

Abstract
For the last two decades, most efforts on new drug development to treat Alzheimer's disease have been focused to inhibit the synthesis of amyloid beta (Aβ), to prevent Aβ deposition, or to clear up Aβ plaques from the brain of Alzheimer's disease (AD) patients. Other pathogenic mechanisms such as the hyperphosphorylation of the microtubular tau protein (that forms neurofibrillary tangles) have also been addressed as, for instance, with inhibitors of the enzyme glycogen synthase-3 kinase beta (GSK3β). However, in spite of their proven efficacy in animal models of AD, all these compounds have so far failed in clinical trials done in AD patients. It seems therefore desirable to explore new concepts and strategies in the field of drug development for AD. We analyze here our hypothesis that a trifunctional chemical entity acting on the L subtype of voltage-dependent Ca2+ channels (VDCCs) and on the mitochondrial Na+/Ca2+ exchanger (MNCX), and having additional antioxidant properties, may efficiently delay or stop the death of vulnerable neurons in the brain of AD patients. In recent years, evidence has accumulated indicating that enhanced neuronal Ca2+ cycling (NCC) and futile mitochondrial Ca2+ cycling (MCC) are central stage in activating calpain and calcineurin, as well as the intrinsic mitochondrial pathway for apoptosis, leading to death of vulnerable neurons. An additional contributing factor to neuronal death is the excess free radical production linked to distortion of Ca2+ homeostasis. We propose that an hybrid compound containing a dihydropyridine moiety (to block L channels and mitigate Ca2+ entry) and a benzothiazepine moiety (to block the MNCX and slow down the rate of Ca2+ efflux from the mitochondrial matrix into the cytosol), as well as a polyphenol moiety (to sequester excess free radicals) could break down the pathological enhanced NCC and MCC, thus delaying the initiation of apoptosis and the death of vulnerable neurons. In so doing, such a trifunctional compound could eventually become a neuroprotective medicine capable of delaying disease progression in AD patients.
Keywords: Alzheimer's disease, neuronal calcium cycling, mitochondrial calcium cycling, calcium and cell death, L-type calcium channel, mitochondrial sodium−calcium exchanger, functional tetrad, multitarget compounds, neurotoxicity, neuroprotection
The calcium ion (Ca2+) acts as an ubiquitous intracellular messenger to regulate a pleiad of physiological functions. Being a divalent cation that binds to multiple proteins, ion channels, and receptors, together with the existence of a four order magnitude gradient from the extracellular to the intracellular space, makes Ca2+ a suitable messenger. In excitable cells and particularly in neurons of the central nervous system that are continuously firing action potentials at various frequencies, Ca2+ ions undergo an endless cycling of Ca2+ influx through plasmalemmal Ca2+ channels, its intracellular buffering by Ca2+ binding proteins (CBPs) and organelles, particularly the endoplasmic reticulum (ER) and mitochondria, Ca2+ release from these organelles into the cytosol, and Ca2+ efflux through plasmalemmal Ca2+ transporters, namely, the ATPase Ca2+ pump and the Na+/Ca2+ exchanger. Thus, there are two “Ca2+ circuits” that we will refer to in this review as neuronal Ca2+ cycling (NCC) and mitochondrial Ca2+ cycling (MCC). These Ca2+ circuits serve to regulate important neuronal functions such as the synaptic release of neurotransmitters, or the respiration rate of mitochondria by Ca2+-dependent dehydrogenases, that couple bioenergetics through ATP synthesis to neuronal activity. Disruption of NCC and/or MCC will enhance the vulnerability of neurons to various stressors, leading to necrotic and/or apoptotic death of the vulnerable neurons in neurodegenerative diseases and stroke. To better understand how NCC and MCC are affected in those vulnerable neurons, in this review we should first describe the fine-tuning of the ion channels and transporters that maintain the equilibrium of cell Ca2+ homeostasis under physiological conditions. We will then focus on the implications of Ca2+ dysregulation in Alzheimer's disease (AD). Finally, we will describe our hypothesis for the development of a novel multitarget neuroprotective medicine for AD.
Calcium Signaling and Calcium Cycling in Neurons
The increase of local cytosolic Ca2+ concentrations ([Ca2+]c) during cell activation is determined by Ca2+ entry through plasmalemmal Ca2+ channels, by its sequestration into and its subsequent release from organelles, and by Ca2+ efflux through plasmalemmal pumps (Figure 1). Ca2+ signaling becomes quantal at the molecular level as very high [Ca2+]c are required for triggering certain Ca2+ dependent processes for instance, fast neurotransmitter release. Although the [Ca2+]c peaks measured with Ca2+ probes are usually underestimated, it is accepted that highly localized and transient high [Ca2+]c microdomains occurs underneath the plasmalemma, nearby the exocytotic machinery.1−4 The likelihood for generation of a high Ca2+ microdomain augments drastically with the coincidence in time and space of the opening of various Ca2+ channels upon action potential firing. These localized [Ca2+]c transients may also be favored by Ca2+-induced Ca2+ release (CICR) from the endoplasmic reticulum (ER), through both ryanodine receptor (RyR) and inositol tris-phosphate receptor (InsP3R) channels.5 Furthermore, the formation of Ca2+ microdomains may also be favored by the geometric disposition of ER, mitochondria, nucleus, secretory vesicles, or dendritic spines.6−10 Thus, the Ca2+ signaling system is organized to favor the generation of large [Ca2+]c microdomains that are highly localized in space and time. This allows the regulation of several functions using the same triggering signal, but with different subcellular locations and time patterns.
Figure 1.
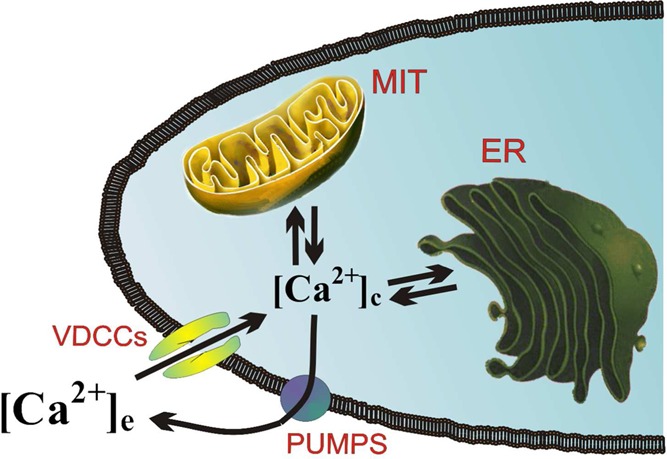
Changes of cytosolic Ca2+ concentrations ([Ca2+]c) occurring during cell activation are determined by Ca2+ entry through voltage-dependent Ca2+ channels (VDCCs) or other plasmalemmal Ca2+ channels, its redistribution into endoplasmic reticulum (ER) and mitochondria (MIT), and its ejection to the extracellular space through Ca2+ pumps. The rate of this Ca2+ cycling is a function of cell activity and its bioenergetic demands, which are controlled by mitochondrial Ca2+ cycling.
At each moment of cell activity, Ca2+ homeostasis is defined by fluxes between three compartments, that is, the extracellular milieu, the cytosol, and the Ca2+-storing organelles. Much information on Ca2+ fluxes occurring during activation of neurosecretory cells and their correlation with vesicle flow and exocytosis have been obtained in adrenal medullary chromaffin cells, that are excellent models of catecholaminergic neurons.11,12 At rest, these fluxes are small, with exchange rates in the range of 1–10 μmol/L cells/s, both at the plasma membrane and ER membrane. Mitochondrial Ca2+ uptake through the uniporter (MCU) is very slow because of its low Ca2+ affinity and its exponential kinetics. The [Ca2+]c at steady state is in the range of 10–7 M in the cytosol and the mitochondrial matrix, and around 10–3 M at the extracellular milieu and at the ER lumen; thus, very high gradients of about 104-fold exist favoring Ca2+ diffusion into the cytosol.
When voltage-dependent calcium channels (VDCCs) open during an action potential, peak inward Ca2+ currents of about 800 pA develop (equivalent to 2000–3000 μmol of Ca2+/L cells/s). At low stimulation, the rates of diffusion through the cytosol and binding by endogenous buffers are the main determinants of the Ca2+ signal;3,4 under these low stimulation conditions, the [Ca2+]c reaches the level of 10–6 M and clearance by the high-affinity Ca2+ pumps (plasma membrane and SERCA) dominates. At high stimulation rates, [Ca2+]c may reach levels high enough to stimulate transport through MCU; under this condition, most of the Ca2+ load is taken up by mitochondria.13−16 For depolarizations with 10 s pulses of high K+, more than 90% of the Ca2+ load is taken up by mitochondria during the stimulation period; once stimulation ceases, mitochondrial Ca2+ is released into the cytosol during a period of seconds or minutes.14,15 In this manner, the [Ca2+]c remains discretely elevated during this period and this may help to mobilize secretory vesicles from the reserve pool toward the membrane, thus becoming ready to be used for the next exocytotic episode.3 In addition, Ca2+ accumulated in mitochondria stimulates respiration and ATP synthesis.17,18 This may help to provide extra energy for maintaining the exocytotic release of neurotransmitters under intense stimulation and to clear up the Ca2+ load, thus restoring cell homeostasis after the activity period.
Since high-Ca2+ microdomains are particularly pronounced nearby the inner mouth of VDCCs, mitochondrial Ca2+ uptake could take place locally at these places during physiological stimulation. This is suggested by the fact that anterior pituitary cells that exhibit spontaneous action potentials and [Ca2+]c oscillations also show parallel oscillations of Ca2+ concentrations inside the mitochondrial matrix ([Ca2+]m).19 Furthermore, during stimulation of chromaffin cells, the Ca2+ entering through VDCCs is taken up by a pool of mitochondria located close to the plasma membrane. This stops the progression of the Ca2+ wave toward the cell core. The rate of Ca2+ uptake into subplasmalemmal mitochondria measured as [Ca2+]c units is higher than 50 μM/s; this indicates that this mitochondrial subpopulation, so-called M1, is seeing local [Ca2+]c underneath the plasmalemma of as much as 30 μM. On the other hand, the inner mitochondria subpopulation, so-called subpopulation M2, takes up Ca2+ at a much lower rate, namely, 0.3 μM/s, equivalent to [Ca2+]c of 2 μM at the cell core.14,19
The CICR may modulate or amplify the Ca2+ signals generated by Ca2+ entry through VDCCs. Thus, measurements of the variations of the [Ca2+] at the ER ([Ca2+]er) during stimulation with high K+ shows net decreases of 60–100 μM, about 10–15% of the total ER Ca2+ content.20 This may seem a small amount of Ca2+; however, this averaged value may be composed of strong liberation in some cell compartments that is compensated by strong uptake in others. In addition, the strength of CICR may be under regulation, since it was sensitized by low caffeine concentrations or by increasing the load of ER Ca2+. On the other hand, fast confocal measurements showed that the wave of Ca2+ induced by 100 ms depolarizing pulses was delayed and reduced in ryanodine-treated chromaffin cells.20 CICR seems to colocalize with plasmalemmal VDCCs and the mitochondrial M1 pool that undergoes large [Ca2+]m changes during depolarization.
On the basis of experiments performed on chromaffin cells, we raised the hypothesis for the existence of complex functional units clustering together the VDCCs, the cytosolic CBPs, the MCU, and the ER RyR (Figure 2). This functional tetrad could be responsible for the generation of the local high [Ca2+]c transients that control the rate and extent of catecholamine release. These transients may reach concentrations of about 50 μM at the subplasmalemmal region where the secretory vesicles are docked. The Ca2+ channel will act as the trigger, the RyR as the signal amplifier, and the mitochondrion as a contention wall that avoids propagation of the high-Ca2+ tide to the cell core, where such a large Ca2+ signal is not required. In addition, Ca2+ taken up by mitochondria stimulates respiration, thus tuning up energy production to support the increased requeriments of the exocytotic activity. The respiratory activity will lag behind the cessation of cell activity until the mitochondrial Ca2+ load is completely cleared. Mitochondria from the M2 subpopulation may play an additional role in redistributing Ca2+ to deep regions of the cytosol. Thus, much of the Ca2+ that enters mitochondria at subplasmalemmal sites (pool M1) may diffuse through the mitochondrial matrix to other cell locations and be eventually extruded from mitochondria near the cell core. In so doing, this mitochondrial Ca2+ release would contribute to keep [Ca2+]c discretely increased during the poststimulus period, perhaps facilitating the transport of new vesicles from a reserve pool to a ready-release vesicle pool and an immediate-release vesicle pool to refill the ready-release vesicle pool underneath the plasma membrane.21
Figure 2.
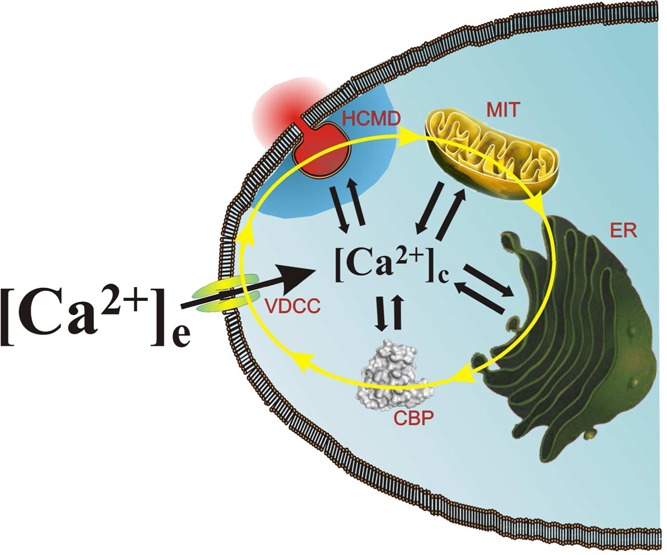
Chromaffin cell Ca2+ cycling and its cytosolic concentration ([Ca2+]c) at a given moment during cell activation are determined by a functional tetrad composed of voltage-dependent Ca2+ channels (VDCCs), Ca2+-binding proteins (CBPs), the endoplasmic reticulum (ER), and mitochondria (MIT). This functional tetrad controls the generation of high-Ca2+ microdomains (HCMDs) at subplasmalemmal exocytotic sites, to trigger the exocytotic release of catecholamine during cell activation.
We can speculate that if either the location or the Ca2+ uptake properties of mitochondria in those functional tetrads could be modulated, this would be an effective strategy to regulate the exocytotic process and, hence, the release of neurotransmitters and synaptic plasticity. Thus, under pathological conditions, that is, excitatory neurotoxicity, ischemia–reperfusion in stroke, aging, or neurodegenerative diseases, mitochondrial damage may reduce their ability to take up Ca2+. This could lead to increased secretion of excitatory neurotransmitters and increased neuronal activation, a vicious circle that may trigger Ca2+-dependent processes leading to necrosis or apoptosis. Changes in CICR could also modulate the synaptic efficacy under physiological or pathophysiological conditions.
Calcium Dysregulation in Alzheimer's Disease
AD is characterized by the progressive impairment of higher cognitive function, memory loss, and altered behavior.22 The pathological hallmarks were already described in the original report of the disease;23 they are characterized by the presence of senile plaques composed of extracellular amyloid beta (Aβ) peptide aggregates, intracellular neurofibrillary tangles formed by hyperphosphorylated tau protein deposits, and the shrinkage of the cerebral cortex due to extensive neuronal loss.24
The mechanisms involved in the formation of Aβ and hyperphosphorylated tau, and the relationship between them are not yet clear. It is known that Aβ aggregation and neurotoxicity augment in the presence of acetylcholinesterase (AChE).25 On the other hand, when hyperphosphorylated by glycogen synthase kinase-3-beta (GSK3β), tau detaches from the microtubulules, disrupts axonal transport, and contributes to Aβ neurotoxicity.26 In addition to GSK3β, Aβ also elicits the activation of cyclin-dependent kinase 5 (CDK5) and extracellular signal-regulated kinase 2 (ERK2), leading to tau hyperphosphorylation and ultimately to apoptosis.27
Most efforts on new drug development are being made in the context of the dominant model to explain the pathogenesis of AD, the amyloid hypothesis, which attributes the increased production of Aβ42 (or increase in Aβ42–Aβ40 ratio), the major cause of neural and synaptic loss.28 In support of the hypothesis are the following findings: (1) accumulation of amyloid plaques in the brain of AD patients; (2) the familial AD cases that result from missense mutations in amyloid precursor protein (APP); and (3) the familial cases resulting from missense mutations in presenilins, which form a catalytic subunit of the APP-cleaving enzyme γ-secretase. The amyloid-targeting therapies have been the main focus of AD drug development for the last 20 years. However, clinical trials with compounds targeting Aβ have repeatedly failed; thus, additional targets beyond Aβ need to be seriously considered for developing new medicines to treat AD patients.29
Since the initial proposal of a Ca2+ imbalance as a cause of neuronal degeneration in AD,30 much evidence has accumulated to substantiate this hypothesis.31−36 One potential connection between Ca2+ and AD pathogenesis comes from the observation that Aβ oligomers can form Ca2+-permeable channels in membranes37 (Figure 3). Vulnerable cells in conditions of energy deficit, enhance the ability of Aβ to associate with membranes.38 This is consistent with the fact that neurons with reduced cytosolic ATP levels are particularly vulnerable to Aβ toxicity.39 This is in line with recent data obtained with in vivo Ca2+ imaging experiments performed with APP transgenic mice. This study shows that resting [Ca2+]c is significantly elevated in 35% of neurites located in the immediate vicinity of Aβ plaques.40 This is probably due to the formation of Ca2+-permeable channels in the area of the neuronal plasma membrane close to Aβ plaques, due to the high concentration of Aβ oligomers. The neurites with elevated [Ca2+]c lack spines and display an abnormal morphology, that is reduced by the calcineurin inhibitor FK-506. Aβ oligomers also affect neuronal Ca2+ homeostasis by modulating the activity of NMDA receptors.41,42
Figure 3.
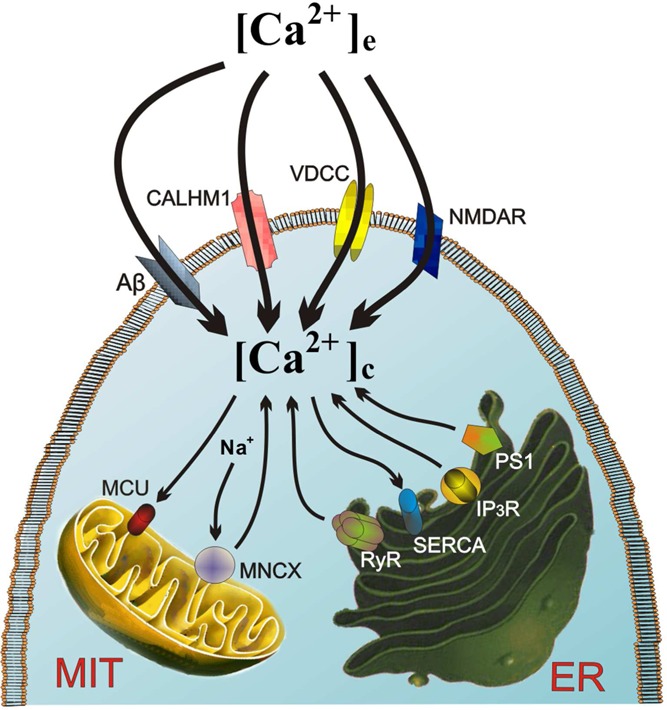
Ca2+ dysregulation is central stage in neuronal vulnerability and death in AD. Various distorted Ca2+ entry pathways in the plasmalemma can give rise to neuronal Ca2+ overload, that is, amyloid beta (Aβ), the newly discovered Ca2+ channel CALHM1, NMDA, and AMPA receptors for glutamate (NMDAR and AMPAR), and the neuronal L subtype of voltage-dependent Ca2+ channels (VDCCs). Ca2+ release from the endoplasmic reticulum (ER) may also contribute to [Ca2+]c elevation by enhanced Ca2+-induced Ca2+ release (CICR) through ryanodine receptor channels (RyR) or IP3 channels, as well as through Ca2+ leak channels formed by mutated presenilins. Finally, this excess [Ca2+]c is taken up by the mitochondrial Ca2+ uniporter (MCU) and released back into the cytosol through the mitochondrial Na+/Ca2+ exchanger (MNCX), thus creating and endless futile mitochondrial Ca2+ cycling (MCC) that eventually will cause energy depletion, opening of the mitochondrial transition pore (MPTP), apoptosis, and neuronal death.
Recently, presenilin-1 (PS1) has emerged as central stage in the regulation of ER Ca2+.43−45 PS1 was originally identified in a genetic screen for causative factors of familial AD.46 Since then, over 150 mutations of PS1 have been identified. The involvement of PS1 in AD pathology has been primarily ascribed to altered processing of APP by γ-secretase, a multiprotein complex containing PS1.47 Mutations in PS1 have been suggested to cause both overloading and underfilling of ER Ca2+ stores, and to both increase and decrease Ca2+ release from the ER.43,45,48−52
Fibroblasts from familial AD patients release supranormal amounts of ER Ca2+.53 This was reproduced in oocytes expressing mutant presenilins52 and in cortical neurons of knock-in mice.48,54 The mechanism responsible for this enhanced Ca2+ release has been associated to effects of mutated presenilins on Ca2+ influx through store-operated Ca2+ channels (SOCs),51,55 increased activity and/or expression of RyR channels54,56,57 and InsP3R channels.45,58 Also, presenilins themselves may function as ER Ca2+-leak channels and their mutations cause the loss of such channel function, leading to ER Ca2+ overload and supranormal ER Ca2+ release.43,49
Various Ca2+-dependent neurotoxic effects may be derived from [Ca2+]c augmentation as a result of enhanced Ca2+ entry through Aβ channels or excessive ER Ca2+ release associated presenilin mutations. This is the case for the activation of calcineurin and neurite atrophy described above,40 or the calpain activation and degradation of signaling enzymes involved in learning and memory.59,60 On the other hand, a tight correlation exists between the reduction in expression of CBPs in the hippocampal dentate gyrus and onset of cognitive deficits in AD.61
Recently, novel Ca2+ influx channel CALHM1 (Ca2+ homeostasis modulator 1) has been associated to increased late-onset AD;62 however, other studies did not find such association.63 Expression of CALHM1 mutation P86L in HeLa cells led to an impaired capacity of mitochondria to clear up a [Ca2+]c load.64 Furthermore, this mutation impairs Ca2+ handling by neurons.62 This, together with the supranormal [Ca2+]c signals described above, could lead to mitochondrial Ca2+ overload and activation of apoptotic neuronal death. Thus, the neuroprotective actions of nonsteroidal anti-inflammatory drugs could be related to their ability to reduce mitochondrial Ca2+ uptake.65
Aβ Ca2+ channels are an attractive target to develop inhibitory compounds.66 On the other hand, Aβ increases Ca2+ influx elicited upon glutamate activation of NMDARs.67 This is in line with the observation that NMDAR blocker memantine (the first noncholinergic drug approved to treat AD)68,69 blocks Aβ-induced Ca2+ influx, indicating that drug restoring the Ca2+ balance in neurons might indeed generate therapeutic options for the disease.70 Thus, NMDAR blockers more specific than memantine such as nitromemantines are being developed.71 On the other hand, the NR2B-specific antagonist E-VT-101 was recently developed by Evotec. Furthermore, the L VDCC blocker MEM-1003 has completed phase II clinical trials in AD patients. Other potential largely unexplored targets include RyR and InsP3R channels, SERCA, calcineurin and the mitochondrial transporters to regulate Ca2+ cycling and bioenergetics.
Multitarget Stabilizers of Neuronal and Mitochondrial Calcium Cycling
To communicate between them, neurons are in a continuous state of action potential firing at different frequencies. So, Ca2+ influx from the extracellular space into the cytosol is an endless activity that must be counterbalanced by a continuous Ca2+ efflux through plasmalemmal Ca2+ pump and NCX. This NCC is connected with a second intracellular MCC mechanism that is mainly controlled by mitochondrial Ca2+ uptake through the MCU and Ca2+ release through its MNCX; this intracellular Ca2+ circulation is tightly regulated by the Ca2+ buffering capacity of each neuron, linked to their CBP contents, as well as by the intimate association of ER InsP3R and RyR channels with the MCU. The rates of both Ca2+ cycling mechanisms are interdependent in such a manner that slowing down the NCC by decreasing action potential firing will result in smaller [Ca2+]c transients and, hence, diminished mitochondrial Ca2+ uptake through the low-Ca2+ affinity of the MCU. Conversely, a higher discharge rate of action potentials will enhance the rates of both NCC and MCC. This harmonious coordination between the two Ca2+ circuits permits the coupling of mitochondrial rate of respiration and ATP synthesis to the requirements of the degree of neuronal activity under different physiological and pathological situations.
As discussed above, in the pathogenesis of AD two main mechanisms have been considered: (i) alteration of specific protein functions that in the case of familial disease are linked to various mutations; and (ii) a pathway downstream of such protein alterations that involves a disruption of the delicate coupling between the rates of neuronal and intracellular Ca2+ circuits, leading to a deficit in mitochondrial bioenergetics and to a state of greater susceptibility for these vulnerable neurons to enter in apoptosis and die.
It is interesting that although damage of specific proteins constitutes the hallmarks of AD, disruption of Ca2+ homeostastic mechanisms and mitochondrial damage are critical pathogenic mechanisms, as recently reviewed.72 Given the complexity of these pathogenic pathways, it seems understandable that, during the last two decades, clinical trials with single-target drugs generally directed to disease-specific pathological proteins, enzymes, or receptors have repeatedly failed to show a therapeutic effect. Therefore, the hypothesis that a multifunctional compound may be more effective as a neuroprotective, disease-modifying medicine is gaining increased interest. This medicine should target the neuronal and/or intracellular Ca2+ circuits to preserve mitochondrial functioning of vulnerable neurons as long as possible. We can call this class of compounds as multitarget stabilizers of neuronal Ca2+ cycling.
Slow-inactivating L-type VDCCs have been the focus of attention in the design and synthesis of multitarget neuroprotective compounds. Hybrid compounds having the moieties of tacrine and 1,4-dihydropyridines (DHPs) inhibit both AChE and Ca2+ entry through L channels in neuroblastoma cells; furthermore, they exhibit neuroprotection on in vitro models of neurotoxicity elicited by Ca2+ overload and oxidative stress.73 Another naftiridine family, exemplified by compound ITH4012, caused AChE inhibition but mildly enhanced Ca2+ influx through L channels; the compound afforded neuroprotection against oxidative stress, Ca2+ overload, and Aβ toxicity and induced the expression of antiapoptotic Bcl-2.74 In a third generation of these multitarget tacripyrines, it was found that they behaved as dual inhibitors of AChE, at catalytic and peripheral sites, decreased AChE and Aβ-self-aggregation, inhibited L channels, and caused neuroprotection against oxidative stress and Ca2+ overload toxicity.75
A family of dicarboxylic amino acid derivatives was synthesized taking l-glutamic acid as a linker for different bioactive moieties. They are potent inhibitors of catalytic and peripheral sites of AChE, with potential Aβ antiaggregating effects and exhibit neuroprotective properties on various in vitro models of oxidative stress.76 Compound ITH33/IQM9.21 from this family reduces infarct volume in a photothrombotic model of permanent focal cerebral ischemia in mice (Silvia Lorrio, personal communication) and causes a gradual, slow-developing reversible blockade of VDCCs.77
Multitarget compounds acting on NMDAR Ca2+ channels have also been synthesized, inspired by carvedilol, a vasodilator beta-blocker having antioxidant and neuroprotective properties.78 In this report, the tetrahydroacridine moiety from tacrine and the carbazole core from carvedilol were linked. The resulting dimeric molecules were expected to inhibit Aβ fibril formation, since carbazols are efficient inhibitors of Aβ aggregation.79 The compound carbacrine from this family blocked both the catalytic and PAS sites of AChE and inhibited both AChE- and self-induced Aβ aggregation. Its potency to inhibit the NMDAR was higher than that of the reference compound carvedilol, behaving as a noncompetitive open-channel blocker; as the case is for memantine, this property may result in blockade of pathological excessive NMDAR activity without affecting normal synaptic glutamatergic transmission. Finally, carbacrine was more potent than trolox as an antioxidant and affords neuroprotection against oxidative stress.70 Another family of NMDAR blockers was inspired in the monovalent β-carbolines structure, known to be potent AChE inhibitors.80 Bivalent β-carboline compound blocked AChE as well as the [Ca2+]c transients elicited by glutamate by inhibiting NMDARs.81
Destabilization of microctubules through tau hyperphosphorylation distorts axonal transport and contributes to neuronal death in AD.82 Such hyperphosphorylation is carried out by various kinases, i.e. cyclin-dependent kinase 5 (CDK5), glycogen synthase 3β (GSK3β), extracellular signal regulated kinase 2 (ERK2) and casein kinase 1 (CK1). Roscovitine is a purine-based inhibitor of CDKs and other kinases.83−85 In addition to this AD-specific target, roscovitine also targets the N and PQ VDCCs, delaying their deactivation.86−88 Whether this dual action leads to activation of neuroprotection signaling pathways remains to be elucidated.
Other Ca2+ targets are InsP3R, RyR, SERCA pump, and store-operated Ca2+ influx channels (SOC). Development of more specific inhibitors of those targets combined with a disease-specific target could possibly be achieved in the next years. Another question is whether the MCU, the MPTP, or the MNCX is a drugable target for AD. Finally, the pharmacological manipulation of CBPs and hence the neuronal Ca2+ buffering capacity, or the intracellular Ca2+ clearance mechanisms, should also be considered as potential targets for pharmacological neuroprotection.
Regulation of Mitochondrial Calcium Cycling as a Strategy to Develop Novel Multitarget Neuroprotective Compounds
As discussed before, Ca2+ uptake into mitochondria is mediated by the electrophoretic MCU, that uses the large electrochemical gradient across the inner membrane (negative on the inside) to drive Ca2+ from the cytosol into the mitochondrial matrix; Ca2+ ejection from the mitochondrial matrix into the cytosol occurs through an electrochemical MNCX that uses a large driving force for Na+ influx in exchange for Ca2+ efflux.89 The existence of these two independent pathways for Ca2+ uptake and release permits Ca2+ cycling to occur across the inner mitochondrial membrane (Figure 4). The discovery that several intramitochondrial dehydrogenases are regulated by the [Ca2+]m suggested that MCC could regulate NADH production and an increase in the overall rate of oxidative ATP synthesis. This led to the proposition that the role of the mitochondrial Ca2+ transport processes is to regulate the [Ca2+]m.90 Hence, this MCC provides a mechanism by which increases and decreases of [Ca2+]c during cell activity can be translated into parallel changes in [Ca2+]m. This allows the activities of the Ca2+-sensitive dehydrogenases to be sensitive to changes of [Ca2+]c and hence to couple cell activity to ATP synthesis. In this manner, neurons may use Ca2+ as a dual signal, for instance, stimulating neurotransmitter release, neuronal firing of action potentials, and ATP utilization in the cytosol, while stimulating oxidative ATP synthesis in mitochondria; this will balance the demand and supply of ATP.
Figure 4.
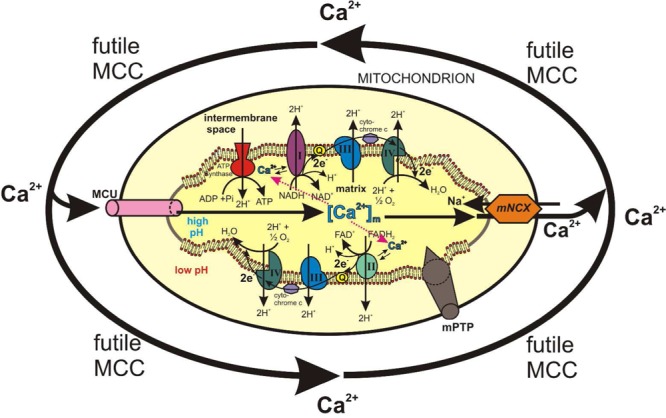
Futile mitochondrial Ca2+ cycling (MCC) may lead to energy waste and ATP depletion. Under physiological conditions, the rate of MCC is coupled to neuronal activity; in this manner, the Ca2+-dependent dehydrogenases of the respiratory chain and ATP synthesis will be tightly coupled to cell activity to cope with its bioenergetic needs. Under pathological conditions such as excess glutamate-activated NMDA channels or other plasmalemmal Ca2+ channels, excess Ca2+ entry, accompanied by augmented ER Ca2+ release will cause an acceleration of mitochondrial Ca2+ uptake through its uniporter (MCU) and excess mitochondrial Ca2+ release through the Na+/Ca2+ exchanger (MNCX). This would lead to a futile MCC (arrows) with ATP waste, ATP depletion, and apoptotic death of vulnerable neurons.
Since long, the pharmacological regulation of MCC has been attempted as a strategy to augment ATP synthesis and to improve the cell functioning. This was exemplified by pioneering experiments showing that [Ca2+]m levels increase in response to inotropic stimulation of perfused hearts91 and isolated myocytes.92 This has been associated with a stimulation of Ca2+-sensitive dehydrogenase activity.91 Furthermore, the MCU blocker ruthenium red inhibited these responses,93 emphasizing the relevance of the Ca2+ uniporter in transmitting this signal. But the [Ca2+]m may also be increased by mitigating the rate of Ca2+ efflux via the MNCX. Since the primary role of this exchanger is to maintain a low [Ca2+]m by extruding Ca2+, it follows that a MNCX blocker could be expected to increase the [Ca2+]m and, as a result, possibly stimulate the rate of ATP synthesis. By delaying mitochondrial Ca2+ efflux into the cytosol, this MNCX blocker could also decrease the rate of ATP breakdown that is associated to [Ca2+]c increases during cell activation. Thus, interest in developing blockers of the MNCX arose near three decades ago.94
For instance, some Ca2+ antagonists were found to inhibit the MNCX,95 with d-cis-diltiazem being the most potent.96 Also, certain benzodiazepines such as clonazepam inhibited the MNCX in isolated heart mitochondria, with the same potency as d-cis-diltiazem.96,97 In contrast to d-cis-diltiazem, clonazepam did not block the heart L VDCCs.98 Although this may be taken as clonazepam exhibiting more specificity to block the MNCX, it still requires more than 100 μM to completely block the transporter.98 Screening of a series of compounds with structures similar to diltiazem and clonazepam led to the more potent MNCX blocker CGP37157.99 It was later on shown that this compound enhances the [Ca2+]m, as well as the rate of NADH formation by the Krebs cycle and the overall rate of oxidative ATP synthesis in isolated heart mitochondria; CGP37157 was 20-fold more potent than clonazepam and d-cis-diltiazem in inhibiting the MNCX.100
From the standpoint of mitochondrial Ca2+ overload as a signal to produce the MPTP opening and the activation of the apoptotic cascade, it may seem paradoxical to look for a therapeutic application of MNCX inhibition. In fact, certain mutations or PINK1 knockout (a gene associated with familial PD) show impaired MNCX activity; this results in mitochondrial Ca2+ overload that sensitizes mitochondria to MPTP opening, impairing respiration and renderig neurons vulnerable to undergo cell death.101 Hence, the extent of inhibition of the MNCX under in vivo conditions is a relevant consideration. Complete blockade will prevent any efflux of Ca2+ once it has entered into the mitochondrial matrix, and may convert mitochondria into Ca2+ sinks. However, partial MNCX inhibition would lead to [Ca2+]m augmentation to a higher physiological steady-state level that could stimulate Ca2+-sensitive dehydrogenase activity and the rate of ATP synthesis. This may be particularly true in vulnerable neurons where mitochondrial dysfunction causes a decreased ATP synthesis.
In evaluating the role of Ca2+ in activating cell survival or cell death signaling pathways, we should consider the overall neuronal Ca2+ cycling as well as the local MNCX. The precise coupling between these two Ca2+ circuits is essential for functioning and adaptation of ATP production to neuronal activity. Ca2+ may behave both as a supporter of cell survival or as an inducer of cell death. For instance, mild cell depolarisation and subsequent Ca2+ entry into the cytosol helps to sustain the survival of cerebellar granule cells102 and bovine chromaffin cells.103 Conversely, by reducing the [Ca2+]c, Ca2+ antagonists may also cause neuronal death104 and chromaffin cell death.105 These apparent contradictory findings may be explained in the frame of the hypothesis suggesting that the [Ca2+]c changes occurring during cell activation must move within a critical set point; beyond this point, a cytoprotective signal might turn into a cytotoxic one.106 In this context, the suggestion of Nicholls107 and White and Reynolds108 that Ca2+ accumulation into mitochondria could play a neuroprotective role by removing Ca2+ from the cytoplasm, fits well in the hypothesis.
The question on whether the inhibition of the MNCX by CGP37157 could produce neuroprotection was first explored in bovine chromaffin cells stressed with veratridine, a model of neuronal death associated to Na+ and Ca2+ overload.109 By slowing down Na+ channel inactivation, veratridine causes Na+ accumulation in the cytosol,110,111 cell depolarization,112 opening of VDCCs, and increased [Ca2+]c.112−114 This will cause mitochondrial Ca2+ overload and activation by enhanced cytosolic [Na+] of the MNCX, thereby producing a futile MCC with a concomitant excessive production of ROS; this could be the signal to initiate the process of neuronal death and neurodegeneration.115 Slowing down this futile MCC with CGP37157 could partially lead to delay or prevention of cell death; this supposition was demonstrated to be true in bovine chromaffin cells114 and rat hippocampal slices116 stressed with veratridine.
One limitation to clearly explain the neuroprotective action of CGP37157 is its capacity to block the voltage-dependent Na+ and Ca2+ channels.114 However, the compound seems to be more efficacious than various blockers of L, N or PQ VDCCs in hippocampal slices stressed with veratridine;116 this suggests that, in eliciting neuroprotection, CGP37157 could in fact be mitigating the rates of both Ca2+ circuits, namely, the NCC and the MCC. We have previously commented that, under physiological conditions, the two Ca2+ cycling mechanisms are tightly coupled in order to secure neuronal functioning and bioenergetics. In vulnerable neurons such as those stressed by veratridine, a dual-acting compound such as CGP37157 could restore the coupling between NCC (by partially blocking the VDCCs) and MCC (by partial blockade of MNCX), thereby enhancing ATP synthesis and improving the viability of neurons. Whether this hypothesis can be extrapolated to relevant in vitro and in vivo models of neurodegenerative diseases and stroke remains to be explored. Nevertheless, it could be interesting to design and synthesize multitarget compounds that could add to the Ca2+-stabilizing properties of CGP37157 if endowed with additional disease-specific properties and/or antioxidant effects. Some of those compounds are described next.
Multitarget Neuroprotective Compounds to Correct the Disruption of Calcium Homeostasis and Oxidative Stress
As discussed above, disruption of Ca2+ homeostasis enhances oxidative stress and leads to death of vulnerable neurons in AD. Therefore, we raise the hypothesis that a multitarget compound having the capacity to slow down futile NCC and MCC, and endowed with an additional antioxidant activity, could have the potential to rescue from death those vulnerable neurons.
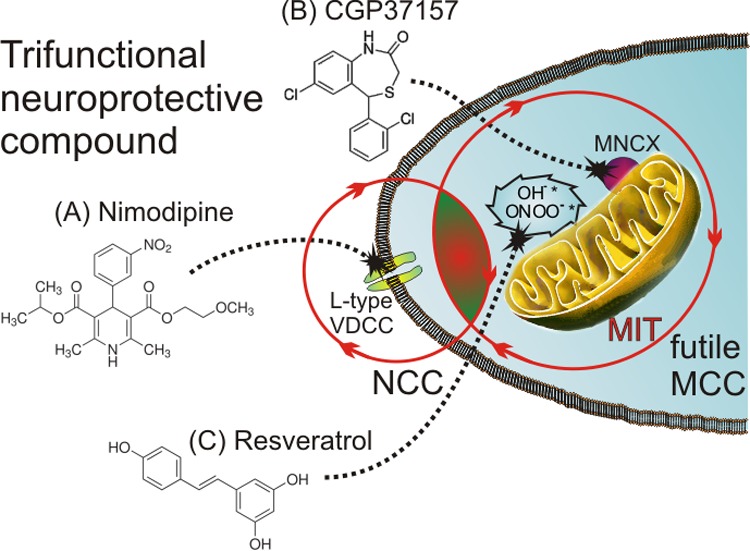
We have previously used l-glutamic acid as a linker for different bioactive moieties.76,77 Here we propose a similar approach, also using l-glutamic acid as a linker for the following bioactive moieties: (1) a DHP group (i.e., nimodipine-like) to target the L-type VDCCs, thus mitigating the excess Ca2+ entry into the soma of vulnerable neurons; (2) a benzothiazepine group (i.e., CGP37157-like) to target the MNCX and abort the futile MCC in vulnerable neurons; and (3) a polyphenolic group (i.e., resveratrol-like) to combat excess free radical production in vulnerable neurons (Figure 5). An aliphatic group linked with an ester bond could facilitate the crossing of the blood-brain barrier (BBB) of this multifunctional compound.
Figure 5.
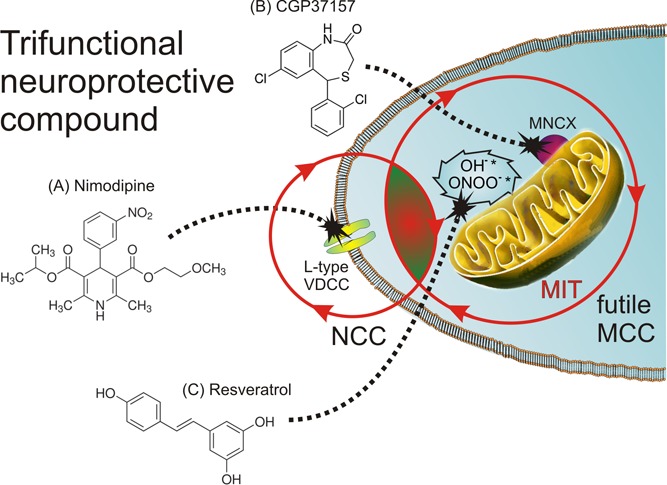
Multitarget compounds acting on L-type VDCCs of soma of vulnerable neurons (A, a DHP-like moiety to mitigate excess Ca2+ entry) and on the MNCX (B, a benzothiazepine moiety to abort the futile MCC) as well as on oxidative stress linked to distorted Ca2+ homeostasis (C, a polyphenol-like moiety to sequester excess free radicals) could be a more efficacious strategy to rescue neurons from death in AD. In this scheme, we propose that a trifunctional compound (i.e., a hybrid molecule with dihydropyridine, benzothiazepine, and polyphenol moieties) could effectively mitigate the augmented neuronal (NCC) and mitochondrial Ca2+ cycling (MCC), thereby exerting a protective survival effects on vulnerable neurons in AD.
Conclusions and Perspectives
Single-target ligands for specific disease-linked proteins and altered signaling pathways have shown neuroprotection efficacy on in vitro and in vivo models of AD; however, they have repeatedly failed in clinical trials. Thus, multitarget compounds acting on specific disease proteins and pathways, but also on distal pathways (i.e., dysregulation of neuronal and mitochondrial Ca2+ cycling, oxidative stress, and impairment of mitochondrial bioenergetics), could be more efficacious to rescue vulnerable neurons from death in AD (Figure 5). Whether this supposition may be true requires much research efforts in the coming years, both in preclinical and clinical setups.
Glossary
Abbreviations
- Aβ
amyloid beta
- AChE
acetylcholinesterase
- AD
Alzheimer's disease
- AMPAR
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor
- APP
amyloid precursor protein
- [Ca2+]c
cytosolic Ca2+ concentrations
- [Ca2+]m
mitochondrial matrix Ca2+ concentrations
- CALHM1
Ca2+ homeostasis modulator 1
- CBPs
Ca2+ binding proteins
- CDK5
cyclin-dependent kinase 5
- CICR
Ca2+-induced Ca2+ release
- CK1
casein kinase 1
- DHPs
1,4-dihydropyridines
- ER
endoplasmic reticulum
- ERK2
extracellular signal-regulated kinase 2
- GSK3β
glycogen synthase kinase-3-beta
- InsP3R
inositol tris-phosphate receptor channels
- MCC
mitochondrial Ca2+ cycling
- MCU
mitochondrial Ca2+ uniporter
- MNCX
mitochondrial Na+/Ca2+ exchanger
- MPTP
mitochondrial permeability transition pore
- NCC
neuronal Ca2+ cycling
- NMDAR
N-methyl-d-aspartate receptor
- PAS
peripheral anionic site
- PD
Parkinson's disease
- PS1
presenilin-1
- ROS
reactive oxygen species
- RyR
ryanodine receptor
- SERCA
sarco(endo)plasmic reticulum Ca2+-ATPase
- SOCs
store-operated Ca2+ channels
- VDCC
voltage-dependent Ca2+ channels.
The work from the authors' laboratory mentioned in this review was supported by the following grants to A.G.G.: (1) FUNDACION CIEN, I.S. Carlos III; (2) SAF 2010-21795 (Ministerio de Economía y Competitividad); (3) RETICS RD06/0009, I.S. Carlos III; (4) Fundación Eugenio Rodríguez Pascual; (5) Cátedra de Patrocinio, UAM/Bioibérica on Chronic Inflammation and Cytoprotection (CABICYC). We also thank the continued support of Fundación Teófilo Hernando.
The authors declare no competing financial interest.
Author Contributions
⊥ These authors are equal contributors.
Special Issue
References
- Chad J. E.; Eckert R. (1984) Calcium domains associated with individual channels can account for anomalous voltage relations of Ca-dependent responses. Biophys. J. 45(5), 993–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon S. M.; Llinas R. R. (1985) Compartmentalization of the submembrane calcium activity during calcium influx and its significance in transmitter release. Biophys. J. 48(3), 485–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neher E. (1998) Vesicle pools and Ca2+ microdomains: new tools for understanding their roles in neurotransmitter release. Neuron 20(3), 389–399. [DOI] [PubMed] [Google Scholar]
- Neher E. (1998) Usefulness and limitations of linear approximations to the understanding of Ca+2 signals. Cell Calcium 24(5–6), 345–357. [DOI] [PubMed] [Google Scholar]
- Berridge M. J.; Lipp P.; Bootman M. D. (2000) The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 1(1), 11–21. [DOI] [PubMed] [Google Scholar]
- Csordas G.; Thomas A. P.; Hajnoczky G. (1999) Quasi-synaptic calcium signal transmission between endoplasmic reticulum and mitochondria. EMBO J. 18(1), 96–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csordas G.; Hajnoczky G. (2003) Plasticity of mitochondrial calcium signaling. J. Biol. Chem. 278(43), 42273–42282. [DOI] [PubMed] [Google Scholar]
- Rizzuto R.; Pinton P.; Carrington W.; Fay F. S.; Fogarty K. E.; Lifshitz L. M.; Tuft R. A.; Pozzan T. (1998) Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science 280(5370), 1763–1766. [DOI] [PubMed] [Google Scholar]
- Szabadkai G.; Simoni A. M.; Rizzuto R. (2003) Mitochondrial Ca2+ uptake requires sustained Ca2+ release from the endoplasmic reticulum. J. Biol. Chem. 278(17), 15153–15161. [DOI] [PubMed] [Google Scholar]
- Thomas A. P.; Bird G. S.; Hajnoczky G.; Robb-Gaspers L. D.; Putney J. W. Jr. (1996) Spatial and temporal aspects of cellular calcium signaling. FASEB J. 10(13), 1505–1517. [PubMed] [Google Scholar]
- Garcia A. G.; Garcia-De-Diego A. M.; Gandia L.; Borges R.; Garcia-Sancho J. (2006) Calcium signaling and exocytosis in adrenal chromaffin cells. Physiol. Rev. 86(4), 1093–1131. [DOI] [PubMed] [Google Scholar]
- de Diego A. M.; Gandia L. (2008) A physiological view of the central and peripheral mechanisms that regulate the release of catecholamines at the adrenal medulla. Acta Physiol. 192(2), 287–301. [DOI] [PubMed] [Google Scholar]
- Herrington J.; Park Y. B.; Babcock D. F.; Hille B. (1996) Dominant role of mitochondria in clearance of large Ca2+ loads from rat adrenal chromaffin cells. Neuron 16(1), 219–228. [DOI] [PubMed] [Google Scholar]
- Montero M.; Alonso M. T.; Carnicero E.; Cuchillo-Ibanez I.; Albillos A.; Garcia A. G.; Garcia-Sancho J.; Alvarez J. (2000) Chromaffin-cell stimulation triggers fast millimolar mitochondrial Ca2+ transients that modulate secretion. Nat. Cell Biol. 2(2), 57–61. [DOI] [PubMed] [Google Scholar]
- Villalobos C.; Nunez L.; Montero M.; Garcia A. G.; Alonso M. T.; Chamero P.; Alvarez J.; Garcia-Sancho J. (2002) Redistribution of Ca2+ among cytosol and organella during stimulation of bovine chromaffin cells. FASEB J. 16(3), 343–353. [DOI] [PubMed] [Google Scholar]
- Xu T.; Naraghi M.; Kang H.; Neher E. (1997) Kinetic studies of Ca2+ binding and Ca2+ clearance in the cytosol of adrenal chromaffin cells. Biophys. J. 73(1), 532–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunter T. E.; Gunter K. K.; Sheu S. S.; Gavin C. E. (1994) Mitochondrial calcium transport: physiological and pathological relevance. Am. J. Physiol. 267(2 Pt 1), C313–C339. [DOI] [PubMed] [Google Scholar]
- Rizzuto R.; Bernardi P.; Pozzan T. (2000) Mitochondria as all-round players of the calcium game. J. Physiol. 529, 37–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villalobos C.; Nunez L.; Chamero P.; Alonso M. T.; Garcia-Sancho J. (2001) Mitochondrial [Ca2+] oscillations driven by local high [Ca2+] domains generated by spontaneous electric activity. J. Biol. Chem. 276(43), 40293–40297. [DOI] [PubMed] [Google Scholar]
- Alonso M. T.; Barrero M. J.; Michelena P.; Carnicero E.; Cuchillo I.; Garcia A. G.; Garcia-Sancho J.; Montero M.; Alvarez J. (1999) Ca2+-induced Ca2+ release in chromaffin cells seen from inside the ER with targeted aequorin. J. Cell Biol. 144(2), 241–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Ruden L.; Neher E. (1993) A Ca-dependent early step in the release of catecholamines from adrenal chromaffin cells. Science 262(5136), 1061–1065. [DOI] [PubMed] [Google Scholar]
- Goedert M.; Spillantini M. G. (2006) A century of Alzheimer’s disease. Science 314(5800), 777–781. [DOI] [PubMed] [Google Scholar]
- Alzheimer A. (1906) Uber eine einegartigen sweren Erkrank ungsprozeb der Hirnrinde. Neurobiologisches Centralblatt 23, 1129–1136. [Google Scholar]
- Giannakopoulos P.; Kovari E.; Gold G.; von Gunten A.; Hof P. R.; Bouras C. (2009) Pathological substrates of cognitive decline in Alzheimer’s disease. Front. Neurol. Neurosci. 24, 20–29. [DOI] [PubMed] [Google Scholar]
- Inestrosa N. C.; Alvarez A.; Perez C. A.; Moreno R. D.; Vicente M.; Linker C.; Casanueva O. I.; Soto C.; Garrido J. (1996) Acetylcholinesterase accelerates assembly of amyloid-beta-peptides into Alzheimer’s fibrils: possible role of the peripheral site of the enzyme. Neuron 16(4), 881–891. [DOI] [PubMed] [Google Scholar]
- Grimes C. A.; Jope R. S. (2001) The multifaceted roles of glycogen synthase kinase 3beta in cellular signaling. Prog. Neurobiol. 65(4), 391–426. [DOI] [PubMed] [Google Scholar]
- Takashima A.; Honda T.; Yasutake K.; Michel G.; Murayama O.; Murayama M.; Ishiguro K.; Yamaguchi H. (1998) Activation of tau protein kinase I/glycogen synthase kinase-3beta by amyloid beta peptide (25–35) enhances phosphorylation of tau in hippocampal neurons. Neurosci. Res. 31(4), 317–323. [DOI] [PubMed] [Google Scholar]
- Hardy J.; Selkoe D. J. (2002) The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297(5580), 353–356. [DOI] [PubMed] [Google Scholar]
- Seabrook G. R.; Ray W. J.; Shearman M.; Hutton M. (2007) Beyond amyloid: the next generation of Alzheimer’s disease therapeutics. Mol. Interventions 7(5), 261–270. [DOI] [PubMed] [Google Scholar]
- Khachaturian Z. S. (1989) Calcium, membranes, aging, and Alzheimer’s disease. Introduction and overview. Ann. N.Y. Acad. Sci. 568, 1–4. [DOI] [PubMed] [Google Scholar]
- LaFerla F. M. (2002) Calcium dyshomeostasis and intracellular signalling in Alzheimer’s disease. Nat. Rev. Neurosci. 3(11), 862–872. [DOI] [PubMed] [Google Scholar]
- Marx J. (2007) Alzheimer’s disease. Fresh evidence points to an old suspect: calcium. Science 318(5849), 384–385. [DOI] [PubMed] [Google Scholar]
- Bezprozvanny I. (2009) Calcium signaling and neurodegenerative diseases. Trends Mol. Med. 15(3), 89–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezprozvanny I.; Mattson M. P. (2008) Neuronal calcium mishandling and the pathogenesis of Alzheimer’s disease. Trends Neurosci. 31(9), 454–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson M. P. (2010) ER calcium and Alzheimer’s disease: in a state of flux. Sci. Signaling 3(114), pe10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demuro A.; Parker I.; Stutzmann G. E. (2010) Calcium signaling and amyloid toxicity in Alzheimer disease. J. Biol. Chem. 285(17), 12463–12468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arispe N.; Pollard H. B.; Rojas E. (1993) Giant multilevel cation channels formed by Alzheimer disease amyloid beta-protein [A beta P-(1–40)] in bilayer membranes. Proc. Natl. Acad. Sci. U.S.A. 90(22), 10573–10577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee G.; Pollard H. B.; Arispe N. (2002) Annexin 5 and apolipoprotein E2 protect against Alzheimer’s amyloid-beta-peptide cytotoxicity by competitive inhibition at a common phosphatidylserine interaction site. Peptides 23(7), 1249–1263. [DOI] [PubMed] [Google Scholar]
- Simakova O.; Arispe N. J. (2007) The cell-selective neurotoxicity of the Alzheimer’s Abeta peptide is determined by surface phosphatidylserine and cytosolic ATP levels. Membrane binding is required for Abeta toxicity. J. Neurosci. 27(50), 13719–13729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuchibhotla K. V.; Goldman S. T.; Lattarulo C. R.; Wu H. Y.; Hyman B. T.; Bacskai B. J. (2008) Abeta plaques lead to aberrant regulation of calcium homeostasis in vivo resulting in structural and functional disruption of neuronal networks. Neuron 59(2), 214–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Felice F. G.; Velasco P. T.; Lambert M. P.; Viola K.; Fernandez S. J.; Ferreira S. T.; Klein W. L. (2007) Abeta oligomers induce neuronal oxidative stress through an N-methyl-D-aspartate receptor-dependent mechanism that is blocked by the Alzheimer drug memantine. J. Biol. Chem. 282(15), 11590–11601. [DOI] [PubMed] [Google Scholar]
- Shankar G. M.; Bloodgood B. L.; Townsend M.; Walsh D. M.; Selkoe D. J.; Sabatini B. L. (2007) Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J. Neurosci. 27(11), 2866–2875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu H.; Nelson O.; Bezprozvanny A.; Wang Z.; Lee S. F.; Hao Y. H.; Serneels L.; De Strooper B.; Yu G.; Bezprozvanny I. (2006) Presenilins form ER Ca2+ leak channels, a function disrupted by familial Alzheimer’s disease-linked mutations. Cell 126(5), 981–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green K. N.; Demuro A.; Akbari Y.; Hitt B. D.; Smith I. F.; Parker I.; LaFerla F. M. (2008) SERCA pump activity is physiologically regulated by presenilin and regulates amyloid beta production. J. Cell Biol. 181(7), 1107–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung K. H.; Shineman D.; Muller M.; Cardenas C.; Mei L.; Yang J.; Tomita T.; Iwatsubo T.; Lee V. M.; Foskett J. K. (2008) Mechanism of Ca2+ disruption in Alzheimer’s disease by presenilin regulation of InsP3 receptor channel gating. Neuron 58(6), 871–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherrington R.; Rogaev E. I.; Liang Y.; Rogaeva E. A.; Levesque G.; Ikeda M.; Chi H.; Lin C.; Li G.; Holman K.; Tsuda T.; Mar L.; Foncin J. F.; Bruni A. C.; Montesi M. P.; Sorbi S.; Rainero I.; Pinessi L.; Nee L.; Chumakov I.; Pollen D.; Brookes A.; Sanseau P.; Polinsky R. J.; Wasco W.; Da Silva H. A.; Haines J. L.; Perkicak-Vance M. A.; Tanzi R. E.; Roses A. D.; Fraser P. E.; Rommens J. M.; St George-Hyslop P. H. (1995) Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature 375(6534), 754–760. [DOI] [PubMed] [Google Scholar]
- Duff K.; Eckman C.; Zehr C.; Yu X.; Prada C.-M.; Perez-Tur J.; Hutton M.; Buee L.; Harigaya Y.; Yager D.; Morgan D.; Gordon M. N.; Holcomb L.; Refolo L.; Zenk B.; Hardy J.; Younkin S. (1996) Increased amyloid-42(43) in brains of mice expressing mutant presenilin 1. Nature 383, 710–713. [DOI] [PubMed] [Google Scholar]
- Stutzmann G. E.; Caccamo A.; LaFerla F. M.; Parker I. (2004) Dysregulated IP3 signaling in cortical neurons of knock-in mice expressing an Alzheimer’s-linked mutation in presenilin1 results in exaggerated Ca2+ signals and altered membrane excitability. J. Neurosci. 24(2), 508–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson O.; Tu H.; Lei T.; Bentahir M.; de Strooper B.; Bezprozvanny I. (2007) Familial Alzheimer disease-linked mutations specifically disrupt Ca2+ leak function of presenilin 1. J. Clin. Invest. 117(5), 1230–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zatti G.; Burgo A.; Giacomello M.; Barbiero L.; Ghidoni R.; Sinigaglia G.; Florean C.; Bagnoli S.; Binetti G.; Sorbi S.; Pizzo P.; Fasolato C. (2006) Presenilin mutations linked to familial Alzheimer’s disease reduce endoplasmic reticulum and Golgi apparatus calcium levels. Cell Calcium 39(6), 539–550. [DOI] [PubMed] [Google Scholar]
- Leissring M. A.; Akbari Y.; Fanger C. M.; Cahalan M. D.; Mattson M. P.; LaFerla F. M. (2000) Capacitative calcium entry deficits and elevated luminal calcium content in mutant presenilin-1 knockin mice. J. Cell Biol. 149(4), 793–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leissring M. A.; Paul B. A.; Parker I.; Cotman C. W.; LaFerla F. M. (1999) Alzheimer’s presenilin-1 mutation potentiates inositol 1,4,5-trisphosphate-mediated calcium signaling in Xenopus oocytes. J. Neurochem. 72(3), 1061–1068. [DOI] [PubMed] [Google Scholar]
- Ito E.; Oka K.; Etcheberrigaray R.; Nelson T. J.; McPhie D. L.; Tofel-Grehl B.; Gibson G. E.; Alkon D. L. (1994) Internal Ca2+ mobilization is altered in fibroblasts from patients with Alzheimer disease. Proc. Natl. Acad. Sci. U.S.A. 91(2), 534–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stutzmann G. E.; Smith I.; Caccamo A.; Oddo S.; Laferla F. M.; Parker I. (2006) Enhanced ryanodine receptor recruitment contributes to Ca2+ disruptions in young, adult, and aged Alzheimer’s disease mice. J. Neurosci. 26(19), 5180–5189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo A. S.; Cheng I.; Chung S.; Grenfell T. Z.; Lee H.; Pack-Chung E.; Handler M.; Shen J.; Xia W.; Tesco G.; Saunders A. J.; Ding K.; Frosch M. P.; Tanzi R. E.; Kim T. W. (2000) Presenilin-mediated modulation of capacitative calcium entry. Neuron 27(3), 561–572. [DOI] [PubMed] [Google Scholar]
- Chan S. L.; Mayne M.; Holden C. P.; Geiger J. D.; Mattson M. P. (2000) Presenilin-1 mutations increase levels of ryanodine receptors and calcium release in PC12 cells and cortical neurons. J. Biol. Chem. 275(24), 18195–18200. [DOI] [PubMed] [Google Scholar]
- Rybalchenko V.; Hwang S. Y.; Rybalchenko N.; Koulen P. (2008) The cytosolic N-terminus of presenilin-1 potentiates mouse ryanodine receptor single channel activity. Int. J. Biochem. Cell Biol. 40(1), 84–97. [DOI] [PubMed] [Google Scholar]
- Cai C.; Lin P.; Cheung K. H.; Li N.; Levchook C.; Pan Z.; Ferrante C.; Boulianne G. L.; Foskett J. K.; Danielpour D.; Ma J. (2006) The presenilin-2 loop peptide perturbs intracellular Ca2+ homeostasis and accelerates apoptosis. J. Biol. Chem. 281(24), 16649–16655. [DOI] [PubMed] [Google Scholar]
- Vosler P. S.; Brennan C. S.; Chen J. (2008) Calpain-mediated signaling mechanisms in neuronal injury and neurodegeneration. Mol. Neurobiol. 38(1), 78–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinchese F.; Fa M.; Liu S.; Zhang H.; Hidalgo A.; Schmidt S. D.; Yamaguchi H.; Yoshii N.; Mathews P. M.; Nixon R. A.; Arancio O. (2008) Inhibition of calpains improves memory and synaptic transmission in a mouse model of Alzheimer disease. J. Clin. Invest. 118(8), 2796–2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palop J. J.; Jones B.; Kekonius L.; Chin J.; Yu G. Q.; Raber J.; Masliah E.; Mucke L. (2003) Neuronal depletion of calcium-dependent proteins in the dentate gyrus is tightly linked to Alzheimer’s disease-related cognitive deficits. Proc. Natl. Acad. Sci. U.S.A. 100(16), 9572–9577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreses-Werringloer U.; Lambert J. C.; Vingtdeux V.; Zhao H.; Vais H.; Siebert A.; Jain A.; Koppel J.; Rovelet-Lecrux A.; Hannequin D.; Pasquier F.; Galimberti D.; Scarpini E.; Mann D.; Lendon C.; Campion D.; Amouyel P.; Davies P.; Foskett J. K.; Campagne F.; Marambaud P. (2008) A polymorphism in CALHM1 influences Ca2+ homeostasis, Abeta levels, and Alzheimer’s disease risk. Cell 133(7), 1149–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertram L.; Schjeide B. M.; Hooli B.; Mullin K.; Hiltunen M.; Soininen H.; Ingelsson M.; Lannfelt L.; Blacker D.; Tanzi R. E. (2008) No association between CALHM1 and Alzheimer’s disease risk. Cell 135(6), 993–994author reply 994–996.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno-Ortega A. J.; Ruiz-Nuno A.; Garcia A. G.; Cano-Abad M. F. (2010) Mitochondria sense with different kinetics the calcium entering into HeLa cells through calcium channels CALHM1 and mutated P86L-CALHM1. Biochem. Biophys. Res. Commun. 391(1), 722–726. [DOI] [PubMed] [Google Scholar]
- Sanz-Blasco S.; Valero R. A.; Rodriguez-Crespo I.; Villalobos C.; Nunez L. (2008) Mitochondrial Ca2+ overload underlies Abeta oligomers neurotoxicity providing an unexpected mechanism of neuroprotection by NSAIDs. PLoS One 3(7), e2718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arispe N.; Diaz J. C.; Simakova O. (2007) Abeta ion channels. Prospects for treating Alzheimer’s disease with Abeta channel blockers. Biochim. Biophys. Acta 1768(8), 1952–1965. [DOI] [PubMed] [Google Scholar]
- Li S.; Hong S.; Shepardson N. E.; Walsh D. M.; Shankar G. M.; Selkoe D. (2009) Soluble oligomers of amyloid Beta protein facilitate hippocampal long-term depression by disrupting neuronal glutamate uptake. Neuron 62(6), 788–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons C. G.; Danysz W.; Quack G. (1999) Memantine is a clinically well tolerated N-methyl-D-aspartate (NMDA) receptor antagonist--a review of preclinical data. Neuropharmacology 38(6), 735–767. [DOI] [PubMed] [Google Scholar]
- Parsons C. G.; Stoffler A.; Danysz W. (2007) Memantine: a NMDA receptor antagonist that improves memory by restoration of homeostasis in the glutamatergic system--too little activation is bad, too much is even worse. Neuropharmacology 53(6), 699–723. [DOI] [PubMed] [Google Scholar]
- Rosini M.; Simoni E.; Bartolini M.; Cavalli A.; Ceccarini L.; Pascu N.; McClymont D. W.; Tarozzi A.; Bolognesi M. L.; Minarini A.; Tumiatti V.; Andrisano V.; Mellor I. R.; Melchiorre C. (2008) Inhibition of acetylcholinesterase, beta-amyloid aggregation, and NMDA receptors in Alzheimer’s disease: a promising direction for the multi-target-directed ligands gold rush. J. Med. Chem. 51(15), 4381–4384. [DOI] [PubMed] [Google Scholar]
- Lipton S. A. (2006) Paradigm shift in neuroprotection by NMDA receptor blockade: memantine and beyond. Nat. Rev. Drug Discovery 5(2), 160–170. [DOI] [PubMed] [Google Scholar]
- Berridge M. J. (2010) Calcium hypothesis of Alzheimer’s disease. Pflugers Arch. 459(3), 441–449. [DOI] [PubMed] [Google Scholar]
- Leon R.; Marco-Contelles J.; Garcia A. G.; Villarroya M. (2005) Synthesis, acetylcholinesterase inhibition and neuroprotective activity of new tacrine analogues. Bioorg. Med. Chem. 13(4), 1167–1175. [DOI] [PubMed] [Google Scholar]
- Marco J. L.; de los Rios C.; Garcia A. G.; Villarroya M.; Carreiras M. C.; Martins C.; Eleuterio A.; Morreale A.; Orozco M.; Luque F. J. (2004) Synthesis, biological evaluation and molecular modelling of diversely functionalized heterocyclic derivatives as inhibitors of acetylcholinesterase/butyrylcholinesterase and modulators of Ca2+ channels and nicotinic receptors. Bioorg. Med. Chem. 12(9), 2199–2218. [DOI] [PubMed] [Google Scholar]
- Marco-Contelles J.; Leon R.; de los Rios C.; Samadi A.; Bartolini M.; Andrisano V.; Huertas O.; Barril X.; Luque F. J.; Rodriguez-Franco M. I.; Lopez B.; Lopez M. G.; Garcia A. G.; Carreiras Mdo C.; Villarroya M. (2009) Tacripyrines, the first tacrine-dihydropyridine hybrids, as multitarget-directed ligands for the treatment of Alzheimer’s disease. J. Med. Chem. 52(9), 2724–2732. [DOI] [PubMed] [Google Scholar]
- Arce M. P.; Rodriguez-Franco M. I.; Gonzalez-Munoz G. C.; Perez C.; Lopez B.; Villarroya M.; Lopez M. G.; Garcia A. G.; Conde S. (2009) Neuroprotective and cholinergic properties of multifunctional glutamic acid derivatives for the treatment of Alzheimer’s disease. J. Med. Chem. 52(22), 7249–7257. [DOI] [PubMed] [Google Scholar]
- Maroto M.; de-Diego A. M. G.; Albinana E.; Fernandez-Morales J. C.; Caricati-Neto A.; Jurkiewicz A.; Yanez M.; Rodriguez-Franco M. I.; Conde S.; Arce M. P.; Hernandez-Guijo J. M.; Garcia A. G. (2011) Multi-target novel neuroprotective compound ITH33/IQM9.21 inhibits calcium entry, calcium signals and exocytosis. Cell Calcium 50(4), 359–369. [DOI] [PubMed] [Google Scholar]
- Lysko P. G.; Lysko K. A.; Webb C. L.; Feuerstein G.; Mason P. E.; Walter M. F.; Mason R. P. (1998) Neuroprotective activities of carvedilol and a hydroxylated derivative: role of membrane biophysical interactions. Biochem. Pharmacol. 56(12), 1645–1656. [DOI] [PubMed] [Google Scholar]
- Howlett D. R.; George A. R.; Owen D. E.; Ward R. V.; Markwell R. E. (1999) Common structural features determine the effectiveness of carvedilol, daunomycin and rolitetracycline as inhibitors of Alzheimer beta-amyloid fibril formation. Biochem. J. 343(Pt 2), 419–423. [PMC free article] [PubMed] [Google Scholar]
- Schott Y.; Decker M.; Rommelspacher H.; Lehmann J. (2006) 6-Hydroxy- and 6-methoxy-beta-carbolines as acetyl- and butyrylcholinesterase inhibitors. Bioorg. Med. Chem. Lett. 16(22), 5840–5843. [DOI] [PubMed] [Google Scholar]
- Rook Y.; Schmidtke K. U.; Gaube F.; Schepmann D.; Wunsch B.; Heilmann J.; Lehmann J.; Winckler T. (2010) Bivalent beta-carbolines as potential multitarget anti-Alzheimer agents. J. Med. Chem. 53(9), 3611–3617. [DOI] [PubMed] [Google Scholar]
- Alonso A. C.; Li B.; Grundke-Iqbal I.; Iqbal K. (2008) Mechanism of tau-induced neurodegeneration in Alzheimer disease and related tauopathies. Curr. Alzheimer Res. 5(4), 375–384. [DOI] [PubMed] [Google Scholar]
- Bain J.; McLauchlan H.; Elliott M.; Cohen P. (2003) The specificities of protein kinase inhibitors: an update. Biochem. J. 371(Pt 1), 199–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellmich M. R.; Kennison J. A.; Hampton L. L.; Battey J. F. (1994) Cloning and characterization of the Drosophila melanogaster CDK5 homolog. FEBS Lett. 356(2–3), 317–321. [DOI] [PubMed] [Google Scholar]
- Tsai L. H.; Delalle I.; Caviness V. S. Jr.; Chae T.; Harlow E. (1994) p35 is a neural-specific regulatory subunit of cyclin-dependent kinase 5. Nature 371(6496), 419–423. [DOI] [PubMed] [Google Scholar]
- Yan Z.; Chi P.; Bibb J. A.; Ryan T. A.; Greengard P. (2002) Roscovitine: a novel regulator of P/Q-type calcium channels and transmitter release in central neurons. J. Physiol. 540(Pt 3), 761–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buraei Z.; Anghelescu M.; Elmslie K. S. (2005) Slowed N-type calcium channel (CaV2.2) deactivation by the cyclin-dependent kinase inhibitor roscovitine. Biophys. J. 89(3), 1681–1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeStefino N. R.; Pilato A. A.; Dittrich M.; Cherry S. V.; Cho S.; Stiles J. R.; Meriney S. D. (2010) (R)-roscovitine prolongs the mean open time of unitary N-type calcium channel currents. Neuroscience 167(3), 838–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crompton M. (1985) The Regulation of Mitochondrial Calcium-Transport in Heart. Curr. Top. Membr. Transp. 25, 231–276. [Google Scholar]
- Denton R. M.; McCormack J. G. (1980) On the role of the calcium transport cycle in heart and other mammalian mitochondria. FEBS Lett. 119(1), 1–8. [DOI] [PubMed] [Google Scholar]
- Hansford R. G.; Hogue B.; Prokopczuk A.; Wasilewska E.; Lewartowski B. (1990) Activation of pyruvate dehydrogenase by electrical stimulation, and low-Na+ perfusion of guinea-pig heart. Biochim. Biophys. Acta 1018(2–3), 282–286. [DOI] [PubMed] [Google Scholar]
- Miyata H.; Silverman H. S.; Sollott S. J.; Lakatta E. G.; Stern M. D.; Hansford R. G. (1991) Measurement of mitochondrial free Ca2+ concentration in living single rat cardiac myocytes. Am. J. Physiol. 261(4 Pt 2), H1123–H1134. [DOI] [PubMed] [Google Scholar]
- McCormack J. G.; England P. J. (1983) Ruthenium Red inhibits the activation of pyruvate dehydrogenase caused by positive inotropic agents in the perfused rat heart. Biochem. J. 214(2), 581–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox D. A.; Matlib M. A. (1993) Modulation of intramitochondrial free Ca2+ concentration by antagonists of Na+-Ca2+ exchange. Trends Pharmacol. Sci. 14(11), 408–413. [DOI] [PubMed] [Google Scholar]
- Vaghy P. L.; Johnson J. D.; Matlib M. A.; Wang T.; Schwartz A. (1982) Selective-Inhibition of Na+-Induced Ca-2+ Release from Heart-Mitochondria by Diltiazem and Certain Other Ca-2+ Antagonist Drugs. J. Biol. Chem. 257(11), 6000–6002. [PubMed] [Google Scholar]
- Matlib M. A.; Lee S. W.; Depover A.; Schwartz A. (1983) A Specific Inhibitory-Action of Certain Benzothiazepines and Benzodiazepines on the Sodium-Calcium Exchange Process of Heart and Brain Mitochondria. Eur. J. Pharmacol. 89(3–4), 327–328. [DOI] [PubMed] [Google Scholar]
- Matlib M. A.; Schwartz A. (1983) Selective Effects of Diltiazem, a Benzothiazepine Calcium-Channel Blocker, and Diazepam, and Other Benzodiazepines on the Na+/Ca2+ Exchange Carrier System of Heart and Brain Mitochondria. Life Sci. 32(25), 2837–2842. [DOI] [PubMed] [Google Scholar]
- Matlib M. A.; Doane J. D.; Sperelakis N.; Riccipponeto F. (1985) Clonazepam and Diltiazem Both Inhibit Sodium-Calcium Exchange of Mitochondria, but Only Diltiazem Inhibits the Slow Action-Potentials of Cardiac Muscles. Biochem. Biophys. Res. Commun. 128(1), 290–296. [DOI] [PubMed] [Google Scholar]
- Chiesi M.; Schwaller R.; Eichenberger K. (1988) Structural Dependency of the Inhibitory-Action of Benzodiazepines and Related-Compounds on the Mitochondrial Na+-Ca2+ Exchanger. Biochem. Pharmacol. 37(22), 4399–4403. [DOI] [PubMed] [Google Scholar]
- Cox D. A.; Matlib M. A. (1993) A Role for the Mitochondrial Na+-Ca2+ Exchanger in the Regulation of Oxidative-Phosphorylation in Isolated Heart-Mitochondria. J. Biol. Chem. 268(2), 938–947. [PubMed] [Google Scholar]
- Gandhi S.; Wood-Kaczmar A.; Yao Z.; Plun-Favreau H.; Deas E.; Klupsch K.; Downward J.; Latchman D. S.; Tabrizi S. J.; Wood N. W.; Duchen M. R.; Abramov A. Y. (2009) PINK1-Associated Parkinson’s Disease Is Caused by Neuronal Vulnerability to Calcium-induced Cell Death. Mol. Cell 33(5), 627–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallo V.; Kingsbury A.; Balazs R.; Jorgensen O. S. (1987) The Role of Depolarization in the Survival and Differentiation of Cerebellar Granule Cells in Culture. J. Neurosci. 7(7), 2203–2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orozco C.; Garcia-De-Diego A. M.; Arias E.; Hernandez-Guijo J. M.; Garcia A. G.; Villarroya M.; Lopez M. G. (2006) Depolarization preconditioning produces cytoprotection against veratridine-induced chromaffin cell death. Eur. J. Pharmacol. 553(1–3), 28–38. [DOI] [PubMed] [Google Scholar]
- Koh J. Y.; Cotman C. W. (1992) Programmed cell death: its possible contribution to neurotoxicity mediated by calcium channel antagonists. Brain Res. 587(2), 233–240. [DOI] [PubMed] [Google Scholar]
- Novalbos J.; Abad-Santos F.; Zapater P.; Cano-Abad M. F.; Moradiellos J.; Sanchez-Garcia P.; Garcia A. G. (1999) Effects of dotarizine and flunarizine on chromaffin cell viability and cytosolic Ca2+. Eur. J. Pharmacol. 366(2–3), 309–317. [DOI] [PubMed] [Google Scholar]
- Koike T.; Martin D. P.; Johnson E. M. (1989) Role of Ca-2+ Channels in the Ability of Membrane Depolarization to Prevent Neuronal Death Induced by Trophic-Factor Deprivation - Evidence That Levels of Internal Ca-2+ Determine Nerve Growth-Factor Dependence of Sympathetic-Ganglion Cells. Proc. Natl. Acad. Sci. U.S.A. 86(16), 6421–6425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls D. G. (1985) A role for the mitochondrion in the protection of cells against calcium overload?. Prog. Brain. Res. 63, 97–106. [DOI] [PubMed] [Google Scholar]
- White R. J.; Reynolds I. J. (1995) Mitochondria and Na+/Ca2+ exchange buffer glutamate-induced calcium loads in cultured cortical neurons. J. Neurosci. 15(2), 1318–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauwels P. J.; Vanassouw H. P.; Leysen J. E.; Janssen P. A. J. (1989) Ca-2+-Mediated Neuronal Death in Rat-Brain Neuronal Cultures by Veratridine - Protection by Flunarizine. Mol. Pharmacol. 36(4), 525–531. [PubMed] [Google Scholar]
- Deri Z.; Adamvizi V. (1993) Detection of Intracellular Free Na+ Concentration of Synaptosomes by a Fluorescent Indicator, Na+-Binding Benzofuran Isophthalate - the Effect of Veratridine, Ouabain, and Alpha-Latrotoxin. J. Neurochem. 61(3), 818–825. [DOI] [PubMed] [Google Scholar]
- Rose C. R.; Ransom B. R. (1997) Regulation of intracellular sodium in cultured rat hippocampal neurones. J. Physiol. 499(Pt 3), 573–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez M. G.; Artalejo A. R.; Garcia A. G.; Neher E.; Garcia-Sancho J. (1995) Veratridine-induced oscillations of cytosolic calcium and membrane potential in bovine chromaffin cells. J. Physiol. 482(Pt 1), 15–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maroto R.; De la Fuente M. T.; Artalejo A. R.; Abad F.; Lopez M. G.; Garcia-Sancho J.; Garcia A. G. (1994) Effects of Ca2+ channel antagonists on chromaffin cell death and cytosolic Ca2+ oscillations induced by veratridine. Eur. J. Pharmacol. 270(4), 331–339. [DOI] [PubMed] [Google Scholar]
- Nicolau S. M.; de Diego A. M.; Cortes L.; Egea J.; Gonzalez J. C.; Mosquera M.; Lopez M. G.; Hernandez-Guijo J. M.; Garcia A. G. (2009) Mitochondrial Na+/Ca2+-exchanger blocker CGP37157 protects against chromaffin cell death elicited by veratridine. J. Pharmacol. Exp. Ther. 330(3), 844–854. [DOI] [PubMed] [Google Scholar]
- Dykens J. A. (1994) Isolated cerebral and cerebellar mitochondria produce free radicals when exposed to elevated Ca2+ and Na+: implications for neurodegeneration. J. Neurochem. 63(2), 584–591. [DOI] [PubMed] [Google Scholar]
- Nicolau S. M.; Egea J.; Lopez M. G.; Garcia A. G. (2010) Mitochondrial Na+/Ca2+ exchanger, a new target for neuroprotection in rat hippocampal slices. Biochem. Biophys. Res. Commun. 400(1), 140–144. [DOI] [PubMed] [Google Scholar]