

Abstract
The M1 muscarinic acetylcholine receptor is thought to play an important role in memory and cognition, making it a potential target for the treatment of Alzheimer’s disease (AD) and schizophrenia. Moreover, M1 interacts with BACE1 and regulates its proteosomal degradation, suggesting selective M1 activation could afford both palliative cognitive benefit as well as disease modification in AD. A key challenge in targeting the muscarinic acetylcholine receptors is achieving mAChR subtype selectivity. Our lab has previously reported the M1 selective positive allosteric modulator ML169. Herein we describe our efforts to further optimize this lead compound by preparing analogue libraries and probing novel scaffolds. We were able to identify several analogues that possessed submicromolar potency, with our best example displaying an EC50 of 310 nM. The new compounds maintained complete selectivity for the M1 receptor over the other subtypes (M2–M5), displayed improved DMPK profiles, and potentiated the carbachol (CCh)-induced excitation in striatal MSNs. Selected analogues were able to potentiate CCh-mediated nonamyloidogenic APPsα release, further strengthening the concept that M1 PAMs may afford a disease-modifying role in the treatment of AD.
Keywords: Muscarinic, acetylcholine, positive allosteric modulator (PAM), ML169, Alzheimer’s disease (AD), medium spiny neurons (MSNs), MLPCN
The muscarinic acetylcholine receptors (mAChRs) are members of the family A G-protein-coupled receptors (GPCRs) and comprise five subtypes (M1–M5) that mediate a broad range of actions of the neurotransmitter acetylcholine (ACh, 1) in the central nervous system and other tissues (Figure 1) and play key roles in memory and attention mechanisms, motor control, nociception, regulation of sleep-wake cycles, cardiovascular function, renal function, and GI function.1 mAChR subtypes M1, M3, and M5 are coupled to Gq and stimulate phospholipase C and intracellular Ca2+ release, while M2 and M4 are coupled to Gi and block adenylyl cyclase.2 Data from brain expression, cellular localization, and knockout mice have implicated that M1 plays a vital role in both memory and cognition,3 and numerous studies have shown efficacy in preclinical cognition models with selective M1 activators.4−7 Recently, studies have conclusively shown that M1 interacts with BACE1 to regulate its protesomal degradation and that M1 activation activates α-secretase, which leads to an increase in sAPPα thereby preventing the formation of Aβ via MAPK- and PKC-dependent pathways.8 In addition, M1 activation decreases τ phosphorylation; therefore, M1 activation affects the major pathological hallmarks of AD.8,9 Beyond the cholinergic and NMDA hypofunction hypotheses of schizophrenia where M1 could play a critical therapeutic role,10 BACE1 has also been linked to schizophrenia, via neuregulin 1; thus, the ability of M1 activation to regulate BACE1 suggests that pharmacological agents that can selectivity activate M1 represent a novel treatment for both Alzheimer’s disease (AD) and schizophrenia.8,9 Indeed, clozapine’s major metabolite (10–90% of dose), N-desmethylclozapine (6), is an M1 allosteric agonist, in sharp contrast to the typical orthosteric agonists 2 and 3, and is regarded as the most effective antipsychotic agent available today.10,11
Figure 1.

Structures of orthosteric mAChR agonists and M1-selective allosteric agonists and positive allosteric modulators (PAMs).
Numerous orthosteric mAChR agonists have demonstrated both palliative and disease-modifying efficacy in both preclinical animal studies and clinical trials.12 Notably, xanomeline (4), an M1/M4 preferring orthosteric agonist demonstrated efficacy in both AD and schizophrenic patients, further igniting the field. However, despite demonstrating efficacy, xanomeline failed in phase III due to an intolerable side effect profile which included decreased heart rate and GI distress due to off-target effects at peripheral M2 and M3 receptors.12,13 One of the key challenges to the development of ligands targeting these receptors is the highly conserved nature of the orthosteric binding site between the five receptor subtypes.1−7,14,15 Our group has addressed this challenge by developing positive allosteric modulators (PAMs) and allosteric agonists that selectively activate M1 by engaging allosteric sites with less evolutionary pressure for conservation across mAChR subtypes. However, M1 allosteric agonists, such as 5(4d) and 7,6 are more accurately coined bitopic agonists and suffer from receptor reserve-dependent, brain-region-specific pharmacology.7 Thus, our lab and others have shifted efforts toward M1 PAMs. Despite a decade of concerted effort, only three chemotypes have been reported as M1 PAMs: BQCA (8)16 and congeners, ML137 (9),17a and ML169 (10).17b In this Letter, we detail our efforts on further chemical optimization and pharmacological characterization of the ML169 series of M1 PAMs.
ML169 (M1 EC50 = 1.38 μM, 84% AChMax, 49-fold shift, >30 μM vs M2-M5, and brain/plasma ratio = 0.3) was a potent and efficacious M1 PAM optimized from a weak (EC50 ∼ 13 μM) MLPCN high-throughput screening hit.17b While ML169 was important, as it was a novel M1 PAM chemotype with a profile rivaling BQCA,16 ML169 suffered from both high predicted clearance (rat CLINT/CLHEP = 3341/68.6 mL/min/kg) and human CYP450 inhibition (IC50: 2C9 = 1.4 μM, 2D6 = 15 μM, and 3A4 = 1.5 μM), possibly owing to its high molecular weight and lipophilicity (cLogP = 4.8). Our initial efforts were directed at elucidating the minimum pharmacophore by deletion of various regions of the molecule, which we hoped would address the metabolic and pharmacodynamic liabilities of ML169. Truncation of various regions of the molecule, including the phenyl ring of the indole (11), the heteroaryl moiety (12), the benzyl moiety (13), and the sulfone moiety (14) resulted in a complete loss of activity at hM1 (Figure 2). Similarly, attempts to introduce polarity by incorporation of an azaindole (15) or benzimidazole (16) core also resulted in a complete loss of M1 PAM activity as did heterocyclic replacements, 17 and 18, for the northern amide. With marginally more success, we next investigated analogues in which the sulfone linker was replaced with both a methylene and an amide linker. We hoped that these analogues would not only address some of our metabolic and pharmacokinetic issues but also offer a simplified synthetic approach as well. While the methylene linker led exclusively to inactive compounds, replacement of the sulfone with an amide linker did afford some weak M1 PAMs (19–21).
Figure 2.

Initial optimization efforts aimed at ML169. All attempts to identify a minimum pharmacophore or replacement of multiple regions of ML169 with heterocycles led to a complete loss of M1 PAM activity. Amide replacements for the sulofone led to weak, low efficacy M1 PAMs (19–20) or inactive compounds.
Thus, like many other allosteric ligands, SAR was steep, and we would need to employ our fluorine walk methodology14a,17a,18 to identify positions on ML169 that would be tolerant of substitution. After surveying many fluorine substitutions about the ML169 core, we found that 4,6-difluorination of the indole core provided productive avenues for SAR. Thus, our synthesis of ML169 analogues commenced with an electrophilic sulfenylation of 4,6-difluoroindole (22) with methyl thioglycolate (Scheme 1). Alkylation of compound 23 with a variety of bromo-containing benzyl bromides proceeded smoothly to afford derivatives 24, which upon saponification with LiOH yielded carboxylic acids 25. Amide coupling to access congeners 26 was accomplished using either HATU or PyClu conditions. Oxidation of the sulfide to the sulfone was carried out with Oxone to give sulfones 27. Biaryl and heteroaryl analogue libraries 28 were generated from the corresponding aryl bromides 27 via Suzuki couplings. The synthesis route employed was quite flexible and could be adapted based on the reaction requirements of various analogues. During library synthesis, it was desirable to conduct the Suzuki coupling last to generate diversity in the southern region. However, for scale-ups of individual compounds, it was generally more advantageous to alkylate with the desired functionality already installed on a fully elaborated benzyl bromide. When the Suzuki reaction was employed in the final step, lower yields were observed, which were partially attributed to hydrolysis/decarboxylation of the methyl sulfone, which was especially prevalent when the isoxazole amide was present. This side reaction could be suppressed using Pd(OAc)2 and S-Phos as the catalyst system.19 Although this protocol often proceeded more slowly and failed to reach completion, it provided an overall higher yield of the desired product. Additionally, we were unable to find conditions that allowed us to perform Buchwald–Hartwig couplings on the aryl bromide to install N-bound heterocycles. In these cases, it was necessary to either conduct the cross coupling on compound 24 prior to the introduction of the amide or to perform the alkylation last with fully functionalized benzyl bromides.
Scheme 1. Synthesis of Fluorinated Analogues 29 of ML169.

One of the first areas we surveyed was heterocyclic amide replacements for the isoxazole moiety due to stability concerns. We prepared libraries of aromatic (not shown) and heterocyclic substituents in the 3-position of the benzyl ring (Table 1), and initially focused our efforts on analogues bearing the 2-pyridyl amide, which had also shown promising potencies in initial optimization studies on ML169. All of the biphenyl-derived compounds were inactive (data not shown). Introduction of heterocyclic substituents proved to be more fruitful; however, the SAR for this series was quite steep, as is common with allosteric modulators. Thiophene, pyrrole, and indole derivatives were inactive (29a–d). Indazole substituents attached at the 6-position showed weak potency (29e–f), while other attachment points proved inactive (29g–i). A number of promising analogues bearing a pyridine or pyrimidine ring were prepared (29j–p), with hM1 EC50’s ranging from 3 to 5 μM and efficacies ranging from 39 to 57% AChMax. An unadorned benzyl group (29x) was also tolerated (EC50 = 1.42 μM for 29x versus EC50 = 2.2 μM for ML169). 1,2,3-Triazole (29t) was tolerated at the 3-position, as was the precursor azide (29u), which could be a useful photoaffinity tool in studies to identify the allosteric site at which these compounds bind, provided this compound possesses high affinity at the hM1 receptor but shows micromolar potency due to a low cooperativity (not yet determined). The most potent compound prepared in this series was the methyl pyrazole 29s, which exhibited an hM1 EC50 of 360 nM potency, albeit with very low efficacy (33% AChMax).
Table 1. Structures and Activities of Analogues with meta-Substituted Benzylic Rings and the 2-Pyridyl Amidea.
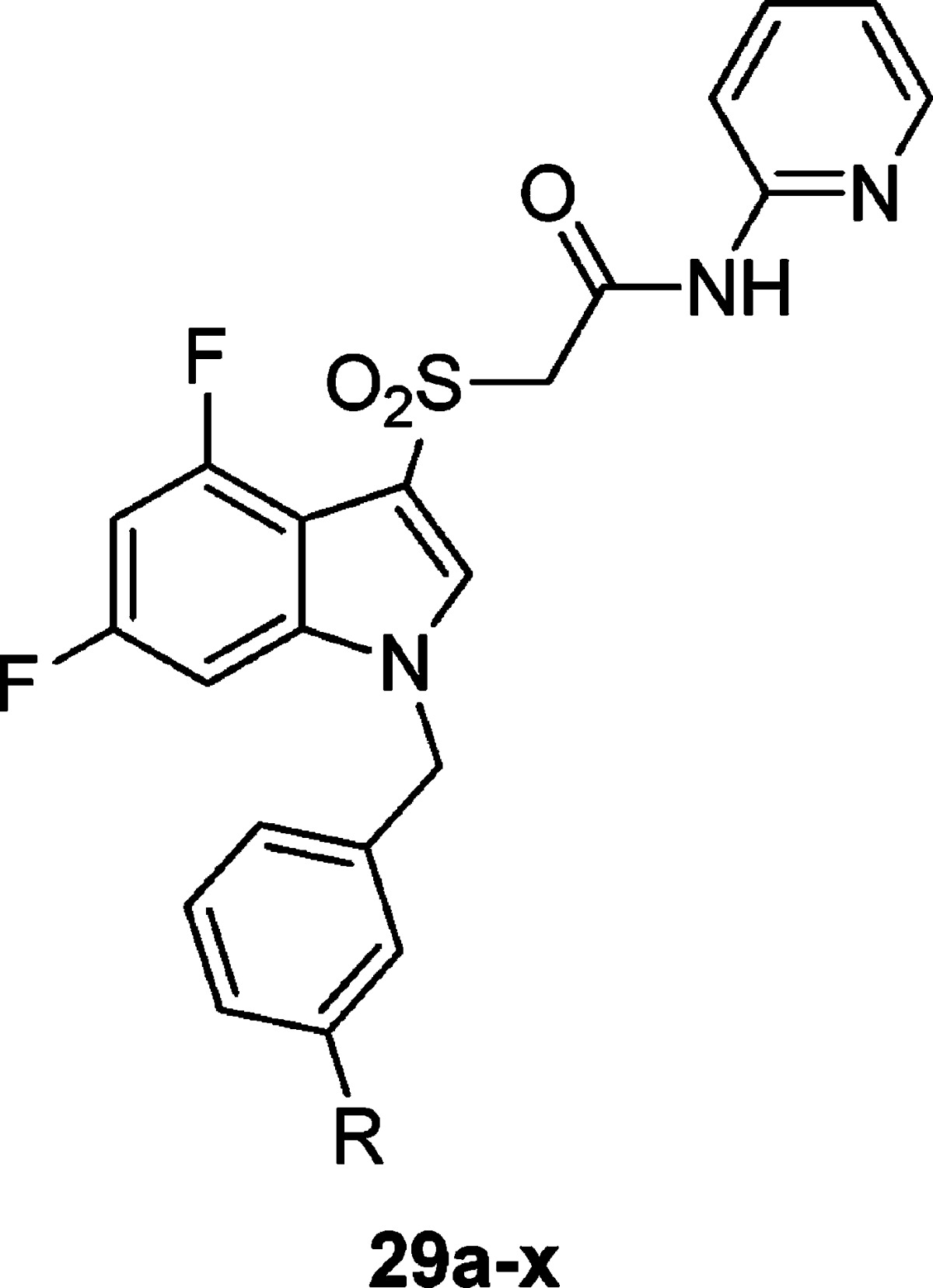

Values obtained are the average of at least three independent determinations performed in triplicate ± standard deviations.
In a subsequent round of SAR, we examined analogues bearing the isoxazole amide (Table 2). While we had hoped that the 2-pyridyl amide would improve the metabolic stability of the compounds as compared to the isoxazole amides, analogues from both amide series suffered from high hepatic intrinsic clearance and were potent inhibitors of multiple cytochrome P450s in vitro. As the 2-pyridyl amide did not seem to address these metabolic and clearance issues, as of yet, but still presented an active series with potentially divergent SAR, both amides were evaluated in additional libraries. Comparing Tables 1 and 2, potencies tracked reasonably well between the two amides. The isoxazole tended to give modestly higher potency and efficacy. A key divergence in SAR was noted within the N-H and N-methyl pyrazoles (30e–f). In the 2-pyridyl amide series, the methyl pyrazole 29s was highly potent (EC50 = 0.36 μM), and deletion of the N-methyl group abolished hM1 activity. In the isoxazole amide series, however, the N-H-pyrazole 30e is more potent than the methyl derivative 30f. While the methyl pyrazole is ∼6-fold less potent in the isoxazole amide series than its comparator in the 2-pyridyl series (29s), it is almost twice as efficacious.
Table 2. Structures and Activities of Analogues with meta-Substituted Benzylic Rings and Isoxazole Amidea.
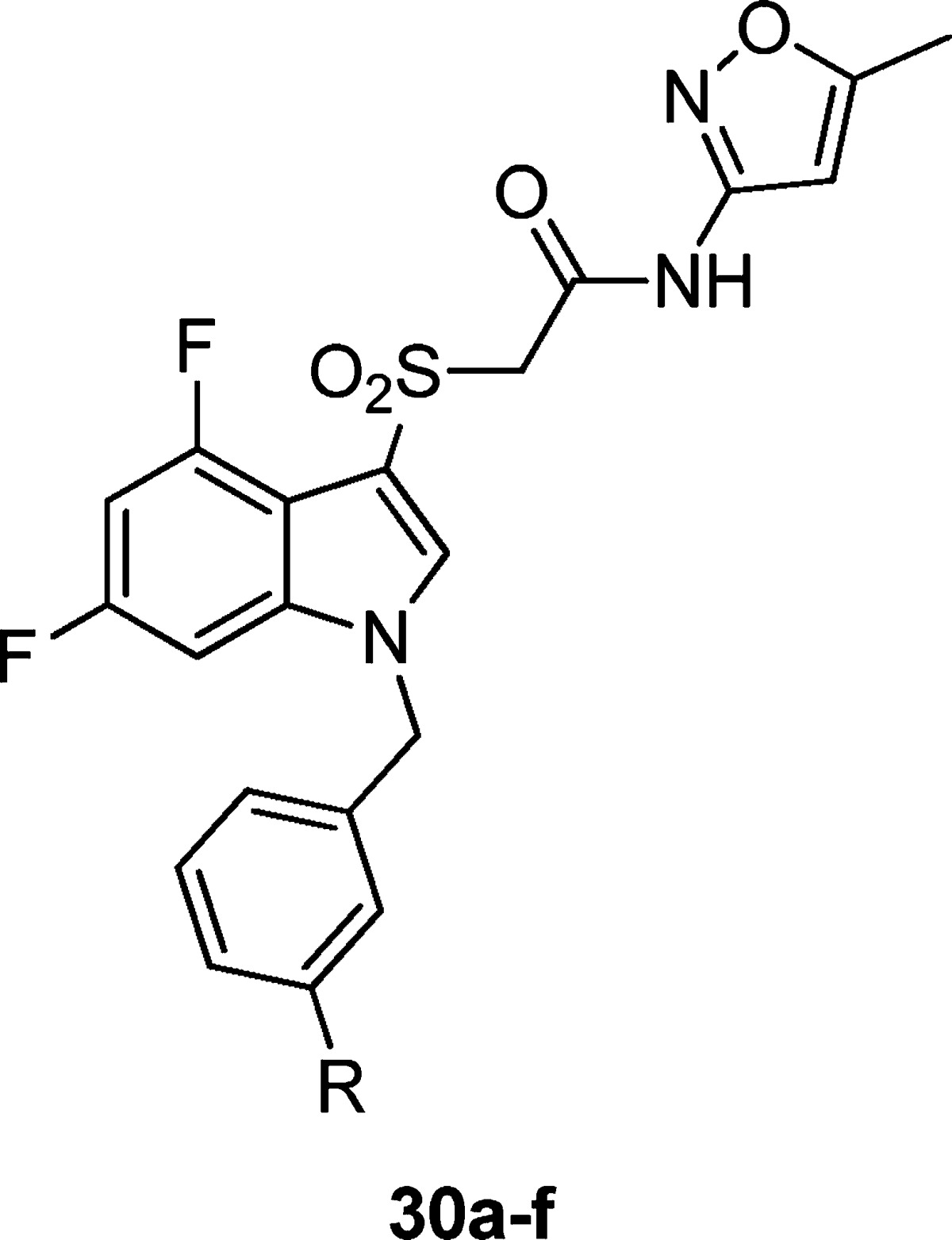

Values obtained are the average of at least three independent determinations performed in triplicate ± standard deviations.
While substitution at the 3-position of the benzyl ring was optimal for simple substituents (halogen, -OMe) during the initial optimization of ML169, we wished to see if this trend tracked for the heteroaryl-substituted benzyl groups (Table 3). To our surprise, we saw a deviation from the ML169 series, as well as differences between the 2-pyridyl and isoxazole amide series. For the 2-pyridyl amides, moving the substituent to the 4-position provided a minimal improvement over the 3-position (31a–d). For the isoxazole amides, however, three of the derivatives (31e–g) had sharply improved potency (∼3.5-fold, 2.8-fold, and ∼5-fold, respectively). Gratifyingly, compounds 31e–g displayed both submicromolar EC50’s and good efficacy (67–75% of AChMax responses) as well.
Table 3. Structures and Activities of Analogues with para-Substituted Benzylic Rings and 2-Pyridyl and Isoxazole Amidesa.
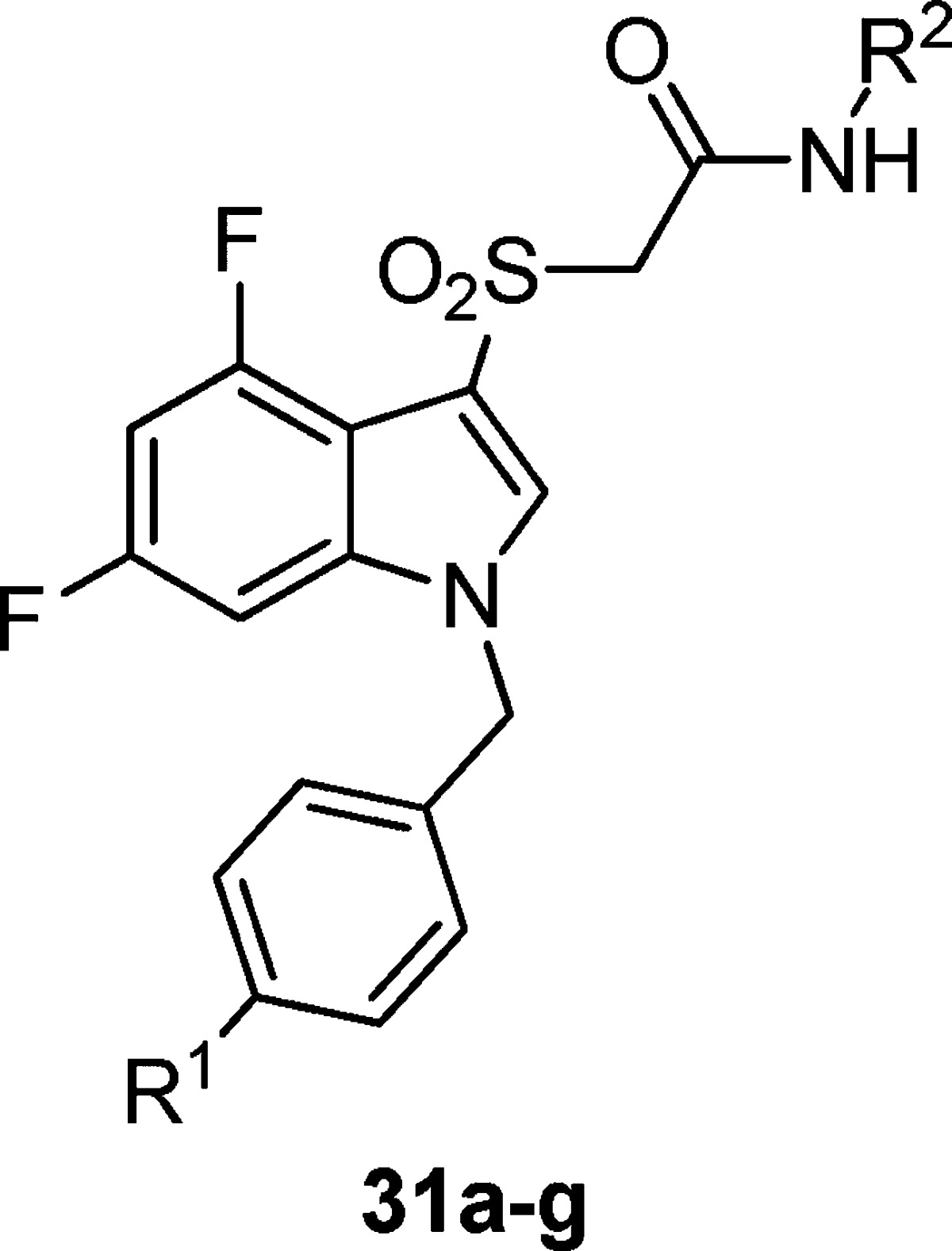

Values obtained are the average of at least three independent determinations performed in triplicate ± standard deviations.
On the basis of our experience in the chemical optimization of other M1 PAM scaffolds, we looked at subtly changing the electronics of the benzyl ring by introducing a fluorine (i.e., the fluorine walk) at various locations and found the 2-position to be the most beneficial (Table 4). For the first time, we were able to obtain a compound (32a) with submicromolar potency (EC50 = 950 nM) and robust efficacy (70% AChmax) in the 2-pyridyl amide series. We were also able to obtain two compounds (32j and 32o) with ∼500 nM potency in the isoxazole amide series. Again, the SAR was exceedingly steep; deletion of the pyrazole N-Me group resulted in a drastic loss in efficacy while maintaining potency (32o), while replacement of the N-methyl group with an N-propyl group resulted in a loss of both potency and efficacy (not shown). A variety of both pyrazole and pyridine substituents were prepared and further illustrate how subtle changes affect both potency and efficacy. At this point, we revisited the possibility of truncating portions of the molecule. Gratifyingly, we found that when the 2-fluoro-4-(1-N-methyl-pyrazole) benzyl group was employed, the simple methyl sulfone 33 maintained measurable activity (hM1 EC50 = 3.62 ± 0.36 μM, AChMax = 55.33 ± 12.9%). This truncated version offered improved metabolic properties including lower intrinsic and predicted hepatic clearance in both rats and humans. Further efforts to explore these analogues are under way and will be reported in due course. This agrees with the prior observation that the optimized 2-fluoro-4-(1-N-methyl-pyrazole) benzyl group was necessary to arrive at active analogues 31d–f, where the amide for sulfone replacement had been explored.
Table 4. Structures and Activities of Analogues with 2-Fluoro para-Substituted Benzylic Rings and 2-Pyridyl and Isoxazole Amidesa.


Values obtained are the average of at least three independent determinations performed in triplicate ± standard deviations.
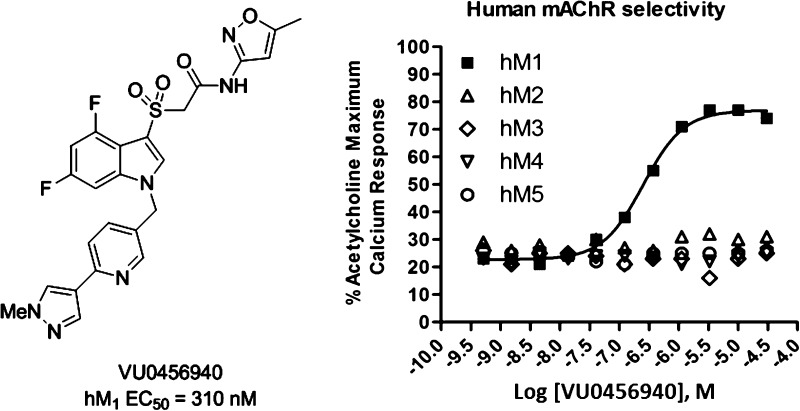
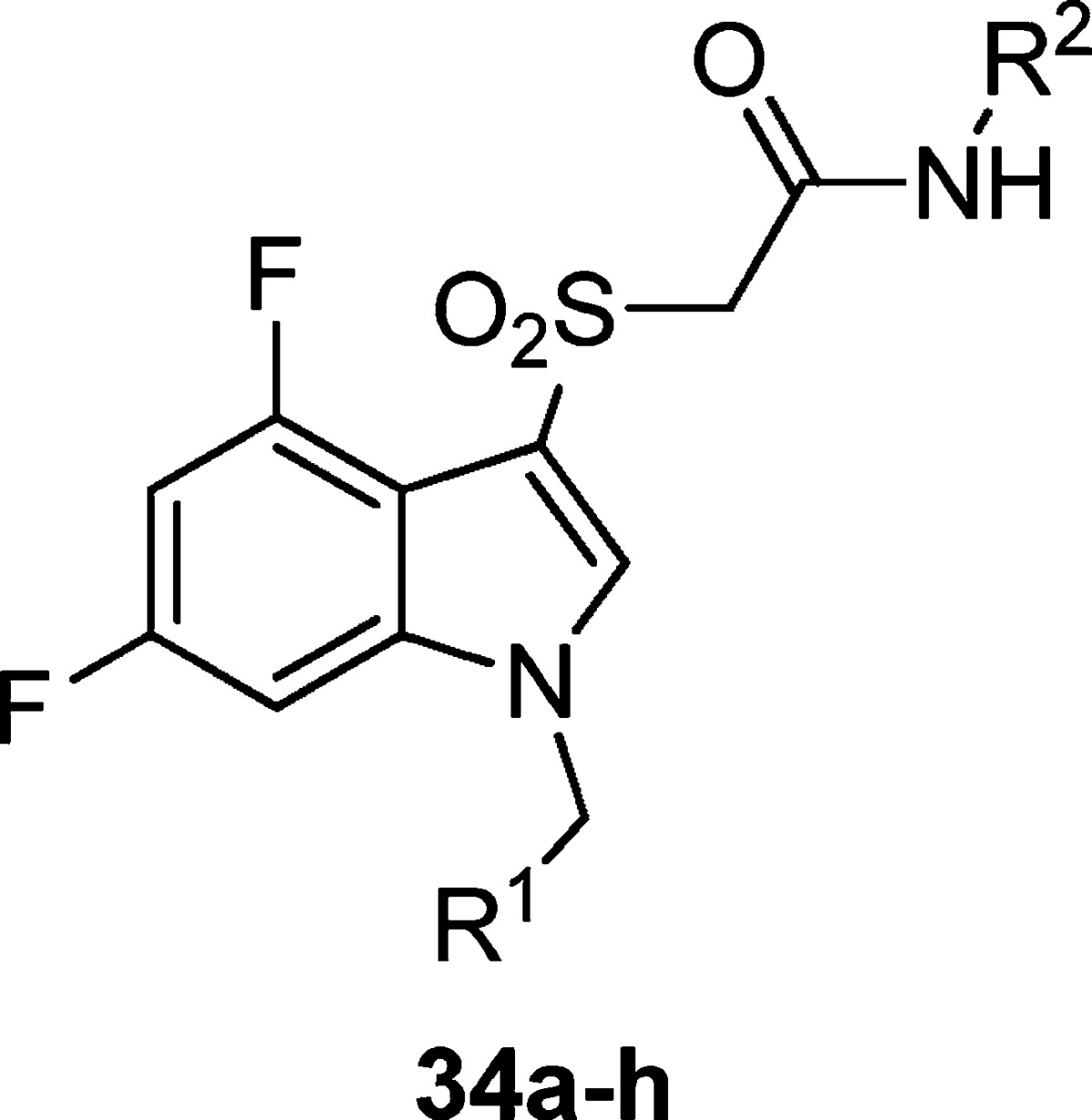
In a further attempt to improve physical properties for this series of M1 PAMs, we incorporated polarity by replacing the central benzyl ring with a pyridyl ring (Table 5). Unlike the unsubstituted benzyl ring in 29x, which maintained potency, the unsubstituted pyridyl rings resulted in inactive or only weakly active compounds (34a and 34b). However, incorporation of the N-methyl pyrazole at the 3- or 4-position of the central pyridyl ring proved to be quite beneficial. The N-oxides of 34c and 34d were tested and showed a 2-fold decrease in potency. Again, the isoxazole derivatives proved to be both more potent and efficacious than the 2-pyridyl compounds. Incorporation of the pyrazole at the 3-position with the isoxazole amide (34g) provided the highest efficacy observed in this series. When the N-methyl pyrazole was placed in the 4-position (34h), we arrived at our most potent M1 PAM (EC50 = 310 nM, 69% AChMax) to date in this series. With submicromolar compounds in hand, we screened compounds 34h, 32j, and 31e against all five mAChR subtypes. We were gratified to see that all three compounds maintained their M1 selectivity, with no measurable activity across the M2–M5 receptors. Only the results for 34h are shown in Figure 3, but 32j and 31e provided identical mAChR selectivity profiles. While ML169 showed a robust left-ward shift of the ACh concentration–response-curve, 34h showed an 14.3 ± 0.9 fold-shift at 1.25 μM; however, it is not clear if potency, efficacy (%ACh max and fold-shift), or both are required to potentiate M1 in native systems.
Table 5. Structures and Activities of Analogues with Substituted Pyridyl Ringsa.

Values obtained are the average of at least three independent determinations performed in triplicate ± standard deviations.
Figure 3.

Concentration–response curves for 34h (VU0456940) for human mAChR subtypes M1–5. Data are an average of three determinations.
In addition to optimizing the potency and efficacy of our lead compounds, we also evaluated the metabolic and pharmacodynamic properties of the new analogues. The predicted hepatic clearance in rat for ML169 was at or near hepatic blood flow (CLHEP = 68.6 mL/min/kg). Additionally, ML169 was also a moderately potent CYP450 inhibitor (IC50: 2C9 = 1.4 μM, 2D6 = 15 μM, and 3A4 = 1.5 μM) and possesses a very high cLogP (4.8). Many of our initial analogues such as 29k not only suffered from very high clearance but were even more potent CYP450 inhibitors (IC50: 2C9 < 0.1 μM, 2D6 = 0.3 μM, and 3A4 < 0.1 μM for 29k). While 32j offered a significant increase in potency from ML169, it still suffered high intrinsic hepatic clearance and potent CYP450 inhibition (IC50: 2C9 < 0.1 μM, 2D6 > 30 μM, and 3A4 = 0.77 μM). Compound 34h showed slightly lower predicted hepatic clearance in rats (CLHEP = 59.4 mL/min/kg); however, we were able to substantially decrease the intrinsic clearance as compared to ML169 (392 mL/min/kg for 34h, 3341 mL/min/kg for ML169). We were also able to decrease cLogP (3.41) by the addition of the pyridine core. Additionally, the CYP450 inhibition profile of 34h was moderately improved over ML169 (IC50 for 34h: 2C9 = 4.6 μM, 2D6 > 30 μM, and 3A4 = 3.3 μM). We also observed a good fraction unbound in human plasma protein binding (fu = 0.093) and brain homogenate binding (fu = 0.054) in rat. Compound 34h also possessed a brain/plasma level >1, similar to ML169, but due to rapid clearance in rat plasma (plasma, 10.9 ng/mL and brain 14.1 ng/mL), a definitive number is not warranted.20 While compound 34g was ∼10-fold less potent than 34h, we did however see lower predicted clearance in both rat and human (CLHEP = 50.9 and 11.9 mL/min/kg, respectively). Similarly, methyl sulfone 33, which only displayed modest potency (3.6 μM), provided the lowest predicted hepatic clearance observed in rat for any of the analogues studied (CLHEP = 42.8 mL/min/kg), highlighting the β-sulfone/amide as a potential metabolic soft spot.
Next, we evaluated 34h (VU0456940) for its ability to potentiate M1 in a native system. Because of the in vivo PK profile, we elected to explore activity in native systems using an electrophysiology approach in brain slice preparations. One of the well-established physiological effects of M1 in the CNS is modulation of excitability of medium spiny neurons (MSNs) in the striatum,21,22 a key structure of the basal ganglia that is critically involved in motor control and cognitive functions.23,24 Disturbance of cholinergic signaling in the striatum has been implicated in a variety of neurological and neuropsychiatric disorders such as Parkinson’s disease, Alzheimer’s disease, and schizophrenia.25−28 We tested the hypothesis that M1 PAM 34h would potentiate the M1-mediated excitation in striatal MSNs. Bath application of a low concentration of muscarinc agonist CCh (0.5 μM) slightly increased the excitability of MSNs as indicated by an increase in number of action potential discharge in response to a near threshold depolarization current pulse (change in the number of spikes/pulse) by 2.1 ± 0.5 (n = 7, Figure 4A and C). By contrast, in the presence of 1 μM 34h, 0.5 μM CCh induced a robust increase in the number of spikes/pulse (13.8 ± 2.1, n = 6, compared to 2.1 ± 0.5 when 0.5 μM CCh was applied alone, n = 7, p < 0.002; Figure 4B and C). As noted, application of 34h alone also caused a slight excitation of MSNs (Figure 4B and C), and the number of spikes/pulse increased by 1.7 ± 0.3 (from 2.3 ± 0.5 in control to 4.0 ± 0.7 in VU0456940, p < 0.005, n = 6), which might be due to the fact that 34h potentiated endogenous ambient acetylcholine action on M1 receptors in MSNs. These data demonstrate that 34h is efficacious in native tissue and able to potentiate M1-mediated response in the CNS, despite possessing a modest fold-shift.
Figure 4.

Compound 34h potentiates the CCh-induced excitation in MSNs. (A) Membrane potential responses to a depolarization current step in control and after application of 0.5 μM CCh in a typical experiment, showing that low concentration of CCh causes slight excitation in a MSN. (B) Membrane potential responses to a depolarization current step in the control, during application of 1 μM 34h and coapplication of 34h and 0.5 μM CCh in another MSN, showing 34h potentiates CCh-induced excitation of the MSN. (C) Bar graph summarizes group data showing the change in number of action potential discharges in response to the depolarization current step following application of 0.5 μM CCh, 1 μM, and 0.5 μM CCh and 1 μM 34h combined. *p < 0.002.
Our lab has previously demonstrated that both M1-selective allosteric agonists alone (ML071 (7) and TBPB (5)),4d,6 as well as M1 PAMs (BQCA and ML169)6,17 co-dosed with an orthosteric agonist (CCh) can shift amyloid protein precursor (APP) processing toward a nonamyloidogenic pathway, which is measured by an increase in soluble APP (APPsα) production.29 It is believed that this shift in APP processing may prevent the accumulation of β-amyloid plaques and provide a therapy with a disease-modifying role in the treatment of Alzheimer’s disease. Maximum response in APPsα production was observed in the hM1-overexpressing cells treated with 10 μM carbachol (CCh), whereas a 100 nM concentration of CCh elicited minimal response (Figure 5). However, when M1 PAMs 34 h, 32j, and 32o were co-treated at 2 μM with 100 nM of CCh, the minimal APPsα release by 100 nM CCh was dramatically potentiated to 97%, 78%, and 100% response of the 10 μM dose of CCh, respectively. Compound 34h, the most potent of those tested, was able to achieve a potentiation of 100 nM of CCh to 88% of the 10 μM CCh dose at just 200 nM. When BQCA, which possesses dual agonist-PAM activity, was examined in this assay at 2 μM, we did not observe the PAM activity of BQCA because BQCA alone showed nearly the same maximum response as that of 10 μM CCh (data not shown). In contrast, 34h and 32j displayed almost no activity in the absence of CCh. While 32o did display modest activity in the absence of CCh (30%), when 32o was coincubated with CCh at 100 nM, there was a significantly higher response.
Figure 5.
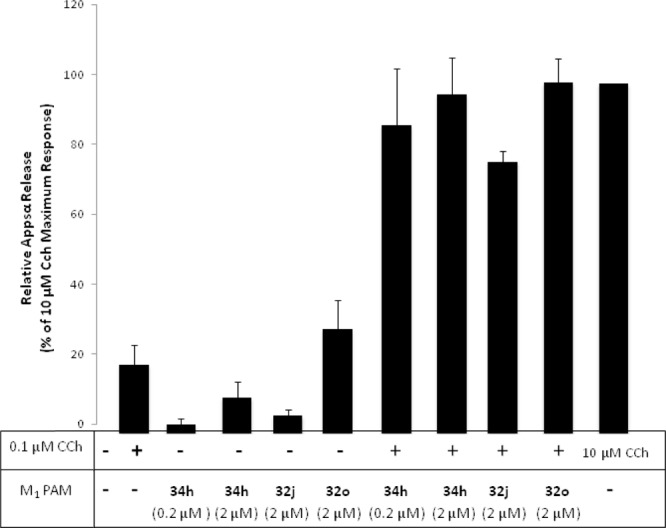
M1 PAMs 32j, 32o, and 34h potentiate the CCh-mediated nonamyloidogenic APPsα release in TREx293-hM1 cells.
In summary, we have developed a potent and selective M1 PAM by further optimization of ML169. We have increased potency ∼7-fold from ML169 to 34h (310 nM), while still maintaining exclusive subtype selectivity for M1. We have also achieved modest reductions in CYP450 inhibition at 2C9, 2D6, and 3A4 (2–3-fold), and significantly lowered cLogP. Compounds 34h and 32j were shown to potentiate CCh-mediated APP processing, suggesting that M1 PAMs may be able to play a disease-modifying role in the treatment of Alzheimer’s disease. Despite a modest fold-shift, 34h robustly potentiated the M1-mediated excitation in striatal MSNs, providing efficacy in a native system. New chemical scaffolds, including the methyl sulfone 33 and amides 28d–f, have also been shown to be viable analogues, and subsequent studies on these entities will be reported in due course.
Methods
General Medicinal Chemistry Methods
All NMR spectra were recorded on a 400 MHz Bruker NMR spectrometer. 1H chemical shifts are reported in δ values in parts per million downfield from TMS as the internal standard in CDCl3. Data are reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, dd = doublet of doublets, and dm = doublet of multiplets), integration, and coupling constant (Hz). Low resolution mass spectra were obtained on an Agilent 1200 LCMS with electrospray ionization. High resolution mass spectra were recorded on a Waters QToF-API-US plus Acquity system. Analytical thin-layer chromatography was performed on 250 mm silica gel 60 F254 plates. Analytical HPLC was performed on an Agilent 1200 analytical LCMS with UV detection at 214 and 254 nm along with ELSD detection. Preparative purification was performed on a custom Agilent 1200 LCMS with collection triggered by UV detection. Solvents for extraction, washing, and chromatography were HPLC grade. All reagents were commercially available and used without purification. All tested compounds were ≥95% purity.
General Procedure for the Synthesis of Analogue Libraries
Electrophilic Sulfenylation (22)
To a solution of 4,6-difluoro-indole 22 (10.0 g, 65.3 mmol) and methyl glycolate (5.84 mL, 65.3 mmol) in methanol/water (300 mL, 3:1) was added iodine (16.5 g, 65.3 mmol) and potassium iodide (10.9 g, 65.3 mmol). The reaction mixture was stirred at ambient temperature for 60 h. The methanol was removed in vacuo, and the aqueous layer was then diluted with a saturated solution of sodium bicarbonate and extracted with ethyl acetate. The organic layer was dried over magnesium sulfate, evaporated in vacuo, and the resulting residue was purified on a silica gel column (0–100% ethyl acetate/hexanes) to afford methyl 2-((4,6-difluoro-1H-indol-3-yl)thio)acetate (23) (5.5 g, 32% yield). LCMS >98% at 254 nm, m/z = 258 (M+H). 1H NMR in CDCl3: δ 8.83 (s, 1H), 7.23–7.21 (s, 1H), 6.81 (dd, J = 8.8 Hz, 2.0 Hz, 1H), 6.66 (t, J = 10.4 Hz, 1H), 3.70 (s, 3H), 3.53 (s, 2H); 13C NMR δ 170.9, 159.4 (dd, JC–F = 240 Hz, 12 Hz), 156.0 (dd, JC–F = 249 Hz, 15 Hz), 138.2 (t, JC–F = 13 Hz), 130.5, 113.8 (d, JC–F = 18 Hz), 102.4, 96.5 (t, JC–F = 24 Hz), 94.3 (dd, JC–F = 26 Hz, 5 Hz), 52.3, 38.9.
Indole Alkylation (24)
In a dry 40-mL scintillation vial, methyl 2-((4,6-difluoro-1H-indol-3-yl)thio)acetate 23 (300 mg, 1.17 mmol), potassium carbonate (400 mg, 2.90 mmol), and 3-bromobenzyl bromide (325 mg, 1.31 mmol) were massed. The vial was charged with a stir bar, and acetonitrile (10 mL) was added. The reaction was capped and heated to 70 °C for 4 h, at which time the reaction was determined to be complete by LCMS. The reaction was cooled to ambient temperature, then diluted with ethyl acetate (30 mL), and washed with brine (30 mL). The aqueous layer was extracted with ethyl acetate (3 × 30 mL). The organic layers were combined, dried over MgSO4, filtered, and concentrated in vacuo. The crude product was purified by flash column chromatography eluting with 0 to 100% ethyl acetate/hexanes to afford 454 mg (87% yield) of methyl 2-((1-(3-bromobenzyl)-4,6-difluoro-1H-indol-3-yl)thio)acetate (24). LCMS >98% pure at 254 nm, m/z = 426 (M+H). 1H NMR in CDCl3: δ 7.44 (d, J = 8.0 Hz, 1H), 7.22–7.16 (m, 3H), 7.00 (d, J = 7.6 Hz, 1H), 6.74–6.64 (m, 2H), 5.19 (s, 2H), 3.68 (s, 3H), 3.53 (s, 2H); 13C NMR δ 170.3, 159.5 (dd, JC–F = 240 Hz, 12 Hz), 156.3 (dd, JC–F = 251 Hz, 15 Hz), 138.6 (t, JC–F = 13 Hz), 138.0, 134.1, 131.3, 130.5, 129.7, 125.3, 123.1, 114.6 (d, JC–F = 18 Hz), 102.3, 96.8 (dd, JC–F = 28 Hz, 23 Hz), 93.0, (d, JC–F = 31 Hz), 52.1, 49.9, 38.8.
Saponification (25)
In a 40-mL scintillation vial, methyl 2-((1-(3-bromobenzyl)-4,6-difluoro-1H-indol-3-yl)thio)acetate 24 was massed (450 mg, 1.06 mmol). Methanol and water were added (6 mL, 1:1), followed by lithium hydroxide (50.0 mg, 2.08 mmol). The reaction was capped and heated to 70 °C and monitored by LCMS. Upon completion, the reaction was cooled to ambient temperature and diluted into 40 mL of dichloromethane/brine (1:1). The aqueous layer was acidified with 2 M HCl. The organic layer was separated, and an aqueous layer was extracted with dichloromethane (2 × 20 mL). The organic layers were combined, dried over MgSO4, and filtered. The solvent was removed in vacuo to afford 405 mg (93% yield) of 2-((1-(3-bromobenzyl)-4,6-difluoro-1H-indol-3-yl)thio)acetic acid (25), and the residue was used in subsequent reactions without further purification. LCMS: > 98% pure at 254 nm, m/z = 412 (M+H). 1H NMR in CDCl3: δ 7.42 (d, J = 8.0 Hz, 1H), 7.24–7.16 (m, 3H), 6.92 (d, J = 8.0 Hz, 1H), 6.72–6.63 (m, 2H), 5.18 (s, 2H), 3.53 (s, 2H); 13C NMR δ 172.2, 159.4 (dd, JC–F = 238 Hz, 11 Hz), 156.2 (dd, JC–F = 264 Hz, 14 Hz), 138.9, 138.7 (d, JC–F = 13 Hz), 134.3, 130.7, 130.3, 129.6, 125.2, 122.5, 114.4 (d, JC–F = 19 Hz), 102.0, 95.9 (dd, JC–F = 28 Hz, 23 Hz), 92.9 (d, JC–F = 31 Hz), 49.3, 38.7.
HATU Mediated Amide Coupling (i) (26a, R = 2-Aminopyridine and N-aryl = 2-Fluoro-4-bromo)
In a dry 4-dram vial equipped with a stir bar, 2-((1-(4-bromo-2-fluorobenzyl)-4,6-difluoro-1H-indol-3-yl)thio)acetic acid (402 mg, 0.93 mmol) and HATU (550 mg, 1.45 mmol) were massed. DMF (6 mL) was added, followed by DIPEA (450 μL, 2.52 mmol). The reaction was allowed to stir at ambient temperature for 10 min, then 2-aminopyridine (180 mg, 1.92 mmol) was added. The reaction was monitored by LCMS. Upon completion, reaction was diluted into 40 mL of dichloromethane/brine (1:1). The layers were separated, and the aqueous layer was extracted with dichloromethane (2 × 20 mL). The organic layers were combined, dried over MgSO4, and concentrated in vacuo. The crude material was purified on SiO2 gel by flash column chromatography eluting with 0 to 100% hexanes/ethyl acetate to afford 350 mg (74% yield) of 2-((1-(4-bromo-2-fluorobenzyl)-4,6-difluoro-1H-indol-3-yl)thio)-N-(pyridin-2-yl)acetamide (26a, R1 = 2-aminopyridien; R = 2-fluoro-4-bromo). LCMS: > 98% pure at 254 nm, m/z = 506 (M+H). 1H NMR in CDCl3: δ 9.08 (s, 1H), 8.26 (d, J = 4.8 Hz, 1H), 8.10 (d, J = 8.4 Hz, 1H), 7.66 (dt, J = 8.8 Hz, 1 Hz, 1H), 7.38 (d, J = 8.0 Hz, 1H), 7.26–7.20 (m, 2H), 7.09–7.03 (m, 2H), 6.88 (d, J = 7.6 Hz, 1H), 6.71–6.65 (m, 2H), 5.10 (s, 2H), 3.65(s, 2H); 13C NMR δ 167.3, 159.6 (dd, JC–F = 241 Hz, 12 Hz), 156.2 (dd, JC–F = 265 Hz, 15 Hz), 151.0, 147.8, 138.6 (dd, JC–F = 27 Hz, 13 Hz), 138.3, 137.7, 133.5, 131.3, 130.5, 129.7, 125.2, 123.0, 119.9, 114.2 (d, JC–F = 18 Hz), 113.7, 101.7, 96.9 (dd, JC–F = 28 Hz, 23 Hz), 93.4 (dd, JC–F = 26 Hz, 4 Hz), 50.0, 41.6.
PyClu Mediated Amide Coupling (ii) (26b, R = 2-Aminopyridine, N-aryl = 3-bromo)
2-((1-(3-Bromobenzyl)-4,6-difluoro-1H-indol-3-yl)thio)acetic acid (100 mg, 0.243 mmol), 2-aminopyridine (48 mg, 51 mmol), PyClu (165 mg, 50.0 mmol), and DIPEA (120 μL, 68 mmol) were massed into a 5-mL microwave vial. Dichloroethane (5 mL) was added, and the reaction was heated in the microwave at 120 °C for 30 min. The reaction was diluted into dichloromethane/brine (40 mL, 1:1). The organic layer was separated, and the aqueous layer extracted with dichloromethane (2 × 20 mL). The organic layers were combined, dried over MgSO4, filtered, and concentrated in vacuo. The crude material was purified on SiO2 gel by flash column chromatography eluting with 0 to 100% hexanes/ethyl acetate to afford 100 mg (85% yield) of 2-((1-(3-bromobenzyl)-4,6-difluoro-1H-indol-3-yl)thio)-N-(pyridin-2-yl)acetamide (26b, R′ = 2-aminopyridine; R = 3-bromo). LCMS: > 98% pure at 254 nm, m/z = 488 (M+H).
Sulfide Oxidation (27, R = 2-Aminopyridine)
In a 20-mL scintillation vial with a stir bar, 2-((1-(3-bromobenzyl)-4,6-difluoro-1H-indol-3-yl)thio)-N-(pyridin-2-yl)acetamide (100 mg, 0.21 mmol) was massed. Methanol/water (9:1, 5 mL) was added, followed by Oxone (330 mg, 0.54 mmol). The reaction was monitored by LCMS and found to complete after 3 h. The reaction was diluted into 1:1 CH2Cl2/brine (40 mL) and the layers separated. The aqueous layer was extracted with CH2Cl2 (2 × 20 mL). The organic layers were combined, dried over MgSO4, filtered, and concentrated in vacuo to afford 86 mg (81% yield) of 2-((1-(3-bromobenzyl)-4,6-difluoro-1H-indol-3-yl)sulfonyl)-N-(pyridin-2-yl)acetamide. The crude material was used in subsequent reactions without further purification. LCMS >95% at 254 nm, m/z = 520 (M+H). 1H NMR in CDCl3: δ 9.23 (s, 1H), 8.30 (d, J = 3.6 Hz, 1H), 8.02 (d, J = 8.4 Hz, 1H), 7.82 (s, 1H), 7.67 (dt, J = 8.8 Hz, 1 Hz, 1H), 7.45 (d, J = 8.4 Hz, 1H), 7.28–7.26 (m, 1H), 7.15 (t, J = 7.6 Hz, 1H), 7.06 (d, J = 6.8 Hz, 5.2 Hz, 1H), 6.93 (d, J = 7.6 Hz, 1H), 6.87–6.79 (m, 2H), 5.22 (s, 2H), 4.45 (s, 2H). 13C NMR: δ 160.4 (dd, JC–F = 242 Hz, 10 Hz), 159.5, 155.1 (dd, JC–F = 251 Hz, 14 Hz), 150.6, 148.0, 138.8 (dd, JC–F = 25 Hz, 11 Hz), 138.3, 136.2, 131.9, 130.8, 129.9, 125.3, 123.3, 120.5, 114.2, 112.4, 109.7 (d, JC–F = 20 Hz), 99.3 (dd, JC–F = 28 Hz, 24 Hz), 94.2 (dd, JC–F = 27 Hz, 5 Hz), 63.0, 50.7.
Pd(dppf)Cl2-Catalyzed Suzuki Coupling
In a microwave vial, 2-((1-(3-bromobenzyl)-4,6-difluoro-1H-indol-3-yl)sulfonyl)-N-(pyridin-2-yl)acetamide (10 mg, 0.019 mmol), pyridin-4-ylboronic acid (5 mg, 0.041 mmol), cesium carbonate (20 mg, 0.062 mmol), and Pd(dppf)Cl2 (2 mg, 0.0025 mmol) were massed. The vessel was charged with a stir bar and THF/H2O (5:1, 3 mL), and then sealed with a crimped-cap. The reaction was heated to 150 °C for 10 min. The reaction was cooled to ambient temperature and determined to be complete by LCMS. The reaction mixture was diluted in 1:1 CH2Cl2/brine (40 mL). The layers were separated, and the aqueous layer was extracted with CH2Cl2 (2 × 10 mL). The organic layers were combined, dried over MgSO4, filtered, and concentrated in vacuo. The crude residue was dissolved in 1 mL of DMSO and purified on reverse phase HPLC eluting with acetonitrile/H2O/TFA to afford 2-((4,6-difluoro-1-(3-(pyridin-4-yl)benzyl)-1H-indol-3-yl)sulfonyl)-N-(pyridin-2-yl)acetamide (29j). The resultant oil was >98% pure by LCMS, m/z = 519 (M+H). 1H NMR in CDCl3: δ 9.35 (s, 1H), 8.67 (s, 2H), 8.27 (d, J = 4.4 Hz, 1H), 7.97 (d, J = 8.4 Hz, 1H), 7.87 (s, 1H), 7.62–7.58 (m, 2H), 7.46–7.38 (m, 4H), 7.11 (d, J = 7.6 Hz, 1H), 7.03 (dd, J = 6.8 Hz, 4.8 Hz, 1H), 6.87–6.80 (m, 2H), 5.33 (s, 2H), 4.45 (s, 2H). 13C NMR: δ 160.2 (dd, JC–F = 242 Hz, 11 Hz), 159.7, 155.0 (dd, JC–F = 251 Hz, 14 Hz), 150.6, 150.3, 147.9, 147.2, 139.3, 138.8 (t, JC–F = 14 Hz), 138.4, 136.1, 135.1, 130.1, 127.5, 127.4, 125.5, 121.7, 120.5, 114.3, 112.2, 109.7 (d, JC–F = 20 Hz), 99.2 (dd, JC–F = 29 Hz, 24 Hz), 94.3 (dd, JC–F = 26 Hz, 4 Hz), 63.0, 51.3.
Pd(OAc)2/S-Phos-Catalyzed Suzuki Coupling
In a dry 4-dram vial equipped with a stir bar, Pd(OAc)2 (2 mg, 0.009 mmol), S-Phos (8 mg, 0.02 mmol), and K3PO4 (35 mg, 0.17 mmol) were massed. The reaction was charged with solvent (THF/H2O, 3 mL, 10:1) and sparged with argon for 5 min. The reaction was then capped and stirred for 30 min. 2-((1-(3-Bromobenzyl)-1H-pyrrolo[2,3-b]pyridin-3-yl)sulfonyl)-N-(5-methylisoxazol-3-yl)acetamide (25 mg, 0.051 mmol) and pyridin-3-ylboronic acid (0.097 mmol) were then added in a minimal amount of degassed THF. The reaction was heated to 60–80 °C until the reaction was determined to be complete, or no further progress was observed by LCMS. The reaction was then cooled to ambient temperature and diluted into dichloromethane/brine (20 mL, 1:1). The layers were separated and the aqueous layer extracted with dichloromethane (2 × 10 mL). The organic layers were combined, dried over MgSO4, and filtered, and the solvent was removed in vacuo. The crude residue was dissolved in 1 mL of DMSO and purified on reverse phase HPLC eluting with acetonitrile/H2O/TFA to afford N-(5-methylisoxazol-3-yl)-2-((1-(3-(pyridin-3-yl)benzyl)-1H-pyrrolo[2,3-b]pyridin-3-yl)sulfonyl)acetamide (30b, R = 3-pyridyl). LCMS: > 98% pure at 254 nm, m/z = 488 (M+H).
Synthesis and Characterization of 32j and 34h
The most effective route to prepare specific analogues, especially those bearing the isoxazole amide, is described in the approach used to resynthesize 32j and 34h. The procedures were identical to those found in the General Procedure for the Synthesis of Analogue Libraries section, with the only difference being the order in which the steps were performed. The route described below avoids conducting the Suzuki coupling in the presence of the isoxazole amide/sulfone, which resulted in significant byproduct formation.
In a 40-mL scintillation vial, methyl 2-((4,6-difluoro-1H-indol-3-yl)thio)acetate (1.0 equiv) was massed. Methanol and water were added (6 mL, 1:1), followed by lithium hydroxide (2.0 equiv). The reaction was capped and heated to 70 °C and monitored by LCMS. Upon completion, the reaction was cooled to ambient temperature and diluted into dichloromethane/brine (1:1). The aqueous layer was acidified with 2 M HCl. The organic layer was separated, and the aqueous layer was extracted with dichloromethane. The organic layers were combined, dried over MgSO4, and filtered. The solvent was removed in vacuo, and the residue acid was used in subsequent reactions without further purification. LCMS: > 98% at 254 nm, m/z = 244 (M+H).
In a dry 4-dram vial equipped with a stir bar, 2-((4,6-difluoro-1H-indol-3-yl)thio)acetic acid (1.0 equiv) and HATU (1.5 equiv) were massed. DMF (3 mL) was added, followed by DIPEA (3.0 equiv). The reaction was allowed to stir at ambient temperature for 10 min, then 5-methylisoxazol-3-amine (2.0 equiv) was added. The reaction was monitored by LCMS. Upon completion, the reaction was diluted into dichloromethane/brine (1:1). The layers were separated, and the aqueous layer was extracted with dichloromethane. The organic layers were combined, dried over MgSO4, and concentrated in vacuo. The crude material was purified on SiO2 gel by flash column chromatography eluting with 0 to 100% hexanes/ethyl acetate to afford 2-((4,6-difluoro-1H-indol-3-yl)thio)-N-(5-methylisoxazol-3-yl)acetamide. LCMS: > 98% at 254 nm, m/z = 324 (M+H).
In a 20-mL scintillation vial with a stir bar, 2-((4,6-difluoro-1H-indol-3-yl)thio)-N-(5-methylisoxazol-3-yl)acetamide (1.0 equiv) was massed. Methanol/water (10:1, 5 mL) was added, followed by Oxone (2.5 equiv). The reaction was monitored by LCMS, and found to be complete after 3 h. The reaction was diluted into 1:1 CH2Cl2/brine and the layers separated. The aqueous layer was extracted with CH2Cl2. The organic layers were combined, dried over MgSO4, filtered, and concentrated in vacuo. The crude material (2-((4,6-difluoro-1H-indol-3-yl)sulfonyl)-N-(5-methylisoxazol-3-yl)acetamide) was used in subsequent reactions without further purification.
2-((4,6-Difluoro-1-(2-fluoro-4-(1-methyl-1H-pyrazol-4-yl)benzyl)-1H-indol-3-yl)sulfonyl)-N-(5-methylisoxazol-3-yl)acetamide (32j)
In a dry 40-mL scintillation vial, 2-((4,6-difluoro-1H-indol-3-yl)sulfonyl)-N-(5-methylisoxazol-3-yl)acetamide (15.0 mg, 0.0423 mmol), potassium carbonate (20.0 mg, 0.145 mmol), and 4-(4-(chloromethyl)-3-fluorophenyl)-1-methyl-1H-pyrazole (12.0 mg, 0.053 mmol) were massed. The vial was charged with a stir bar and DMF (3 mL) was added. The reaction was capped and heated to 70 °C for 4 h, at which time the reaction was determined to be complete by LCMS. The reaction was cooled to ambient temperature, then diluted with ethyl acetate (30 mL), and washed with brine (30 mL). The aqueous layer was extracted with ethyl acetate (3 × 30 mL). The organic layers were combined, dried over MgSO4, filtered, and concentrated in vacuo. The crude product was purified on reverse phase HPLC eluting with acetonitrile/H2O/TFA to afford 8 mg (35% yield) of 2-((4,6-difluoro-1-(2-fluoro-4-(1-methyl-1H-pyrazol-4-yl)benzyl)-1H-indol-3-yl)sulfonyl)-N-(5-methylisoxazol-3-yl)acetamide (32j). LCMS >98% at 254 nm; LRMS m/z = 544 (M+H). 1H NMR in CDCl3: δ 9.28 (s, 1H), 7.84 (s, 1H), 7.73 (s, 1H), 7.61 (s, 1H), 7.19–7.17 (m, 2H), 7.02–6.96 (m, 2H), 6.85 (t, J = 10.4 Hz, 1H), 6.53 (s, 1H), 5.27 (s, 2H), 4.41 (s, 2H), 3.95 (s, 3H), 2.37 (s, 3H); 13C NMR δ 170.7, 160.7 (d, JC–F = 245 Hz), 161.4–159.3 (dm), 159.0, 157.0, 155.0 (dd, JC–F = 251.3 Hz, 25.0), 138.8 (t, JC–F = 13 Hz), 136.7, 136.1–136.0 (m), 135.9, 129.9, 127.3, 121.7, 121.3, 118.5 (d, JC–F = 12 Hz), 112.7 (d, JC–F = 18 Hz), 111.9, 109.5 (d, JC–F = 17 Hz), 99.4–99.0 (m), 96.4, 94.2 (d, JC–F = 24 Hz), 62.3, 45.2, 39.1, 12.5. HRMS: Calculated for C25H21N5O4F3S, 544.1266; found, 544.1271.
2-((4,6-Difluoro-1-((6-(1-methyl-1H-pyrazol-4-yl)pyridin-3-yl)methyl)-1H-indol-3-yl)sulfonyl)-N-(5-methylisoxazol-3-yl)acetamide (34h)
In a dry 40-mL scintillation vial, 2-((4,6-difluoro-1H-indol-3-yl)sulfonyl)-N-(5-methylisoxazol-3-yl)acetamide (150 mg, 0.423 mmol), potassium carbonate (150 mg, 1.09 mmol), and 5-(chloromethyl)-2-(1-methyl-1H-pyrazol-4-yl)pyridine (100 mg, 0.480 mmol) were massed. The vial was charged with a stir bar, and DMF (10 mL) was added. The reaction was capped and heated to 60 °C for 6 h, at which time the reaction was determined to be complete by LCMS. The reaction was cooled to ambient temperature, then diluted with ethyl acetate (30 mL) and washed with brine (30 mL). The aqueous layer was extracted with ethyl acetate (3 × 30 mL). The organic layers were combined, dried over MgSO4, filtered, and concentrated in vacuo. The crude product was purified on reverse phase HPLC eluting with acetonitrile/H2O/TFA to afford 120 mg (54% yield) of 2-((4,6-difluoro-1-((6-(1-methyl-1H-pyrazol-4-yl)pyridin-3-yl)methyl)-1H-indol-3-yl)sulfonyl)-N-(5-methylisoxazol-3-yl)acetamide (34h). LCMS > 98% at 254 nm; LRMS m/z = 527 (M+H). 1H NMR in CDCl3: δ 9.65 (s, 1H), 8.55 (s, 1H), 8.05 (s, 1H), 7.94 (s, 1H), 7.90 (s, 1H), 7.48–4.42 (m, 2H), 6.88–6.80 (m, 2H), 6.51 (s, 1H), 5.32 (s, 2H), 4.44 (s, 2H), 3.97 (s, 3H), 2.34 (s, 3H). 13C NMR in DMSO-d6: δ 170.2, 160.5, 159.6 (dd, JC–F = 191 Hz, 9 Hz), 158.0, 154.7 (dd, JC–F = 200 Hz, 12 Hz), 151.5, 148.5, 139.0–138.8 (m), 137.8, 137.6, 136.8, 130.1, 129.4, 122.2, 119.7, 112.7, 109.8 (d, JC–F = 21 Hz), 98.7, 96.4, 95.5 (d, JC–F = 25 Hz), 62.6, 49.0, 47.7, 12.4. HRMS: Calculated for C24H21N6O4SF2, 527.1313; found, 527.1314.
Calcium Mobilization Assay
CHO-K1 cells stably expressing human M1 receptors were maintained in F-12 supplement (Invitrogen, Carlsbad, CA) with 10% fetal bovine serum (Invitrogen), antibiotic/antimycotic (Invitrogen), 20 mM HEPES (Invitrogen), and 500 μg/mL G418 (Mediatech, Manassas, VA) at 37 °C in the presence of 5% CO2. Cells were plated at 15,000 cells/20 μL/well in black-walled, clear-bottomed, tissue culture-treated, 384-well plates (Greiner Bio-One, Monroe, NC) in growth media lacking selection antibiotics; cells were grown overnight. The next day, media were washed off and replaced with 20 μL of assay buffer (Hanks’ balanced salt solution (Invitrogen), 20 mM HEPES, and 2.5 mM probenecid (Sigma, St. Louis, MO). Then, 2.3 mM Fluo-4 AM calcium dye (Invitrogen) was prepared in DMSO, mixed with equal amounts of 10% (w/v) pluronic acid (Invitrogen), and then diluted in assay buffer 2× the final concentration. Twenty microliters of dye solution was added to cells for a final concentration of 1 μM. Cells were incubated with dye at 37 °C for 1 h; at 10 min prior to assay, cells were thoroughly washed with assay buffer and incubated with 20 μL of assay buffer at 37 °C. Compounds were serially diluted 1:3 into 11-point concentration–response curves in DMSO and transferred to daughter plates by an Echo acoustic plate reformatter (Labcyte, Sunnyvale, CA) and diluted to a 2× final concentration in assay buffer. To measure Ca2+ flux, cell plates were loaded onto a Functional Drug Screening System 6000 (FDSS6000; Hamamatsu Photonics Systems, Hamamatsu, Japan). Baseline fluorescence was established over 3 s (1 Hz; excitation, 470 ± 20 nm; emission, 540 ± 30 nm), followed immediately be the addition of 20 μL test compound serial dilutions to the cells; the response was measured for 140 s to determine agonist activity. Immediately afterward, 10 μL of an EC20 concentration of acetylcholine (5× final concentration) was added to cells; the response was measured for 86 s to determine PAM activity. Then, 12 μL of an EC80 concentration of acetylcholine (5× final concentration) was immediately added to cells; the response was measured for 70 s to determine NAM or antagonist activity. Fluorescence was recorded as fold over basal fluorescence within the same well. Calcium fluorescence traces were reduced to maximal response to acetylcholine per assay window. Potency (EC50) and maximum response (percentage of 10 μM acetylcholine) for compounds were determined using a four-parameter logistical equation in XlFit (ID Business Solutions, Guildford, UK), plugin for Excel (Microsoft, Redmond, WA).
APP Processing
In order to test the effect of M1 PAMs on M1-stimulated APPsα release, a human M1 overexpressing stable cell line was generated in TREx293 cells (Invitrogen). Cells were plated at 0.3 × 106 cells in a 6-well plate 2 days prior to experiments. Cells were pretreated with 2 μM of the M1 PAM being tested or DMSO for 15 min. Immediately, 100 nM or 10 μM carbachol was added, and the medium was then conditioned for 1 h at 37 °C. Western blot analysis of the endogenous APPsα in conditioned media was performed as described.4d
In Vitro Electrophysiology
All experiments were conducted in accordance with a protocol approved by the Animal Care and Use Committee of Vanderbilt University. Coronal brain slices (300 μm thick) containing the striatum were prepared from C57BL/6J mice (21–26 days old) using the procedures described previously.21 Animals were deeply anesthetized with isoflorane and decapitated. The brain was rapidly removed from skulls and submerged in ice-cold modified artificial cerebrospinal fluid (ACSF) oxygenated with 95% O2/5% CO2. The modified ACSF was composed of (in mM) 210 glucose, 2.5 KCl, 8 MgSO4, 0.5 CaCl2, 1.25 NaH2PO4, 26 NaHCO3, and 10 d-glucose. Brain slices were cut using a Compresstome VF-700 (Precisionary Instruments, San Jose, CA). Slices were incubated in oxygenated ACSF at 32 °C for 30 min and then maintained at room temperature (20–22 °C) afterward until they were transferred to a recording chamber. The recording chamber was continuously perfused with oxygenated ACSF containing (mM) 126 NaCl, 2.5 KCl, 2.0 CaCl2, 1.5 MgSO4, 1.25 NaH2PO4, 26 NaHCO3, and 10 d-glucose.
Whole cell recordings were made from visually identified striatal median spiny neurons (MSNs) under an Olympus BX50WI upright microscope (Olympus, Lake Success, NY). A low-power objective (4×) was used to identify brain region, and a 40× water immersion objective coupled with Hoffman optics was used to visualize the individual neurons. Whole cell current-clamp signal was amplified using Axon Multiclamp 700B amplifiers (Molecular Devices, Sunnyvale, CA) with appropriate electrode-capacitance compensation and bridge balance. Patch pipettes were prepared from borosilicate glass (Sutter Instruments, Novato, CA) with the resistance of 3–5 MΩ when filled with the following intracellular solution (in mM): 120 K-MeSO4, 1 MgCl2, 0.1 CaCl2, 10 HEPES, 1 EGTA, 12 phosphocreatine, 0.3 GTP, and 4 ATP). The pH of the pipet solution was adjusted to 7.3 with 1 M KOH, and osmolarity was adjusted to 285–290. Striatal MSNs were identified based on previously established electrophysiological characteristics (Wilson, 2004). The change of excitability of MSN was assessed in current clamp mode by monitoring the change in the number of spike discharges in response to near rheobase depolarization current injection. The electrical signal was low-pass-filtered at 5 kHz, digitized at 20 kHz, and acquired using a Clampex9.2/Digidata 1332 system (Molecular Devices, Sunnyvale, CA). ClampFit (Molecular Devices, Sunnyvale, CA) and Prism 4 software (GraphPad Software) were used for data analysis. Group data were presented as the mean ± SEM, statistical analysis was performed using the Student’s t-test, and p-values less than 0.05 (p < 0.05) were considered to be significant.
Acknowledgments
We thank the NIMH for support or our M1 program (R01MH082867 and U01MH087965). Vanderbilt University is a Specialized Chemistry Center within the MLPCN (U54MH084659), and ML169 is an MLPCN probe, freely available upon request.
Glossary
Abbreviations
- APP
amyloid protein precursor
- BQCA
benzylquinoline carboxylic acid
- CCh
carbachol
- CLHEP
predicted hepatic clearance
- CNS
central nervous system
- CYP450
cytochrome P-450
- DIPEA
diisopropylethyl amine
- DMF
dimethyl formamide
- DMPK
drug metabolism and pharmacokinetics
- GI
gastrointestinal
- GPCR
G-protein coupled receptor
- HATU
2-(1H-7-azabenzotriazol-1-yl)-1,1,3,3-tetramethyl uranium hexafluoro-phosphate methanaminium
- hM1
human M1 receptor
- mAChR
muscarinic acetylcholine receptor
- PAM
positive allosteric modulator
- PyClu
chlorodipyrrolidinocarbenium hexafluorophosphate
- SAR
structure–activity relationship
- TBPB
[1-(1′-2-methylbenzyl)-1,4′-bipiperidin-4-yl)-1H-benzo[d]imidazol-2(3H)-one]
- THF
tetrahydrofuran
Supporting Information Available
Experimental procedures for metabolism and pharmacokinetic experiments. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
# U.S. Army Research Office, Research Triangle Park, NC 27709.
Author Contributions
C.W.L., M.R.W., P.J.C., and C.M.N. conceived and directed the medicinal chemistry and molecular pharmaocology. J.C.T., M.L.T., P.R.R. synthesized compounds and J.C.T. and C.W.L. wrote the manuscript. T.J.U. and D.J.S. perfomred all the mAChR pharmacology. H.P.C. and R.K. perfromed the A-beta experiments. T.P., M.T.K., and Z.X. performed the electrophysiology experiments. T.M.B., R.D.M., A.L.B., and J.S.D performed all DMPK experiments.
The authors declare no competing financial interest.
Supplementary Material
References
- a Langmead C. J.; Watson J.; Reavill C. (2008) Muscarinic acetylcholine receptors as CNS drug targets. Pharmacol. Ther. 117, 232–243. [DOI] [PubMed] [Google Scholar]; b Digby G. J.; Shirey J. K.; Conn P. J. (2010) Allosteric activators of muscarinic receptors as novel approaches for treatment of CNS disorders. Mol. Biosyst. 6, 1345–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caulfield M. P. (1993) Muscarinic receptors – characterization coupling and function. Pharmacol. Ther. 58, 319–379. [DOI] [PubMed] [Google Scholar]
- a Levey A. I.; Kitt C. A.; Simonds W. F.; Price D. L.; Brann M. R. (1991) Identification and localization of muscarinic acetylcholine receptor proteins in brain with subtype-specific antibodies. J. Neurosci. 11, 3218–3226. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Levey A. I.; Edmunds S. M.; Koliatsos V.; Wiley R. G.; Heilman C. J. (1995) Expression of M1-M4 muscarinic acetylcholine receptor proteins in rat hippocampus and regulation by cholinergic innervations. J. Neurosci. 15, 4077–4092. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Levey A. I. (1996) Muscarinic acetylcholine receptor expression in memory circuits: implications for treatment of Alzheimer disease. Proc. Natl. Acad. Sci. U.S.A. 93, 13541–13546. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Felder C. C.; Porter A. C.; Skillman T. L.; Zhang L.; Bymaster F. P.; Nathanson N. M.; Hamilton S. E.; Gomeza J.; Wess J.; McKinzie D. L. (2001) Elucidating the role of muscarinic receptors in psychosis. Life Sci. 68, 2605–2613. [DOI] [PubMed] [Google Scholar]; e Anagnostaras S. G.; Murphy G. G.; Hamilton S. E.; Mitchell S. L.; Rahnama N. P.; Nathanson N. M.; Silva A. J. (2003) Selective cognitive dysfunction in acetylcholine M1 muscarinic receptor in mutant mice. Nat. Neurosci. 6, 51–58. [DOI] [PubMed] [Google Scholar]
- a Bodick N. C.; Offen W. W.; Levey A. I.; Cutler N. R.; Gauthier S. G.; Satlin A.; Shannon H. E.; Tollefson G. D.; Rasmussen K.; Bymaster F. P.; Hurley D. J.; Potter W. Z.; Paul S. M. (1997) Effects of xanomeline, a selective muscarinic receptor agonsist, on cognitive and behavioral symptoms in Alzheimer disease. Arch. Neurol. 54, 465–473. [DOI] [PubMed] [Google Scholar]; b Caccamo A.; Oddo S.; Billings L. M.; Green K. N.; Martinez-Coria H.; Fisher A.; LaFerla F. M. (2006) M1 receptors play a central role in modulating AD-like pathology in transgenic mice. Neuron 49, 671–682. [DOI] [PubMed] [Google Scholar]; c Caccamo A.; Fisher A.; LaFerla F. M. (2009) M1 agonists as a potential disease-modifying therapy for Alzheimer’s disease. Curr. Alzheimer Res. 6, 112–117. [DOI] [PubMed] [Google Scholar]; d Jones C. K.; Brady A. E.; Davis A. A.; Xiang Z.; Bubser M.; Tantawy M. N.; Kane A. S.; Bridges T. M.; Kennedy J. P.; Bradley S. R.; Peterson T. E.; Ansari M. S.; Baldwin R. M.; Kessler R. M.; Deutch A. Y.; Lah J. J.; Levey A. I.; Lindsley C. W.; Conn P. J. (2008) Novel selective allosteric activator of the M1 muscarinic acetylcholine receptor regulates amyloid processing and produces antipsychotic-like activity in rats. J. Neurosci. 28, 10422–10433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirey J. K.; Brady A. E.; Jones P. J.; Davis A. A.; Bridges T. M.; Jadhav S. B.; Menon U.; Christain E. P.; Doherty J. J.; Quirk M. C.; Snyder D. H.; Levey A. I.; Watson M. L.; Nicholle M. M.; Lindsley C. W.; Conn P. J. (2009) A selective allosteric potentiator of the M1 muscarinic acetylcholine receptor increases activity of medial prefrontal cortical neurons and restores impairments in reversal learning. J. Neurosci. 29, 14271–14286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebois E. P.; Bridges T. M.; Dawson E. S.; Kennedy J. P.; Xiang Z.; Jadhav S. B.; Yin H.; Meiler J.; Jones C. K.; Conn P. J.; Weaver C. D.; Lindsley C. W. (2010) ‘Discovery and development of novel subtype-selective M1 allosteric agonists for the investigation of M1 receptor function. ACS Chem. Neurosci. 1, 104–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Digby G. J.; Noetzel M. J.; Bubser M.; Utley T. J.; Walker A. G.; Byun N. B.; LeBois E. P.; Xiang Z.; Sheffler D. J.; Niswender C. M.; Plumley H. C.; Davis A. A.; Nemirovsky N. E.; Mennenga S.; Camp B. W.; Bimonte-Nelson H. A.; Morrison R.; Daniels S.; Lindsley C. W.; Olive M. F.; Conn P. J. (2012) Novel allosteric agonists of the M1 muscarinic acetylcholine receptor induce brain region specific responses and correspond with behavioral effects in animal models. J. Neurosci. 32, 8532–8544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jian S.; Wang Y.; Ma Q.; Zhou A.; Zhang Y. W. (2012) M1 muscarinic acetylcholine receptor interacts with BACE1 and regulates its proteosomal degradation. Neurosci. Lett. 515, 125–130. [DOI] [PubMed] [Google Scholar]
- Zuchner T.; Perez-Polo J. R.; Schliebs R. (2004) Beta-secretase BACE1 is differentially controlled through muscarinic acetylcholine receptor signaling. J. Neurosci. Res. 77, 250–257. [DOI] [PubMed] [Google Scholar]
- a Lindsley C. W.; Shipe W. D.; Wolkenberg S. E.; Theberge C. R.; Williams D. L. Jr.; Sur C.; Kinney G. G. (2006) Progress towards validating the NMDA receptor hypofunction hypothesis of schizophrenia. Curr. Topics Med. Chem. 8, 771–784. [DOI] [PubMed] [Google Scholar]; b Conn P. J.; Tamminga C.; Schoepp D. D.; Lindsley C. W. (2008) Schizophrenia: moving beyond monoamine antagonists. Mol. Interventions 8, 99–105. [DOI] [PubMed] [Google Scholar]; c Bridges T. M.; LeBois E. P.; Hopkins C. R.; Wood M. R.; Jones J. K.; Conn P. J.; Lindsley C. W. (2010) Antipsychotic potential of muscarinic allosteric modulation. Drug News Perspect. 23, 229–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sur C.; Mallogora P. J.; Wittman M.; Jacobson M. A.; Pascarella D.; Williams J. B.; Brandish P. E.; Pettibone D. J.; Scolnick E. M.; Conn P. J. (2003) N-desmethylclozapine, an allosteric agonist at muscarinic 1 receptor, potentiates N-methyl-D-aspartate activity. Proc. Natl. Acad. Sci. U.S.A. 100, 13674–13679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bymaster F. P.; Whitesitt C. A.; Channon H. E.; DeLapp N.; Ward J. S.; Calligaro D. O.; Shipley L. A.; Buelke-Sam J. L.; Bodick N. C.; Farde L.; Sheardown M. J.; Olesen P. H.; Hansen K. T.; Suzdak P. D.; Swedberg M. D. B.; Sauerberg P.; Mitch C. H. (1997) Xanomeline: a selective muscarinic agonist for the treatment of Alzheimers disease. Drug Dev. Res. 40, 158–170. [Google Scholar]
- Shekhar A.; Potter W. Z.; Lightfoot J.; Lienemann J.; Dube S.; Mallinckrodt C.; Bymaster F. P.; McKinzie D. L.; Felder C. C. (2008) Selective muscarinic receptor agonist xanomeline as a novel treatment for schizophrenia. Am. J. Psychiatry 165, 1033–1039. [DOI] [PubMed] [Google Scholar]
- a Melancon B. J.; Hopkins C. R.; Wood M. R.; Emmitte K. A.; Niswender C. M.; Christopoulos A.; Conn P. J.; Lindsley C. W. (2012) Allosteric modulation of 7 transmembrane spanning receptors: theory, practice and opportunities for CNS drug discovery. J. Med. Chem. 55, 1445–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Conn P. J.; Christopolous A.; Lindsley C. W. (2009) Allosteric modulators of GPCRs as a novel approach to treatment of CNS disorders. Nat. Rev. Drug Discovery 8, 41–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Kuduk S. D.; Chang R. K.; Di Marco C. N.; Ray W. J.; Ma L.; Wittmann M.; Seager M. A.; Koeplinger K. A.; Thompson C. D.; Hartman G. D.; Bilodeau M. T. (2010) Quinolizidinone carboxylic acids as CNS penetrant, selective M1 allosteric muscarinic receptor modulators. ACS Med. Chem. Lett. 1, 263–267. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Brady A. E.; Jones C. K.; Bridges T. M.; Kennedy J. P.; Thompson A. D.; Heimann J. U.; Breininger M. L.; Gentry P. R.; Yin H.; Jadhav S. B.; Shirey J. K.; Conn P. J.; Lindsley C. W. (2008) Centrally active allosteric potentiators of the M4 muscarinic acetylcholine receptor reverse amphetamine-induced hyperlocomotor activity in rats. J. Pharmacol. Exp. Ther. 327, 941–953. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Bridges T. M.; Kennedy J. P.; Hopkins C. R.; Conn P. J.; Lindsley C. W. (2010) Heterobiaryl and heterobiaryl ether derived M5 positive allosteric modulators. Bioorg. Med. Chem. Lett. 20, 5617–5622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L.; Seager M. A.; Wittmann M.; Jacobson M.; Bickel D.; Burno M.; Jones K.; Graufelds V. K.; Xu G.; Pearson M.; McCampbell A.; Gaspar R.; Shughrue P.; Danziger A.; Regan C.; Flick R.; Pascarella D.; Garson S.; Doran S.; Kreatsoulas C.; Veng L.; Lindsley C. W.; Shipe W.; Kuduk S.; Sur C.; Kinney G.; Seabrook G. R.; Ray W. J. (2009) Selective activation of the M1 muscarinic acetylcholine receptor achieved by allosteric potentiation. Proc. Natl. Acad. Sci. U.S.A. 106, 15950–15955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Bridges T. M.; Kennedy J. P.; Noetzel M. J.; Breininger M. L.; Gentry P. R.; Conn P. J.; Lindsley C. W. (2010) Chemical lead optimization of a pan Gq mAChR M1, M3, M5 positive allosteric modulator (PAM) lead. Part II: Development of a potent and highly selective M1 PAM. Bioorg. Med. Chem. Lett. 20, 1972–1975. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Reid P. R.; Bridges T. M.; Sheffler D. J.; Cho H. P.; Lewis L. M.; Days E.; Daniels J. S.; Jones C. K.; Niswender C. M.; Weaver D. C.; Conn P. J.; Lindsley C. W.; Wood M. R. (2011) Discovery and optimization of a novel, selective and brain penetrant M1 positive allosteric modulator (PAM): the development of ML169, and MLPCN probe. Bioorg. Med. Chem. Lett. 21, 2697–2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang F. V.; Shipe W. D.; Bunda J. L.; Nolt M. B.; Wisnoski D. D.; Zhao Z.; Barrow J. C.; Ray W. J.; Ma L.; Wittman M.; Seager M.; Koeplinger K.; Hartman G. D.; Lindsley C. W. (2010) Parallel synthesis of N-birayl quinolone carboxylic acids as selective M1 positive allosteric modulators. Bioorg. Med. Chem. Lett. 20, 531–536. [DOI] [PubMed] [Google Scholar]
- Billingsley K.; Buchwald S. L. (2007) Highly efficient monophosphine-based catalyst for the palladium-catalyzed Suzuki-Miyaura reaction of heteroaryl halides and heteroaryl boronic acids and esters. J. Am. Chem. Soc. 129, 3358–3366. [DOI] [PubMed] [Google Scholar]
- The determination of the brain/plasma ratio for compound 34h was complicated by rapid clearance from rat plasma. The observed value was greater than 1.
- Xiang Z.; Thompson A. D.; Jones C. K.; Lindsley C. W.; Conn P. J. (2012) Roles of the m1 muscarinic acetylcholine receptor subtype in the regulation of Basal Ganglia function and implications for the treatment of Parkinson’s disease. J. Pharmacol. Exp. Ther. 340, 595–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen W.; Tian X.; Day M.; Ulrich S.; Tkatch T.; Nathanson N. M.; Surmeier D. J. (2007) Cholinergic modulation of Kir2 channels selectively elevates dendritic excitability in striatopallidal neurons. Nat. Neurosci. 10, 1458–1466. [DOI] [PubMed] [Google Scholar]
- Graybiel A. M.; Aosaki T.; Flaherty A. W.; Kimura M. (1994) The basal ganglia and adaptive motor control. Science 265, 1826–1831. [DOI] [PubMed] [Google Scholar]
- Nicola S. M.; Surmeier J.; Malenka R. C. (2000) Dopaminergic modulation of neuronal excitability in the striatum and nucleus accumbens. Annu. Rev. Neurosci. 23, 185–215. [DOI] [PubMed] [Google Scholar]
- Holt D. J.; Herman M. M.; Hyde T. M.; Kleinman J. E.; Sinton C. M.; German D. C.; Hersh L. B.; Graybiel A. M.; Saper C. B. (1999) Evidence for a deficit in cholinergic interneurons in the striatum in schizophrenia. Neuroscience 94, 21–31. [DOI] [PubMed] [Google Scholar]
- Lehericy S.; Hirsch E. C.; Cervera P.; Hersh L. B.; Hauw J. J.; Ruberg M.; Agid Y. (1989) Selective loss of cholinergic neurons in the ventral striatum of patients with Alzheimer disease. Proc. Natl. Acad. Sci. U.S.A. 86, 8580–8584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisani A.; Bernardi G.; Ding J.; Surmeier D. J. (2007) Re-emergence of striatal cholinergic interneurons in movement disorders. Trends. Neurosci. 30, 545–553. [DOI] [PubMed] [Google Scholar]
- Simpson E. H.; Kellendonk C.; Kandel E. (2010) A possible role for the striatum in the pathogenesis of the cognitive symptoms of schizophrenia. Neuron 65, 585–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher A. (2008) Cholinergic treatments with emphasis on M1 muscarinic agonists as potential disease-modifying agents for Alzheimer’s disease. Neurotherapeutics 5, 433–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.