin induces morphological alterations which are protected by PLD-specific inhibitor (FIPI) in ECs. Bovine lung microvascular ECs ([BLMVECs] 5 × 105 cells/35-mm dish) were subjected to treatment with MEM alone or MEM containing bleomycin (5, 10, and 100 μg) for 4 to 24 hours to examine the bleomycin-induced cell morphology alterations (A). Bovine lung microvascular ECs (5 × 105 cells/35-mm dish) were pretreated with complete MEM or complete MEM containing FIPI (1 μmol/L) for 12 hours and then subjected to co-treatment with MEM alone or MEM containing bleomycin (5, 10, and 100 μg) for 4 to 24 hours to determine the protective effect of FIPI on bleomycin-induced cell morphology alterations (B). At the end of the incubation period, the cell morphology was examined under light microscope (as an index of cytotoxicity). Each micrograph is a representative picture obtained from 3 independent experiments conducted under identical conditions. PLD indicates phospholipase D; FIPI, 5-fluoro-2-indolyl des-chlorohalopemide; MEM, minimal essential medium; ECs, endothelial cells.
Bleomycin Induces Cytotoxicity in ECs
As bleomycin in this study was shown to cause morphological alterations in ECs, here, another measure of cytotoxicity, the release of LDH by the BLMVECs exposed to bleomycin was determined. Bleomycin induced the release of LDH in a dose- and time-dependant fashion. Bleomycin (5, 10, and 100 μg) caused significant LDH release (0.2-, 0.2-, and 1-fold, 2-, 2-, and 3-fold, and 2-, 2-, and 3-fold increase at 8, 12, and 24 hours, respectively) as compared with the same in the control untreated cells (Figure 9A, B, and C). These results indicated that bleomycin induced cytotoxicity in ECs as evidenced by LDH release.
Figure 9.
Bleomycin induces cytotoxicity in ECs. Bovine lung microvascular ECs ([BLMVECs] 2.5 × 105 cells/17.5-mm dish) were subjected to treatment with MEM alone or MEM containing bleomycin (5, 10, and 100 μg) for 8 (A), 12 (B), and 24 (C) hours. At the end of the incubation period, the release of LDH into the medium (as an index of cytotoxicity) was determined spectrophotometrically. Data represent mean ± SD calculated from 3 independent experiments. *Significantly different at P < .05 as compared to cells treated with MEM alone. LDH, indicates lactate dehydrogenase; MEM, minimal essential medium; ECs, endothelial cells.
PLD-Specific Inhibitor, FIPI, Protects Against Bleomycin-Induced Cytotoxicity and Attenuates Bleomycin-Induced PLD Activation in ECs
As the PA generated from PC through PLD action has been implicated in many physiological responses, here, we investigated whether the cytotoxic effects of bleomycin were caused by the PLD-generated PA in ECs. Therefore, we investigated whether the PLD-specific inhibitor, FIPI, would offer protection against the bleomycin-induced cytotoxicity as assayed by the LDH release from the cells. ECs were pretreated with FIPI (250, 500, and 1000 nmol/L) for 12 hours, prior to exposure to bleomycin (5 μg) for 12 hours. 5-Fluoro-2-indolyl des-chlorohalopemide (250, 500, and 1000 nmol/L) significantly caused dose-dependent attenuation of the bleomycin-induced LDH release (23%, 36%, and 45%) from BLMVECs (Figure 10A). These results suggested the role of PLD and PLD-generated PA in the bleomycin-induced cytotoxicity in ECs. We speculated that the cytotoxic effects of bleomycin were mediated by the PLD-generated PA. To test this premise, we wanted to inhibit the PLD activity using the novel PLD inhibitor, FIPI.5,21 Bovine lung microvascular ECs were pre-treated with FIPI (250, 500, and 1000 nmol/L) for 12 hours while they were being labeled with [32P]orthophospate to ensure the effective uptake of the inhibitor. Following that the ECs were then co-treated with FIPI and bleomycin (5 μg) for 12 hours. The results revealed that FIPI significantly attenuated the bleomycin-induced PLD activation (25%, 42%, and 68% at 250, 500, and 1000 nmol/L, respectively). 5-Fluoro-2-indolyl des-chlorohalopemide was also found to inhibit the basal normal PLD activity (27%, 47%, and 62% at 250, 500, and 1000 nmol/L, respectively; Figure 10B). 5-Fluoro-2-indolyl des-chlorohalopemide by itself was not observed to cause any adverse or cytotoxic effects in BLMVEC as revealed fromthe cell morphology examination (Figure 8B) and LDH release assay (Figure 10A). Therefore, these results clearly demonstrated that FIPI caused effective inhibition of the bleomycin-induced activation of PLD in ECs in a dose-dependent manner.
Figure 10.
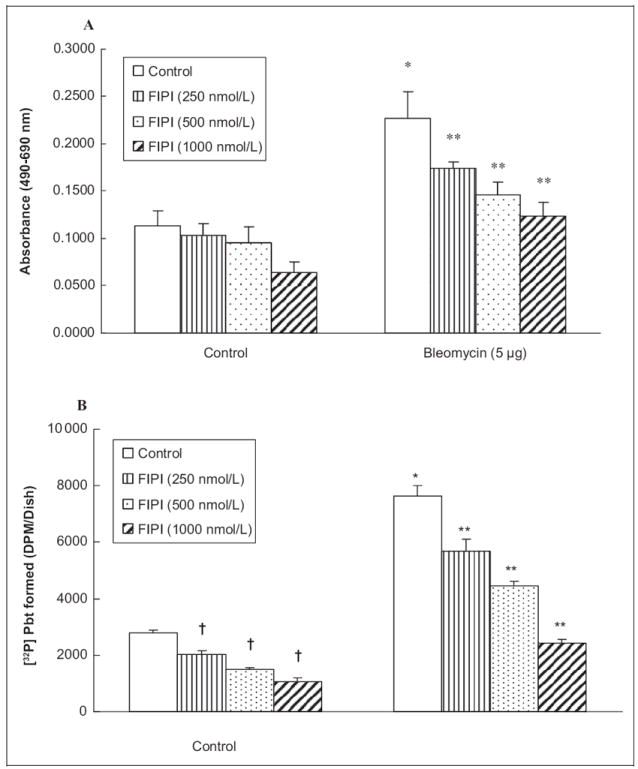
PLD-specific inhibitor, FIPI, protects against bleomycin-induced cytotoxicity and attenuates bleomycin-induced PLD activation in ECs. Bovine lung microvascular ECs ([BLMVECs] 2.5 × 105 cells/17.5-mm dish) were pretreated with MEM alone or MEM containing FIPI (250, 500, 1000 nmol/L) for 12 hours. Following the pretreatment, cells were subjected to treatment with MEM alone or MEM containing bleomycin (5 μg) alone or MEM containing FIPI alone or MEM containing FIPI and bleomycin (5 μg) for 12 hours. At the end of the incubation period, the release of LDH into the medium (as an index of cytotoxicity) was determined spectrophotometrically (A). To determine the inhibitory effect of FIPI on the bleomycin-induced PLD activation, BLMVECs (5 × 105 cells/35-mm dish) were labeled with [32P]orthophosphate in DMEM phosphate-free medium along with pretreatment of MEM alone or MEM containing FIPI (250, 500, 1000 nmol/L) for 12 hours. Following the [32P]orthophosphate labeling and FIPI pretreatment, cells were subjected to co-treatment with MEM alone or MEM containing bleomycin (5 μg) for 12 hours in presence of 0.05% (vol/vol) 1-butanol. At the end of the incubation period, [32P]PBt formed (B) was determined. Data represent mean ± SD calculated from 3 independent experiments. *Significantly different at P < .05 as compared to cells treated with MEM alone. **Significantly different at P < .05 as compared to cells treated with MEM containing bleomycin alone. LDH, indicates lactate dehydrogenase; MEM, minimal essential medium; ECs, endothelial cells. FIPI, 5-fluoro-2-indolyl des-chlorohalopemide; [32P]PBt, [32P]-labeled phosphatidylbutanol; PLD, phospholipase D.
Bleomycin Induces PA Formation in ECs
As PA is the lipid signal mediator generated by PLD action on cellular membrane phospholipids, here, we investigated the bleomycin-induced formation of PA in BLMVECs. Bleomycin (5, 10, and 100 μg), following 12 hours of treatment of cells induced significant formation of PA in BLMVECs as compared with the same in the control untreated cells (125%, 122%, and 133% of increase at 5, 10, and 100 μg, respectively; Figure 11A and B). Upon increasing the dose up to 100 μg, bleomycin did not appear to alter/enhance the extent of PA formation that was observed in BLMVECs treated with the lowest dose of bleomycin (5 μg). These results revealed that bleomycin induced the formation of PA in BLMVECs.
Figure 11.
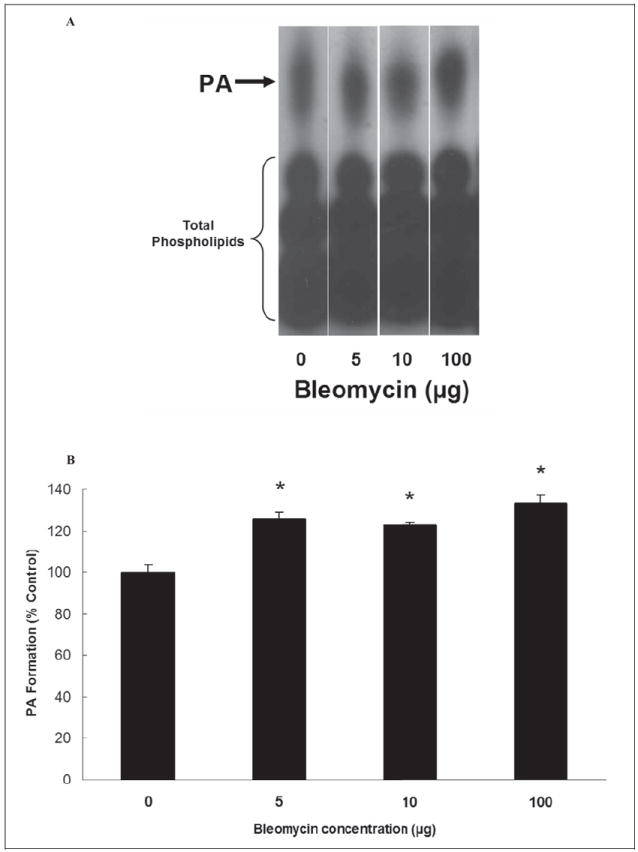
Bleomycin induces phosphatidic acid (PA) formation in ECs. Bovine lung microvascular ECs ([BLMVECs] 5 × 105 cells/35-mm dish) were labeled with [32P]orthophosphate in DMEM phosphate-free medium for 12 hours. Following [32P]orthophosphate labeling, cells were treated with MEM alone or MEM containing different concentrations (5, 10, and 100 μg) of bleomycin for 12 hours. At the end of the incubation period, [32P]PA formed was determined from the autoradiogram (A) and quantified from intensity (B). Data represent mean ± SD calculated from 3 independent experiments. *Significantly different at P < .05 as compared to cells treated with MEM alone. DMEM, Dulbecco modified Eagle medium; MEM, minimal essential medium; ECs, endothelial cells.
Phosphatidic Acid Induces Cytotoxicity in ECs
As shown earlier in this study, the PLD-generated PA was associated with cytotoxicity, here we investigated whether exogenously added PA would cause cytotoxicity in BLMVECs. Lactate dehydrogenase cytotoxicity release and MTT reduction by cells treated with varying concentrations of 1,2-dilinoleoyl-sn- glycero-3-phosphate (C18:2 PA), and 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphate (C16:0-18:1 PA) were determined as the indices of cytotoxicity. C18:2 PA (1, 10, and 25 μg/mL) caused enhanced extent of LDH release by 234%, 167%, and 309%, respectively, at 12 hours of treatment of BLMVECs as compared to the same in the control untreated cells (Figure 12A). C16:0-18:1 PA (1, 5, and 10 μg/mL) caused significant decrease in MTT reduction by 7%, 43%, and 40%, respectively, at 12 hours of treatment of BLMVECs, as compared to the same in the control untreated cells (Figure 12B). These results revealed that exogenous PA caused cytotoxicity in ECs.
Figure 12.
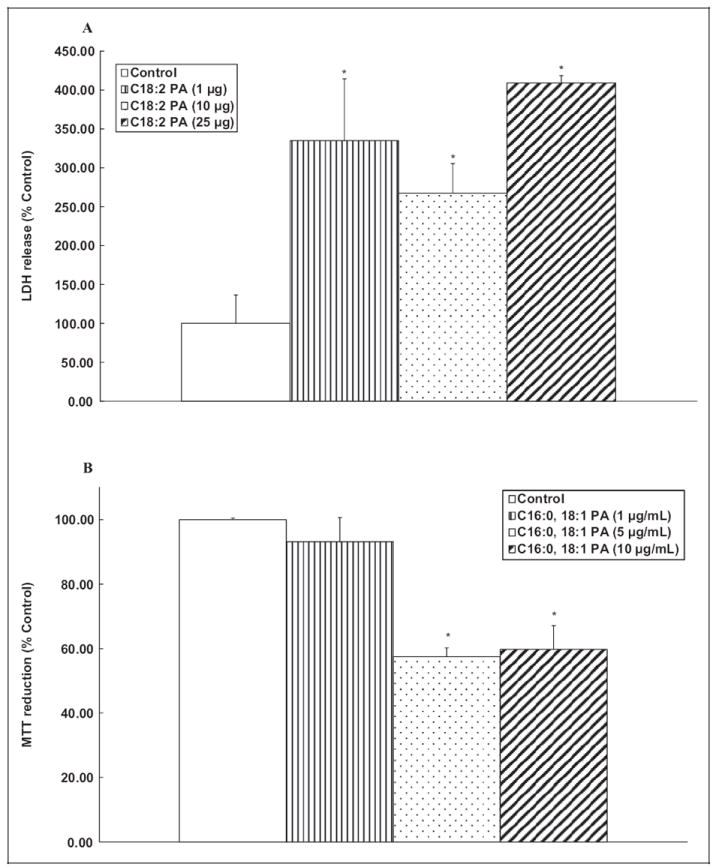
Phosphatidic acid (PA) induces cytotoxicity in ECs. Bovine lung microvascular ECs ([BLMVECs] 2.5 × 105 cells/17.5-mm dish) were subjected to treatment with MEM alone or MEM containing C18:2 PA (1, 10, and 25 μg/mL) or MEM containing C16:0-18:1 PA (1, 5, and 10 μg/mL) for 12 hours. At the end of the incubation period, the release of LDH into the medium (A) and decrease in MTT reduction (B, as an index of cytotoxicity) were determined spectrophotometrically. Data represent mean ± SD calculated from 3 independent experiments. *Significantly different at P < .05 as compared to cells treated with MEM alone. LDH, indicates lactate dehydrogenase; MTT, 3-[4,5-dimethylthiazol-2-yl]-2, 5-diphenyl tetrazolium; MEM, minimal essential medium; ECs, endothelial cells.
Phosphatidic Acid is Taken up and Localized in ECs
Earlier in this study, we showed that bleomycin caused the production of PA and exogenously added PA caused cytotoxicity in ECs. To observe the intracellular targets of PA, BLMVECs were treated with fluorescent C6:0-18:1 NBD-PA (5 μmol/L) for 24 hours, fixed, and stained with nuclear-specific stain, DAPI. Using the confocal fluorescence microscopy, fluorescent C6:0-18:1 NBD-PA, cellular uptake was noticed and localized heavily in the cytosol and the perinuclear regions with scanty localization in the nucleus of BLMVECs. Control cells treated with vehicle alone showed no fluorescence (Figure 13A), as opposed to the cells treated with the fluorescent C6:0-18:1 NBD-PA exhibiting intracellular distribution of PA (Figure 13B, C, and D). These results clearly demonstrated that the lipid signal mediator PA exhibited intracellular localization at targeted specific sites.
Figure 13.
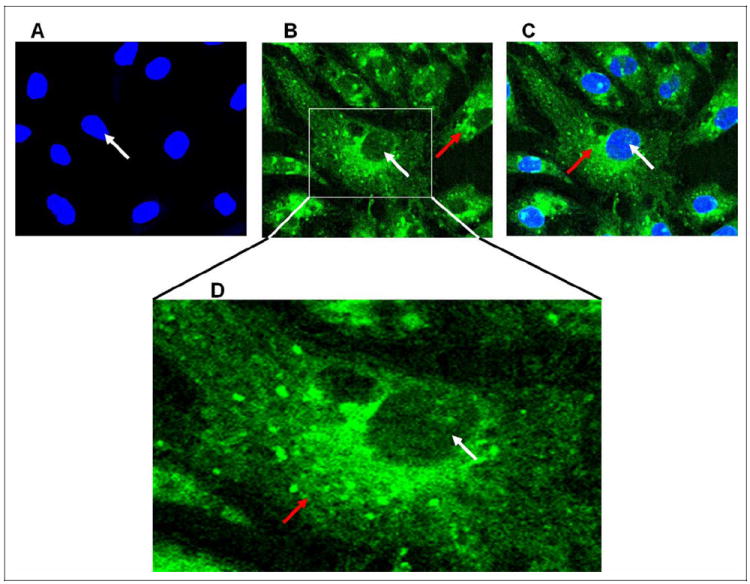
Phosphatidic acid (PA) is taken up and localized in ECs. Bovine lung microvascular ECs (BLMVECs) on coverslips (5 × 105 cells/35-mm dish) were subjected to treatment with MEM alone or fluorescent C6:0-18:1 NBD-PA, 1-oleoyl-2-{6-[(7-nitro-2-1, 3-benzoxadiazol-4-yl)amino]hexanoyl}-sn-glycero-3-phosphate (5 μmol/L) for 24 hours. At the end of the incubation period, the cells were fixed, stained for nuclei, and visualized using confocal fluorescence microscopy. The red arrow indicates the PA distribution in the cytosol, while the white arrow indicates the nuclear distribution (B and C). A selected region of a micrograph was enlarged for clarity showing distribution of PA in cytosol, perinuclear region, and nucleus (D). Each micrograph is a representative picture obtained from 3 independent experiments conducted under identical conditions. MEM indicates minimal essential medium; ECs, endothelial cells.
Discussion
The results of the current study revealed that bleomycin at μg doses induced the activation of PLD, which was attenuated by antioxidants, thiol protectants, and iron chelators, suggesting the upstream role of oxidants and altered redox signaling in the activation of PLD in BLMVECs. This was further confirmed by the enhanced generation of ROS and decrease in thiol levels upon treatment of BLMVECs with bleomycin. Phospholipase D-specific inhibitor, FIPI, caused complete inhibition of the bleomycin-induced activation of PLD and also offered protection against the cytotoxic effects of bleomycin in BLMVECs. Furthermore, the current study also revealed that bleomycin caused enhanced formation of PA and exogenous PA treatment resulted in cytotoxicity in BLMVECs. Using the fluorescent NBD-PA, we also demonstrated the uptake and subcellular localization of PA in BLMVECs. Taken together, these results demonstrated the role of PLD-generated PA in the bleomycin-induced lung microvascular EC damage (Figure 14).
Figure 14.
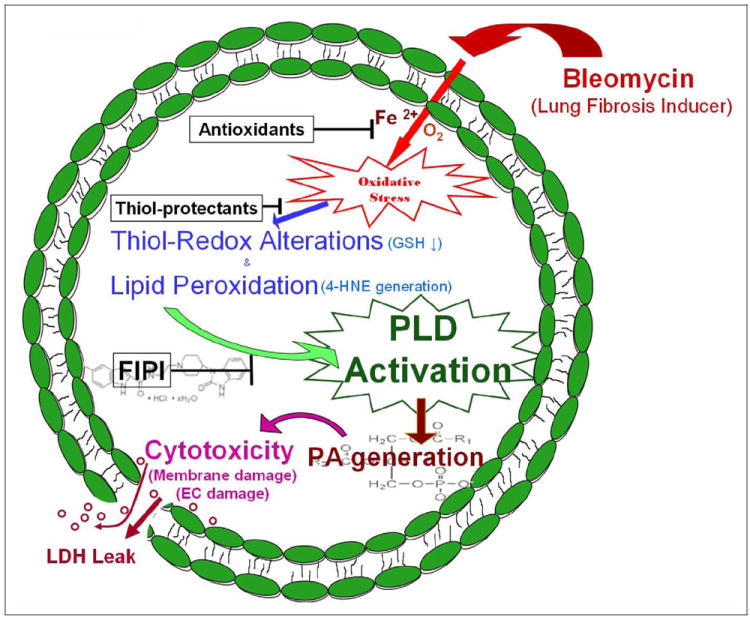
Proposed mechanism. Schematic representation of bleomycin-induced PLD activation and generation of lipid signal mediator, PA, and the subsequent lung endothelial damage. PLD indicates phospholipase D; PA, phosphatidic acid.
Phospholipases are enzymes capable of hydrolyzing the membrane phospholipids at specific sites and generating bioactive lipid second messengers, which may play vital roles in cell signaling.28 Phospholipase D is one such lipid-signaling enzyme ubiquitously present in all mammalian cells that preferentially hydrolyzes phosphatidylcholine (PC), thus generating PA and choline.29 Phosphatidic acid is further metabolized to either DAG by phosphatidate phosphohydrolase (lipid phosphate phosphohydrolase) or LPA by phospholipase A1/A2.30-32 Agonist-mediated activation of PLD plays a pivotal role in signal transduction in mammalian cells.33,34 Several functions, including the promotion of mitogenesis in fibroblasts, stimulation of oxidative stress in neutrophils, increase of intracellular calcium, activation of protein kinases, and phospholipases have been attributed to the signaling actions of PA and LPA.29 Two major forms of PLD, hPLD1 and hPLD2 have been cloned in mammalian cells.35-38 Even though the 2 isoforms of PLD catalyze hydrolysis of PC to PA, they are selectively activated by various cofactors such as Arf, Rho, Cdc42, and detergents.39,40 Phosphatidylinositol 4,5-bisphosphate (PIP2), a lipid mediator, activates both hPLD1 and hPLD2 in cell-free preparations. The results of the current work showed that bleomycin activated PLD in the BLMVECs, which resulted in the enhanced generation of the lipid signal mediator, PA, and this could be possible through the actions of both PLD1 and PLD2 isoforms as the presence of both the isoforms in ECs has been detected.22 However, the relative activity of both the isoforms in the bleomycin-induced formation of PA is yet to be determined.
Earlier studies have shown that oxidants and ROS such as hydrogen peroxide, fatty acid hydroperoxide, and 4- hydroxynonenal simulate PLD in vascular ECs, smooth muscle cells, and fibroblasts.41-45 The ROS-mediated PLD activation is attenuated by tyrosine kinase inhibitors in ECs, suggesting a role for protein tyrosine phosphorylation in PLD activation.22 In addition to tyrosine kinase inhibitors, PLD activation by oxidants is also attenuated by antioxidants, indicating redox regulation of PLD.2 Recently, we have reported that mercury activates PLD in the vascular ECs through thiol redox alteration and generation of ROS.23 Antioxidants including the thiol protectants have been shown to attenuate the pro-oxidantinduced activation of PLD in lung microvascular ECs, suggesting the role of ROS and thiol redox in the enzyme activation.1 Along these lines, the results of the current study also revealed that the bleomycin-induced PLD activation was regulated by the ROS, thiol redox (GSH) perturbation, and lipid peroxidation (4-HNE formation) in BLMVECs. The current study also demonstrated the involvement of oxidant-induced and thiol-regulated signaling in the bleomycin-induced activation of PLD, generation of PA, and cytotoxicity in the lung microvascular ECs. However, the regulation of PLD activation by different stimuli is complex and involves changes in intracellular Ca2+, PKC, heterotrimeric G proteins, small molecular weight G proteins, and protein tyrosine kinases/protein tyrosine phosphatases. 6,22,46 The precise signaling node responsible for mediating the oxidant-induced and thiol-regulated activation of PLD caused by bleomycin in BLMVECs needs to be established.
Bleomycin is an oxidant drug and has been commonly used to induce pulmonary fibrosis in animal models.47,48 Bleomycin is well known for its action of inducing oxidative stress and redox alteration both in vivo and in vitro.19,49 Iron binds to bleomycin to generate oxidants which damage DNA.50 The bleomycin-induced DNA damage is protected by iron chelators such as EDTA.51 Oxidation of extracellular cysteine and alteration of redox state in lung, induction of apoptosis and senescence in lung cells, and disruption of iron homeostasis due to bleomycin exposure have been noticed in relation to the bleomycin-induced pulmonary fibrosis.26,49,52,53 Formation and role of lipid-derived free radicals have been observed in bleomycin-induced lung injury, indicating the role of lipid peroxidation in the pulmonary toxicity of bleomycin.54 Free radical quencher and lipid peroxidation chain-breaking antioxidant such as α-tocopherol has also been reported to protect against the bleomycin-induced lung injury, suggesting the role of lipid peroxidation and lipid radicals in the pulmonary toxicity of the drug.55 Resveratrol, a phytochemical and antioxidant, has been shown to alleviate the bleomycin-induced lung injury in rats in vivo.56 The thiol protectant, NAC, and the iron chelator, deferoxamine, have been observed to attenuate the bleomycin-induced oxidative stress and lung injury.57 Thus, these reports support our current findings that bleomycin induced oxidative stress, altered thiol redox status, induced lipid peroxidation, activated PLD, and caused cytotoxicity in a redox-dependent pathway in BLMVECs. Our current study also provided evidence in favor of the protection of the bleomycin-induced adverse effects in the ECs offered by anti-oxidants, thiol protectants, and iron chelator.
One of the key findings of the current study was the role of PLD-generated PA in the cytotoxicity of bleomycin in BLMVECs. This was further confirmed by the use of the novel PLD-specific inhibitor, FIPI,5,21 which not only completely inhibited the bleomycin-induced activation of PLD but also attenuated the cytotoxicity (altered cell morphology and LDH release) of bleomycin in the lung microvascular ECs. This observation further established a link between the upstream activation of PLD and downstream cytotoxicity exerted by bleomycin in BLMVECs, thus offering first evidence in favor of the role of the PLD-generated bioactive lipid mediator, PA therein. As the PLD-generated PA can further undergo enzymatic conversion into DAG and/or LPA, it was not clear whether the cytotoxicity-inducing bioactive lipid, generated by PLD through bleomycin action, was PA, DAG, LPA, or combination of those. Phospholipase D-generated PA has been identified as the precursor of LPA formation through the actions of PLA1/PLA2.58,59 Lysophosphatidic acid receptors, a member of the G protein-coupled receptors, have been shown to be involved in several physiological and pathophysiological states.58,60 Lysophosphatidic acid receptors 1 and 2 have been identified to play roles in the regulation of vascular injury.61 The role of LPA receptor 1 linking pulmonary fibrosis to lung injury toward causing fibroblast recruitment and vascular leak in the bleomycin mouse model of pulmonary fibrosis has been unequivocally elucidated.62 Nevertheless, the sources of LPA in these studies have not been identified at least in the bleomycin pulmonary fibrosis model. On the other hand, the current study indicated PLD as a possible source of PA that could be converted into LPA, another potent bioactive lipid mediator, which could be responsible for the observed bleomycin-induced vascular EC damage. Also, it should be emphasized that PLD-derived PA could have also exerted its cellular effects through PA-dependent or DAG-mediated signaling cascades.
The bleomycin-induced cytotoxicity in BLMVECs as observed in the current study might be due to both PA and other PA-derived signal mediators such as LPA and DAG. Nevertheless, our current study demonstrated that when BLMVECs were treated with exogenous PA, (i) the lipid was taken up and localized in cellular compartments including cytosol, perinuclear, and nuclear region (Figure 13) and (ii) induced cytotoxicity. This observation reinforced the role of PA in causing cytotoxicity of bleomycin through the activation of PLD in ECs.
Overall, the current study offered a novel signaling pathway of bleomycin-induced lung microvascular EC damage, involving the oxidative stress-mediated and thiol redox-regulated PLD activation and generation of the bioactive lipid mediator, PA (Figure 14). Furthermore, the findings of the current study also identified PLD as a possible target for the therapeutic intervention of IPF.
Acknowledgments
Funding
The author(s) disclosed receipt of the following financial support for the research and/or authorship of this article: Funds from the Dorothy M. Davis Heart and Lung Research Institute and the Division of Pulmonary, Allergy, Critical Care, and Sleep Medicine of the Ohio State University, and the National Institute of Health (HL093463).
Footnotes
Declaration of Conflicting Interests
The author(s) declared no conflicts of interest with respect to the authorship and/or publication of this article.
References
- 1.Steinhour E, Sherwani SI, Mazerik JN, et al. Redox-active antioxidant modulation of lipid signaling in vascular endothelial cells: Vitamin C induces activation of phospholipase D through phospholipase A2, lipoxygenase, and cyclooxygenase. Mol Cell Biochem. 2008;315(1-2):97–112. doi: 10.1007/s11010-008-9793-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Parinandi NL, Scribner WM, Vepa S, Shi S, Natarajan V. Phospholipase D activation in endothelial cells is redox sensitive. Antioxid Redox Signal. 1999;1(2):193–210. doi: 10.1089/ars.1999.1.2-193. [DOI] [PubMed] [Google Scholar]
- 3.Cummings R, Parinandi N, Wang L, Usatyuk P, Natarajan V. Phospholipase D/phosphatidic acid signal transduction: role and physiological significance in lung. Mol Cell Biochem. 2002;234-235(1-2):99–109. [PubMed] [Google Scholar]
- 4.Tappia PS, Dent MR, Dhalla NS. Oxidative stress and redox regulation of phospholipase D in myocardial disease. Free Radic Biol Med. 2006;41(3):349–361. doi: 10.1016/j.freeradbiomed.2006.03.025. [DOI] [PubMed] [Google Scholar]
- 5.Su W, Chen Q, Frohman MA. Targeting phospholipase D with small-molecule inhibitors as a potential therapeutic approach for cancer metastasis. Future Oncol. 2009;5(9):1477–1486. doi: 10.2217/fon.09.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Varadharaj S, Steinhour E, Hunter MG, et al. Vitamin C-induced activation of phospholipase D in lung microvascular endothelial cells: regulation by MAP kinases. Cell Signal. 2006;18(9):1396–1407. doi: 10.1016/j.cellsig.2005.10.019. [DOI] [PubMed] [Google Scholar]
- 7.Kinnula VL, Fattman CL, Tan RJ, Oury TD. Oxidative stress in pulmonary fibrosis: a possible role for redox modulatory therapy. Am J Respir Crit Care Med. 2005;172(4):417–422. doi: 10.1164/rccm.200501-017PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zisman DA, Keane MP, Belperio JA, Strieter RM, Lynch JP., 3rd Pulmonary fibrosis. Methods Mol Med. 2005;117:3–44. doi: 10.1385/1-59259-940-0:003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Taskar V, Coultas D. Exposures and idiopathic lung disease. Semin Respir Crit Care Med. 2008;29(6):670–679. doi: 10.1055/s-0028-1101277. [DOI] [PubMed] [Google Scholar]
- 10.Wilson MS, Wynn TA. Pulmonary fibrosis: pathogenesis, etiology and regulation. Mucosal Immunol. 2009;2(2):103–121. doi: 10.1038/mi.2008.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ryu JH, Krowka MJ, Pellikka PA, Swanson KL, McGoon MD. Pulmonary hypertension in patients with interstitial lung diseases. Mayo Clin Proc. 2007;82(3):342–350. doi: 10.4065/82.3.342. [DOI] [PubMed] [Google Scholar]
- 12.Cordier JF. Pulmonary hypertension in chronic pulmonary respiratory and cardiac diseases. Rev Prat. 2008;58(18):2019–2023. [PubMed] [Google Scholar]
- 13.Behr J, Ryu JH. Pulmonary hypertension in interstitial lung disease. Eur Respir J. 2008;31(6):1357–1367. doi: 10.1183/09031936.00171307. [DOI] [PubMed] [Google Scholar]
- 14.Calabrese F, Giacometti C, Rea F, Loy M, Valente M. Idiopathic interstitial pneumonias: Primum movens: epithelial, endothelial or whatever. Sarcoidosis Vasc Diffuse Lung Dis. 2005;22(suppl 1):S15–S23. [PubMed] [Google Scholar]
- 15.Magro CM, Allen J, Pope-Harman A, et al. The role of microvascular injury in the evolution of idiopathic pulmonary fibrosis. Am J Clin Pathol. 2003;119(4):556–567. doi: 10.1309/0B06-Y93E-GE6T-Q36Y. [DOI] [PubMed] [Google Scholar]
- 16.Gosens R, Mutawe M, Martin S, et al. Caveolae and caveolins in the respiratory system. Curr Mol Med. 2008;8(8):741–753. doi: 10.2174/156652408786733720. [DOI] [PubMed] [Google Scholar]
- 17.Garneau-Tsodikova S, Thannickal VJ. Protein kinase inhibitors in the treatment of pulmonary fibrosis. Curr Med Chem. 2008;15(25):2632–2640. doi: 10.2174/092986708785908969. [DOI] [PubMed] [Google Scholar]
- 18.Chopra P, Kanoje V, Semwal A, Ray A. Therapeutic potential of inhaled p38 mitogen-activated protein kinase inhibitors for inflammatory pulmonary diseases. Expert Opin Investig Drugs. 2008;17(10):1411–1425. doi: 10.1517/13543784.17.10.1411. [DOI] [PubMed] [Google Scholar]
- 19.Kinnula VL, Myllarniemi M. Oxidant-antioxidant imbalance as a potential contributor to the progession of human pulmonary fibrosis. Antioxid Redox Signal. 2008;10(4):727–738. doi: 10.1089/ars.2007.1942. [DOI] [PubMed] [Google Scholar]
- 20.Monovich L, Mugrage B, Quadros E, et al. Optimization of halopemide for Phospholipase D2 inhibition. Bioorg Med Chem Lett. 2007;17(8):2310–2311. doi: 10.1016/j.bmcl.2007.01.059. [DOI] [PubMed] [Google Scholar]
- 21.Su W, Yeku O, Olepu S, et al. 5-Fluoro-2-indolyl des-chlorohalopemide (FIPI), a phospholipase D pharmacological inhibitor that alters cell spreading and inhibits chemotaxis. Mol Pharmacol. 2009;75(3):437–446. doi: 10.1124/mol.108.053298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Parinandi NL, Roy S, Shi S, et al. Role of Src kinase in diperoxovanadate-mediated activation of phospholipase D in endothelial cells. Arch Biochem Biophys. 2001;396(2):231–243. doi: 10.1006/abbi.2001.2609. [DOI] [PubMed] [Google Scholar]
- 23.Hagele TJ, Mazerik JN, Gregory A, et al. Mercury activates vascular endothelial cell phospholipase D through thiols and oxidative stress. Int J Toxicol. 2007;26(1):57–69. doi: 10.1080/10915810601120509. [DOI] [PubMed] [Google Scholar]
- 24.Mazerik JN, Hagele T, Sherwani S, et al. Phospholipase A2 activation regulates cytotoxicity of methylmercury in vascular endothelial cells. Int J Toxicol. 2007;26(6):553–569. doi: 10.1080/10915810701707759. [DOI] [PubMed] [Google Scholar]
- 25.Valko M, Morris H, Cronin MT. Metals, toxicity and oxidative stress. Curr Med Chem. 2005;12(10):1161–1208. doi: 10.2174/0929867053764635. [DOI] [PubMed] [Google Scholar]
- 26.Ghio AJ. Disruption of iron homeostasis and lung disease. Biochim Biophys Acta. 2009;1790(7):731–739. doi: 10.1016/j.bbagen.2008.11.004. [DOI] [PubMed] [Google Scholar]
- 27.Catala A. Lipid peroxidation of membrane phospholipids generates hydroxyl-alkenals and oxidized phospholipids active in physiological and/or pathological conditions. Chem Phys Lipids. 2009;157(1):1–11. doi: 10.1016/j.chemphyslip.2008.09.004. [DOI] [PubMed] [Google Scholar]
- 28.Devecha N, Irvine RF. Phospholipid signaling. Cell. 1995;80(2):269–278. doi: 10.1016/0092-8674(95)90409-3. [DOI] [PubMed] [Google Scholar]
- 29.Exton JH. Regulation of phospholipase D. Biochim Biophys Acta. 1999;1439(2):121–133. doi: 10.1016/s1388-1981(99)00089-x. [DOI] [PubMed] [Google Scholar]
- 30.Natarajan V, Garcia JG. Agonist-induced activation of phospholipase D in bovine pulmonary artery endothelial cells: regulation by protein kinase C and calcium. J Lab Clin Med. 1993;121(2):337–347. [PubMed] [Google Scholar]
- 31.Brindley DN, Waggoner DW. Phosphatidate phosphohydrolase and signal transduction. Chem Phys Lipids. 1996;80(1-2):45–57. doi: 10.1016/0009-3084(96)02545-5. [DOI] [PubMed] [Google Scholar]
- 32.Nanjundan M, Possmayer F. Pulmonary phosphatidic acid phosphatase and lipid phosphate phosphohydrolase. Am J Physiol Lung Cell Mol Physiol. 2003;284(1):L1–L23. doi: 10.1152/ajplung.00029.2002. [DOI] [PubMed] [Google Scholar]
- 33.Exton JH. New developments in phospholipase D. J Biol Chem. 1997;272(25):15579–15582. doi: 10.1074/jbc.272.25.15579. [DOI] [PubMed] [Google Scholar]
- 34.Singer WD, Brown HA, Jiang X, Sternweis PC. Regulation of phospholipase D by protein kinase C is synergistic with ADP-ribosylation factor and independent of protein kinase activity. J Biol Chem. 1996;271(8):4504–4510. doi: 10.1074/jbc.271.8.4504. [DOI] [PubMed] [Google Scholar]
- 35.Hammond SM, Altshuller YM, Sung TC, et al. Human ADP-ribosylation factor-activated phosphatidylcholine-specific phospholipase D defines a new and highly conserved gene family. J Biol Chem. 1995;270(50):29640–29643. doi: 10.1074/jbc.270.50.29640. [DOI] [PubMed] [Google Scholar]
- 36.Colley WC, Sung TC, Roll R, et al. Phospholipase D2, distinct phospholipase D isoform with novel regulatory properties that provokes cytoskeletal reorganization. Curr Biol. 1997;7(3):191–201. doi: 10.1016/s0960-9822(97)70090-3. [DOI] [PubMed] [Google Scholar]
- 37.Lopez I, Arnold RS, Lambeth JD. Cloning and initial characterization of a human phospholipase D2 (hPLD2). ADP-ribosylation factor regulates hPLD2. J Biol Chem. 1998;273(21):12846–12852. doi: 10.1074/jbc.273.21.12846. [DOI] [PubMed] [Google Scholar]
- 38.Morris AJ. Regulation of phospholipase D activity, membrane targeting, and intracellular trafficking by phosphoinositides. Biochem Soc Symp. 2007;74:247–257. doi: 10.1042/BSS0740247. [DOI] [PubMed] [Google Scholar]
- 39.Liscovitch M, Chalifa-Caspi V. Enzymology of mammalian phospholipases D: in vitro studies. Chem Phys Lipids. 1996;80(1-2):37–44. doi: 10.1016/0009-3084(96)02544-3. [DOI] [PubMed] [Google Scholar]
- 40.Houle MG, Bourgoin S. Regulation of phospholipase D by phosphorylation dependent mechanisms. Biochim Biophys Acta. 1999;1439(2):135–149. doi: 10.1016/s1388-1981(99)00090-6. [DOI] [PubMed] [Google Scholar]
- 41.Natarajan V, Scribner WM, Al-Hassani M, Vepa S. Reactive oxygen species signaling through regulation of protein tyrosine phosphorylation in endothelial cells. Environ Health Perspect. 1998;106(5):1205–1212. doi: 10.1289/ehp.98106s51205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Natarajan V, Scribner WM, Taher MM. 4-Hydroxynonenal, a metabolite of lipid peroxidation, activates phospholipase D in vascular endothelial cells. Free Radic Biol Med. 1993;15(4):365–375. doi: 10.1016/0891-5849(93)90036-t. [DOI] [PubMed] [Google Scholar]
- 43.Natarajan V, Taher MM, Roehm B, et al. Activation of endothelial cell phospholipase D by hydrogen peroxide and fatty acid hydroperoxide. J Biol Chem. 1993;268(2):930–937. [PubMed] [Google Scholar]
- 44.Kiss Z, Anderson WH. Hydrogen peroxide regulates phospholipase D-mediated hydrolysis of phosphatidylethanolamine and phosphatidylcholine by different mechanisms in NIH 3T3 fibroblasts. Arch Biochem Biophys. 1994;311(2):430–436. doi: 10.1006/abbi.1994.1258. [DOI] [PubMed] [Google Scholar]
- 45.Min DS, Kim EG, Exton JH. Involvement of tyrosine phosphorylation and protein kinase C in the activation of phospholipase D by H2O2 in Swiss 3T3 fibroblasts. J Biol Chem. 1998;273(45):29986–29994. doi: 10.1074/jbc.273.45.29986. [DOI] [PubMed] [Google Scholar]
- 46.Peltz A, Sherwani SI, Kotha SR, et al. Calcium and calmodulin regulate mercury-induced Phospholipase D activation in vascular endothelial cells. Int J Toxicol. 2009;28(3):190–206. doi: 10.1177/1091581809338077. [DOI] [PubMed] [Google Scholar]
- 47.Moeller A, Ask K, Warhurton D, Gauldie J, Kolb M. The bleomycin animal model: a useful tool to investigate treatment option for idiopathic pulmonary fibrosis? Int J Biochem Cell Biol. 2008;40(3):362–382. doi: 10.1016/j.biocel.2007.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Moore BB, Hogaboam CM. Murine models of pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2008;294(2):L152–L160. doi: 10.1152/ajplung.00313.2007. [DOI] [PubMed] [Google Scholar]
- 49.Iyer SS, Ramirez AM, Ritzenthaler JD, et al. Oxidation of extracellular cysteine/cystine redox state in bleomycin-induced lung fibrosis. Am J Physiol Lung Cell Mol Physiol. 2009;296(1):L37–L45. doi: 10.1152/ajplung.90401.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hay J, Shahzeidi S, Laurent G. Mechanisms of bleomycin-induced lung damage. Arch Toxicol. 1991;65(2):81–94. doi: 10.1007/BF02034932. [DOI] [PubMed] [Google Scholar]
- 51.Lim LO, Neims AH. Mitochondrial DNA damage by bleomycin. Biochem Pharmacol. 1987;36(17):2769–2774. doi: 10.1016/0006-2952(87)90263-2. [DOI] [PubMed] [Google Scholar]
- 52.Kasper M, Barth K. Bleomycin and its role in inducing apoptosis and senescence in lung cells-modulating effects of caveolin-1. Curr Cancer Drug Targets. 2009;9(3):341–353. doi: 10.2174/156800909788166501. [DOI] [PubMed] [Google Scholar]
- 53.Mungunsukh O, Griffin AJ, Lee YH, Day RM. Bleomycin induces the extrinsic apoptotic pathway in pulmonary endothelial cells. Am J Physiol Lung Cell Mol Physiol. 2010 doi: 10.1152/ajplung.00322.2009. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sato K, Tashiro Y, Chibana S, Yamashita A, Karakawa T, Kohrogi H. Role of lipid-derived free radical in bleomycin-induced lung injury in mice: availability for ESR spin trap method with organic phase extraction. Biol Pharm Bull. 2008;31(10):1855–1859. doi: 10.1248/bpb.31.1855. [DOI] [PubMed] [Google Scholar]
- 55.Suntres ZE, Shek PN. Protective effect of liposomal alpha-tocopherol against bleomycin-induced lung injury. Biomed Environ Sci. 1997;10(1):47–59. [PubMed] [Google Scholar]
- 56.Sener G, Topalogu N, Sehirli AO, Ercan F, Gedik N. Resveratrol alleviates bleomycin-induced lung injury in rats. Pulm Pharmacol Ther. 2007;20(6):642–649. doi: 10.1016/j.pupt.2006.07.003. [DOI] [PubMed] [Google Scholar]
- 57.Teixeira KC, Soares FS, Rocha LG, et al. Attenuation of bleomycin-induced lung injury and oxidative stress by N-acetylcysteine plus deferoxamine. Pulm Pharmacol Ther. 2008;21(2):309–316. doi: 10.1016/j.pupt.2007.07.006. [DOI] [PubMed] [Google Scholar]
- 58.Aoki J, Inoue A, Okudaira S. Two pathways for lysophosphatidic acid production. Biochim Biophys Acta. 2008;1781(9):513–518. doi: 10.1016/j.bbalip.2008.06.005. [DOI] [PubMed] [Google Scholar]
- 59.Aoki J. Mechanisms of lysophosphatidic acid production. Semin Cell Dev Biol. 2004;15(5):477–489. doi: 10.1016/j.semcdb.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 60.Hama K, Aoki J. LPA(3), a unique G protein-coupled receptor for lysophosphatidic acid. Prog Lipid Res. 2010 doi: 10.1016/j.plipres.2010.03.001. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 61.Panchatcharam M, Miriyala S, Yang F, et al. Lysophosphatidic acid receptors 1 and 2 play roles in regulation of vascular injury responses but not blood pressure. Circ Res. 2008;103(6):662–670. doi: 10.1161/CIRCRESAHA.108.180778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tager AM, LaCamera P, Shea SB, et al. The lysophosphatidic acid receptor LPA1 links pulmonary fibrosis to leak injury by mediating fibroblast recruitment and vascular leak. Nat Med. 2008;14(1):45–54. doi: 10.1038/nm1685. [DOI] [PubMed] [Google Scholar]