

Abstract
Chemoreceptor-based signaling is a central mechanism in bacterial signal transduction. Receptors are classified according to the size of their ligand-binding region. The well-studied cluster I proteins have a 100- to 150-residue ligand-binding region that contains a single site for chemoattractant recognition. Cluster II receptors, which contain a 220- to 300-residue ligand-binding region and which are almost as abundant as cluster I receptors, remain largely uncharacterized. Here, we report high-resolution structures of the ligand-binding region of the cluster II McpS chemotaxis receptor (McpS-LBR) of Pseudomonas putida KT2440 in complex with different chemoattractants. The structure of McpS-LBR represents a small-molecule binding domain composed of two modules, each able to bind different signal molecules. Malate and succinate were found to bind to the membrane-proximal module, whereas acetate binds to the membrane-distal module. A structural alignment of the two modules revealed that the ligand-binding sites could be superimposed and that amino acids involved in ligand recognition are conserved in both binding sites. Ligand binding to both modules was shown to trigger chemotactic responses. Further analysis showed that McpS-like receptors were found in different classes of proteobacteria, indicating that this mode of response to different carbon sources may be universally distributed. The physiological relevance of the McpS architecture may lie in its capacity to respond with high sensitivity to the preferred carbon sources malate and succinate and, at the same time, mediate lower sensitivity responses to the less preferred but very abundant carbon source acetate.
Keywords: sensor domain, four-helix bundle
The ability to sense and respond to extracellular signals is of crucial importance for microorganisms. Bacteria have several types of signal-transduction systems that sense environmental signals and trigger a corresponding response. Genome analyses indicate a dominant role for one-component systems, two-component systems, and chemoreceptor-based mechanisms in bacterial signal transduction (1, 2).
Chemoreceptors have been initially described in the context of chemotactic signaling. However, more recent studies reveal that chemoreceptors are also involved in the regulation of different cellular processes, such as the synthesis of second messengers (3) or the control of gene expression during development (4). Typically, chemoreceptors are composed of a ligand-binding region (LBR) and a signaling domain. Signal recognition by the LBR creates a molecular stimulus that is conveyed to the signaling domain, which forms a complex with CheA and CheW. This molecular stimulus modulates CheA autophosphorylation and, subsequently, transphosphorylation toward the response regulator (5).
The enterobacterial chemoreceptors, and in particular Tar and Tsr, have been studied extensively (5, 6). The Tar-LBR forms a four-helix bundle structure (7), and there are two mechanisms by which Tar-LBR recognizes chemoattractants. One mode consists of the direct recognition of chemoattractants like aspartate (7). The other mode is based on the interaction of the chemoattractant with a periplasmic binding protein, followed by the binding of the resulting complex to the Tar-LBR (8, 9). Enterobacterial chemoreceptors contain an LBR composed of around 150 aa (10). However, genome analyses revealed that many chemoreceptors have a larger LBR (10). This study allowed the classification of chemoreceptors according to the size of the LBR into cluster I (120–210 residues) and cluster II (220–300 residues). Interestingly, cluster II receptors account for around 40% of all chemoreceptors (10).
Although very abundant, there is currently very limited information available on the structure, mechanism, and physiological relevance of cluster II receptors. Based on the observation that cluster II LBRs are approximately double the size of that of cluster I proteins, we have proposed that cluster II LBRs may be composed of two structural modules, each of which recognizes a different type of ligand (10). Another recent bioinformatic analysis has revealed that the predominant family of sensor kinases also contains larger LBRs that are composed of two structural modules (11).
To gain insight into the structure and action of cluster II receptors, we studied the McpS chemoreceptor of Pseudomonas putida KT2440, which was previously shown to mediate chemotaxis toward tricarboxylic acid (TCA) cycle intermediates and butyrate (12). The LBR of McpS (McpS-LBR) was predicted to contain two long and four short helices (12), which could not be shown to conform to the existing structural information on small-molecule sensor domains. Recombinant McpS-LBR was found to recognize chemoattractants with affinities from 8 to 300 μM (12).
We report high-resolution structures of McpS-LBR in complex with malate, succinate, and acetate. The protein is composed of two modules that each contain a ligand-binding site. We show that ligand binding to both structural modules causes a chemotactic response. We report on a chemoreceptor with a bimodular LBR containing two sites for the direct recognition of ligands.
Results
Overall Structure of the McpS-LBR.
The secondary structure prediction of McpS-LBR is not compatible with existing structural information on small-molecule binding domains, which prompted the resolution of its 3D structure. Among the chemoattractants identified for McpS, malate and succinate caused the strongest response (12). Therefore, McpS-LBR was crystallized in complex with these ligands, and the structures were solved using single-wavelength anomalous dispersion of selenomethionine derivatives. Both structures were refined to resolutions below 2 Å with crystallographic R values of 20.4 and 20.9% and Rfree values of 25.1 and 26.8% for the malate and succinate structures, respectively. Ramachandran plots of the final models revealed a satisfactory stereochemistry (Table 1). The asymmetric unit contains two molecules related by dyad symmetry, and the final models contain residues 47–282 from molecule A and 48–282 from molecule B.
Table 1.
Properties of the final models
| McpS-LBR + malate | McpS-LBR + succinate | |
| Resolution (Å) | 25.0–1.8 (1.85–1.80) | 20.0–1.9 (1.97–1.90) |
| No. of reflections | 47,278 | 40,558 |
| Rwork/Rfree (%) | 20.9/25.1 | 20.4/27.2 |
| No. of atoms | 4,234 | 4,325 |
| Protein | 3,658 | 3,787 |
| Ligand/ion | 43 | 40 |
| Water | 533 | 498 |
| B factors (Å2) | 27.6 | 37.9 |
| Protein | 27.3 | 37.7 |
| Ligand/ion | 23.0 | 33.6 |
| Water | 29.8 | 40.3 |
| rmsds | ||
| Bond lengths (Å) | 0.006 | 0.007 |
| Bond angles (°) | 0.884 | 0.940 |
| Ramachandran plot favored/outliers (%) | 99.1/0 | 99.4/0 |
| PDB ID code | 2YFA | 2YFB |
The malate and succinate structures are almost identical (rmsd of 0.2 Å) and differ only slightly in the mode of ligand binding as discussed below. The McpS-LBR structure is composed of six helices linked by loops (Fig. 1). Two short helices (α1 and α2) at the membrane-proximal part of the structure are followed by the long helix α3. This segment is followed by another couple of short helices (α4 and α5) and a second long helix, α6, which is predicted to continue as a transmembrane helix. A sequence alignment of the fragment comprising α1-α3 with the fragment of α4-α6 shows 20.5% sequence identity, which is significantly above the sequence identity of two random sequences (SI Appendix, Fig. 1). The structure can, therefore, be understood as a duplication of a structural element made of two short and a long helix.
Fig. 1.

Three-dimensional structure of the LBR of the McpS chemoreceptor. Ribbon representation in which each monomer is colored differently. The helices and loops are numbered. The binding sites for malate, succinate, and acetate are indicated.
The six helices of McpS-LBR arrange into two modules, each forming a four-helix bundle. The membrane proximal module (Fig. 1) is composed of α1, α2, the N-terminal segment of α3, and the C-terminal segment of α6. The membrane distal module is composed of the C-terminal segment of α3, helices α4 and α5, and the N-terminal part of α6. Both modules are joined by the long helices α3 and α6. The modules are positioned 180° with respect to each other. Malate and succinate bind to the membrane proximal module, whereas acetate, present in the crystallization buffer, was present at the membrane distal module (Fig. 1) in both structures. As shown in Fig. 2, the proximal and distal modules can be aligned closely using the DALI algorithm. In this alignment, the malate/succinate-binding pocket (proximal module) coincides with the acetate-binding pocket (distal module). McpS crystallized as a dimer, which is consistent with ultracentrifugation studies that demonstrated the existence of stable dimers in the presence of bound ligand (12). The dimer interface involves interactions between α1 and α4 of both monomers. The dimer interface is established by 11 hydrogen bonds and 3 salt bridges (SI Appendix, Table 1) and is further stabilized by ligand binding as detailed below.
Fig. 2.
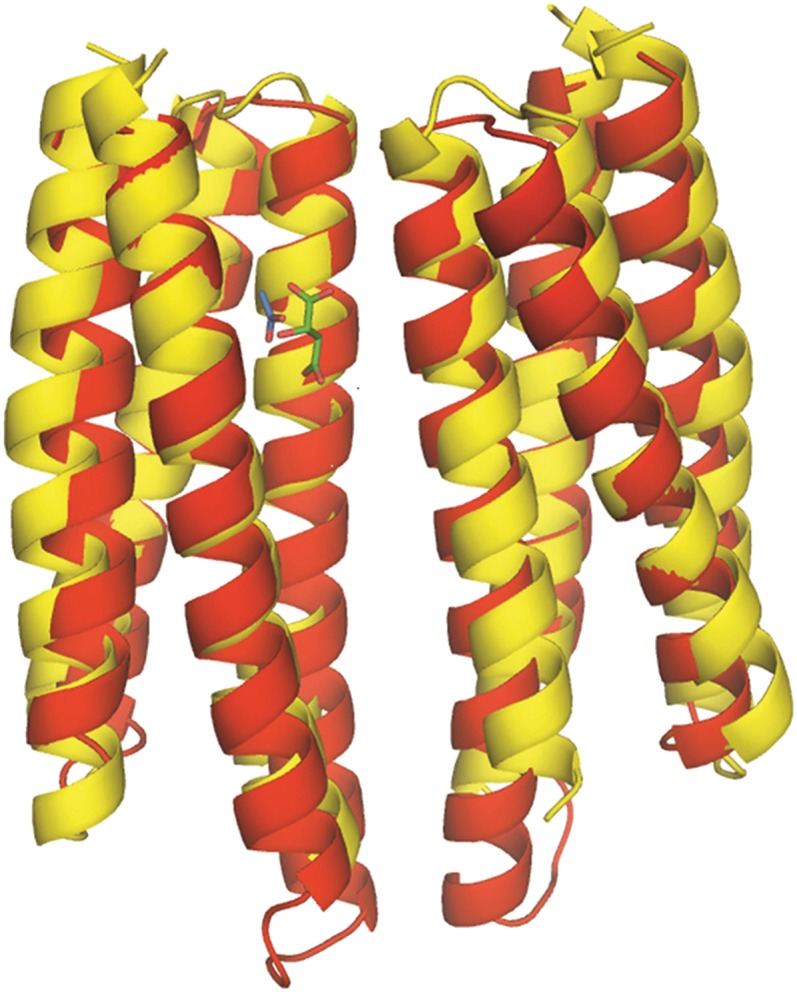
Superimposition of the proximal module with the distal module of McpS-LBR. This alignment was made with the DALI pairwise alignment algorithm. Bound acetate is shown in blue and bound malate in green. Red, distal module; yellow, proximal module.
Ligand-Binding Site at the Proximal Module of McpS-LBR.
McpS-LBR was crystallized in complex with either malate or succinate. Both ligands could be easily identified at the proximal module (Figs. 1 and 3A). Malate binds to a cavity formed by α1 and the C-terminal segment of α6, and its coordination is achieved through interactions with R60, R63, R254, and T258 from one monomer and Q65 from the other monomer (Fig. 3B). Residues R60, R63, and Q65 form part of helix α1, whereas R254 and T258 are part of α6. The five oxygen atoms of malate are involved in eight hydrogen bonds with these five amino acids. The fact that malate is bound by amino acids from both monomers of the dimer provides an explanation for our previous observation that McpS-LBR monomers are unable to bind malate and that malate binding stabilizes the protein dimer (12).
Fig. 3.

Structural basis for ligand recognition at McpS-LBR. (A) Representative section of the final 2Fo−Fc electron density map including data from 25 to 1.8 Å contoured at 2.5 σ above the mean of the map. The region shown corresponds to the malate-binding site with a molecule of malate shown. (B and C) View of the interaction of malate (B) and succinate (C) with the protein. Q65 is from the A monomer, whereas the remaining interactions are with the B monomer. (D) View of the interaction of acetate with McpS-LBR. (E) Superimposition of structures containing malate (green) and succinate (orange).
Succinate binds to McpS-LBR in an almost identical manner as malate (Fig. 3 C and E). Unlike malate, succinate has no hydroxyl group at C2. Therefore, the two hydrogen bonds that are established between the malate hydroxyl group, and the protein cannot be formed in the succinate structure. The remaining interactions of succinate with R60, R63, R254, and Q65 are analogous to the malate structure where they establish six hydrogen bonds. The rmsd between the malate and succinate cocrystal structure is only 0.2 Å indicating that differences in the nature of the ligand do not significantly alter the protein structure. In a sequence alignment of McpS-LBR homologs (SI Appendix, Fig. 2), the amino acids involved in malate and succinate binding are conserved. R60 is almost fully conserved, whereas the remaining residues are conserved to between 30–80%.
To evaluate the contribution of individual amino acids in ligand binding, three site-directed mutants were generated in which R60, R63, or R254 were replaced by alanine. The circular dichroism spectra of the mutant protein could be closely superimposed onto that of the wild-type protein, indicating that the amino acid changes did not significantly alter the secondary structure (SI Appendix, Fig. 3). The mutant proteins were submitted to microcalorimetric titration with each of the seven McpS ligands. Mutation of any of these three amino acids abolished binding of malate, fumarate, oxaloacetate, succinate, and isocitrate (SI Appendix, Table 2). This is exemplified in Fig. 4, which shows titrations of McpS-LBR and the R60A and R254A mutants with malate. The heat changes observed for the titration of mutants correspond to the dilution heats of ligands into buffer, indicating an absence of binding. In contrast, citrate was found to bind to all of these mutants (SI Appendix, Fig. 4) with affinities reduced by a factor of 4.5–7.5 (SI Appendix, Table 2). Among the seven ligands, butyrate is the only monocarboxylic acid, whereas the remaining compounds are all C4-dicarboxylic acids. Butyrate bound to R254A with a similar affinity to the native protein (SI Appendix, Table 2) but failed to bind to the R60A and R63A mutants.
Fig. 4.

Microcalorimetric titrations of native and mutant McpS-LBR with malate. (Upper) Raw titration data of McpS-LBR, McpS-LBR R60A, and McpS-LBR R254A. Protein was at 33 μM, and aliquots of 1 mM malate were injected. (Lower) Integrated, concentration-normalized, and dilution heat-corrected raw data of the titration of McpS-LBR with malate.
Evidence for a Ligand-Binding Site at the Distal Module of McpS-LBR.
Both the malate and succinate structures contain acetate bound to the distal module, which is attributable to the presence of Na acetate in the crystallization buffer. It should be noted that the presence of malate or succinate and acetate was essential for crystallization (13). Acetate is bound at the interface between α6 and α4 in both monomers (Fig. 1). The electron density for acetate in the malate structure is shown in SI Appendix, Fig. 5. R183 and R187 of α4 and Y236 of α6 are involved in establishing five hydrogen bonds with acetate (Fig. 3D). All three amino acids are conserved to a high degree in the alignment of McpS homologs (SI Appendix, Fig. 2); R183 and R187 are conserved in more than 80% of the sequences, whereas Y236 is almost fully conserved. As stated above, the segment comprising α1-α3 can be aligned with that of α4-α6 (SI Appendix, Fig. 1). Interestingly, in this alignment R183 aligns with R60, which is involved in the recognition of malate and succinate (Fig. 3 B and C).
Isothermal titration calorimetry was used to determine the thermodynamic parameters for acetate binding to McpS-LBR. Binding was driven by favorable enthalpy (ΔH = −2.6 kcal/mol) and entropy changes (−TΔS = 1.8 kcal/mol) and was characterized by a KD of 574 μM (SI Appendix, Fig. 6). Because R183 is involved in acetate binding (Fig. 3), the corresponding alanine mutant was constructed and submitted to ITC studies. Initial experiments involved its titration with malate to verify whether this mutation impacts the membrane proximal site. Malate binding to McpS-LBR R183A (SI Appendix, Fig. 7) resulted in an enthalpy change of −20.5 kcal/mol and a KD of 9.1 μM. These values are almost identical to the parameters for malate binding to the native protein (ΔH = −23 kcal/mol; KD = 8.5 μM) (12). The titration of R183A with acetate (SI Appendix, Fig. 6) resulted in a lower affinity (KD of 3.3 mM) and less favorable enthalpy (ΔH = −2 kcal/mol) and entropy changes (−TΔS = 1.3 kcal/mol). The mutation of R183 resulted thus in a decrease of ΔG by 1.04 kcal/mol, causing an approximate sixfold reduction in affinity, which is consistent with its role in ligand binding.
Binding of Acetate to McpS Causes a Chemotactic Response.
Chemoattraction to acetate has been observed for a number of bacteria, including R. sphaeroides (14), P. putida (15), or Methanospirillum (16). Because acetate was bound to McpS, we wanted to establish whether this translates into a chemotactic response. To assess the role of McpS, quantitative capillary chemotaxis assays were carried out using the wild-type strain, the McpS mutant, and the complemented mutant strain. Initial control experiments involved the assessment of chemotaxis of these three strains toward alanine. In P. putida, taxis toward alanine is mediated by the PctA chemoreceptor (17). Assays show that the mutation of McpS and its complementation do not alter taxis toward alanine (Fig. 5A). Experiments using acetate as chemoattractant showed that the wild-type strain shows concentration-dependent taxis, with a clear response detectable at 1 mM (Fig. 5B). A decrease in the tactic response at 100 mM was noted, indicating that prolonged exposure to high acetate concentrations may be toxic. However, the McpS mutant strain was almost devoid of acetate taxis (Fig. 5B), with only a minor response at 10 and 25 mM, which may indicate the existence of a secondary acetate receptor. Complementation of the McpS mutant restored acetate taxis to almost wild-type levels. Subsequently, a series of control experiments were conducted to exclude the possibility that potential acetate-mediated pH changes may be the cause for the taxis observed. To this end, experiments similar to those described in Ingham and Armitage (14) were conducted. Quantitative capillary assays were used to measure taxis of the wild-type strain to acetate at pH values of 6.5, 7.0, and 8.2. The corresponding results were then compared with those obtained for taxis toward buffer solutions at these 3 pH values. As shown in SI Appendix, Fig. 8, the response to acetate at the different pH values was, in all cases, superior to the taxis toward the buffer solutions, which indicates that pH-induced effects may, if at all, have only a marginal contribution to acetate taxis. Because we observed a modest pH taxis, we aimed to establish whether these responses are mediated by McpS. To this end, the chemotaxis assays to buffer at different pH values were repeated with the McpS mutant (SI Appendix, Fig. 9). However, the obtained responses with the mutant were very similar to those of the type strain, indicating that McpS is not involved in pH taxis. Lastly, experiments were conducted to establish whether cells saturated with succinate respond to acetate and, vice versa, whether acetate saturated cells move toward succinate. Before the assays, acetate (final concentration of 50 mM) or succinate (final concentration of 10 mM) was added to the cells. Acetate-containing cells showed no taxis to a 50 mM acetate solution but responded well to a mixture of 50 mM acetate/10 mM succinate (SI Appendix, Fig. 10). Similar results were obtained for the analogous experiment: succinate-containing cells showed no taxis to a 10 mM succinate solution but were attracted by the acetate/succinate mixture (SI Appendix, Fig. 10). Taken together, these data demonstrate that McpS is the primary chemoreceptor for acetate in P. putida KT2440 and, combined with the experiments reported earlier (12), show that ligand binding to the membrane proximal and distal module of McpS-LBR triggers chemotactic responses. These data are consistent with the notion that the responses toward ligands that bind to one module occur irrespective of the presence of ligands that bind to the other module.
Fig. 5.

Quantitative capillary chemotaxis assay of P. putida strains toward alanine (A) and acetate (B). Data are means of at least three independent experiments, each conducted in triplicate. The average number of cells that swam into the capillary has been subtracted.
Discussion
The chemoreceptors studied so far were found to have a single site for the direct recognition of chemoattractants. Here, we report on a chemoreceptor (McpS) that has two distinct sites for the direct recognition of different chemoattractants. Structurally, McpS-LBR contains six helices that arrange into two four-helix bundle modules, each of which contains a ligand-binding site. TCA-cycle intermediates interact with the membrane proximal module and acetate with the membrane distal module. Further evidence for acetate binding comes from microcalorimetric experiments (SI Appendix, Fig. 6) and the demonstration that the mutation of an amino acid in the acetate binding site reduces acetate affinity. The screening of crystallization conditions for McpS-LBR revealed that crystals appear exclusively in the presence of malate or succinate and acetate, suggesting that the occupation of both binding sites causes structural alterations that are essential for protein crystallization. We have reported previously that McpS mediates a chemotactic response toward TCA-cycle intermediates (12). Here, we show that McpS also mediates taxis to acetate and that the mutation of the McpS gene almost abolishes acetate taxis. Taken together, we propose a mechanistic model for McpS in which ligand binding to each of the two modules causes a chemotactic response. Chemotaxis assays of cells saturated with either acetate or succinate indicated that the presence of a ligand that binds to one module does not impede taxis toward ligands that bind to the other module. This suggests that response to ligands that bind to one module occur irrespectively of ligands that bind to the other module.
In this respect, clear parallels exist to the Tar chemoreceptor, for which it was shown that responses to aspartate and the maltose/maltose binding protein complex occur in an additive manner (8). The Tar and McpS receptors share the common element that the signal input occurs at two different sites permitting responses to two different types of ligand. However, the structural and functional bases for this dual response are different because Tar has a single-module LBR recognizing ligands in a direct manner or in complex with a periplasmic binding protein, whereas McpS has a sensor region composed of two modules, which are each able to bind ligands in a direct fashion and to trigger a response. Thus, there appears to be two different strategies that permit chemoreceptors to integrate multiple signal molecules.
The inspection of the structures may provide an initial insight into the mechanism of action of McpS. Signal transmission by chemoreceptors containing a four-helix bundle LBR, such as Tar, have been studied in detail. Ligand binding causes a piston-like shift of around 1 Å of the final helix toward the membrane. This stimulus is then transmitted across the membrane, where it alters the activity of CheA (18, 19). Helix α6 is the terminal helix of McpS-LBR. Interestingly, amino acids that make direct contact with ligands bound to the proximal and distal module are located on this terminal helix (Fig. 6). These amino acids are R254 and T258, which are involved in malate binding, and Y236, that interacts with acetate. In a sequence alignment these residues are highly conserved. It appears likely that ligand recognition at both modules causes a piston-like shift of α6 in a similar manner to that of Tar.
Fig. 6.
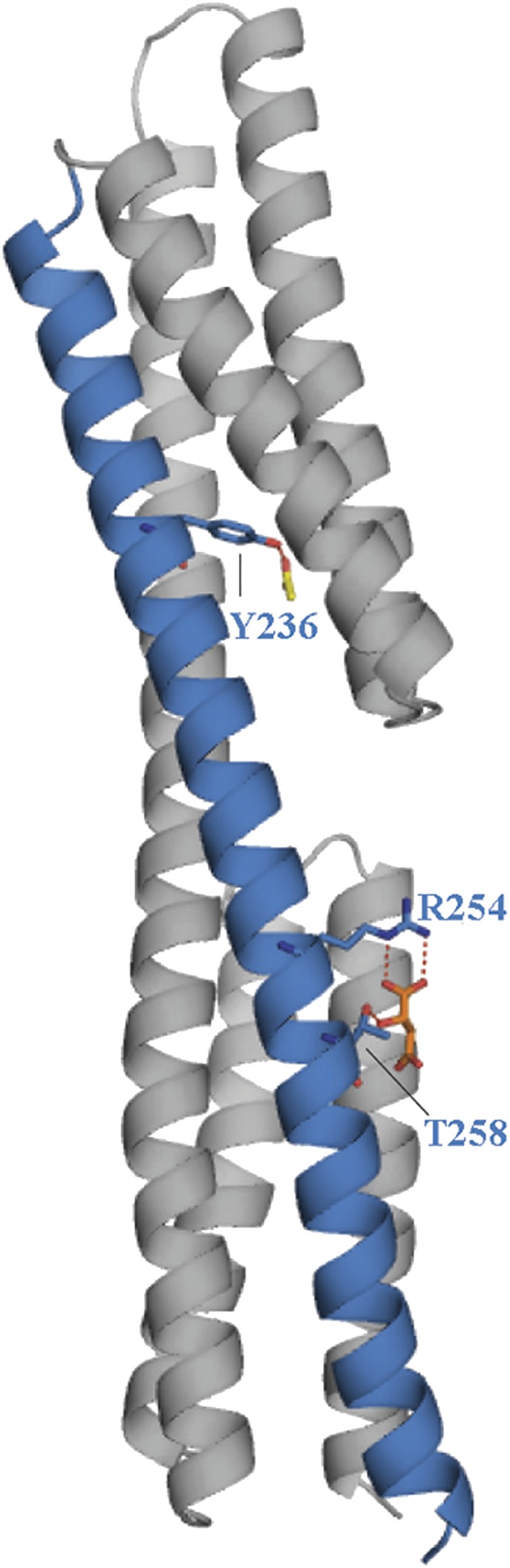
Terminal helix α6 is the proposed signaling helix. Shown is a monomer of McpS-LBR and helix α6, proposed to be the signaling helix (highlighted). Bound malate and acetate are shown in orange and yellow, respectively. R254, T258, and Y236, involved in ligand binding, are shown in stick mode.
What, therefore, is the physiological relevance of the McpS architecture? As reported in more detail in SI Appendix, Table 3, acetate is present in millimolar concentrations in natural habitats of P. putida. In addition, acetate diffuses freely across cell membranes (20) and can, therefore, serve as a carbon and energy source for many bacteria. We have conducted growth experiments of P. putida in a minimal medium supplemented with glucose, succinate, malate, or acetate (SI Appendix, Fig. 11) that show that all four compounds are growth substrates. The results indicated that succinate and malate were optimal growth substrates, followed by glucose; acetate was the least optimal substrate. McpS-LBR binds succinate and malate much tighter than acetate (12) (SI Appendix, Fig. 6), which is also reflected by a higher sensitivity of P. putida for malate and succinate in chemotaxis assays compared with acetate (12) (Fig. 5). The physiological relevance of the McpS architecture may, therefore, lie in its capacity to respond with high sensitivity to the preferred carbon sources malate and succinate while, at the same time, mediate with lower sensitivity the response to the less preferred but very abundant carbon source acetate.
The major classes of bacterial-sensing and signal-transduction proteins share the same types of sensor domains (21). The input into these signal transduction systems is frequently mediated by PAS, GAF, CACHE, TarH, and CHASE domains (21, 22), which all share a common size of 100–150 aa. A DALI search revealed that McpS-LBR shares the same fold with the sensor region of the TorS sensor kinase (23). In contrast to McpS, TorS does not recognize small molecules but interacts with the periplasmic ligand-binding protein TorT (24). This indicates, firstly, that the McpS/TorS fold is found in two different protein superfamilies and, secondly, that this fold has evolved to recognize small-molecule ligands, as well as proteins. It has been suggested that TorS, as well as the double PhoQ/DcuS/CitA (PDC) domains, are the result of an insertion of a domain into another sensor domain (22).
Almost 40% of chemoreceptors belong to cluster II, characterized by a larger LBR of around 250 aa (10). What, therefore, is the physiological relevance of cluster II receptors, and how abundant are McpS homologs within this cluster? Because of the elevated sequence divergence of the McpS LBR, this is a question that cannot be addressed by solely analyzing sequence similarities. We have chosen an alternative approach in which we also take into account information from secondary structure predictions and 3D homology modeling. Fifty sequences of cluster II receptors were selected randomly and the secondary and 3D structure of their respective LBRs were predicted (SI Appendix, Analysis 1). Based on this information 23 proteins could be classified as McpS-like proteins and the remaining 27 as receptors with double PDC-like domains (11) (SI Appendix, Analysis 1). None of the sequences analyzed was incompatible with the secondary structure pattern or 3D structure of any of the two domain types. The 23 McpS-like proteins were present in α, β, and γ proteobacteria, as well as in deinococci, which, of course does not exclude that McpS-like proteins are found in other taxa. This analysis also suggests that the cluster II receptor family is composed of receptors containing either a McpS-like or a double PDC-like ligand-binding domain. Most interestingly, the common feature of double PDC- and McpS-like domains is their bimodular architecture: the McpS-LBR is composed of two four-helix bundles, whereas the double PDC-like domains are composed of two CACHE domain–like α/β modules (11). Here, we show that each module of the McpS-like fold contains a ligand-binding domain and demonstrate that ligand binding causes a response. It appears plausible that chemoreceptors with a double PDC domain operate by a similar mode, but the current model for the function for double PDC domains involves ligand binding at the membrane-distal module, with the membrane-proximal module proposed to be involved in transmitting conformational changes arising from ligand binding to the transmembrane helices (11). It should be noted that receptors with multiple sensor modules are frequently found in eukaryotes, as exemplified by LDL receptors (25) or protein tyrosine kinases (26). The increasing number of genome sequences has shown that cluster II chemoreceptors are almost as abundant as cluster I receptors. This study is an important initial step in closing the gap in knowledge that exists in the field of cluster II receptors.
Materials and Methods
Protein Expression and Purification.
Native and mutant McpS-LBR was purified as described in ref. 12. Selenomethionine-derivatized McpS-LBR was produced as described (27) and purified using the protocol of the native protein.
Crystallization, Data Collection, and Structure Resolution.
Crystallization and data collection were carried out as reported in ref. 13. Based on an initial analysis of the data, the maximum resolution for substructure determination and initial phase calculation was set to 2.5 Å. Fifteen heavy atoms out of the maximum number of 16 were found using SHELXD (28). The correct hand for the substructure was determined using ABS (29) and SHELEX (30). The twofold noncrystallographic symmetry (NCS) operator was determined using RESOLVE (31). Density modification, phase extension, and NCS-averaging were performed using DM (32). The model was built using Coot (33), and refinement was initially done with REFMAC (CCP4) and later with PHENIX (34). Iterative cycles of phase recombination, model building, and refinement led to the final models with satisfactory stereochemistry (Table 1).
Site-Directed Mutagenesis.
Site-directed mutants were prepared in which either R60, R63, or R254 were replaced by alanine. The site-directed mutations were introduced into plasmid pETMcpS using the QuikChange Mutagenesis Kit (Stratagene). Mutant R183A was prepared using the overlapping PCR fragment approach. The oligonucleotides used are listed in SI Appendix, Table 4.
Isothermal Titration Calorimetry.
Measurements were performed on a VP microcalorimeter (MicroCal) at 20 °C. McpS-LBR was dialyzed against 5 mM Tris-HCl, 5 mM Hepes, and 5 mM Mes (pH 6.0). Protein was introduced into the sample cell and titrated with aliquots of ligand solution. The ligand solutions were prepared by dissolving the compounds in dialysis buffer. Thermograms were corrected for dilution heats and concentration-normalized before data analysis.
Chemotaxis Assay.
Chemicals used were of the highest purity available. A modified version of the capillary assay was used (35). Mineral salt medium (MS) supplemented with 10 mM glucose was inoculated with P. putida KT2440R, KT2440RmcpS::Tn5, and KT2440RmcpS::Tn5 (pRK415-mcpS) and grown to early stationary phase and then diluted in MS medium to an OD600 = 0.08. Capillaries (Microcaps; Drummond Scientific) were sealed at one end and warmed over the flame, and the open end was inserted into the chemoattractant solution, of which the pH had been adjusted to that measured for the bacterial suspension. The medium lacking effectors was used as a control. The bacterial suspension was placed into a small chamber formed by placing a V-shaped needle onto a microscope slide. The system was then closed with a glass coverslip. The capillary was immersed into the cell suspension at its open end. After incubation for 10 min (alanine) or 30 min (acetate), the open end of the capillary was rinsed and placed into a microfuge tube containing 1 mL of medium. The sealed end was broken, and the contents were emptied into the tube. A volume of 0.1 mL of the resulting cell suspension was plated out and incubated at 30 °C. Colonies were counted after growth at 30 °C for 48 h.
Supplementary Material
Acknowledgments
We thank C. García-Fontana for technical assistance and C. Fernandez-Tornero for his support during crystallographic data analysis. We thank the BM16 and ID14-4 Grenoble beam-line staff. This study supported by the Banco Bilbao Vizcaya Argentaria (BBVA) Foundation, the Andalusian regional government (Grant P09-RNM-4509), and FEDER- supported grants from the Spanish Ministry for Science and Innovation (MICINN) (Grant BIO2010-16937) and Consolider-Ingenio “The Iberian Metagenome” (MICINN CSD2010-0005). This is a product of the “Factoría Española de Cristalización,” Consolider-Ingenio 2010 project (MICINN). E.P.-M. is supported by a “Ramón y Cajal” contract.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.pdb.org (PDB ID code 2YFA).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1201400109/-/DCSupplemental.
References
- 1.Galperin MY. A census of membrane-bound and intracellular signal transduction proteins in bacteria: Bacterial IQ, extroverts and introverts. BMC Microbiol. 2005;5:35–53. doi: 10.1186/1471-2180-5-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ulrich LE, Koonin EV, Zhulin IB. One-component systems dominate signal transduction in prokaryotes. Trends Microbiol. 2005;13(2):52–56. doi: 10.1016/j.tim.2004.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hickman JW, Tifrea DF, Harwood CS. A chemosensory system that regulates biofilm formation through modulation of cyclic diguanylate levels. Proc Natl Acad Sci USA. 2005;102(40):14422–14427. doi: 10.1073/pnas.0507170102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zusman DR, Scott AE, Yang Z, Kirby JR. Chemosensory pathways, motility and development in Myxococcus xanthus. Nat Rev Microbiol. 2007;5(11):862–872. doi: 10.1038/nrmicro1770. [DOI] [PubMed] [Google Scholar]
- 5.Hazelbauer GL, Falke JJ, Parkinson JS. Bacterial chemoreceptors: High-performance signaling in networked arrays. Trends Biochem Sci. 2008;33(1):9–19. doi: 10.1016/j.tibs.2007.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sourjik V, Armitage JP. Spatial organization in bacterial chemotaxis. EMBO J. 2010;29(16):2724–2733. doi: 10.1038/emboj.2010.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Milburn MV, et al. Three-dimensional structures of the ligand-binding domain of the bacterial aspartate receptor with and without a ligand. Science. 1991;254(5036):1342–1347. doi: 10.1126/science.1660187. [DOI] [PubMed] [Google Scholar]
- 8.Mowbray SL, Koshland DE., Jr Additive and independent responses in a single receptor: Aspartate and maltose stimuli on the tar protein. Cell. 1987;50(2):171–180. doi: 10.1016/0092-8674(87)90213-3. [DOI] [PubMed] [Google Scholar]
- 9.Gardina PJ, Bormans AF, Hawkins MA, Meeker JW, Manson MD. Maltose-binding protein interacts simultaneously and asymmetrically with both subunits of the Tar chemoreceptor. Mol Microbiol. 1997;23(6):1181–1191. doi: 10.1046/j.1365-2958.1997.3001661.x. [DOI] [PubMed] [Google Scholar]
- 10.Lacal J, García-Fontana C, Muñoz-Martínez F, Ramos JL, Krell T. Sensing of environmental signals: Classification of chemoreceptors according to the size of their ligand binding regions. Environ Microbiol. 2010;12(11):2873–2884. doi: 10.1111/j.1462-2920.2010.02325.x. [DOI] [PubMed] [Google Scholar]
- 11.Zhang Z, Hendrickson WA. Structural characterization of the predominant family of histidine kinase sensor domains. J Mol Biol. 2010;400(3):335–353. doi: 10.1016/j.jmb.2010.04.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lacal J, et al. Identification of a chemoreceptor for tricarboxylic acid cycle intermediates: Differential chemotactic response towards receptor ligands. J Biol Chem. 2010;285(30):23126–23136. doi: 10.1074/jbc.M110.110403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gavira JA, et al. Crystallization and crystallographic analysis of the ligand-binding domain of the Pseudomonas putida chemoreceptor McpS in complex with malate and succinate. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2012;68(Pt 4):428–431. doi: 10.1107/S1744309112004940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ingham CJ, Armitage JP. Involvement of transport in Rhodobacter sphaeroides chemotaxis. J Bacteriol. 1987;169(12):5801–5807. doi: 10.1128/jb.169.12.5801-5807.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Toepfer JA, Ford RM, Metge D, Harvey RW. Impact of fluorochrome stains used to study bacterial transport in shallow aquifers on motility and chemotaxis of Pseudomonas species. FEMS Microbiol Ecol. 2012;81(1):163–171. doi: 10.1111/j.1574-6941.2012.01355.x. [DOI] [PubMed] [Google Scholar]
- 16.Migas J, Anderson KL, Cruden DL, Markovetz AJ. Chemotaxis in Methanospirillum hungatei. Appl Environ Microbiol. 1989;55(1):264–265. doi: 10.1128/aem.55.1.264-265.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kuroda A, et al. Molecular cloning and characterization of a chemotactic transducer gene in Pseudomonas aeruginosa. J Bacteriol. 1995;177(24):7019–7025. doi: 10.1128/jb.177.24.7019-7025.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yu EW, Koshland DE., Jr Propagating conformational changes over long (and short) distances in proteins. Proc Natl Acad Sci USA. 2001;98(17):9517–9520. doi: 10.1073/pnas.161239298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ottemann KM, Xiao W, Shin YK, Koshland DE., Jr A piston model for transmembrane signaling of the aspartate receptor. Science. 1999;285(5434):1751–1754. doi: 10.1126/science.285.5434.1751. [DOI] [PubMed] [Google Scholar]
- 20.Wolfe AJ. The acetate switch. Microbiol Mol Biol Rev. 2005;69(1):12–50. doi: 10.1128/MMBR.69.1.12-50.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhulin IB, Nikolskaya AN, Galperin MY. Common extracellular sensory domains in transmembrane receptors for diverse signal transduction pathways in bacteria and archaea. J Bacteriol. 2003;185(1):285–294. doi: 10.1128/JB.185.1.285-294.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cheung J, Hendrickson WA. Sensor domains of two-component regulatory systems. Curr Opin Microbiol. 2010;13(2):116–123. doi: 10.1016/j.mib.2010.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moore JO, Hendrickson WA. Structural analysis of sensor domains from the TMAO-responsive histidine kinase receptor TorS. Structure. 2009;17(9):1195–1204. doi: 10.1016/j.str.2009.07.015. [DOI] [PubMed] [Google Scholar]
- 24.Baraquet C, et al. TorT, a member of a new periplasmic binding protein family, triggers induction of the Tor respiratory system upon trimethylamine N-oxide electron-acceptor binding in Escherichia coli. J Biol Chem. 2006;281(50):38189–38199. doi: 10.1074/jbc.M604321200. [DOI] [PubMed] [Google Scholar]
- 25.Lillis AP, Van Duyn LB, Murphy-Ullrich JE, Strickland DK. LDL receptor-related protein 1: Unique tissue-specific functions revealed by selective gene knockout studies. Physiol Rev. 2008;88(3):887–918. doi: 10.1152/physrev.00033.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hubbard SR, Till JH. Protein tyrosine kinase structure and function. Annu Rev Biochem. 2000;69:373–398. doi: 10.1146/annurev.biochem.69.1.373. [DOI] [PubMed] [Google Scholar]
- 27.Doublié S. Preparation of selenomethionyl proteins for phase determination. Methods Enzymol. 1997;276:523–530. [PubMed] [Google Scholar]
- 28.Schneider TR, Sheldrick GM. Substructure solution with SHELXD. Acta Crystallogr D Biol Crystallogr. 2002;58(Pt 10 Pt 2):1772–1779. doi: 10.1107/s0907444902011678. [DOI] [PubMed] [Google Scholar]
- 29.Hao Q. ABS: A program to determine absolute configuration and evaluate anomalous scatterer substructure. J Appl Cryst. 2004;37(pt 3):498–499. [Google Scholar]
- 30.Sheldrick GM. Macromolecular phasing with SHELEX. Z Kristallogr. 2002;217(12):644–665. [Google Scholar]
- 31.Terwilliger TC. Maximum-likelihood density modification. Acta Crystallogr D Biol Crystallogr. 2000;56(Pt 8):965–972. doi: 10.1107/S0907444900005072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cowtan K. ‘dm’: An automated procedure for phase improvement by density modification. Joint CCP4 and ESF-EACBM Newsletter on Protein Crystallography. 1994;31:34–38. [Google Scholar]
- 33.Emsley P, Cowtan K. Coot: Model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60(Pt 12 Pt 1):2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 34. Afonine PV, Grosse-Kunstleve RW, Adams PD (2005) The Phenix refinement framework. CCP4 Newsletter on Protein Crystallography 42:8.
- 35.Adler J. A method for measuring chemotaxis and use of the method to determine optimum conditions for chemotaxis by Escherichia coli. J Gen Microbiol. 1973;74(1):77–91. doi: 10.1099/00221287-74-1-77. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.