

Abstract
Cortical spreading depression (CSD) is a key pathogenetic step in migraine with aura. Dysfunctions of voltage-dependent and receptor-operated channels have been implicated in the generation of CSD and in the pathophysiology of migraine. Although a known correlation exists between migraine and release of the calcitonin gene-related peptide (CGRP), the possibility that CGRP is involved in CSD has not been examined in detail. We analyzed the pharmacological mechanisms underlying CSD and investigated the possibility that endogenous CGRP contributes to this phenomenon. CSD was analyzed in rat neocortical slices by imaging of the intrinsic optical signal. CSD was measured as the percentage of the maximal surface of a cortical slice covered by the propagation of intrinsic optical signal changes during an induction episode. Reproducible CSD episodes were induced through repetitive elevations of extracellular potassium concentration. AMPA glutamate receptor antagonism did not inhibit CSD, whereas NMDA receptor antagonism did inhibit CSD. Blockade of voltage-dependent sodium channels by TTX also reduced CSD. CSD was also decreased by the antiepileptic drug topiramate, but not by carbamazepine. Interestingly, endogenous CGRP was released in the cortical tissue in a calcium-dependent manner during CSD, and three different CGRP receptor antagonists had a dose-dependent inhibitory effect on CSD, suggesting a critical role of CGRP in this phenomenon. Our findings show that both glutamate NMDA receptors and voltage-dependent sodium channels play roles in CSD. They also demonstrate that CGRP antagonism reduces CSD, supporting the possible use of drugs targeting central CGRP receptors as antimigraine agents.
Keywords: cortex, epilepsy, headache, pain
Migraine is a common episodic headache disorder characterized by attacks that include various combinations of headache and neurologic, gastrointestinal, and autonomic symptoms. It is among the most common neurologic conditions, affecting ∼12–20% of the population. The International Headache Society classifies migraine into migraine without aura and migraine with aura, with aura defined as the complex of focal neurologic symptoms that most often precedes or accompanies an attack (1–3).
Cortical spreading depression (CSD) is thought to represent a key pathogenetic step in migraine with aura (4, 5). First reported by Leão in 1944 (6), CSD is a wave of electrical activity (excitation followed by depression) that moves across the cerebral cortex after electrical or chemical stimulation. The pivotal event in the generation and propagation of CSD is a marked decrease in neuronal membrane resistance associated with massive increases in extracellular K+ and neurotransmitters, as well as increases in intracellular Na+ and Ca2+ (7–9). Genetic and environmental factors modulate individual susceptibility by lowering the CSD threshold, and cortical excitation can cause sufficient elevation in extracellular K+ and glutamate to initiate CSD (5, 7, 8, 10–14). Conversely, the CSD threshold also may be increased by factors that reduce the susceptibility to migraine, such as postmenopausal age and male gonadal hormones (15, 16).
Although knowledge of the main molecular players in migraine pathophysiology remains incomplete, recent preclinical and clinical findings indicate a clear correlation between migraine and the release of the calcitonin gene-related peptide (CGRP) (17–19). Moreover, the possibility that CGRP antagonists might offer advantages over existing symptomatic therapies (e.g., nonsteroidal anti-inflammatory drugs, ergot derivatives, triptans) for patients suffering from migraine has been investigated recently (20, 21). Relief of migraine corresponds to a reduction of blood CGRP, and thus migraine pharmacotherapy is aimed at decreasing CGRP. A CGRP receptor (CGRP-R) antagonist can block vasodilatation and prevent vasoconstriction in the meninges, as well as alter CGRP action in the trigeminal ganglion and reduce pain transmission (19). Although modulation of CSD could represent a possible central mechanism of action of CGRP antagonists in migraine (19), the possible effects of these drugs on CSD have not been addressed experimentally.
In this study, using imaging of intrinsic optical signals (IOS) in rat neocortical slices, we analyzed the physiological and pharmacological features of CSD triggered by increasing extracellular K+ ion concentration. We also explored the possibility that antagonism of CGRP-Rs could reduce CSD induction and propagation.
Results
High Potassium Concentration Evokes Reproducible CSD in Rat Brain Slices.
A single rat neocortical slice was placed in the recording chamber in continuously flowing Krebs solution and visualized with an upright microscope equipped with a 2× objective (Fig. 1A). Reproducible CSD episodes were induced by repetitively elevating the extracellular K+ concentration through 2- to 3-min bath applications of 26 mM KCl solution (9, 10). These transient increases in K+ concentration induced increases in IOS that began within cortical layers II and III, expanded radially, and then propagated along all cortical layers (Fig. 1B).
Fig. 1.

Reproducible effects of high potassium-induced CSD. (A) Representation of the acquisition system for CSD analysis. The IOS emitted by the slice are acquired by a CCD camera and transferred to a personal computer. A KCl application delivered by the perfusion system can trigger a CSD episode that originates near the KCl input. (B) A neocortical slice showing the effect of KCl application on IOS responses. (C and D) Acquisition of cortical slice images before (Pre) and after KCl application at different time points illustrating CSD propagation after a first induction (first episode) (C), as well as after a second and then a third KCl application (second and third episodes) (D). Note that multiple KCl applications in the same slice can induce reproducible CSD episodes. (E) Time course of three consecutive KCl applications showing reproducible IOS changes measured in one of three different ROIs (ROI 2) in the same slice presented in C. (F) Histogram revealing no differences across the parameters chosen to measure IOS—early peak, delayed phase, and slice area—after three consecutive KCl applications. (G) Time course of reproducible effects on cortical field potential (FP) amplitude during three episodes of CSD induced by repetitive KCl applications. (Upper) Representative traces showing a cortical field potential evoked under control conditions (a), during a CSD episode (b), and immediately after the return of field potential amplitude to baseline level (c).
CSD was analyzed by measuring IOS changes in control neocortical slices. A CSD episode induced in the control condition propagated normally along the slice (Fig. 1 B–D). The time course of IOS during a CDS episode showed a biphasic transient increase consisting of an early peak (amplitude 65.3 ± 3.9%) followed by a delayed, long-lasting phase (amplitude 79.3 ± 6.5%; six slices) (Fig. 1E). IOS changes terminated within 10 min of the onset of KCl application. Two additional applications of 26 mM KCl induced two successive CSD episodes of similar amplitude (six slices from three rats; P > 0.05) (Fig. 1 C–F). In most cases, at least 10–15 min between applications was required to induce CSD episodes with similar features.
CSD and Cortical Field Potentials.
Cortico-cortical field potentials were recorded from frontal cortical slices (six slices from three rats). Field potentials measured 1.7 ± 0.24 mV in amplitude. After a stable baseline was achieved (10 min), 26 mM KCl was bath-applied for 2–3 min to induce a CSD episode. CSD strongly affected field potentials; in fact, field potentials were completely abolished within 5 min of KCl application, and returned to baseline values approximately 8 min after KCl washout (Fig. 1G). After an additional 10 min, a second CSD episode and then a third CSD episode were able to induce reproducible effects on field potential amplitude (Fig. 1G).
Effect of Ionotropic Glutamate Receptor Antagonists on CSD.
Propagation of CSD is accompanied by the release and diffusion of several chemical mediators, including excitatory amino acids, and the varying manifestations of CSD indicate that different receptors may play important roles in this phenomenon (22). NMDA glutamate receptor activation could in fact contribute to CSD initiation and propagation (23).
A group of 10 slices (from five rats) was exposed to KCl for 2–3 min to induce CSD. After a first CSD episode, followed by a 10-min recovery period, 50 μM NMDA glutamate receptor antagonist l-2-amino-5-phosphonovaleric acid (l-APV) was bath-applied for 10 min, then coapplied with KCl (Fig. 2 A and B). In this condition, l-APV reduced the CSD area to 32.3 ± 7.2% (P < 0.001) and reduced the relative IOS intensity to 18.7 ± 7.3% for the early peak (P < 0.001) and 67.5 ± 8.5% for the delayed phase (P < 0.05) (Fig. 2 B and D).
Fig. 2.

Effects of NMDA and AMPA glutamate receptor antagonists, TTX, and AEDs on CSD. (A) Schematic representation of two CSD episodes propagating on a cortical slice before (first episode; blue region) and after drug administration (second episode; red region). (B) (Upper) Images of a cortical slice showing CSD propagation before KCl application (Left), in a predrug condition (Center), and after incubation with 50 μM of the NMDA receptor antagonist l-APV (Right). (Lower) Time course graphs of IOS changes in three different ROIs during CSD, in the predrug condition, and after incubation with l-APV. (C) Time course graphs of IOS changes in three different ROIs during CSD, in the predrug condition, and after incubation with 10 μM of the AMPA receptor antagonist CNQX. (D) Histogram illustrating the effects of l-APV and CNQX on IOS early peak, delayed phase, and slice area measured during KCl-induced CSD. (E–G) Time course graphs of IOS changes in three different ROIs during KCl-induced CSD propagation in the predrug condition and after incubation of the slice with 1 μM TTX (E), 30 μM CBZ (F), and 100 μM TPM (G). (H) Histogram illustrating the effects of TTX, CBZ, and TPM on IOS early peak, delayed phase, and slice area measured during KCl-induced CSD. *P < 0.05; ***P < 0.001, Student t test.
Conversely, pretreatment with the AMPA glutamate receptor antagonist 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX; 10 μM) failed to affect CSD in six slices (from three rats). In fact, the CSD area and the relative IOS intensities (both early peak and delayed phase) remained unchanged (P > 0.05) (Fig. 2 C and D). Thus, AMPA receptor antagonism does not inhibit CSD, whereas NMDA receptor antagonism significantly reduces both IOS area and intensity.
TTX-Sensitive Na+ Channel Blockade Affects CSD.
Sodium ion channels may be involved in the initiation of CSD (9). Consequently, in a subset of experiments performed on eight cortical slices (from four rats), CSD was induced first in control conditions and then in the presence of the voltage-sensitive Na+ channel blocker TTX (Fig. 2 E and H). In the presence of 1 μM TTX, CSD area was reduced to 1.9 ± 2.5% and IOS intensity was reduced to 2.9 ± 0.5% (early peak) and 19.2 ± 4.7% (delayed phase) (Fig. 2 E and H). In some experiments, a third episode of CSD was induced at an additional 15 min after drug washout. This third episode produced a CSD similar to the control response. These experiments show that propagation of membrane potential changes through voltage-dependent Na+ channels, as in the case of action potentials, is involved in the spreading CSD, in line with previous studies using tissue slices from the hippocampus (9).
CSD Is Reduced by Topiramate, but Not by Carbamazepine.
Antiepileptic drugs (AEDs) have been suggested to be useful in migraine with aura, and controlled trials have shown that some AEDs are effective in migraine prevention, whereas others have limited efficacy. Given these drugs’ multiple modes of action, it is of critical importance to establish whether the efficacy of various AEDs in migraine prophylaxis depends on their capability to block CSD, as has been suggested by previous in vivo experimental studies (7, 12).
To address this issue, we analyzed CSD in control slices and in slices treated with the AEDs carbamazepine (CBZ; 30 μM) and topiramate (TPM; 100 μM). Although CSD area was not significantly modified in the presence of CBZ (six slices from three rats), being reduced only to 74.7 ± 11.4% (IOS: early peak, 86.7 ± 15.7%; delayed phase, 82.0 ± 6.7%; P > 0.05) (Fig. 2 F and H), it was significantly affected in the presence of TPM (eight slices from four rats), decreasing to 47.3 ± 9.8% (IOS: early peak, 29.4 ± 6.3%; delayed phase, 49.4 ± 8.1%; P < 0.001) (Fig. 2 G and H). These findings support the hypothesis that CSD is reduced only by AEDs effective in migraine prevention, such as TPM, and not by those that do not prevent migraine attacks, such as CBZ (24).
CGRP Receptor Antagonism Reduces CSD.
The possibility of raising the CSD threshold to prevent migraine aura and related pain provides a rationale for testing drugs that might be clinically tolerable at prophylactic doses. Activation of CGRP-Rs is a critical step in migraine attacks, and CSD has been implicated as a possible phenomenon linked to this condition. Consequently, we investigated the role of endogenous CGRP in mediating CSD in neocortical slices. It was recently suggested that CGRP-R antagonists could be effective in treating migraine (19). To address this issue, we used three selective CGRP-R antagonists: MK-8825, a new and selective CGRP antagonist with a good oral bioavailability in rodents (25); BIBN4096BS (olcegepant), a high-affinity antagonist with established clinical efficacy in migraine (26-28); and CGRP 8-37, the first peptidic CGRP fragment used as a CGRP-R antagonist (29, 30). We first induced a CSD episode in the control condition by bath application of 26 mM KCl. We then induced a second CSD episode in the same slice after bath-application of MK-8825 in the external medium for 10 min. The effect of MK-8825 (0.1–10 μM) was analyzed in 22 neocortical slices obtained from 12 rats. In nine of these slices, MK-8825 (10 μM) significantly reduced the extent of maximal IOS area, to 46.1 ± 8.6% (P < 0.01), and IOS intensity, to 7.3 ± 1.2% for the early peak and 26.8 ± 9.3% for the delayed phase compared with the control condition (P < 0.001) (Fig. 3 A, C, and D). In the same experimental paradigm, in 14 neocortical slices from four rats, BIBN4096BS (0.01–1 μM) significantly reduced maximal IOS area to 25.8 ± 5.5% and IOS intensity to 25.2 ± 5.9% for the early peak and 40.5 ± 7.2% for the delayed phase compared with control (P < 0.001) (Fig. 3 B–D). Both MK-8825 and BIBN4096BS reduced CSD in a dose-dependent manner, with an IC50 of 1.8 μM and 0.07 μM, respectively (Fig. 3C). We also measured IOS maximal area in control slices and CGRP 8-37–treated slices (six slices per group from four rats). CGRP 8-37 treatment significantly reduced CSD area from 19.7 ± 1.5 mm2 in control slices to 11.8 ± 1.6 mm2 (P < 0.05) in slices incubated with 3 μM CGRP 8-37 and 3.4 ± 1 mm2 (P < 0.001) in slices incubated with 10 μM CGRP 8-37 (Fig. 4A).
Fig. 3.

CGRP-R antagonism reduces CSD. (A) (Upper) Images of a cortical slice before CSD induction (Left), in the predrug condition (Center), and after incubation with 10 μM CGRP-R antagonist MK-8825 (Right). (Lower) Time course graphs of KCl-induced changes in IOS in the predrug condition and after incubation with MK-8825. (B) Time course graphs of KCl-induced changes in IOS in the predrug condition and after incubation with 0.1 μM BIBN4096BS. (C) Dose–response curves of the effects of MK-8825 (Left) and BIBN4096BS (Right) on CSD area. (D) Histogram comparing IOS changes in the presence of 10 μM MK-8825 or 0.1 μM BIBN4096BS. **P < 0.01; ***P < 0.001, Student t test.
Fig. 4.
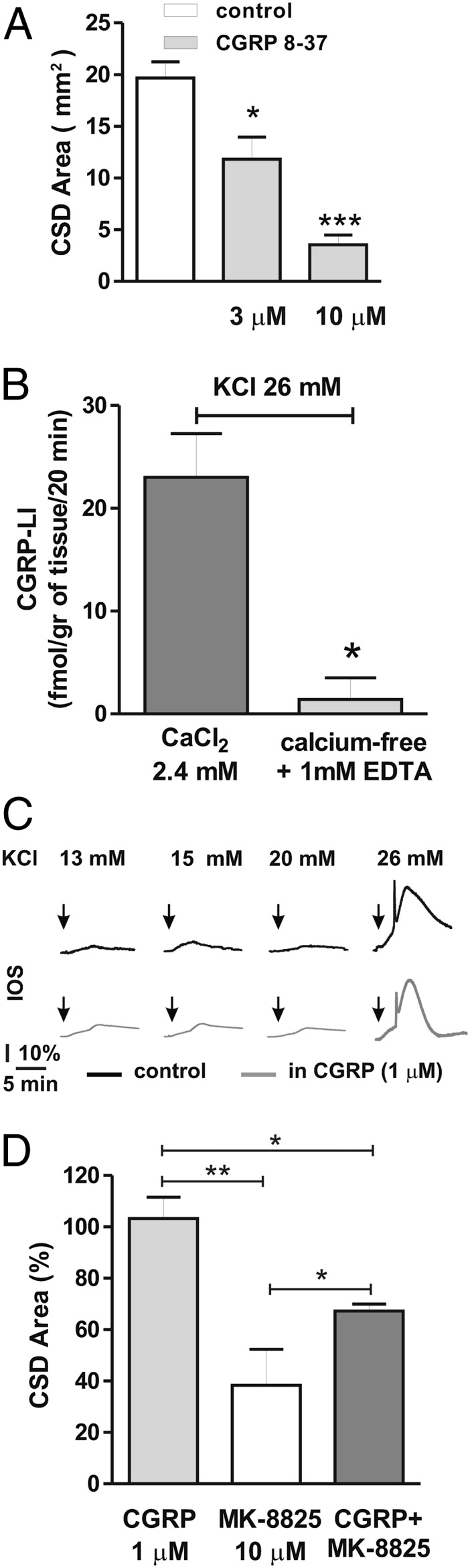
Characterization of the roles of endogenous and exogenous CGRP in CSD. (A) Histogram showing IOS area during a CSD episode in control slices (white bar) and in slices incubated in the presence of 3 μM and 10 μM CGRP-R antagonist CGRP 8-37 (gray bars). (B) The increase in CGRP-LI outflow from rat cortical slices evoked by 26 mM KCl under control conditions (gray bar) was absent in experiments performed in a calcium-free medium (light-gray bar). (C) Time course of IOS measurements in control slices (black traces) and in slices incubated with 1 μM CGRP (gray traces) in the presence of increasing extracellular K+ concentrations (13–26 mM KCl). Note that CSD is induced in the presence of 26 mM KCl, but not at lower KCl concentrations both in control conditions and in the presence of 1 μM CGRP. (D) Histogram showing the area of slices covered by IOS changes during CSD episodes in the presence of 1 μM CGRP (light-gray bar), 10 μM MK-8825 (white bar), and CGRP plus MK-8825 (gray bar). *P < 0.05; **P < 0.01, Student t test.
Roles of Endogenous and Exogenous CGRP in CSD.
To explore whether endogenous CGRP is released from cortical tissue during CSD, we measured the outflow of CGRP from rat cortical slices after KCl application (Materials and Methods). Exposure to 26 mM KCl evoked an increase in CGRP outflow from rat cortical slices (Fig. 4B), an effect that was almost fully abolished by extracellular calcium removal, indicating a calcium-dependent neurosecretory process underlying the CGRP release (P < 0.05).
We next tested whether exogenous CGRP (1 μM) exacerbates CSD. CGRP did not increase CSD area measured in slices treated with the vehicle (six slices from three rats) (P > 0.05; Fig. 4 C and D). The failure of CGRP per se to evoke CSD could be related to the fact that CGRP-R might be saturated by endogenous CGRP released from cortical slices during CSD. To uncover a possible CGRP-induced amplification in this phenomenon, we also used lower KCl concentrations to induce submaximal CSD; however, these lower KCl concentrations failed to induce CSD, even in the presence of exogenous CGRP (Fig. 4C). Finally, we analyzed whether the dose of CGRP, at a concentration (1 μM) unable to affect the amplitude of CSD triggered by 26 mM KCl, could reverse the inhibitory effects of MK-8825. As expected for a competitive antagonist, exogenous CGRP partially reverted the inhibition evoked by MK-8825 (six slices; P < 0.05) (Fig. 4D).
Discussion
Various pathological conditions can trigger CSD, including trauma, ischemia, and seizures (9, 31). CSD also can be experimentally induced by exposure to high concentrations of excitatory amino acids or K+, direct electrical stimulation, inhibition of Na+/K+-ATPase, and energy failure (9, 23, 31). The depolarization of a minimum critical mass of brain tissue and the presence of a critical level of extracellular K+ are necessary for this induction (9).
A link between CSD and migraine pathogenesis was hypothesized several decades ago. Clinical studies using neurophysiological approaches and imaging techniques have established a link between migraine aura and CSD. Auras most often anticipate headache onset, and experimental data indicate that CSD activates the trigeminovascular system, possibly provoking headache (5, 10, 12).
In the present study, CSD was analyzed in rat neocortical slices by imaging the IOS. CSD was evaluated as percentage of the maximal surface of a cortical slice covered by the propagation of IOS changes during an induction episode. Reproducible CSD episodes were induced by repetitively elevating the extracellular K+ concentration. This in vitro approach allowed precise analysis of the physiological and pharmacological mechanisms modulating this event. In particular, we found that CSD is coupled with a complete and reversible suppression of intracortical synaptic transmission, as reflected by the transient loss of field potential amplitude.
In line with previous in vitro (10, 22, 32) and in vivo (14) studies, we found that activation of the NMDA glutamate receptor is required for CSD, as demonstrated by the inhibition of CSD by the NMDA receptor antagonist l-APV. Conversely, CSD is not affected by blockade of the AMPA glutamate receptor. The possible affect of AMPA receptors in the induction and propagation of CSD remains a matter of debate, with some studies reporting that CSD is modulated by AMPA receptors (8) and others not observing this modulation (10). Experimental differences, such as in vitro versus in vivo approaches, might account for this discrepancy.
Our experiments using TTX clearly show that activation of voltage-dependent Na+ channels is involved in CSD, in line with previous studies using tissue slices from the hippocampus (9) and in vivo preparations (33). Interestingly, we also found that CSD was prevented by TPM, but not by CBZ, suggesting that only AEDs effective in migraine prophylaxis are able to prevent CSD. In line with our findings, previous in vivo studies have reported blockage of CSD by TPM and valproic acid, AEDs active in migraine prevention (7, 12), but not by oxcarbazepine (34), a drug that, like CBZ, is not effective in migraine prophylaxis (24).
Inhibition of CSD by the blockade of brain CGRP-Rs using a well-defined in vitro model of migraine is a significant finding of the present study. Previous experimental data support the central effects of CGRP antagonists, such as telcagepant (MK-0974), in migraine treatment, and central blockade of CGRP seems to be necessary for a clinical effect (19). In particular, we found that three chemically unrelated and selective CGRP-R antagonists—MK-8825 (25), BIBN4096BS (olcegepant) (26-28), and CGRP 8-37 (29, 30)—all inhibited CSD, strengthening the hypothesis that CGRP-R activation contributes to CSD.
Although the modulation of CSD represents a possible central mechanism of action of CGRP antagonists in migraine, the possible effects of these drugs on CSD have been little explored to date. In vivo animal models have shown that CGRP is released by dilatation of cortical arterioles during CSD (35, 36). In the present study, we have demonstrated a calcium-dependent CGRP release in cortical slices during the application of extracellular K+ at elevated concentrations able to trigger CSD. The calcium dependency of CGRP release from cortical slices during CSD supports the hypothesis that this phenomenon is a neurosecretory process occurring within the brain.
Although CGRP was per se unable to potentiate CSD triggered by low KCl concentrations, exogenous CGRP partially reduced the inhibitory effect of MK-8825, as expected for a competitive antagonist. Our findings in the present study, together with the known distribution of CGRP-Rs in discrete areas of the cerebral cortex (37), further support the hypothesis that CGRP antagonists could act at a central level as well as at the classical vascular sites to inhibit CSD.
In conclusion, CSD inhibition can be achieved through treatments that block CGRP-Rs, by drugs that act on NMDA-mediated transmission and TTX-sensitive Na+ channels, and by AEDs such as TPM. Thus, our model might provide a useful tool for studying the mechanisms underlying migraine and for developing preventive drugs.
Materials and Methods
Preparation and Maintenance of Neocortical Slices.
Wistar male rats (Harlan), 3–4 wk of age, were used for the experiments. All experiments were conducted in accordance with the European Communities Council Directive (86/609/EEC) and with a protocol approved by the University of Perugia’s Animal Care and Use Committee. Every effort was made to minimize animal suffering. Rats were killed under deep halothane anesthesia by cervical dislocation. The brain was promptly removed, and coronal slices (300 µm thick) were cut in Krebs solution (126 mM NaCl, 2.5 mM KCl, 1.3 mM MgCl2, 1.2 mM NaH2PO4, 2.4 mM CaCl2, 10 mM glucose, 18 mM NaHCO3) from the frontal region of the neocortex using a vibratome (38). The slices were maintained in Krebs solution, bubbled with a 95% O2–5% CO2 (vol/vol) gas mixture at room temperature. For treatment with CGRP and CGRP 8-37, slices were incubated in the presence of the drug for at least 2 h before the start of the experiment. Recordings of slices incubated with CGRP or CGRP 8-37 were interspersed with control experiments using untreated slices.
Imaging of IOS in Neocortical Slices.
IOS are produced by changes in light transmittance from cortical slices (10). Optical imaging of IOS maps the brain by measuring intrinsic activity-related changes in tissue reflectance. Functional physiological changes result in intrinsic tissue reflectance changes that are exploited to map functional brain activity. Optical imaging has been selected to study CSD because this technique offers good spatial and temporal resolution simultaneously, making it ideal for studying pathophysiological events occurring at the cortical level. In particular, IOS in the submerged brain slice preparation provides insight into brain activity if it involves significant water movement between intracellular and extracellular compartments (39).
A cortical slice was transferred to the recording chamber submerged in Krebs solution, bubbled with a 95% O2–5% CO2 gas mixture flowing at 2.8–3 mL/min, at room temperature. Transillumination of the slice was provided by a halogen lamp filtered for IR light on a BX51WI microscope equipped with a 2× objective and a 0.35× lens (Olympus). Normoxic CSD was evoked by increasing the extracellular K+ concentration with 26 mM bath-applied KCl. Images were acquired at 2–6 Hz with a CCD camera, digitized, and stored on a personal computer (Fig. 1A). To evaluate changes in IOS over time, the first image in a series (IOSt0) was subtracted from each subsequent image of the same sequence (IOSt), and IOS changes were calculated by (IOSt − IOSt0)/ IOSt0 × 100. During offline analysis, three regions of interest (ROIs) were selected to quantify IOS changes, expressed as percentage of control values (Fig. 1 B and C). Drug application was done by dissolving the drug to the desired final concentration in Krebs solution and then switching from the standard control solution to the drug-containing solution.
Electrophysiology.
The recording electrodes for extracellular field potential measurements were borosilicate glass capillaries (GC150F-10; Harvard Apparatus) filled with 2 M NaCl (resistance, 10–15 MΩ). Under visual control, a stimulating bipolar electrode was positioned 0.5 ± 3 mm distant from the recording electrode in the direction of deep cortical layers (38). Field potential amplitude was defined as the average of the amplitude from the peak of early positivity to the peak of negativity and the amplitude from the peak of negativity to the peak of late positivity. Testing stimuli of 0.1 Hz evoked field potentials that were filtered at 3 KHz, digitized at 10 KHz, and stored on a personal computer. An Axoclamp 2B amplifier (Molecular Devices) was used for extracellular recordings. The onset of each experiment was established as the point at which field potentials of stable amplitude were recorded for 10 min.
CGRP-Like Immunoreactivity from Rat Cortex.
Cortical rat slices (∼100 mg) were placed in 1-mL chambers, superfused at 0.4 mL/min with oxygenated (95% O2, 5% CO2) Krebs solution containing 0.1% BSA, 1 µM phosphoramidon, and 1 µM captopril to prevent peptide degradation, and maintained at 37 °C. In some experiments, slices were perfused with a nominally calcium-free medium containing 1 mM EDTA. At the end of the experiment, tissues were blotted and weighed. Two prestimuli samples were obtained at 10-min intervals, followed by a third set of samples during stimulation with 26 mM KCl. A final sample was also collected at 10 min poststimulus. Fractions were freeze-dried, reconstituted with assay buffer, and analyzed by enzyme immunoassays for CGRP-like immunoreactivity (CGRP-LI) (SPI-Bio) according to the manufacturer’s instructions (40). The detection limit was 2 pg/mL. Increases in CGRP-LI were calculated by subtracting the mean prestimulus value from values obtained during and after stimulation. Results are expressed as fmol of peptide/g of tissue/20 min.
Drugs.
Powders were dissolved in water or DMSO and then stored in aliquots at −20 °C. Drugs to be applied were dissolved to the desired final concentration in external Krebs solution. l-APV, CGRP 8-37, CNQX, and TTX were purchased from Tocris Bioscience. BIBN4096BS was a gift from H. Doods (Boehringer Ingelheim). CGRP and CBZ were obtained from Sigma-Aldrich, TPM was obtained from Johnson & Johnson, and MK-8825 was obtained from Merck Sharp & Dohme.
Data Analysis.
CSD was evaluated in terms of percentage of the maximal surface of the cortical slice covered by the propagation of IOS changes on the slice during an induction episode in the presence of a given drug compared with that in control conditions. Changes in this parameter were considered the main factor in evaluating CSD changes. In some experiments, area values are given in mm2. IOS amplitude was measured at the first early peak and at the subsequent delayed phase of increase, in the presence of a drug and in control conditions. Values are expressed as percentage of IOS amplitude (Figs. 1 and 2).
Data analysis was performed using Clampfit 10 and MetaVue 7 (Molecular Devices), and Prism 5 (GraphPad). Statistical analysis was performed with the Student t test, with values presented as mean ± SE. Significance levels were established at *P < 0.05, **P < 0.01, and ***P < 0.001.
Acknowledgments
We thank C. Spaccatini for excellent technical support. This work was supported by grants from Merck Sharp & Dohme (Contract 36583), the European Community [Contract 222918 (Restorative Plasticity at Corticostriatal Excitatory Synapses) Seventh Framework Programme thematic priority HEALTH (to P.C.)], Progetto di Ricerca di Interesse Nazionale (PRIN) 2008 (to P.C.), and Fondazione Cassa di Risparmio di Perugia (to P.C.).
Footnotes
Conflict of interest statement: P.C. is a member of the editorial boards of Lancet Neurology, Journal of Neuroscience, Movement Disorders, and Synapse and receives research support from Bayer Schering, Biogen, Boehringer Ingelheim, Eisai, Lundbeck, Merck Sharp & Dohme, Novartis, Sanofi-Aventis, Sigma Tau, UCB, Fondazione Santa Lucia, Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS), European Community Grant “Restorative Plasticity At Corticostriatal Excitatory Synapses” (REPLACES), Ministero della Salute, and Agenzia Italiana del Farmaco. P.G. is a member of the editorial boards of Physiological Reviews, Pain and Molecular Pain, and receives research support from Chiesi Farmaceutici, Merck Sharp & Dohme, Italian Institute of Technology, Regione Toscana, Italian Ministry of University and Research, and Ente Cassa di Risparmio di Firenze. All other authors reported no biomedical financial interests or potential conflicts of interest.
This article is a PNAS Direct Submission.
References
- 1.Goadsby PJ. Recent advances in understanding migraine mechanisms, molecules and therapeutics. Trends Mol Med. 2007;13(1):39–44. doi: 10.1016/j.molmed.2006.11.005. [DOI] [PubMed] [Google Scholar]
- 2.Silberstein SD. Practice parameter: Evidence-based guidelines for migraine headache (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2000;55(6):754–762. doi: 10.1212/wnl.55.6.754. [DOI] [PubMed] [Google Scholar]
- 3.Silberstein SD. Treatment recommendations for migraine. Nat Clin Pract Neurol. 2008;4(9):482–489. doi: 10.1038/ncpneuro0861. [DOI] [PubMed] [Google Scholar]
- 4.Ayata C. Cortical spreading depression triggers migraine attack: Pro. Headache. 2010;50(4):725–730. doi: 10.1111/j.1526-4610.2010.01647.x. [DOI] [PubMed] [Google Scholar]
- 5.Moskowitz MA. Pathophysiology of headache—past and present. Headache. 2007;47(Suppl 1):S58–S63. doi: 10.1111/j.1526-4610.2007.00678.x. [DOI] [PubMed] [Google Scholar]
- 6.Leão AAP. Spreading depression of activity in the cerebral cortex. J Neurophysiol. 1944;7:359–390. doi: 10.1152/jn.1947.10.6.409. [DOI] [PubMed] [Google Scholar]
- 7.Akerman S, Goadsby PJ. Topiramate inhibits cortical spreading depression in rat and cat: Impact in migraine aura. Neuroreport. 2005;16(12):1383–1387. doi: 10.1097/01.wnr.0000175250.33159.a9. [DOI] [PubMed] [Google Scholar]
- 8.Holland PR, Akerman S, Goadsby PJ. Cortical spreading depression-associated cerebral blood flow changes induced by mechanical stimulation are modulated by AMPA and GABA receptors. Cephalalgia. 2010;30(5):519–527. doi: 10.1111/j.1468-2982.2009.02021.x. [DOI] [PubMed] [Google Scholar]
- 9.Somjen GG. Mechanisms of spreading depression and hypoxic spreading depression-like depolarization. Physiol Rev. 2001;81(3):1065–1096. doi: 10.1152/physrev.2001.81.3.1065. [DOI] [PubMed] [Google Scholar]
- 10.Anderson TR, Andrew RD. Spreading depression: Imaging and blockade in the rat neocortical brain slice. J Neurophysiol. 2002;88(5):2713–2725. doi: 10.1152/jn.00321.2002. [DOI] [PubMed] [Google Scholar]
- 11.Ayata C. Spreading depression: From serendipity to targeted therapy in migraine prophylaxis. Cephalalgia. 2009;29(10):1095–1114. doi: 10.1111/j.1468-2982.2009.01982.x. [DOI] [PubMed] [Google Scholar]
- 12.Ayata C, Jin H, Kudo C, Dalkara T, Moskowitz MA. Suppression of cortical spreading depression in migraine prophylaxis. Ann Neurol. 2006;59(4):652–661. doi: 10.1002/ana.20778. [DOI] [PubMed] [Google Scholar]
- 13.Eikermann-Haerter K, et al. Enhanced subcortical spreading depression in familial hemiplegic migraine type 1 mutant mice. J Neurosci. 2011;31(15):5755–5763. doi: 10.1523/JNEUROSCI.5346-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Peeters M, et al. Effects of pan- and subtype-selective N-methyl-D-aspartate receptor antagonists on cortical spreading depression in the rat: Therapeutic potential for migraine. J Pharmacol Exp Ther. 2007;321(2):564–572. doi: 10.1124/jpet.106.117101. [DOI] [PubMed] [Google Scholar]
- 15.Eikermann-Haerter K, et al. Androgenic suppression of spreading depression in familial hemiplegic migraine type 1 mutant mice. Ann Neurol. 2009;66(4):564–568. doi: 10.1002/ana.21779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eikermann-Haerter K, et al. Genetic and hormonal factors modulate spreading depression and transient hemiparesis in mouse models of familial hemiplegic migraine type 1. J Clin Invest. 2009;119(1):99–109. doi: 10.1172/JCI36059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brain SD. Calcitonin gene-related peptide (CGRP) antagonists: Blockers of neuronal transmission in migraine. Br J Pharmacol. 2004;142(7):1053–1054. doi: 10.1038/sj.bjp.0705806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ho TW, Edvinsson L, Goadsby PJ. CGRP and its receptors provide new insights into migraine pathophysiology. Nat Rev Neurol. 2010;6(10):573–582. doi: 10.1038/nrneurol.2010.127. [DOI] [PubMed] [Google Scholar]
- 19.Tepper SJ, Stillman MJ. Clinical and preclinical rationale for CGRP-receptor antagonists in the treatment of migraine. Headache. 2008;48(8):1259–1268. doi: 10.1111/j.1526-4610.2008.01214.x. [DOI] [PubMed] [Google Scholar]
- 20.Doods H, Arndt K, Rudolf K, Just S. CGRP antagonists: Unravelling the role of CGRP in migraine. Trends Pharmacol Sci. 2007;28(11):580–587. doi: 10.1016/j.tips.2007.10.005. [DOI] [PubMed] [Google Scholar]
- 21.Hoffmann J, Goadsby PJ. New agents for acute treatment of migraine: CGRP receptor antagonists, iNOS inhibitors. Curr Treat Options Neurol. 2012;14(1):50–59. doi: 10.1007/s11940-011-0155-4. [DOI] [PubMed] [Google Scholar]
- 22.Gorji A, et al. Spreading depression in human neocortical slices. Brain Res. 2001;906(1-2):74–83. doi: 10.1016/s0006-8993(01)02557-4. [DOI] [PubMed] [Google Scholar]
- 23.Smith JM, Bradley DP, James MF, Huang CL. Physiological studies of cortical spreading depression. Biol Rev Camb Philos Soc. 2006;81(4):457–481. doi: 10.1017/S1464793106007081. [DOI] [PubMed] [Google Scholar]
- 24.Calabresi P, Galletti F, Rossi C, Sarchielli P, Cupini LM. Antiepileptic drugs in migraine: From clinical aspects to cellular mechanisms. Trends Pharmacol Sci. 2007;28(4):188–195. doi: 10.1016/j.tips.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 25.Bell IM, et al. MK-8825: A potent and selective CGRP receptor antagonist with good oral activity in rats. Bioorg Med Chem Lett. 2012;22(12):3941–3945. doi: 10.1016/j.bmcl.2012.04.105. [DOI] [PubMed] [Google Scholar]
- 26.Doods H, et al. Pharmacological profile of BIBN4096BS, the first selective small molecule CGRP antagonist. Br J Pharmacol. 2000;129(3):420–423. doi: 10.1038/sj.bjp.0703110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Olesen J, et al. BIBN 4096 BS Clinical Proof of Concept Study Group Calcitonin gene-related peptide receptor antagonist BIBN 4096 BS for the acute treatment of migraine. N Engl J Med. 2004;350(11):1104–1110. doi: 10.1056/NEJMoa030505. [DOI] [PubMed] [Google Scholar]
- 28.Trocóniz IF, Wolters JM, Schaefer HG, Roth W. Population pharmacokinetic modelling of BIBN 4096 BS, the first compound of the new class of calcitonin gene-related peptide receptor antagonists. Eur J Pharm Sci. 2004;22(4):287–295. doi: 10.1016/j.ejps.2004.03.018. [DOI] [PubMed] [Google Scholar]
- 29.Chiba T, et al. Calcitonin gene-related peptide receptor antagonist human CGRP-(8-37) Am J Physiol. 1989;256(2 Pt 1):E331–E335. doi: 10.1152/ajpendo.1989.256.2.E331. [DOI] [PubMed] [Google Scholar]
- 30.Reuter U, et al. Perivascular nerves contribute to cortical spreading depression-associated hyperemia in rats. Am J Physiol. 1998;274(6 Pt 2):H1979–H1987. doi: 10.1152/ajpheart.1998.274.6.H1979. [DOI] [PubMed] [Google Scholar]
- 31.Aitken PG, Tombaugh GC, Turner DA, Somjen GG. Similar propagation of SD and hypoxic SD-like depolarization in rat hippocampus recorded optically and electrically. J Neurophysiol. 1998;80(3):1514–1521. doi: 10.1152/jn.1998.80.3.1514. [DOI] [PubMed] [Google Scholar]
- 32.Peters O, Schipke CG, Hashimoto Y, Kettenmann H. Different mechanisms promote astrocyte Ca2+ waves and spreading depression in the mouse neocortex. J Neurosci. 2003;23(30):9888–9896. doi: 10.1523/JNEUROSCI.23-30-09888.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Akerman S, Holland PR, Goadsby PJ. Mechanically-induced cortical spreading depression associated regional cerebral blood flow changes are blocked by Na+ ion channel blockade. Brain Res. 2008;1229:27–36. doi: 10.1016/j.brainres.2008.07.014. [DOI] [PubMed] [Google Scholar]
- 34.Hoffmann U, Dileköz E, Kudo C, Ayata C. Oxcarbazepine does not suppress cortical spreading depression. Cephalalgia. 2011;31(5):537–542. doi: 10.1177/0333102410388433. [DOI] [PubMed] [Google Scholar]
- 35.Colonna DM, Meng W, Deal DD, Busija DW. Calcitonin gene-related peptide promotes cerebrovascular dilation during cortical spreading depression in rabbits. Am J Physiol. 1994;266(3 Pt 2):H1095–H1102. doi: 10.1152/ajpheart.1994.266.3.H1095. [DOI] [PubMed] [Google Scholar]
- 36.Wahl M, Schilling L, Parsons AA, Kaumann A. Involvement of calcitonin gene-related peptide (CGRP) and nitric oxide (NO) in the pial artery dilatation elicited by cortical spreading depression. Brain Res. 1994;637(1–2):204–210. doi: 10.1016/0006-8993(94)91234-3. [DOI] [PubMed] [Google Scholar]
- 37.Ma W, et al. Localization and modulation of calcitonin gene-related peptide-receptor component protein-immunoreactive cells in the rat central and peripheral nervous systems. Neuroscience. 2003;120(3):677–694. doi: 10.1016/s0306-4522(03)00159-3. [DOI] [PubMed] [Google Scholar]
- 38.Calabresi P, et al. A field potential analysis on the effects of lamotrigine, GP 47779, and felbamate in neocortical slices. Neurology. 1996;47(2):557–562. doi: 10.1212/wnl.47.2.557. [DOI] [PubMed] [Google Scholar]
- 39.Andrew RD, Jarvis CR, Obeidat AS. Potential sources of intrinsic optical signals imaged in live brain slices. Methods. 1999;18(2):185–196. doi: 10.1006/meth.1999.0771. [DOI] [PubMed] [Google Scholar]
- 40.Nassini R, et al. The “headache tree” via umbellulone and TRPA1 activates the trigeminovascular system. Brain. 2012;135(Pt 2):376–390. doi: 10.1093/brain/awr272. [DOI] [PubMed] [Google Scholar]