

Abstract
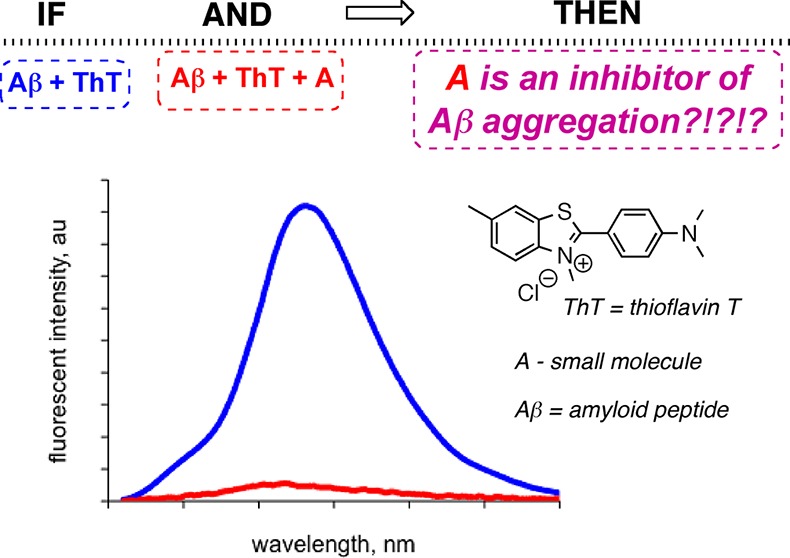
Dye-binding assays, such as those utilizing Congo red and thioflavin T, are among the most widely used tools to probe the aggregation of amyloidogenic biomolecules and for the evaluation of small molecule inhibitors of amyloid aggregation and fibrillization. A number of recent reports have indicated that these dye-binding assays could be prone to false positive effects when assessing inhibitors’ potential toward Aβ peptides, species involved in Alzheimer’s disease. Specifically, this review focuses on the application of thioflavin T for determining the efficiency of small molecule inhibitors of Aβ aggregation and addresses potential reasons that might be associated with the false positive effects in an effort to increase reliability of dye-binding assays.
Keywords: Alzheimer’s disease, amyloid peptide, small molecule inhibitor, dye-binding assay, thioflavin T, fluorescence
Amyloid is a general term for typically ordered, β-sheet rich, often insoluble, misfolded protein aggregates that are associated with certain pathological events. Amyloid β-proteins (Aβ), which are implicated in the occurrence and progression of Alzheimer’s disease (AD), are among the primary examples of amyloids. AD is an age related cognitive disorder associated with the decline of learning and memory brought on by a loss of neuronal function, and is characterized by the formation of neurofibrillary tangles and amyloid β-protein based deposits, that is, plaques.1 Several hypotheses have been proposed regarding the pathogenesis of the disease, which eventually leads to neurodegeneration and other clinical manifestations, such as memory impairment and confusion.2−4 This review, however, will only deal with the so-called amyloid cascade hypothesis that states that the accumulation of the amyloid β-protein drives the occurrence and progression of AD.5−8 The mechanism of the neurotoxicity associated with these peptides is complex and the exact pathways remain to be elucidated. However, it is known that one of the primary neurotoxic species involves the 40–42 amino acid long Aβ, that is, Aβ1–40 and Aβ1–42. Once formed from the cleavage of amyloid precursor protein (APP) by the β- and γ-secretases, Aβ1–40 and Aβ1–42 undergo an aggregation process, which ultimately leads to the insoluble aggregates and plaques.9−11 It has long been suggested that these insoluble aggregates, that is, Aβ fibrils, were the pathogenic culprits in AD.12 Yet, recent studies have shown that soluble oligomeric aggregates are indeed responsible for disruption of neuronal communication which can lead to cell death.13−15 Additionally, the conformational transition from a soluble unordered conformation to an ordered β-sheet rich species is linked to this neurotoxicity.16−20 Inhibition, reversal, and reduction of the aggregation of the Aβ peptides should constitute viable approaches to therapy and represent a significant area of AD related research. However, the nature of the soluble oligomers is complex and their recognition presents an enormous challenge21 and to date no cure has been found.
Due to the accessibility and ease of modification, small molecules with inhibitory ability on the oligomerization or other self-assembly processes that lead to conformational changes and/or the fibrillization of amyloid peptides should provide viable leads for therapeutic intervention.22 A few representative classes of known molecular inhibitors include polyphenols, anthracyclines, benzothiazoles, lignans, phenothiazines, polyene macrolides, rifamycins, steroids, terpenoids, porphyrins, and camphors.23,24 In addition, these motifs might be applicable for structural studies on protein folding, in general. Yet, it can be argued that compounds that are capable of interacting with a particular secondary structure and/or fold of the peptide and peptide assemblies would be a suitable starting point for understanding the behavior of both oligomeric and fibrillar amyloid-like species. There are many compounds with diverse structures and properties, which have been identified as having inhibitory properties on some part of the aggregation process. Inhibition could occur by stabilization of the monomeric or low molecular weight oligomeric forms of the peptide and thus decrease the rate of the conformational change to the β-sheet structure,25 or by direct inhibition of the fibrillization process which might happen independently.26
From the chemical point of view, it is imperative to unambiguously identify small molecules that could alter the amyloid self-assembly. This review is not intended to indicate whether the compounds mentioned herein are or are not inhibitors of amyloid aggregation or fibrillization, but rather to highlight the discrepancies that one might encounter when the antiaggregation ability of a small molecule is evaluated by using dye-binding assays.
Dye-Binding Assays to Examine Amyloid Aggregation
The use of small molecular probes (dyes) to monitor biological processes is well established as fluorescent dye binding assays have been used considerably for in vitro recognition of protein misfolding,27,28 and have important, practical implications for the understanding of the many human diseases that involve protein aggregation. In particular, dye-binding assays are often used to examine the amyloid self-assembly processes. Congo red (CR) and thioflavin T (ThT), shown in Figure 1, are the most commonly used dyes to study amyloid aggregation and fibril formation.29−31 Upon binding to the peptide assemblies, these dyes must exhibit distinct spectral properties (spectral shift and/or intensity change) as compared to their unbound state to be viable reporters of peptide aggregation processes.
Figure 1.

Structures of UV/vis and fluorescent dyes used for monitoring amyloid aggregation.
Due to practical considerations, specifically the ease of manipulation and high throughput screening possibilities, dye-binding assays often serve as the initial test of antiaggregation ability of small molecules. Primarily, two types of dye-binding assays have been utilized: (i) the dye is continuously present in the assay mixture (so-called continuous or in situ dye-binding assays);32 (ii) the dye is added to an aliquot of the amyloid-containing mixture at certain time intervals (so-called single time-point dilution method).32 From the experimental point of view, a typical assay is performed as follows: a given concentration of the peptide is incubated with and without a small molecule inhibitor in the presence or in the absence of the dye. In the case of the single time–point dilution assay, periodically, aliquots from peptide solutions are withdrawn and a solution of the dye is added, incubated for a given period of time. The corresponding spectra are measured and the fluorescence intensities of the dye are plotted as a function of time (Figure 2). Any deviation from the control sample along the time scale (a), that is, peptide aggregation in the absence of any additive, could be indicative of inhibition (b–d) or acceleration (e) of the aggregation processes (Figure 2). The corresponding compounds could be viewed as kinetic (W), as thermodynamic and kinetic (X), or as ideal (Y) inhibitors of Aβ aggregation, while compound Z will be a promoter of the Aβ aggregation process.
Figure 2.

Schematic representation of the effect of small molecule inhibitors on the kinetics of amyloid aggregation using a fluorescent dye.
Arguably, molecules that do not show any inhibition of Aβ aggregation in such an assay (Figure 2) would not be considered for any subsequent testing. Conversely, those compounds that show some inhibition will be scrutinized further, and in many cases might not show a significant inhibitory ability or may lack activity altogether. Therefore, the dye-binding assays must be adapted for a given system to become unambiguous and/or the potential pitfalls must be identified in a set of control experiments. General aspects of both CR and ThT dyes, as examples of the most widely utilized dyes, and their use in the evaluation of inhibitors of amyloid aggregation are presented in the subsequent sections.
Congo Red
The use of CR (Figure 1) spectral shift assays are routinely employed as a technique to quantify fibril content and inhibitory ability of small molecules toward amyloids. On the structural level, it is known that CR has two binding sites in amyloid, parallel to the β-sheet and antiparallel to the β-sheet, and the binding ratio may depend on the type of amyloid under investigation.33−36 It should also be pointed out that computational studies could suggest alternative modes of CR binding to amyloids,37−39 albeit caution should be exercised since some important empirical components are usually not accounted for in calculations. CR binding to the extensive β-sheet structures results in an enhanced absorption as well as a bathochromic shift in its absorption spectrum (from approximately 480 nm unbound to 540 nm bound), which is accompanied by a change in color from orange-red to rose and depends on the aggregation state of the proteins.30 Green birefringence in polarized light (when crossed polarizers are used) is observed upon binding to the fibrils and has been typically used as a qualitative measure of fibril formation.27,31 On the other hand, analysis of the absorption spectra of the unbound CR, fibrils, and the bound CR can provide a quantification of fibril concentration.30 Considering that CR is known to form micelle-like assemblies at concentrations above 5 μM, it was proposed that this dye interacts with the fibrils much like a detergent40 and its inhibitory ability may be related to its colloidal form.41
Several reports have demonstrated that CR is an inhibitor of amyloid aggregation using a number of spectroscopic and microscopic tools as well as cell culture studies.25,42,43 In some cases, the inhibitory ability of small molecules is even compared to CR, thus highlighting the potential complications of using CR as a reporter dye in inhibition assays.44 Primarily, CR was shown to stabilize the monomeric species, while disaggregating larger oligomers.42 Not surprisingly, several compounds which structurally resemble CR, such as RS-0406 (Figure 3)45 and chrysamine G (Figure 3),30,46 have also been noted to be inhibitors of amyloid β-protein aggregation. Interestingly, it has also been shown that CR can disrupt ThT binding to Aβ,28,47 and these interactions between the dyes might be preventing an accurate determination of Aβ quantitation.
Figure 3.

Structures of RS-0406 and chrysamine G.
On the contrary to the aforementioned inhibitory ability of CR, a recent circular dichroism (CD) study demonstrated that CR promoted both a β-sheet formation and peptide aggregation of Aβ1–40.40 This result complements other reports that demonstrated CR’s ability to induce ordered, β-sheet-rich conformations in other amyloidogenic peptides.48 Despite conflicting reports, that is, whether CR is or is not an inhibitor of Aβ aggregation, the utilization of CR as a reporter dye for the evaluation of small molecule inhibitory ability on Aβ aggregation is questionable. Obviously, a reporter that affects the aggregation of amyloids in any way would give results that might not be precise for the small molecule and would require additional experiments to differentiate the effects of the reporter versus the small molecule.
Thioflavin T
The benzothiazole dye, ThT (Figure 1), is widely used for the identification and quantification of amyloid fibrils as well as for the exploration of fibrillization kinetics of Aβ in the presence of small molecules using fluorescence. Specific interactions of ThT with amyloid fibrils have been the subject of numerous studies,49−51 although the exact binding mechanism, stoichiometry, and binding location are still debated and will not be reviewed here.
This dye is only weakly fluorescent in an aqueous environment, with excitation and emission maxima at approximately 350 and 440 nm, respectively. However, upon interacting with β-sheet-rich amyloid fibrils, a bathochromic shift of both excitation and emission maxima to 440 and 490 nm, respectively, are observed. In general, an ca. 10-fold fluorescence enhancement can be expected upon ThT binding to amyloid fibrils. Emission at 490 nm is assumed to be directly proportional to the quantity of amyloid fibrils present, and therefore, the kinetics of fibril formation can easily be followed by measuring the time-dependent increase in fluorescence. Conversely, a reduction in the ThT fluorescence is often taken as an indication of inhibition of the macromolecular amyloid self-assembly process.28,32 However, it was shown that in some cases even the kinetics of the amyloid aggregation might not be unambiguously accessed by ThT fluorescence.52 Specifically, after 8 h, ThT fluorescence reached a plateau and remained unchanged up to 30 h, which is indicative of a constant β-sheet content. However, 2D IR measurements indicated that changes of the β-sheet structure and content were taking place throughout a 24 h period.52 Similar to CR, ThT is also known to form micelles at low a micromolar concentration range.32,53
It should also be pointed out that thioflavin S (ThS) is often used as an alternative or a complement to ThT. However, considering the poorly defined nature of ThS (this is a mixture of compounds which results from a reaction of dehydrothiotoluidine with sulfonic acid), as well as the high intrinsic fluorescence of ThS, it might not be the most suitable probe for amyloid dye binding assays.
A number of other ThT-derivatives as well as other ThT-inspired motifs have been prepared (Figure 4).54−57 Considering that their accessibility is limited as most require multistep syntheses, ThT remains the fluorescent dye of choice.
Figure 4.

Thioflavin T derivatives; R1, R2, R3 = substituents, X = N, CH, n = 1, 2, etc.
Inherent Shortcomings of the Dye-Binding Assays
Arguably, CR- and ThT-based assays, under certain conditions, might provide an unbiased view of Aβ assembly. However, the situation is drastically complicated when these dyes are used to assess the ability of small molecules to inhibit the amyloid β-protein self-assembly processes. Despite wide utility, several drawbacks are associated with the aforementioned dye-binding assays. Primarily, ThT does not recognize soluble unordered oligomers of amyloids (as evident from a characteristic lag phase in the time-dependent studies), but mostly recognizes the insoluble fibrillar aggregates in the β-sheet conformation.29,58−61 However, some reports indicate that ThT is able to distinguish between oligomeric and fibrillar Aβ species.62−64It was suggested that this contradiction originated from differences in the preparation methods for the Aβ oligomers.65
Upon interaction with the fibrillar aggregates, the spectral properties of the dyes are influenced by a number of other factors: (i) the presence of exogenous compounds;30,32 (ii) the composition and properties of the media, such as pH, viscosity, and so forth;31,66 and (iii) protein-to-dye ratios.67
It was suggested that an excess of the dye relative to the peptides is required to ensure a reliable assessment of the amyloid aggregation.29,31,68,69 Arguably, this could enhance the competitive binding of the dye and inhibitor to the peptide. It might also compromise the integrity of the amyloid self-assembly and alter the aggregation profile. Some reports use almost equimolar amounts, but ideally a substoichiometric amount of the dye to peptide should neither perturb the structure of the amyloid self-assembly nor interfere with the amyloid–inhibitor interactions. Furthermore, another report suggested that an excess of ThT can cause self-quenching of the dye.67 It was shown that the ThT concentration accountable for maximal binding fluorescence shifts to higher values with increased amounts of amyloids, suggesting that amyloid to dye ratios are critical to obtain an ideal binding signal and thus special cautions are warranted for universal use.67
It should also be kept in mind that ThT spectral properties depend on fibril morphology, pH, and ionic strength.66 Small molecules might also disrupt the integrity of the amyloid self-assembly as certain molecules have concentration dependent, multiphasic behavior in the modulation of protein aggregation.26 Recent examples highlighted that conflicting results about small molecule inhibitors might be due to differing inhibitor concentrations70 or other experimental conditions such as chemical purity or incubation time.71
Several accounts have suggested that CR and ThT dye-binding assays could be subject to false positive results,24,30,32,72−74and a number of structurally and functionally diverse compounds have been shown to reduce ThT fluorescence without affecting amyloid fibril formation (Figure 5, a few representative examples). However, the seeming simplicity and high-throughput capabilities for carrying out the dye-based assays are the strong driving forces behind the continued use of the dye-binding assays as a first step in the evaluation of the inhibitory ability of small molecules toward amyloid aggregation.
Figure 5.

Some small molecules that alter ThT fluorescence but not amyloid aggregation.
In this light, it appears crucial to emphasize that dye binding assays might be prone to false positive effects by discussing some general considerations that are often overlooked in the literature and by highlighting some representative examples of small molecules which have generated a false positive in a dye-binding assay.
Evaluation of Small Molecule Inhibitors of Amyloid β-Protein Aggregation Using Dye-Binding Assays
General Considerations of Dye–Small Molecule Interactions
When assessing the ability of an inhibitor to affect the aggregation and fibril formation processes of the amyloid peptides, it is important to consider the effect of the inhibitor on the fluorescence of the dye in the absence of the peptide. Because both CR and ThT form micelles or micelle-like assemblies at certain concentrations,32,39 it is plausible that these assemblies of the dyes could interact with hydrophobic small molecules, which may have an influence on the spectral properties of the dyes themselves. It is of interest to note that, at the concentrations often used to monitor the amyloid β-protein fibrillization process (5–20 μM), ThT is able to form micelles.32,53 In addition, it is worth pointing out that a partitioning of an inhibitor into the dye micelles must be taken into consideration since it would affect the effective inhibitor concentration. These factors are often overlooked in the literature, but they may have an influence on the reported claims of many inhibitory small molecules. Additionally, any compounds to be tested for inhibitory properties should not have any spectral activity in the same range as the dyes as it may cause interference with the dyes’ responses.30,32
Another important aspect of dye-binding assays involves competitive binding of the dye and the inhibitors to the amyloidogenic peptides or proteins. If the potential inhibitor binds at the same site of the peptide or protein as that of a dye, the inhibitor will have to compete with the dye for the binding site. This might lead to a less efficient binding of the dye to Aβ aggregates and is likely to alter the intensity of the signal and as a result, artifacts or false positives would be observed.30
The small molecule inhibitors presented in the subsequent sections are diverse in structure, origin, and their effects on the aggregation of amyloid β-protein and they have been the subject of investigation of a number of different research groups. The common theme is the use of ThT-binding assay to evaluate amyloid aggregation inhibition. These examples are not meant to be comprehensive but rather highlight representative cases in an effort to raise awareness about possible issues in using dye-binding assays.
Curcumin
The inhibitory ability of curcumin (Figure 6) toward Aβ peptides has been documented in an array of in vitro and in vivo studies,70,75 albeit not without controversy.76 It has also been described as a very strong inhibitor of a mutated, scaffolded version of Aβ16–37 peptide.24
Figure 6.

Structure of curcumin.
In regard to ThT, an initial study by Ono and co-workers found curcumin to be an inhibitor of both Aβ1–40 and Aβ1–42 fibril formation and accumulation as indicated by an immediate decrease in ThT fluorescence upon addition of curcumin to the mixture of ThT and Aβ.77 At all concentrations tested (10–50 μM), curcumin caused a decrease in ThT fluorescence. It was also found that curcumin could destabilize preformed Aβ fibrils.77
Similarly, Yang et al.44 found that curcumin inhibited Aβ1–40 aggregation, disaggregated fibrillar Aβ1–40, and prevented Aβ1–42 oligomer formation and toxicity at concentrations between 0.1 and 1.0 μM. This group acknowledged the spectroscopic overlap between curcumin and thioflavin derivatives and thus investigated the inhibition of aggregation by curcumin without the use of any dye-based assays.51 They also showed that curcumin could stain amyloid plaques and could be used as a fluorescent probe on its own, with a similar efficiency as compared to ThS, but it was noted that ThS bound more efficiently to the tangles than curcumin.51
On the contrary, Glabe and co-workers26 suggested that curcumin was an oligomer-specific inhibitor (IC50 = 361.11 ± 38.91 μM) and did not inhibit fibril formation, while promoting Aβ1–42 fibril formation when oligomers were present in the reaction mixture in vitro at concentrations between 30 and 300 μM.26
Zovo et al.,73 developed a method to test aggregation and fibrillization of Aβ and inhibitory ability of compounds on fibrillization using MALDI-TOF-MS. The method is based on quantifying the time-dependent decrease of Aβ1–42 monomer in the presence of various inhibitors. Using this method, it was shown that curcumin did not inhibit fibrillization, which contradicts the results of the ThT assays.73 This group also showed that, during the time-dependent increase of ThT fluorescence kinetic curves, addition of curcumin did not affect the kinetic constant of fibril formation.73
Curcumin is a fluorochrome and is known to become highly fluorescent upon binding to hydrophobic regions of proteins such as human and bovine serum albumins. Similar effects were seen upon binding to model amyloid fibril-forming proteins, such as RCMk-CN.32 It was shown that curcumin could affect the fluorescence of ThT: below 10 μM, curcumin increased the fluorescence of ThT, but in the range of 50–100 μM curcumin caused a decrease in ThT fluorescence and a red-shift in the emission spectra. The absorption spectrum of curcumin has a maximum at 426 nm, which is near the excitation wavelength of ThT (λex ≈ 440 nm). Upon excitation of a mixture containing both the dye and the small molecule, possible inner filter effects could occur. Rather than an indication of inhibitory ability of the compound,32 the decrease of ThT fluorescence could be explained by these spectroscopic effects, which ultimately resulted in a false positive. Furthermore, steady-state fluorescence emission spectra of curcumin and protein mixtures were recorded with varying concentrations of curcumin (1–100 μM). At higher concentrations, self-quenching was observed in addition to a red shift in the spectra, offering an additional explanation to the false positive result from the ThT fluorescence assay. Hence, curcumin would be able to affect the ThT fluorescence spectra due to several spectroscopic issues, that is, its overlapping absorption and own intrinsic fluorescence.32
It is arguable that evaluation of the inhibitory ability of compounds that are structurally related to curcumin, such as those shown in Figure 7,78 might also be prone to the same spectral interference effects as those noted for curcumin itself.
Figure 7.

Curcumin related compounds used for antiaggregation studies.
Interestingly, a study by Kim et al.79 reported that curcumin had the strongest inhibitory effect (IC50 = 0.25 μg/mL = 0.679 μM) on Aβ fibril formation of all tested compounds, including curcuminoids, flavons, naphthoquinones, isoflavones, and flavanones among others, tested in an in vitro assay as judged by ThT assay. However, the inhibitory activity of curcuminoids decreased in an in vivo experiment with HT22 murine neuroblastoma cells. Curcuminoids showed cytotoxicity and no inhibitory effect of Aβ against the cells. Although it is possible that the curcuminoids exhibit a different mode of action in a cell culture assay, ThT assays are often the first screen in determining the subsequent fate of the small molecule inhibitor. This highlights the importance of control experiments in the ThT assays, since the difference in inhibition propensity between in vitro and in vivo experiments could be related to a potential false positive result from the initial ThT assays. In addition, the diversity of the conditions used by various research groups arguably suggests that small changes might lead to drastically different outcomes as it has been suggested that “distinct experimental platforms (e.g., ThT, direct binding, etc.) and subtle changes in handling (e.g. different buffers, temperatures, time, etc.) may give rise to different outcomes.”78 In addition, an explanation for the false positive result in the dye-binding assay based on spectroscopic features has been offered.32
Quercetin
Quercetin (Figure 8) is a flavonoid, polyphenol antioxidant found in many fruits, vegetables, Chinese herbs,80 and red wines, and it was suggested to attenuate the toxic effects of amyloid peptides in several cell culture lines and neurons.81,82 In addition, quercetin appeared to affect several cellular pathways related to Aβ-induced neurotoxicity.83It was also shown that pretreating primary hippocampal cultures with small amounts of quercetin could reduce Aβ1–42 cytotoxicity, protein oxidation, lipid peroxidation, and apoptosis.84 It was also suggested that quercetin may have some protective effects in vivo due to its antioxidant ability.82,85 Furthermore, it was shown in a recent report that quercetin could remodel Aβ1–42 oligomers based on the results of dot blot assays.86 However, it should be mentioned that no protective effects of quercetin were observed against synapse damage induced by Aβ1–42.87 Furthermore, in some other in vivo experiments, it was shown that, at 20 and 40 μM, quercetin was unable to reduce the amount of Aβ that was secreted in APP695 transfected HEK293 cells as analyzed by Western blot and ELISA assay.88
Figure 8.

Structure of quercetin.
Several in vitro spectroscopic studies also produced controversial results. One report suggested that quercetin could reduce Aβ25–35 aggregation and fibrillization by ThT assays and electron microscopy. Ono et al. found that quercetin had an EC50 on the order of 0.1–1 μM on the formation, extension, and destabilization of Aβ1–40 and Aβ1–42 fibrils.68 Furthermore, another group showed by ThT assay that quercetin had an inhibitory effect on Aβ1–42 fibril formation, although a significant reduction in ThT fluorescence was only seen upon incubation with 100 μM quercetin.71
By turbidity and ThT assays, it was suggested that, in addition to the fibrillar aggregate, another aggregate was formed in the presence of quercetin, but nevertheless quercetin was suggested to be an inhibitor.80 Using ThT based assays, others showed that quercetin had an IC50 of 2.4 μg/mL = 7.9 μM.79
Similarly, Hudson and co-workers observed a concentration-dependent decrease of ThT fluorescence upon addition of quercetin to ThT and RCMk-CN mixtures,32 and others observed fibril inhibition of quercetin on RCMk-CN.89 However, Zovo and co-workers did not observe any inhibitory ability of quercetin on fibril formation in the MALDI-TOF-MS assay.73 The absorption spectrum of quercetin has a maximum at 374 nm, which somewhat overlaps with the ThT absorption spectra (unbound ThT maximum at 350 nm) and might partially overlap with the emission of the bound ThT (emission maximum at 440 nm). The proximity of the maxima could allow for the possibility of inner filter effects, an alternative explanation of the quenching of ThT fluorescence,32 but lack of inhibitory ability on fibril formation.73
Basic Blue 41
Basic Blue 41 (BB41) (Figure 9) is a known inhibitor of Aβ aggregation as assessed by several groups using ThT based assays.24,68 In a study conducted by Zovo and co-workers,73 the use of MALDI-TOF-MS was implemented and the results were monitored in parallel with the ThT assay. It appeared that the inhibitory constant value of BB41 as determined by ThT analysis (IC50 = 0.1 μM) and the MALDI-TOF-MS analysis (IC50 = 1.6 μM) differed substantially. Furthermore, addition of BB41 in the ThT analysis resulted in a fast and concentration dependent decrease in ThT fluorescence, but it did not affect the kinetic constant of the fibril growth. This could indicate a competitive displacement of ThT from the fibrils or quenching of its fluorescence. It was also shown that BB41 competes with ThT for binding to insulin fibrils.72 However, because BB41 was shown to be a somewhat effective inhibitor of fibrillization by MALDI-TOF-MS, it could be that BB41 interferes with the fluorescence of ThT and thus appears as a more effective inhibitor in ThT assays.73However, in a related study, it was noted that although the presence of BB41 did lower the fluorescence of ThT, no reduction of the fibril formation was noted as compared to the control.23
Figure 9.

Structure of BB41.
Amphotericin B
The polyene macrolide antibiotic amphotericin B, AmB (Figure 10), was shown to bind to fibril-like species of Aβ25–35 (Kd = 6.4 μM) as judged by a red-shift in the AmB monomer absorbance, disassembly of AmB oligomer, as well as the CR dye-binding assay.90,91 AmB was found to bind directly and specifically to the fibrils and not to monomeric, nonaggregated oligomeric forms of the Aβ as indicated by a red-shift in its absorption spectra to ca. 420 nm (AmB, in its monomeric form has maxima at 409, 385, 362, and 345 nm). Additionally, AmB was found to inhibit Aβ1–40 fibril formation by ThS assay.23,91
Figure 10.

Structure of AmB.
However, AmB was shown to directly interact with CR.90 Nonetheless, it was demonstrated that, under the conditions studied, this CR–Aβ interaction could be factored in, and the inhibitory ability of AmB could still be validated.
On the other hand, it was demonstrated using CD that AmB had no measurable impact on either the secondary structure or on the time-dependent conformational transition of soluble Aβ1–42 oligomers from unordered to ordered species.92 Although the inability of AmB to affect the aggregation process of the oligomers does not necessarily imply that AmB cannot inhibit the Aβ fibrillization process (especially considering the differences in the conditions of the aggregation assays), the inhibitory ability of AmB should be taken with caution.
β-Cyclodextrin
β-Cyclodextrin is a naturally occurring and readily available cyclic glucopyranose heptamer (Figure 11). β-Cyclodextrin was shown to reduce Aβ1–40 neurotoxicity toward PC12 cell lines by ca. 40%.93 Importantly, toxicity of Aβ1–40 and Aβ1–42 was reduced in rats via stereotactical injection into the hippocampus.94 Subsequently, several reports demonstrated inhibitory ability of β-cyclodextrin derivatives toward the oligomerization and fibrillization of Aβ peptides using dot-blot and Western-blot assays.95,96
Figure 11.

Structure of β-cyclodextrin.
Spectroscopic studies, on the other hand, created some controversy. In regard to the dye-based assays, the ThT fluorescence assays showed an appreciable inhibitory effect ofβ-cyclodextrin on Aβ1–4097 and Aβ1–4226 fibril formation. Yet application of other techniques, such as dynamic light scattering (DLS), transmission electron microscopy (TEM), and atomic force microscopy (AFM),26,97 strongly indicated that significant fibril/oligomer formation still took place. It was proposed that the β-cyclodextrin was likely to interfere with the binding of ThT to the Aβ fibrils,26 especially considering a recent report concerning ThT complexation with β-cyclodextrin.98 Similarly, it was also shown that at a concentration of 15 mM β-cyclodextrin could directly inhibit the binding of CR to Aβ1–40 fibrils.93 The encapsulation of CR into the β-cyclodextrin could be excluded, due to size mismatch.
Obviously, a direct inhibition of both amyloid β-protein fibril formation91 and oligomerization95 might be considered as a potential mechanism of β-cyclodextrin action. As a proof, nuclear magnetic resonance (NMR) spectroscopy studies indicated the ability of aromatic residues (F19, F20, and/or Y10) in Aβ1–40 and a truncated version, Aβ12–28, interacted with β-cyclodextrin, albeit with millimolar affinities.99,100 Furthermore, CD spectroscopy showed that β-cyclodextrin could inhibit the transition from an unordered to ordered conformation of Aβ12–28.99
It is of interest to note that a recent study indicated that β-cyclodextrin could be a promoter of Aβ aggregation exactly due to these interactions.101 Overall, the inhibitory ability of β-cyclodextrin suggested by the dye-based assays toward amyloid β-protein fibrillization reported in several accounts should be taken with great caution.
Tetracycline
Tetracycline (Figure 12) is an anthracycline that has been shown by several groups, using several different methods, to have inhibitory properties on Aβ fibril formation.24,102 One report suggested that tetracycline could inhibit the formation and extension, and even destabilize, preformed fibrils of Aβ1–40 and Aβ1–42 with modest EC50's, that is, a weak inhibitor, as determined by ThT assays.103
Figure 12.

Structure of tetracycline.
Using CD to follow the secondary structure transition of Aβ1–42 during aggregation, Bartolini et al. showed that tetracycline could slow down the conformational change of Aβ1–42 into the β-sheet,25 indicating an inhibitory effect of tetracycline on Aβ fibril formation.
In a fluorometric ThT analysis, tetracycline decreased the fluorescence of ThT by ca. 45%.25 Other reports suggest similar effects by tetracycline on ThT fluorescene.102 However, in another set of experiments, albeit under distinctly different conditions, it was shown that tetracycline interferes with ThT fluorescence.104 Using several NMR studies, Airoldi et al. showed that tetracycline competes with ThT for binding to Aβ1–40 and Aβ1–42 oligomers, thereby offering a possible explanation for the fluorescence reduction in the ThT assay.104 This verifies the potential for small molecules to give false positive results in the ThT dye binding assay and the mechanism by which it decreases ThT fluorescence could be overlooked if not complemented with additional experiments.
Resveratrol
Resveratrol, a diphenolic compound from grapes (Figure 13), has been shown to have neuroprotective ability against toxicity of amyloid β-proteins, such as Aβ25–35, Aβ1–40, and Aβ1–42.105−107 It also showed some promising results in some other in vivo experiments.88
Figure 13.

Structure of resveratrol.
ThT-based assay demonstrated that resveratrol is capable of modulating the aggregation of Aβ1–42 peptides.108 These results were consistent with the dot-blot, SDS-PAGE, and cell toxicity studies. In addition, resveratrol was demonstrated to drastically suppress the formation of fibrillar Aβ1–42 species in a dose-dependent manner as judged by ThT-assay, albeit failing to affect the formation of β-sheet rich Aβ-oligomers as established by CD experiments.109
It should be pointed out that resveratrol was shown to be a suitable amyloid binding dye on its own with a fluorescence maxima at 395 nm and a shoulder at 440 nm in the presence of amyloids.35 Similar to ThT, resveratrol exhibited sigmoidal binding kinetics to amyloid aggregates. However, at 5 μM concentration of resveratrol, a significantly decreased ThT fluorescence was observed with fibrillar Aβ1–42 in a dilution ThT assay, and it was suggested that resveratrol could be interacting directly with ThT or competitively binding to the fibrils and displacing ThT.32
Guanidiniocarbonyl Pyrrole-Based Inhibitors
In 2005, Schmuck and co-workers reported the inhibition of Aβ1–40 and Aβ1–42 fibril formation by several guanidiniocarbonyl pyrrole-based compounds (Figure 14) using CR and ThT dye binding assays.110 Several compounds (1 and 2, Figure 14) showed an inhibitory effect on fibril formation in the ThT assay as well as a decreased fibril quantity in the CR assay. On the other hand, pyrrole-containing tripeptide 3 showed no effect on fibril formation in the ThT assay while demonstrating a significant reduction in fibril formation in the CR assay. This result appeared to be a false positive as the pyrroles likely interfered with the binding of CR to the fibrils.110 Aside from the value in developing amyloid inhibitors, this account is a rare, but valuable, example that highlighted the idea of using two dyes in the spectroscopic evaluation of the inhibitory ability of small molecules. Arguably, two structurally different dyes are likely to interact at two different sites of the amyloid assembly. Hence, the probability that a small molecule inhibitor will compete with the dyes for those two distinct sites is decreased.
Figure 14.

Structures of guanidiniocarbonyl pyrrole-based inhibitors.
Glycerol
Interestingly, common and structurally simple (as compared to those described above) small molecules have also been demonstrated to have some inhibitory capabilities on amyloid formation. Not surprisingly, similar false positive effects in the dye-binding assays are to be expected.
For example, Ryu and co-workers observed a 40% decrease of the ThT fluorescence when 100 mM glycerol (Figure 15) was tested as an inhibitor for Aβ1–42 fibril formation.111 However, according to AFM analysis, the samples incubated with glycerol had a high fibril content,111 which arguably suggested that the reduction in ThT fluorescence was unrelated to formation of the fibrils. In this case, it is possible that glycerol interferes with ThT fibril binding and thus lowers the fluorescence without inhibiting fibril formation.49
Figure 15.
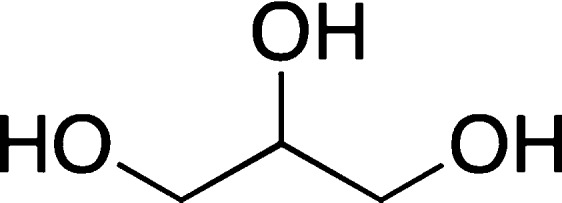
Structure of glycerol.
Dye-Binding Assays beyond Aβ Peptides
The drawbacks of using dye-based assays as the initial determination of inhibitory ability of a compound are not limited to Aβ peptides. In fact, several other studies involving other amyloidogenic, fibril forming proteins showed similar issues with the dye binding assays.
For example, l-arginine (l-Arg), up to 1.4 M, was shown to dramatically reduce ThT fluorescence, but not fibrillation of bovine serum albumin (BSA).74l-Arg has no overlapping emission with ThT, and thus, spectral interference could be excluded. Upon examination of TEM images, no inhibition of fibril formation by l-Arg was observed, indicating that l-Arg most likely interferes with ThT binding to BSA.74
Similarly, it was shown that rifampicin (Figure 16) had an effect on ThT fluorescence when bound to human islet amyloid polypeptide (IAPP).112 Rifampicin lowered ThT fluorescence, but TEM images revealed that it did not inhibit amyloid fibril formation. The cause of this false positive could be spectroscopic in nature, as unoxidized rifampicin has a large absorbance at 483 nm.112
Figure 16.

Structure of rifampicin.
Additionally, amyloid-like fibril formation was investigated with merozoite surface protein 2 (MSP2), a GPI-anchored protein expressed on the surface of Plasmodium falciparum merozoites, which has proved to be a promising malaria vaccine candidate.109 ThT binding assay resulted in a false positive by suggesting inhibition of MSP2 fibril formation by flavonoids, baicalein (Figure 17), and the previously discussed resveratrol (Figure 13), as TEM images and CD revealed the formation of protein aggregates and fibrils.113
Figure 17.

Structure of baicalein.
Recently, disaggregation of β-lactoglobulin fibrils by 4,5-dianilinophthalimide (DAPH) (Figure 18) was suggested by ThT-based assay.114 However, the results of flow-induced birefringence, rheological measurements, and EM indicated the degree of fibrillization as well as the length of the fibrils remained the same despite the treatment with DAPH, thus arguably pointing to a false positive effect of the ThT measurement.
Figure 18.
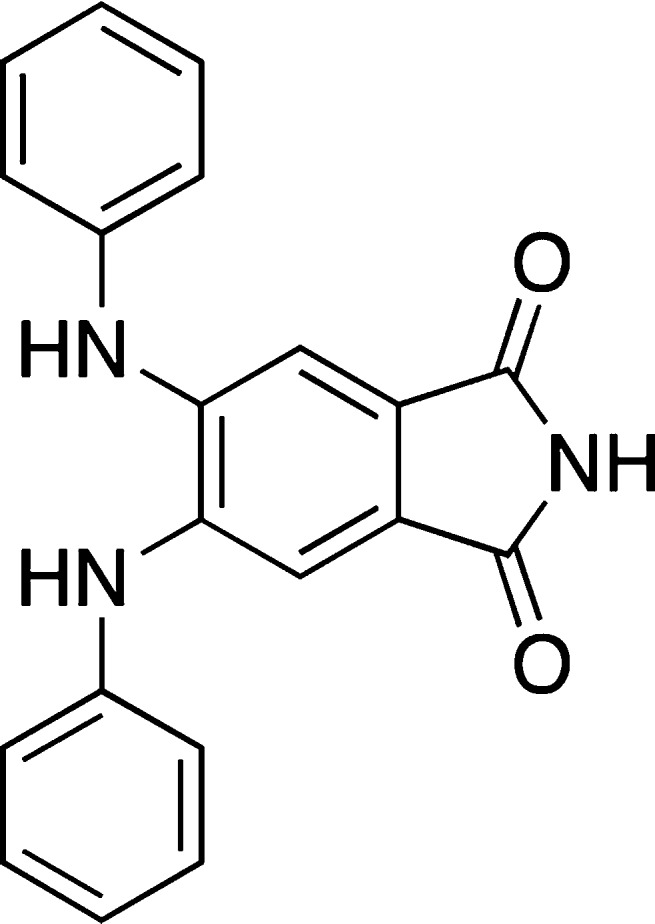
Structure of DAPH.
A recent account, on insulin fibrillization, demonstrated quenching of ThT emission by several small molecules, including Basic Blue 41 (Figure 9) and Basic Blue 12 (Figure 19), azure C (Figure 5), and tannic acid (Figure 19), which may be attributed to competitive binding of the small molecules and ThT.72 In light of other reports describing of inhibition and destabilization of Aβ fibrils by these compounds using ThT based assays,103 these and previously mentioned examples of false positive results with dye-binding assays illustrate that this problem is not specific to a particular peptide or protein.
Figure 19.

Structures of Basic Blue 12 and tannic acid.
Conclusions
The search for potential inhibitors of amyloid fibril formation, and protein aggregation in general, constitutes a practical and viable therapeutic approach, but the dye-binding assays commonly employed to investigate these events might lead to false positive results and unnecessary detours.
As the aforementioned examples have demonstrated, numerous controls are required to eliminate false positive effects when employing dye-binding assays. Specifically, the possibility of intrinsic fluorescence of the small molecule inhibitors to alter the spectral response of the dye should be evaluated. Potential interference from the media components, the concentrations of the dye, inhibitor, and Aβ or other protein should also be considered. In addition, a possibility of a competitive binding between the dye and inhibitor molecule should be assessed.
It is understood that the convenience (small quantities of materials, high sensitivity, capabilities for high-throughput screening, etc.) of using the dye-based methods is one of the strongest driving forces behind a widespread utilization of these assays for assessing aggregation propensity of biomolecules. In this light, dye-free assays should be performed in parallel as well. For example, surface plasmon resonance and quartz microbalances,115 CD,25,92 DLS,13,92 as well as numerous forms of microscopy116,117 have been used alongside the dye-binding assays.118 However, it appears to be a common practice to utilize different conditions (Aβ concentration, identity/composition of the buffer, incubation times, etc.) for each specific technique which hinders the development of suitable rationales, correlations, and universal applications. NMR is a powerful tool in assessing the structure of amyloid species and the inhibitory ability of small molecules;119,120 however, higher concentrations and/or isotope labeling are required for these experiments.
Also, 2D IR might be a viable, sensitive technique for accessing the amyloid structure and potentially the effect of inhibitors on the amyloid self-assembly.52,121 Although, isotope labeling is required, which might pose some experimental constrains.
Another alternative to avoid false positive effects would be the use of more than one dye, with drastically different structural and spectral properties.110 Arguably, structurally diverse dyes are more likely to interact with different parts of the Aβ aggregates, and therefore, the interference with the inhibitor binding could be minimized. Although, this approach is far from being ideal since difference in structure might not necessarily result in a difference in the binding mode. However, a discrepancy between the two binding assays would likely promote a more thorough investigation. Overall, this highlights the need for the development of novel dyes with a greater scope for structural modification. Several recent reports suggested that BODIPY dyes have the ability to recognize oligomeric and fibrillar amyloid assemblies (Figure 20).122−124Considering that modification of the BODIPY scaffolds can be done in a facile and modular manner, these new BODIPY-based fluorescent probes might offer a viable alternative to the currently employed dyes.
Figure 20.

BODIPY dyes that bind to amyloid oligomers and plaques.
In addition, Michler’s hydrol blue (Figure 21) was recently shown to be more sensitive to environmental changes than ThT, as it even allowed for the differentiation between insulin and lysozyme fibrils.125 Also, Bis-ANS (Figure 21) has been shown to bind to oligomers and fibrils of Aβ1–42.126
Figure 21.

Structures of Michler’s hydrol blue and Bis-ANS dyes.
Finally, there might also be a need to reconcile the existing protocols for using the dyes in assessing the ability of small molecules to affect aggregation of Aβ1–40 and Aβ1–42 peptides. As a representative case, Table 1 provides a brief summary of several studies on curcumin–amyloid β-protein interactions, in an attempt to demonstrate the contrasting conditions as potential reasons for contradicting results. Arguably, a great number of procedures for Aβ aggregation and dye assays, which are seemingly similar to each other, have enough small differences, which preclude comparisons and generalizations that are required for the development of efficient anti-Alzheimer’s remedies.
Table 1. Effect of Curcumin on Fibrillization of Aβ1–40 and Aβ1–42 Peptides: Experimental Conditionsa.
| Ono et al.77 | Yang et al.44 | Necula et al.26 | Zovo et al.73 | Reinke et al.78 | Kim et al.79 | |
|---|---|---|---|---|---|---|
| Aβ source | Aβ-TFA salt | Aβb | Aβ-TFA saltc | Aβ-HFIP film | Aβ-HFIP film | Aβe |
| initial [Aβ1–42 or Aβ1–40], μM (media) | 500 (0.02% NH3 in H2O) | 231 (H2O) | 2000 (0.1 M NaOH) | 10–20 (0.02% NH3 in H2O)d | NR (DMSO) | 250 (DMSO) |
| final [Aβ1–42], μM | – | – | 45 | 5 | 25 | 25 |
| final [Aβ1–40], μM | 50 | 11.6 | – | – | – | – |
| aggregation buffer; pH | 50 mM phosphate, 0.1 M NaCl; 7.5 | 0.1 M TBS, 0.02% Tween 20; 7.4 | 10 mM HEPES, 0.1 M NaCl, 0.02% NaN3; 7.4 | 20 mM HEPES, 0.1 M NaCl; 7.3 | PBS (10% DMSO); 7.4 | PBS (10% DMSO); NR |
| time of aggregation, h (temp, °C) | 0–192 (37) | 144 (37) | 96 (rt) | 1 (rt) | 46–48 (rt) | NR |
| [curcumin], μM (stock in) | 0.01, 0.1, 1, 10, 50 (DMSO) | 0.125, 0.25, 0.5, 1, 2, 4, 8 (EtOH) | 30–300 (DMSO) | 5 (NR) | 0.001–1000 (DMSO) | NR |
| assays used | EM, ThT | EM, ELISA | turbidity, ELISA, ThT, TEM | MS, ThT | ThT | ThT |
| ThT solution added to Aβ | 50 mM glycine, pH 8.5 | 0.1 M TBS, 0.02% Tween 20; 7.4 | 10 mM HEPES, 0.1 M NaCl, 0.02% NaN3; 7.4 | 20 mM HEPES, 0.1 M NaCl; 7.3 | 50 mM glycine,pH 8.0 | 50 mM glycine, pH 8.5 |
| ThT (μM)/Aβ (μM) | NR | NA | 2.8:3.5 | 3.3:5 | 4.8:1.2 | 3.8:6.3 |
| inhibits fibrillization: + | + | + | – | – | + | + |
| does not inhibit fibrillization: – |
NR = not reported; NA = ThT assay was not used; rt = room temperature.
Purchased from C. Glabe lab.
Fibrillization conditions; for details, see ref (24).
Aggregation is induced by the addition of 2–10% (v/v) Aβ1–42 fibrillar seed, which was prepared as follows: 5 mM Aβ1–42 in 20 mM HEPES, 0.1 M NaCl, pH 7.3, stirred at rt for 1 h.
Purchased from BaChem California Inc.; HCl salt and neutral Aβ1–42 are sold; the exact one is NR.
Author Contributions
L.P.J., N.W.S., and S.V.D. collected and analyzed the references, wrote and edited the review.
We would like to thank National Institute On Aging (Award Number R15AG038977) for supporting our research on amyloid peptides. N.W.S. acknowledges predoctoral fellowship from the ACS (Wyeth/ACS Division of Medicinal Chemistry).
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute On Aging or the National Institutes of Health.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Galimberti D.; Scarpini E. (2011) Alzheimer’s disease: from pathogenesis to disease-modifying approaches. CNS Neurol. Disord.: Drug Targets 10, 163–174. [DOI] [PubMed] [Google Scholar]
- Smith M. A.; Drew K. L.; Nunomure A.; Takeda A.; Hirai K.; Zhu X.; Atwood C. S.; Raina A. K.; Rottkamp C. A.; Sayer L. M.; et al. (2002) Amyloid-β, tau alterations and mitochondrial dysfanctioun in Alzheimer disease: the chickens or the eggs?. Neurochem. Int. 40, 527–531. [DOI] [PubMed] [Google Scholar]
- Garcia-Ayllon M.-S.; Small D. H.; Avila J.; Saez-Valero J. (2011) Revisiting the role of acetylcholinesterase in Alzheimer’s disease: cross-talk with P-tau and β-amyloid. Front. Mol. Neurosci. 4, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao P.; Reddy H. P. (2011) Aging and amyloid beta-induced oxidative DNA damage and mitochondrial dysfunction in Alzheimer’s disease: Implications for early interventaion and therapeutics. Biochim. Biophys. Acta, Mol. Basis Dis. 1812, 1359–1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy J. A.; Higgins G. A. (1992) Alzheimer’s disease: the amyloid cascade hypothesis. Science 256, 184–185. [DOI] [PubMed] [Google Scholar]
- Tanzi R. E.; Bertram L. (2005) Twenty years of the Alzhemer’s disease amyloid hypothesis: a genetic perspective. Cell 120, 545–555. [DOI] [PubMed] [Google Scholar]
- Jakob-Roetne R.; Jacobsen H. (2009) Alzhemier’s disease: from pathology to therapeutic approach. Angew. Chem., Int. Ed. 48, 3030–3059. [DOI] [PubMed] [Google Scholar]
- Reitz C. (2012) Azlheimer’s disease and the amyloid cascade hypothesis: a critical review. Int. J. Alzheimer’s Dis. 2012, 369808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haass C.; Selkoe D. J. (2007) Soluble protein oligomers in neurodegeneration: lessons from Alzheimer’s amyloid β-peptide. Nat. Rev. Mol. Cell Biol. 8, 101–112. [DOI] [PubMed] [Google Scholar]
- Wang Z.; Yang L.; Zheng H. (2012) Role of APP and Aβ in synaptic physiology. Curr. Alzheimer Res. 9, 217–226. [DOI] [PubMed] [Google Scholar]
- Cole S. L.; Vassar R. (2008) The role of amyloid precursor proteins processing by BACE1, the β-secretase, in Alzheimer disease pathophysiology. J. Biol. Chem. 283, 29621–29625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caughey B.; Lansbury P. T. (2003) Protofibrils, pores, fibrils, and neurodegeneration: separating the responsible protein aggregates from the innocent bystanders. Annu. Rev. Neurosci. 26, 267–298. [DOI] [PubMed] [Google Scholar]
- Larson M. E.; Lesne S. E. (2012) Soluble Aβ oligomer production and toxicity. J. Neurochem. 120(suppl. 1), 125–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krafft G. A.; Klein W. L. (2010) ADDLs and the signalling web that leads to Alzhemier’s disease. Neuropharmacology 59, 230–242. [DOI] [PubMed] [Google Scholar]
- Selkoe D. J. (2008) Soluble oligomers of the amyloid β-protein impair synaptic plasticity and behavior. Behav. Brain Res. 192, 106–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saraiva A. M.; Cardoso I.; Pereira M. C.; Coelho M. A. N.; Saraiva M. J.; Moehwald H.; Brezesinski G. (2010) Controlling amyloid-β peptide(1–42) oligomerization and toxicity by fluorinated nanoparticles. ChemBioChem 11, 1905–1913. [DOI] [PubMed] [Google Scholar]
- Cerf E.; Sarroukh R.; Tiamamizu-Kato S.; Breydo L.; Derclaye S.; Dufrene Y. F.; Narayanaswami V.; Goormaghting E.; Ryusschaert J.-M.; Raussens V. (2009) Antiparallel β-sheet: a signature structure of the oligomeric amyloid β-peptide. Biochem. J. 421, 415–423. [DOI] [PubMed] [Google Scholar]
- Sgourakis N. G.; Yan Y.; McCallum S. A.; Wang C.; Garcia A. E. (2007) The Alzeimer’s Aβ40 and 42 adopt distinct confromations in water: a combined MD/NMR study. J. Mol. Biol. 368, 1448–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao M. Q.; TRzeng Y. J.; Chang L. Y. X.; Huang H. B.; Lin T. H.; Chyan C. L.; Chen Y. C. (2007) The correlation between neurotoxocity, aggregative ability and secondary structure studies by sequence truncated Aβ peptides. FEBS Lett. 581, 1161–1165. [DOI] [PubMed] [Google Scholar]
- Barrow C. J.; Zagorski M. G. (1991) Solution structures of β peptide and it’s constituent fragments: relation to amyloid deposition. Science 253, 179–182. [DOI] [PubMed] [Google Scholar]
- Roychaudhuri R.; Yang M.; Hoshi M. M.; Teplow D. B. (2009) Amyloid-protein assembly and Alzheimer’s disease. J. Biol. Chem. 284, 4749–4753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohamed T.; Rao P. P. N. (2011) Alzhemier’s disease: emerging trends in small molecule therapies. Curr. Med. Chem. 18, 4299–4320. [DOI] [PubMed] [Google Scholar]
- Masuda M.; Suzuki N.; Taniguchi S.; Oikawa T.; Nonaka T.; Iwatsubo T.; Hisagana S.; Goedert M.; Hasegawa M. (2006) Small molecule inhibitors of α-synuclein filament assembly. Biochemistry 45, 6085–6094. [DOI] [PubMed] [Google Scholar]
- Dolphin G. T.; Ouberai M.; Dumy P.; Garcia J. (2007) Designed amyloid β peptide fibril – a tool for high-throughput screening of fibril inhibitors. ChemMedChem 2, 1613–1623. [DOI] [PubMed] [Google Scholar]
- Bartolini M.; Bertucci C.; Bolognesi M. L.; Cavalli A.; Melchiorre C.; Andrisano V. (2007) Insight into the kinetics of amyloid β (1–42) peptide self-aggregation: elucidation of inhibitors’ mechanism of action. ChemBioChem 8, 2152–2161. [DOI] [PubMed] [Google Scholar]
- Necula M.; Kayed R.; Milton S.; Glabe C. G. (2007) Small molecule inhibitors of aggregation indicate that amyloid β oligomerization and fibrillization pathways are independent and distinct. J. Biol. Chem. 282, 10311–10324. [DOI] [PubMed] [Google Scholar]
- Hawe A.; Sutter M.; Jiskoot W. (2008) Extrinsic fluorescent dyes as tools for protein characterization. Pharm. Res. 25, 1487–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buell A. K.; Dobson C. M.; Knowles T. P. J.; Welland M. E. (2010) Interactions between amyloidophilic dyes and their relevnace to studies of amyloid inhibitors. Biophys. J. 99, 3492–3497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson M. R. (2004) Techniques to study amyloid fibril formation in vitro. Methods 34, 151–160. [DOI] [PubMed] [Google Scholar]
- Klunk W. E.; Jacob R. F.; Mason R. P. (1999) Quantifying amyloid β-peptide (Aβ) aggregation using the Congo red-Aβ (CR-Aβ) spectrophotometric assay. Anal. Biochem. 266, 66–76. [DOI] [PubMed] [Google Scholar]
- Eisert R.; Felau L.; Brown L. R. (2006) Methods for enhancing the accuracy and reproducibility of Congo red and thiolavin T assays. Anal. Biochem. 353, 144–146. [DOI] [PubMed] [Google Scholar]
- Hudson S. A.; Ecroyd H.; Kee T. W.; Carver J. A. (2009) The thioflavin T fluorescence assay for amyloid fibril detection can be biased by the presence of exogenous compounds. FEBS J. 276, 5960–5972. [DOI] [PubMed] [Google Scholar]
- Klunk W. E.; Pettegrew J. W.; Abraham D. J. (1989) Quantitative evaluation of Congo red binding amyloid-like proteins with a beta-pleated sheet conformation. J. Histochem. Cytochem. 37, 1273–128. [DOI] [PubMed] [Google Scholar]
- Furumoto S.; Okamura N.; Iwata R.; Yanai K.; Arai H.; Kudo Y. (2007) Recent advances in the development of amyloid imaging agents. Curr. Top. Med. Chem. (Sharjah, United Arab Emirates) 7, 1773–1789. [DOI] [PubMed] [Google Scholar]
- Frid P.; Anisimov S. V.; Popovic N. (2007) Congo red and protein aggregation in neurodegenerative diseases. Brain Res. Rev. 53, 135–160. [DOI] [PubMed] [Google Scholar]
- Howie A. J.; Brewer D. B. (2009) Optical properties of amyloid stained by Congo red: History and mechanisms. Micron 40, 285–301. [DOI] [PubMed] [Google Scholar]
- Wang C.-C.; Huang H-b.; Tsay H.-J.; Shiao M.-S.; Wu W-J. W.; Chen Y.-C.; Lin T.-H. (2012) Characterization of Aβ aggregation mechanism probed by congo red. J. Biomol. Struct. Dyn. 30, 160–169. [DOI] [PubMed] [Google Scholar]
- Cavillon F.; Elhaddaoui A.; Alix A. J. P.; Turrell S.; Dachez M. (1997) Identification of the importance of the secondary structure of Alzheimer’s disease amyloid. J. Mol. Struct. 408–409, 185–189. [Google Scholar]
- Li L.; Darden T. A.; Bartolotti L.; Kominos D.; Pedersen L. G. (1999) An atomic model for the pleated β-sheet structure, of Aβ amyloid protofilaments. Biophys. J. 76, 2871–2878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lendel C.; Bolognesi B.; Wahlstrom A.; Dobson C.; Graslund A. (2010) Detergent-like interaction of Congo red with the amyloid β peptide. Biochemistry 49, 1358–1360. [DOI] [PubMed] [Google Scholar]
- Feng B. Y.; Toyama B. H.; Wille H.; Colby D. W.; Collins S. R.; May B. C. H.; Pruisner S. B.; Weissman J.; Scoichet B. K. (2008) Small-molecule aggregates inhibit amyloid polymerization. Nat. Chem. Biol. 4, 197–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podlisny M. B.; Walsh D. M.; Amarante P.; Ostaszewski B. L.; Stimson E. R.; Maggio J. E.; Teplow D. B.; Selkoe D. J. (1998) Oligomerization of endogenous and synthetic amyloid β-protein at nanomolar levels in cell culture and stabilization of monomer by Congo red. Biochemistry 37, 3602–3611. [DOI] [PubMed] [Google Scholar]
- Hong H.-S.; Rana S.; Barrigan L.; Shi A.; Zhang Y.; Zhou F.; Jin L.-W.; Hua D. H. (2009) Inbibition of Alzheimer’s amyloid toxicity with a tricyclic pyrone molecule in vitro and in vivo. J. Neurochem. 108, 1097–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang F.; Lim G. P.; Begum A. N.; Ubeda O. J.; Simmons M. R.; Ambegaokar S. S.; Chen P.; Kayed R.; Glabe C. G.; Frautschy S. A.; Cole G. M. (2005) Curcumin inhibits formation of amyloid β oligomers and fibrils, binds plaques, and reduces amyloid in vivo. J. Biol. Chem. 280, 5892–5901. [DOI] [PubMed] [Google Scholar]
- Nakagami Y.; Nishimura S.; Murasugi T.; Kaneko I.; Meguro M.; Marumoto S. K., H.; Koyama K.; Oda T. (2002) A novel β-sheet breaker, RS-0406, reverses amyloid β-induced cytotoxicity and impairment of long-term potentiation in vitro. Br. J. Pharmacol. 137, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klunk W. E.; Debnath M. L.; Pettegrew J. W. (1994) Development of small molecule probes for the beta-amyloid protein of Alzhemier’s disease. Neurobiol. Aging 15, 691–698. [DOI] [PubMed] [Google Scholar]
- Patrovi-Nia R.; Beheshti S.; Qin Z.; Mandal H. S.; Long Y.-T.; Girault H. H.; Kraatz H.-H. (2012) Study of amyloid β-peptide (Aβ12–28-Cys) interactions with Congo Red and β-sheet breaker peptides using electrochemical impedance spectroscopy. Langmuir 28, 6377–6385. [DOI] [PubMed] [Google Scholar]
- Kim Y.-S.; Randolph T. W.; Manning M. C.; Stevens F. J.; Carpenter J. F. (2003) Congo red populates partially unfolded states of an amyloidogenic protein to enhance aggregation and amyloid fibril formation. J. Biol. Chem. 278, 10842–10850. [DOI] [PubMed] [Google Scholar]
- Biancalana M.; Koide S. (2010) Molecular mechanism of thioflavin-T binding to amyloid fibrils. Biochim. Biophys. Acta, Proteins Proteomics 1804, 1405–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Rodriguez C.; Rimola A.; Rodriguez-Santiago L.; Ugliengo P.; Alvarez-Larena A.; Gutierrez-de-Teran H.; Sodupe M.; Gonzalez-Duarte P. (2010) Crystal structure of thioflavin-T and binding to amyloid fibrils: insights at the molecular level. Chem. Commun. 46, 1156–1158. [DOI] [PubMed] [Google Scholar]
- Mao X.; Cuo Y.; Wang C.; Zhang M.; Ma X.; Liu L.; Niu L.; Zeng Q.; Yang Y.; Wang C. (2011) Binding modes of thioflavin T molecules to prion peptide assemblies identified by using scanning tunneling microscopy. ACS Chem. Neurosci. 2, 281–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Middleton C. T.; Marek P.; Cao P.; Chiu C-c.; Singh S.; Woys A. M.; de Pablo J. J.; Raleigh D. P.; Zanni M. T. (2012) Two-dimensional infrared spectroscopy reveals the complex behaviour of an amyloid fibril inhibitor. Nat. Chem. 4, 355–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khurana R.; Coleman C.; Ionescu-Zanetti C.; Carter S. A.; Krishna V.; Grover R. K.; Roy R.; Singh S. (2005) Mechanism of thioflavin T binding to amyloid fibrils. J. Struct. Biol. 151, 229–238. [DOI] [PubMed] [Google Scholar]
- Cisek K.; Kuret J. (2012) QSAR studies for prediction of cross-β sheet aggregate binding affinity and selectivity. Bioorg. Med. Chem. 20, 1434–1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono M.; Hayashi S.; Kimura H.; Kawashima H.; Nakayama M.; Saji H. (2009) Push-pull benzothiazole derivatives as probes for detecting β-amyloid plaques in Alzheimer’s brains. Bioorg. Med. Chem. 17, 7002–7007. [DOI] [PubMed] [Google Scholar]
- Yona R. L.; Mazerez S.; Faller P.; Gras E. (2008) Thioflavin derivatives as markers for amyloid-β fibrils: insights into structural features important for high-affinity binding. ChemMedChem 3, 63–66. [DOI] [PubMed] [Google Scholar]
- Inbar P.; Li C. Q.; Takayama S. A.; Bautista M. R.; Yang J. (2006) Oligo(ethylene glycol) derivaitives of thioflavin T as inhibitors of protein-amyloid interactions. ChemBioChem 7, 1563–1566. [DOI] [PubMed] [Google Scholar]
- Bartolini M.; Naldi M.; Fiori J.; Valle F.; Biscarini F.; Nicolau D. V.; Andrisano V. (2011) Kinetic characterization of amyloid-beta 1–42 aggregation with a multimethodological approach. Anal. Biochem. 414, 215–225. [DOI] [PubMed] [Google Scholar]
- Amaro M.; Birch D. J. S.; Rolinski O. J. (2011) Beta-amyloid oligomerization monitored by intrinsic tyrosin fluorescence. Phys. Chem. Chem. Phys. 13, 6434–6441. [DOI] [PubMed] [Google Scholar]
- Benseny-Cases N.; Cocera M.; Cladera J. (2007) Conversion of non-fibrillar β-sheet oligomers into amyloid fibrils in Alzhemier’s disease amyloid peptide aggregation. Biochim. Biophys. Res. Commun. 361, 916–921. [DOI] [PubMed] [Google Scholar]
- LeVine H. III. (1993) Thioflavin T interaction with synthetic Alzhemer’s disease β-amyloid peptides: Detection of amyloid aggregation in solution. Protein Sci. 2, 404–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maezawa I.; Hong H.-S.; Liu R.; Wu C.-Y.; Cheng R. H.; Kung M.-P.; Kung H. F.; Lam K. S.; Oddo S.; LaFerla F. M.; et al. (2008) Congo red and thioflavin-T analogues detect Aβ oligomers. J. Neurochem. 104, 457–468. [DOI] [PubMed] [Google Scholar]
- Ryan D. A.; Narrow W. C.; Federoff H. J.; Bowers W. J. (2010) An improved method for generating consistent soluble amyloid-beta oligomers preparations for in vitro neurotoxicity studies. J. Neurosci. Methods 190, 171–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh D. M.; Hartley D. M.; Kusumoto Y.; Fezoui Y.; Condron M. M.; Lomakin A.; Benedek G. B.; Selkoe D. J.; Teplow (1999) Amyloid beta-fibrillogenesis. Structure and biological activity of protofibrillar intermediates. J. Biol. Chem. 274, 25945–25952. [DOI] [PubMed] [Google Scholar]
- Reinke A. A.; Gestwicki J. E. (2011) Insight into amyloid structure using chemical probes. Chem. Biol. Drug Des. 77, 399–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorbenko G.; Trusova V.; Kirilova E.; Kirilov G.; Kalnina I.; Vasilev A.; Kaloyanova S.; Deligeorgiev T. (2010) New fluorescent probes for detection and characterization of amyloid fibrils. Chem. Phys. Lett. 495, 275–279. [Google Scholar]
- Ahn J. S.; Lee J.-H.; Kim J.-H.; Paik S. R. (2007) Novel method for quantitative determination of amyloid fibrils of α-synuclein and amyloid β/A4 protein by using resveratrol. Anal. Biochem. 367, 259–265. [DOI] [PubMed] [Google Scholar]
- Ono K.; Yoshilke Y.; Takashima A.; Hasegawa K.; Naiki H.; Yamada M. (2003) Potent anti-amyloidogenic and fibril-destabilizing effects of polyphenols in vitro: implications for the prevention and therapeutics of Alzhemier’s disease. J. Neurochem. 87, 172–181. [DOI] [PubMed] [Google Scholar]
- Ahmed M.; Davis J.; Aucoin D.; Sato T.; Ahuja S.; Aimoto S.; Elliott J. I.; Van Nostrand W. E.; Smith S. O. (2010) Structural conversion of neurotoxic amyloid-β1–42 oligomers to fibrils. Nat. Struct. Mol. Bio. 17, 561–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamaguchi T.; Ono K.; Yamada M. (2010) Curcumin and Alzheimer’s disease. CNS Neurosci. Ther. 16, 285–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akaishi T.; Morimoto T.; Shibao M.; Watanabe S.; Sakai-Kato K.; Utsunomiya-Tate N.; Abe K. (2008) Structural requirements for the flavanoid fisetin in inhibiting fibril formation of amyloid β protein. Neurosci. Lett. 444, 280–285. [DOI] [PubMed] [Google Scholar]
- Noormägi A.; Primar K.; Tougu V.; Palumaa P. (2012) Interference of low-molecular substances with the thioflavin-T fluorescence assay of amyloid fibrils. J. Pept. Sci. 18, 59–64. [DOI] [PubMed] [Google Scholar]
- Zovo K.; Helk E.; Karafin A.; Tougu V.; Palumaa P. (2010) Label-free high-throuput screening assay for inhibitors of Alzheimer’s amyloid-β peptide aggregation based on MALDI MS. Anal. Chem. 82, 8558–8565. [DOI] [PubMed] [Google Scholar]
- Liu K.-N.; Wang H.-Y.; Chen C.-Y.; Wang S. S.-S. (2010) L-Argenine reduces thioflavin T fluorescence but not fibrillization of bovine serum albumin. Amino Acids 39, 821–829. [DOI] [PubMed] [Google Scholar]
- Cole G. M.; Yang F.; Lim G. P.; Cummings J. L.; Masterman D. L.; Frautschy S. A. (2003) A rationale for curcuminoids for the prevention or treatment of Alzhemier’s disease. Curr. Med. Chem.: Immunol., Endocr. Metab. Agents 3, 15–25. [Google Scholar]
- Mansuco C.; Siciliano R.; Barone E. (2011) Curcumin and Alzheimer disease: this marriage is not to be performed. J. Biol. Chem. 286, le3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono K.; Hasegawa K.; Naiki H.; Yamada M. (2004) Curcumin has potent anti-amyloidogenic effects for Alzhemier’s β-amyloid fibrils in vitro. J. Neurosci. Res. 75, 742–750. [DOI] [PubMed] [Google Scholar]
- Reinke A. A.; Gestwicki J. E. (2007) Structure-activity relationships of amyloid beta-aggregation inhibitors based on curcumin: influence of linker length and flexibility. Chem. Biol. Drug Des. 70, 206–215. [DOI] [PubMed] [Google Scholar]
- Kim H.; Park B.-S.; Lee K.-G.; Choi C. Y.; Jang S. S.; Kim Y. H.; Lee S. E. (2005) Effects of naturally occuring compounds on fibril formation and oxidative stress of β-amyloid. J. Agric. Food Chem. 53, 8537–8541. [DOI] [PubMed] [Google Scholar]
- Jagota S.; Rajada J. (2012) Effect of phenolic compounds against Aβ aggregation and Aβ-induced toxicity in transgenic C. elegans. Neurochem. Res. 37, 40–48. [DOI] [PubMed] [Google Scholar]
- Shi C.; Zhao L.; Zhu B.; Li Q.; Yew D. T.; Yao Z.; Xu J. (2009) Protective effects of Ginkgo biloba (EGb761) and its constituents quercetin and ginkgolide B against β-amyloid peptide-induced toxicity in SH-SY5Y cell. Chem.-Biol. Interact. 181, 115–123. [DOI] [PubMed] [Google Scholar]
- Jiménez-Aliaga K.; Bermejo-Bescós P.; Benedí J.; Martín-Aragón S. (2011) Quercetin and rutin exhibit antiamyloidogenic and fibril-disaggregating effect in vitro and potent antioxidant activity in APPswe cells. Life Sci. 89, 939–945. [DOI] [PubMed] [Google Scholar]
- Ohta K.; Mizuno A.; Li S.; Itoh M.; Ueda M.; Ohta E.; Hida Y.; Wang M.-x.; Furoi M.; Tsuzuki Y; et al. (2011) Endoplasmic reticulum stress enhances γ-secretase activity. Biochem. Biophys. Res. Commun. 416, 362–366. [DOI] [PubMed] [Google Scholar]
- Ansari M. A.; Abdul H. M.; Joshi G.; Wycliffe O.; Butterfiled D. A. (2009) Protective effect of quercetin in primary neurons against Aβ(1–42): relevance to Alzheimer’s disease. J. Nutr. Biochem. 20, 269–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirohata M.; Hagesawa K.; Tsutsumi-Yasuhara; Ohhashi Y.; Ookoshi T.; Ono K.; Yamada M.; Naiki H. (2007) The anti-amyloidogenic effect is exerted against Alzhemier’s β-amyloid fibrils in vitro by preferential and reversible binding of flavonoids to the amyloid fibril structure. Biochemistry 46, 1888–1899. [DOI] [PubMed] [Google Scholar]
- Ladiwala A. R. A.; More-Pale M.; Lin J. C.; Shyam S.; Fishman Z. S.; Dordick J. S.; Tessier P. M. (2011) Polyphenolic glycosides and aglycones utilize opposing pathways to selectively remodel and inactivate toxic oligomers of amyloid β. ChemBioChem 12, 1749–1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bate C.; Tayebi M.; Williams A. (2008) Ginkgolides protect against amyloid-β1–42-mediated synapse damage in vitro. Mol. Neurodegener. 3, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marambaud P.; Zhao H.; Davies P. (2005) Resveratrol promotes clearance of Alzhemier’s desease amyloid-β peptides. J. Biol. Chem. 280, 37377–37382. [DOI] [PubMed] [Google Scholar]
- Carver J. A.; Duggan P. J.; Ecroyd H. L.; Liu Y.; Meyer A. G.; Tranberg C. E. (2010) Carboxymethylated-κ-casein: a convenient tool for the identification of the polyphenolic inhibitors of amyloid fibril formation. Bioorg. Med. Chem. 18, 222–228. [DOI] [PubMed] [Google Scholar]
- Hartsel S. C.; Weiland T. R. (2003) Amphotericin B binds amyloid fibrils and delays their formation: a therapeutic mechanism?. Biochemistry 42, 6228–6233. [DOI] [PubMed] [Google Scholar]
- Taniguchi S.; Susuki N.; Masuda M.; Hisanaga S.; Iwatsubo T.; Goedert M.; Hasegawa M. (2005) Inhibition of heparin-induced tau filament formation by phenothiazines, polyphenols, and porphyrins. J. Biol. Chem. 280, 7614–7623. [DOI] [PubMed] [Google Scholar]
- Smith N. W.; Annunziata O.; Dzyuba S. V. (2009) Amphotericin B interactions with soluble oligomers of amyloid Aβ1–42 peptide. Bioorg. Med. Chem. 17, 2366–2370. [DOI] [PubMed] [Google Scholar]
- Camilleri P.; Haskins N. J.; Howlett D. R. (1994) β-Cyclodextrin interacts with the Alzhemer amyloid β-A4 peptide. FEBS Lett. 341, 256–258. [DOI] [PubMed] [Google Scholar]
- Waite J.; Cole G. M.; Frautschy S. A.; Connor D. J.; Thakl L. J. (1992) Solvent effects on beta protein toxicity in vivo. Neurobiol. Aging 13, 595–599. [DOI] [PubMed] [Google Scholar]
- Yu J.; Bakhos L.; Chang L.; Holterman M. J.; Klein W. L.; Venton D. L. (2002) Per-6-substituted β-cyclodextrin libraries inhibit formation of β-amyloid-peptide (Aβ)-derived, soluble oligomers. J. Mol. Neurosci. 19, 51–55. [DOI] [PubMed] [Google Scholar]
- Wang Z.; Chang L.; Klein W. L.; Thatcher G. R. J.; Venton D. L. (2004) Per-6-substituted-per-6-deoxy-β-cyclodextrins inhibit the formation of β-amyloid peptide derived soluble oligomers. J. Med. Chem. 47, 3329–3333. [DOI] [PubMed] [Google Scholar]
- Wang M. S.; Boddapati S.; Sierks M. R. (2009) Cyclodextrins promote protein aggregation posing risks for therapeutic applications. Biophys. Biochem. Res. Commun. 386, 526–531. [DOI] [PubMed] [Google Scholar]
- Singh P. K.; Kumbhakar M.; Pal H.; Nath S. (2011) Confined ultrafast torsional dynamics of thiolfavin-T in a nanocavity. Phys. Chem. Chem. Phys. 13, 8008–8014. [DOI] [PubMed] [Google Scholar]
- Qin X.; Abe H.; Nakanishi H. (2002) NMR and CD studies on the interaction of Alzheimer β-amyloid (12–28) with β-cyclodextrin. Biochem. Biophys. Res. Commun. 297, 1011–1015. [DOI] [PubMed] [Google Scholar]
- Danielsson J.; Jarvet J.; Damberg P.; Gräslund A. (2004) Two-site binding of β-cyclodextrin to the Alzheimer Aβ(1–40) peptide measured with combined PFG-NMR diffusion and induced chemical shifts. Biochemistry 43, 6261–6269. [DOI] [PubMed] [Google Scholar]
- Wang H.-Y.; Ying Y.-L.; Li Y.; Kraatz H.-B.; Long Y. T. (2010) Nanopore analysis of β-amyloid peptide aggregation transition induced by small molecules. Anal. Chem. 83, 1746–1752. [DOI] [PubMed] [Google Scholar]
- Forloni G.; Colombo L.; Girola L.; Tagliavini F.; Salmona M. (2001) Anti-amyloidogenic activity of tetracyclines: studies in vitro. FEBS Lett. 487, 404–407. [DOI] [PubMed] [Google Scholar]
- Ono K.; Hasegawa K.; Naiki H.; Yamada M. (2004) Anti-amyloidogenic activity of tannic acidand it’s activity to destabilize Alzhemier’s β-amyloid fibrils in vitro. Biochim. Biophys. Acta, Mol. Basis Dis. 1690, 193–202. [DOI] [PubMed] [Google Scholar]
- Airoldi C.; Colombo L.; Manzoni C.; Sironi E.; Natalello A.; Doglia S. M.; Forloni G.; Tagliavini F.; Del Favero E.; Cantu L.; Nicotra F.; Salmona M. (2011) Tetracycline prevents Aβ oligomer toxicity through an atypical supramolecular interaction. Org. Biomol. Chem. 9, 463–472. [DOI] [PubMed] [Google Scholar]
- Han Y.-S.; Zheng W.-H.; Bastianetto S.; Chabot J.-G.; Quirion R. (2004) Neuroprotective effects of resveratrol against β-amyloid-induced neurotoxicity in rat hippocampal neurons: involvement of protein kinase C. Br. J. Pharmacol. 141, 997–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vingtdeaux V.; Giliberto L.; Zhao H.; Chandakkar P.; Wu Q.; Simon J. E.; Janle E. M.; Lobo J.; Ferruzzi M. G.; Davies P.; Marambaud P. (2010) AMP-activated protein kinase signaling activation by resveratrol modulates amyloid-β peptide mechanism. J. Biol. Chem. 285, 9100–9113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang T.-C.; Lu K.-T.; Wo Y.-Y. P.; Wu Y.-J.; Yang Y.-L. (2011) Resveratrol protect rats from Aβ-induced neurotoxicity by the reduction of iNOS expression and lipid peroxidation. PLoS One 6, e29102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladiwala A. R.; Lin J. C.; Bale S. S.; Marcelin-Cruz A. M.; Bhattacharya M.; Dordick J. S.; Tessler P. M. (2010) Resveratrol selectively remodels soluble oligomers and fibrils of amyloid Aβ into off-pathway conformers. J. Biol. Chem. 285, 24228–24237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y.; Wang X-p.; Yang S-g.; Wang Y-j.; Zhang X.; Du X-t.; Sun X-x.; Zhao M.; Huang L.; Liu R-t. (2009) Resveratrol inhibits beta-amyloid oligomeric cytotoxicity but does not prevent oligomer formation. NeuroToxicology 30, 986–995. [DOI] [PubMed] [Google Scholar]
- Schmuck C.; Frey P.; Heil M. (2005) Inhibition of fibril formation of Aβ by guanidiniocarbonyl pyrrole receptors. ChemBioChem 6, 628–631. [DOI] [PubMed] [Google Scholar]
- Ryu J.; Kanapathipillai M.; Lentzen G.; Park C. B. (2008) Inhibition of β-amyloid peptide aggregation and neurotoxicity by α-D-mannosylglycerate, a natural extremolyte. Peptides 29, 578–584. [DOI] [PubMed] [Google Scholar]
- Meng F.; Marek P.; Potter K. J.; Verchere C. B.; Raleigh D. P. (2008) Rifampicin does not prevent amyloid fibril formation by human islet amyloid polyptide but does inhibit fibril thioflavin-T interactions: implications for mechanistic studies of β-cell death. Biochemistry 47, 6016–6024. [DOI] [PubMed] [Google Scholar]
- Chandrashekaran I. R.; Adda C. G.; MacRaild C. A.; Anders R. F.; Norton R. S. (2010) Inhibition by flavanoids of amyloid-like fibril formation by Plasmodium falciparum merozoite surface protein 2. Biochemistry 49, 5899–5908. [DOI] [PubMed] [Google Scholar]
- Kroes-Nijboer A.; Lubbersen Y. S.; Venema P.; van der Linden E. (2009) Thioflavin T fluorescence assay for β-lactoglobulin fibrils hindered by DAPH. J. Struct. Biol. 165, 140–145. [DOI] [PubMed] [Google Scholar]
- Carrotta R.; Canale C.; Diasporo A.; Trapani A.; San Biagio P. L.; Bulone D. (2012) Inhibiting effect of αs1-casein on Aβ1–40 fibrillogensis. Biochim. Biophys. Acta, Gen. Subj. 1820, 124–132. [DOI] [PubMed] [Google Scholar]
- Goldsbury C.; Baxa U.; Simon M. N.; Steven A. C.; Engel A.; Wall J. S.; Aebi U.; Mueller S. A. (2011) Amyloid structure and assembly: insights from scanning transmission electron microscopy. J. Struct. Biol. 173, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adamcik J.; Jung J.-M.; Flakowski J.; De Los Rios P.; Dietler G.; Mezzenga R. (2010) Understanding amyloid aggregation by statistical analysis of atomic force microscopy images. Nat. Biotechnol. 5, 423–428. [DOI] [PubMed] [Google Scholar]
- Buell A. K.; Esbjoerner E. K.; Riss P. J.; White D. A.; Aibirhio F. I.; Toth G.; Welland M. E.; Dobson C. M.; Knowles T. P. J. (2011) Probing small molecule binding to amyloid fibrils. Phys. Chem. Chem. Phys. 13, 20044–20052. [DOI] [PubMed] [Google Scholar]
- Ono K.; Li L.; Takamura Y.; Yoshiike Y.; Zhu L.; Han F.; Mao X.; Ikeda T.; Takasaki J.-i.; Nishijo H.; Takashima A.; Teplow D. B.; Zagorski M. G.; Yamada M. (2012) Phenolic compounds prevent amyloid β-protein oligomerization and synaptic dysfunction by site-specific binding. J. Biol. Chem. 287, 14631–14643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tycko R. (2011) Solid-state NMR studies of amyloid fibril structure. Annu. Rev. Phys. Chem. 62, 279–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L.; Middleton C. T.; Singh S.; Reddy A. S.; Woys A. M.; Strasfeld D. B.; Marek P.; Raleigh D. P.; de Pablo J. J.; Zanni M. T.; Skinner J. L. (2011) 2DIR spectroscopy of human amylid fibrils reflects stable β-sheet structure. J. Am. Chem. Soc. 133, 16062–16071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith N. W.; Alonso A.; Brown C. M.; Dzyuba S. V. (2010) Triazole-containing BODIPY dyes as novel fluorescent probes for soluble oligomers of amyloid Aβ-142 peptide. Biochem. Biophys. Res. Commun. 391, 1455–1458. [DOI] [PubMed] [Google Scholar]
- Ono M.; Ishikawa M.; Kimura H.; Hayashi S.; Matsumura K.; Watanabe H.; Shimizu Y.; Cheng Y.; Cui M.; Kawashima H.; Saji H. (2010) Development of dual functional SPECT/fluorescent probes for imaging cerebral β-amyloid plaques. Bioorg. Med. Chem. Lett. 20, 3885–3888. [DOI] [PubMed] [Google Scholar]
- Ono M.; Watanabe H.; Kimura H.; Saji H. (2012) BODIPY-based molecular probe for imaging of cerebral β-amyloid plaques. ACS Chem. Neurosci. 3, 319–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitts C. C.; Beke-Somfai T.; Norden B. (2011) Michler’s hydrol blue: a sensitive probe for amyloid fibril detection. Biochemistry 50, 3451–3461. [DOI] [PubMed] [Google Scholar]
- Reinke A. A.; She H. Y.; Gestwicki J. E. (2009) A chemical screening approach reveals that indole fluorescence is quenched by pre-fibrillar but not fibrillar amyloid-β. Bioorg. Med. Chem. Lett. 19, 4952–4957. [DOI] [PMC free article] [PubMed] [Google Scholar]