

Abstract
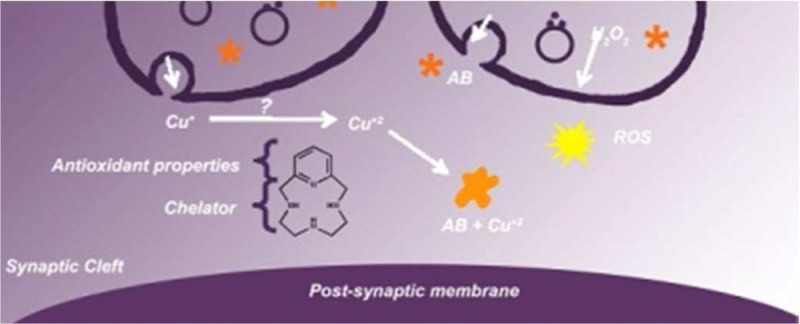
Alzheimer’s disease is a neurodegenerative disorder characterized by the development of intracellular neurofibrillary tangles, deposition of extracellular amyloid beta (Aβ) plaques, along with a disruption of transition metal ion homeostasis in conjunction with oxidative stress. Spectroscopic, transmission electron microscopy, and scanning electron microscopy imaging studies show that 1 (pyclen) is capable of both preventing and disrupting Cu2+ induced AB1–40 aggregation. The pyridine backbone of 1 engenders antioxidant capacity, as shown by cellular DCFH-DA (dichlorodihydrofluorescein diacetate) assay in comparison to other N-heterocyclic amines lacking this aromatic feature. Finally, 1 prevents cell death induced by oxidative stress as shown by the Calcein AM assay. The results are supported using density functional theory studies which show that the pyridine backbone is responsible for the antioxidant capacity observed.
Keywords: Amyloid, Alzheimer’s disease, aggregates, copper, zinc, chelate
Alzheimer’s disease (AD) is a debilitating disease that affects over 5.4 million people currently, at an annual cost exceeding 180 billion dollars in the United States alone.1−6 Physiological and molecular features include the deposition of amyloid beta (Aβ) plaques, elevated levels of transition metals, and oxidative stress.6−11 The exact mechanism leading to AD is still not established, although amyloid is a component in many hypotheses proposed to date.12−15 Recent attention has implicated metal ions in the cascade leading to the physiological and pathological hallmarks of AD, thus forming the “Metal Hypothesis of Alzheimer’s Disease”.8,16−21
Transition metals are trace elements vital for normal biological function because they serve as structural drivers, cofactors, or reactive centers in proteins and enzymes.22,23 Fenton chemistry is defined by the oxidation of redox active metal ions in their reduced from, such as Fe(II) or Cu(I), with H2O2 to produce radicals that are known to cause DNA oxidation, disruption of mitochondrial membrane potentials, and lipid peroxidation.7,18,24,25 Redox chemistry of these elements is tightly regulated throughout biology via regulatory and chaperone systems, so that protein modification, in conjunction with Fenton chemical reactions, producing cellular oxidative stress will be avoided. Disruptions or alterations in the redox potential of metal ion regulatory systems have therefore been implicated in a number of disease states to date which include Huntington’s, Alzheimer’s, Parkinson’s, and Lou Gehrig’s disease, as well as macular degeneration and Freidrich’s ataxia.7,9,18,24,25,28,29,31−37 For example, a histidine rich binding site has been identified in Aβ1–40 or Aβ1–42.12,26 Insoluble amyloid beta plaques have increased levels of copper, zinc, and iron, while intracellular copper stores are deficient in AD patients.6,8 Metal ion chelation by amyloid plaques gives rise to concomitant free radical generation, resulting in neuronal death.7,18,21,38−41 Furthermore, increased levels of oxidative stress have been, in part, attributed to alterations in the expression of superoxide dismutase, as well as protein metal ion chaperones.39−47 Modifications in the levels of metal ion chaperone expression associated with the signal transduction pathway of glutamate receptors, for example, have also been noted with concomitantly higher levels of cleaved amyloid precursor protein to produce amyloid 1–40 and 1–42.48,49 Finally, aged populations naturally exhibit increased levels of ROS due to decreased levels of antioxidants such as melatonin, resulting in higher levels of oxidative stress. However, AD models suggest more exacerbated levels of ROS, thus resulting in AD progression.9,50−56
There is no effective or preventative protocol prescribed for AD, nor have proposed therapies found success in symptom alleviation of neurodegenerative decline associated with AD.12,21,27,57 Many hypothetical pathways of AD have been targeted, with one taking aim at the metal-based hypothesis proposed by Bush and co-workers.27,58−60 Synthetic targets focused on inhibiting the interactions of Aβ with metal ions, along with atypical metal ion homeostasis, are limited by ion specificity, an inability to cross the blood brain barrier (BBB), and/or biological compatibility.12,21,27,57,58 A compound finding exception to these roadblocks has been evaluated in phase II clinical trials.30,61 The chelator clioquinol (CQ) provided improved cognition in mouse models, but its widespread use has been terminated by the adverse side effect of subacute myelo-optic neuropathy. The positive effects exhibited by CQ encourage synthetic chemists to pursue the chelator strategy as a route to potentiating the cognitive decline associated with metal ion misregulation and plaque deposition. A second generation congener of CQ, PBT2, has been produced and is in phase II clinical trials.57,62 Studies of this compound showed improved cognition in AD transgenic mice, and demonstrated positive effects on the learning and memory in AD patients. In contrast to this agent serving as a chelator as utilized in the sense of typical metal-overload disorders, that is, removing excess metal, the authors have shown that these compounds can serve as neuromodulators by restoring the metal ion imbalance for neurochemical communication pathways involved in synaptic activity. When the “lost” metal ions that lead to Aβ deposition are rescued by these synthetic chaperones, their activities in the communication pathways are restored, and Aβ clearance is elevated.57,62 With these results, the pursuit in biologically compatible transition metal ion ligands as therapy for AD is encouraged.27,58 There are a multitude of challenges to overcome when designing a ligand for applications in medicinal use such as biocompatibility, specificity, and efficacy. (60) Utilizing a rational design approach, this work focuses on the use of pyclen (1) shown in Chart 1 as a metal ion passivation and antioxidant agent based on this ligand’s specific metal-ion binding affinity for copper(II) and zinc(II) along with built-in antioxidant functionalities.63 The ligand is the backbone to PCTA, a potential MRI contrast agent explored in recent years and has been repurposed for this work.64−66
Chart 1.

Results and Discussion
Metal ions (copper and zinc) bind to a histidine rich domain in amyloid producing Aβ in the form of insoluble plaques.16−20 This process has been comprehensively studied and described in a number of recent reviews.12,21,27,58 The aggregation of amyloid can be followed by simple spectroscopic techniques such as turbidity and tyrosine fluorescence studies.70−73 These methods were used to investigate the ability of 1 to dissociate preformed amyloid aggregates upon addition of metal ions, as well as prevent amyloid aggregation. As shown in Figure 1, copper(II) or zinc(II) ion addition to a solution of amyloid1–40 results in a visibly turbid solution which scatters light with a consequential increased absorbance signal, using absorption spectrophotometry. The aggregation observed upon metal ion addition can be reversed by coincubation of 1 for 4 h, indicating dissolution of the amyloid aggregates. A visible dissolution of the peptide solution is concomitant to the decrease in absorbance signal. For comparison, we repeated this experiment with 2 and 3 (Chart 1) previously studied by Hindo et al. as Aβ disaggregating agents, which our studies corroborate.74 We used the known open-chain chelator EDTA as a positive control, and our studies demonstrated that all ligands were equally successful in disrupting turbidity of the aggregated amyloids.70−72 In a similar study, the protective capability of 1 was compared to 2–4. The ligands (1–4) were first coincubated with amyloid, and then metal ions were added. The ligands displayed an ability to prevent metal-induced aggregation of amyloid upon exposure to copper(II) or zinc(II) salts (Figure S1, Supporting Information).
Figure 1.

Turbidity assay showing disaggregation of amyloid plaques upon incubation (24 h at 37 °C) with 1–4. Solutions prepared in 15 mM KH2PO4/NaCl buffer. [Aβ1–40] = 200 μM, [CuSO4] or Zn(OAc)2 = 400 μM, [chelator] = 800 μM. n = 3 for each sample. When standard error of the mean are not depicted, they were too small to graph.
To further determine the capability of 1 to prevent or disrupt metal ion induced amyloid aggregation, we studied Tyr fluorescence intensity. Recent reports have also utilized the natural fluorescence of the native Tyr10 in the Aβ1–40 sequence to study the conformational dynamics of amyloid folding as it relates to AD and/or production of H2O2.75−78 As Tyr10 is located within proximity to the metal binding pocket in Aβ1–40, the fluorescence intensity of the phenolic side chain decreases during the folding process due to environmental changes that occur locally. This work was supported by circular dichroism spectra, which verified that fluorescence intensity decreased as the conformation of amyloid changed to β-sheets. Yang and colleagues showed that this spectroscopic marker could be used to follow the folding induced by the addition of copper ions to amyloid, and related the production of H2O2 to this process.78 Our hypothesis was that Tyr fluorescence intensity should be restored to control levels upon addition of 1–4 to a solution of preformed metal aggregates, as shown below in Figure 2. These results are consistent with the turbidity studies presented above. Similar results were also obtained for amyloid samples coincubated with 1–4 prior to metal ion addition (Figure S2). That is, the ligand chelates show a protective capability by preventing the production of aggregates as evidenced by higher Tyr10 fluorescence signal compared to the control of amyloid coincubated with copper(II) or zinc(II), which showed a large decrease in signal intensity after 24 h.
Figure 2.

Addition of ligands 1–4 to aggregated Aβ results in reconstitution of Tyr10 fluorescence which was decreased due to aggregate formation. Solutions prepared in 15 mM KH2PO4/NaCl buffer. [Aβ1–40] = 200 μM, [CuSO4] or [Zn(OAc)2] = 400 μM, [chelator] = 800 μM. n = 3 for each sample.
Transmission electron microscopy (TEM) and scanning electron microscopy (SEM) were utilized to study the morphology of the aggregates with and without chelators, as well.73,74,77,78 The TEM images shown in Figure 3 demonstrate that 1 decreases the amyloid aggregate size by 1–2 orders of magnitude compared to the copper aggregates. Grid (a) shows the large aggregates formed by the addition of metal ions to amyloid (scale 500 nm). Aliquots from the stock used to make grid (a) were incubated with 1 for 12 h and then prepared for microscopy. The sizes of the aggregates upon treatment with 1 are an average of 1–2 orders of magnitude smaller and observably more diffuse, as shown in grid (b). The SEM images in Figure S3 confirm the ability of the chelates to affect aggregate size as well.
Figure 3.

TEM Images showing (a) copper induced aggregation of amyloid and (b) its dissolution by 1.
As increased levels of ROS are associated with AD, we set out to investigate the antioxidant character of 1 compared to the heterocycles 2 and/or 3. Initially, (Figure S5), we showed that 1 was capable of preventing the formation of the ABTS•– radical most effectively compared to 2–4 at ligand concentrations of 100 μM. These results were standardized against Trolox (a known antioxidant) with 1 providing 0.4 Trolox equivalents and 2–4 resulting in values of 0.23, 0.11, and 0.15 respectively. Moreover, many of the pathways proposed to produce ROS leading to AD pathology involve metal ions; therefore, we utilized the Cu-ascorbate redox system, Scheme 1, as a model to determine if the ligands could halt copper based redox activity under aerobic conditions.7,9,18,22,24,25,50,51 This system is a useful model for studying the brain as high levels of oxygen and ascorbate are present, as described by Faller and co-workers, who employed this system to investigate the redox chemistry of amyloid systems with Cu.79 Coumarin-3-carboxylic acid (CCA), which generates fluorescent 7-hydroxy-CCA in the presence of hydroxyl radicals, was used to quantify the reduction of oxygen by copper redox-cycling in the presence of ascorbate. Figure 4 shows that copper and ascorbate generate OH•– as measured by 7-hydroxy-CCA which increases linearly for the first 5 min and then plateaus around 10 min. This process is prevented entirely as shown in the inset of Figure 4 when 1 or 3 is coincubated with the Cu-ascorbate system, indicating these ligands are capable of halting copper redox cycling via metal complexation. Next, the ability of 1 to halt hydroxyl generation was compared to AB1–40, in which Faller reports a decrease the Cu-Ascorbate redox cycle.79 The Cu-ascorbate system was initially coincubated with CCA for 2 min, showing an increase in fluorescent signal as expected (Figure S6). At this point either 1 or AB1–40 were added to the Cu-ascorbate system, resulting in a leveling of the 7-hydroxy-CCA signal. This indicated that the addition of 1 or Aβ1–40 was able to halt the copper redox cycling due chelation of the metal-ion. This redox silencing was verified by the addition of ascorbate to Cu(1) or Cu(Aβ1–40) in the presence of CCA resulting in absence of a fluorescent signal during the 10 min time scale.
Scheme 1. Redox Cycling of Copper in the Presence of Oxygen and Ascorbate to Produce OH·.

Figure 4.

Fluorescence intensity of 7-hydroxy-CCA after incubation of CCA [100 μM] and ascorbate [300 μM] with CuII (■) [40μM]. Compound 1 (◇) and 3 (●) [40 μM] were added at t = 0 s prior to CuII. Asc (▲) is a negative control with buffer and ascorbate only. All solutions except Cu(NO3)2 (Milli-Q water only) were dissolved and diluted in KH2PO4/NaCl [15 mM] buffer containing desferryl [2 μM]. Final volume = 4 mL, n = 3 for each sample.
Cellular studies were then carried out to evaluate the intracellular efficacy and toxicity of 1. Preliminary screens compared the cell viability of 1–4 in an FRDA cell line (fibroblasts from a Friedreich’s ataxia patient) and ability to negate oxidative stress. FRDA cells have higher levels of ROS due to mitochondrial misfunction associated with frataxin expression and therefore serve as a good model for ROS protection. Figure 5 shows the normalized results of cell (FRDA) viability studies with compounds 1–4 at 1 nM final concentration. Calcein AM, a nonfluorescent, hydrophobic compound that easily permeates intact, live cells, was used as an indicator for cell viability. Calcein AM is hydrolyzed by intracellular esterases producing calcein, a hydrophilic, strongly fluorescent compound that is well-retained in the cell cytoplasm. Compared to the control containing untreated cells, incubation with either 1 or 3 exhibited >85% cell viability at the concentration studied, a statistically significant increase from cells treated with BSO (2-amino-4-(butylsulfonimidoyl)butanoic acid) alone, which had a cell viability of ∼45% compared to control cells. Compounds 2 and 4 were less efficatious, but still provided cell viability of greater than 60%. These results confirm that the heterocyclic compounds are compatible for use in cells by providing protection against oxidative stress indcued by BSO. Moreover, the addition of the pyridine ring (1) into the heterocylic core does not induce cell death and, in fact, prevents ROS induced cell death most effectively.
Figure 5.

Calcien AM viability assay of FRDA cells after 48 h exposure to BSO [1 mM] with 1–4 [1 nM final]. n = 4 for each sample. * indicates significance with a p value of <0.05.
The antioxidant activity of 1 compared to 2, 3 was then studied using the cell-permeable fluorophore 2,7′-dichlorodihydrofluorescein diacetate (DCFH-DA) as an indicator for ROS. DCFH-DA diffuses into cells and is deacetylated by cellular esterases to nonfluorescent 2′,7′-dichlorodihydrofluorescein (DCFH). This species is subsequently oxidized to the highly fluorescent 2′,7′-dichlorodihydrofluorescein (DCF) species in the presence of ROS. The fluorescent intensity is directly proportional to the amount of ROS present in cell cytosol.80 BSO was used to inhibit the first step of de novo glutathione synthesis, allowing us to observe the elevated intracellular [ROS] (Figure S4). The results of DCFH-DA cell culture assay shown in Figure 6 indicate that 1 is the most effective antioxidant at low nanomolar concentrations, significantly attenuating the ROS produced by BSO, with only a 15% increase of ROS over the negative control (media) when compared to 2–4. These results are compared to the positive control BSO-treated cells, with a nearly 1.8-fold increase in ROS. Figure 7 shows that 1 is an effective antioxidant in the 1.25 nM to 1.25 μM range for the FRDA cell line as well. These results are consistent with results from the Trolox antioxant assay where we noted that pyclen shows a superior antioxidant activity with and without metal ions present (Figure S5). This was repeated in a neuronal cell line (HT-22), and 1 showed antioxidant nature in the 1 mM to 125 μM range (Figure 8) with a glutamate assault which results in cell death by oxidative glutamate toxicity.81 A comparison of these results with the cell viability studies discussed above shows that the pyridine ring of 1 is responsible for the antioxidant capacity observed. The heterocyclic compounds 2 and 3 which showed cell viability congruent to 1 with BSO assault (Figure 4) are structurally similar to 1 except for the pyridine ring. While 1–3 are all largely capable of preventing cell death induced by BSO, ligand 1 is capable of reducing ROS to the greatest extent, thus resulting in the most effective protective capacity. We attribute this to the pyridine core. We postulate that the ability to prevent BSO induced cell death is a result of metal ion scavenging capacity of these ligands 1–3. Such work is beyond the scope of this study. An interesting feature to be noted from these cellular studies is the fact that 1 is capable of entering cells and does not appear to interupt the vital functions of cytosolic metalloenzymes via extraction metal ions from those functionalities. This permeability and biocompatibility is a characteristic that can be exploited in future studies and is further supported by 1 being consistent with Lipinski’s rules (Figure S7).82,83
Figure 6.

DCFH-DA fluorescent response in FRDA cells after 12 h exposure to BSO [1 mM] with 1–4 [4 nM]. n = 4 for each sample. * indicates significance with a p value of <0.05.
Figure 7.

DCFH-DA fluorescent response in FRDA cells after 12 h exposure to BSO [1 mM] showing dose dependence with 1. n = 4 for each sample. * indicates significance with a p value of <0.05.
Figure 8.

DCFH-DA fluorescent response to chelator 1 in HT-22 cell line. Growth media serves as the positive control and is normalized to 100% while glutamate serves as the negative control. ZYC26 was 1 μM and serves as a positive control for antioxidant ability. *** indicates significance with a p value of <0.001.
We postulate that the observed antioxidant activity of 1 is structurally correlated with the pyridine backbone, as evidenced by the radical studies and incubation of ligands with cells in the DCFH-DA cell culture assays presented above. Orvig and co-workers have reported antioxidant activity with pyridine-like aromatic chelators in separate studies as well.70−72 The Weighardt group has computationally and spectroscopically studied bipyridine-based chelates, and their studies prove that the lowest unoccupied molecular orbital (LUMO) of the metal derivatives are composed of mainly ligand π-character.84 As we observed pyclen to be antioxant in the presence of copper ions, this methodology was applied to 1–3 and their CuII derivatives using density functional theory (DFT) (B3LYP, 6-311G (d,p for copper complexes)) to show that the LUMO of 1 and Cu-12+ is largely centered on the pyridine ring.85 As shown in Figure 9, the highest occuppied molecular orbital (HOMO) of 1 has more electron density on the nitrogen atoms and the LUMO of 1 is composed of >90% π-character as compared to 2 and 3 which have density dispersed throughout. Moreover, the HOMO of 1 is higher in energy compared to 2 as well. The other heterocyclic ligands 2, 3 lack these features, that is, the LUMO being antibonding character alone. Ligands 2 and 3 lack this component and therefore have the LUMO orbital spread throughout the ligand set in an antibonding orbital centered on the nitrogen atoms trans to one another, making reactivity less favored. This is not suprising given that heterocycles containing aromaticity are reported to be highly reactive toward radicals produced via radiolysis of water and naturally react with the heterocycles by addition.86−88 Pyridine containing analogs have long been reported in the literature to be potent antioxidants, which is attributed to the electron deficient nature of the pyridine ring, with potency increasing as electron attracting groups are added onto the ring. Such reactivity is well documented, that is pydridine based compounds are known to produce N-oxides upon incubation with H2O2.89 To further support this, 2 and 3 lack the pyridine ring but retain the secondary amines in the heterocyclic core and show little potency in the assays presented. The secondary amines, therefore, are less prone to produce N-oxides than the pyridine backbone. Interestingly, 2 and 3 do show a degree of antioxidant capacity which we postulate to be an effect of the redox tuning of the ligands on the copper ion, rather than ligand composition itself.23,90
Figure 9.
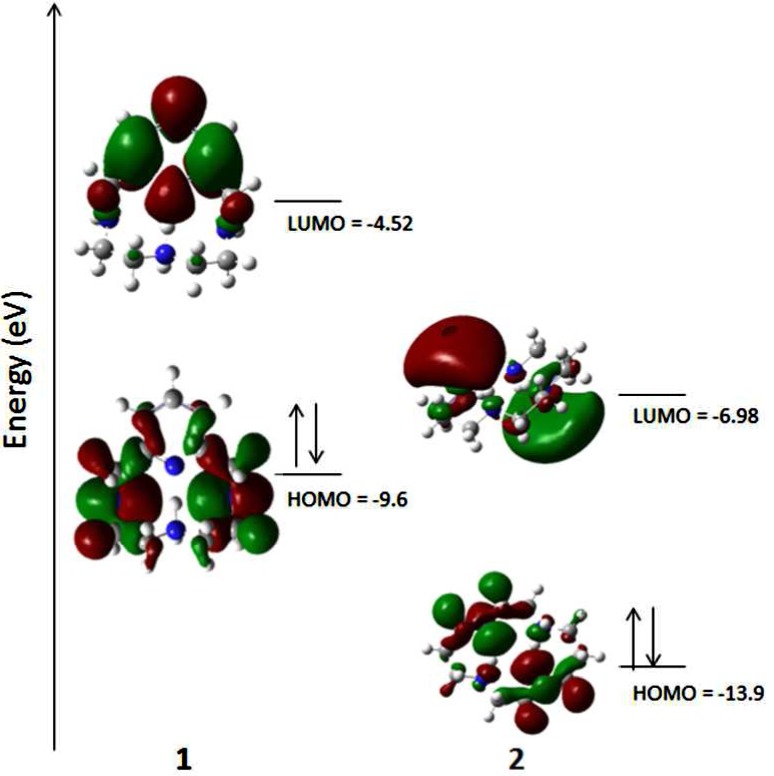
Frontier molecular orbitals of 1 and 2 with the spin density of 1· shown (see Supporting Information for methods and data).
Conclusion
The misregulation of metal ions is known to produce ROS that lead to neurological degradation associated with Alzheimer’s disease in addition to their interaction precipitating amyloid plaques. A molecular system capable of bimodal modulation of the metal ions in amyloid as well as regulation of the increased levels of ROS would prove useful in combating this disease. To address these issues, we have shown that 1, a backbone previously investigated for contrast agent imaging, may be repurposed as a antioxidant chelator for disaggregating amyloid. Spectroscopic and TEM/SEM imaging studies show its ability to protect amyloid from copper ions and also disaggregated amyloid aggregates as well. The antioxidant assays show that 1 has antioxidant capacity in vivo and protective cababilities via Calcein AM studies. The DFT studies and direct reactions with H2O2 show that the pyridine backbone is the key to this ability.
Methods
All reagents were purchased from commercial sources and used as received unless noted. Ligand, Cu(SO4), and Zn(OAc)2 solutions were prepared in Milli-Q water and diluted to the desired concentration using buffer (15 mM/15 mM) KH2PO4/NaCl. Each experiment was performed in triplicate with a peptide to copper to ligand ratio of 0.5:1:2, respectively. Preliminary antioxidants studies (Trolox equivalent assay) were carried out using the Antioxidant Assay Kit available from Sigma-Aldrich.
Physical Methods
A Molecular Devices Spectra Max MS microplate reader was employed to obtain the turbidity results. Fluorescence measurements were recorded on a Varian Cary Eclipse with voltage set to high, and instrument set to record the average of three scans. HR-MS was performed using The Agilent 6224 Accurate-Mass Time-Of-Flight (TOF) MS.
Preparation of Aβ1–40 Stock
Synthetic beta amyloid1–40 peptide was purchased from Twenty-First Century Biochemicals. A peptide stock solution was prepared by dissolving 1.9 mg of Aβ1–40 in 1 mL of buffer, followed by NaOH (20 mM, 300 μL). The solution was sonicated for 1 min then adjusted to pH 7.4 using HCl (0.5 M, ∼10.5 μL). The solution was sonicated for another minute and diluted to 200 μM using MQ water. Any remaining stock was stored at −20 °C for later use.
Turbidity Studies
Two separate turbidity studies (absorbance at 405 nm) were performed using amyloid, copper or zinc, and ligands: (1) to determine disaggregation capability and (2) to determine preventive capability of copper induced beta amyloid formation. Turbidity studies were repeated with HEPES buffer (0.02 HEPES, 0.154 M NaCl, pH = 7.4) giving identical results.
a. Disaggregation
Zinc(II) acetate or copper(II) sulfate solution (400 μM, 40 μL) was added to a solution of Aβ40 (200 μM, 40 μL) and incubated at 37 °C for 1 day. After incubation the ligand stock solutions were added (800 μM, 40 μL) to aggregated peptide solution and incubated for a further 12 h at 37 °C. For analysis of turbidity, 20 μL sample aliquots were diluted with buffer (180 μL) and absorbance recorded using a microplate reader (Softmax Pro M5). The value of the blank absorbance was subtracted from each sample value.
b. Preventative
Ligand stock solutions (800 μM, 40 μL) were added to a solution of Aβ40 (200 μM, 40 μL) and incubated for 5 min at room temperature followed by addition of Zn(OAc)2 or CuSO4 (400 μM, 40 μL). All samples were incubated at 37 °C for 1 day. Absorbance measurements were carried out in the same manner as described above.
Tyrosine Fluorescence
Tyrosine fluorescence studies were carried out on samples used for the turbidity experiments. For sample preparation, 50 μL of sample was diluted with 270 μL of buffer and agitated for 30 s. Excitation and emission values utilized were 278 and 305 nm, respectively.
TEM Imaging
The samples were prepared by incubating amyloid [100 μM, 37°, 24 h] or aggregated amyloid [100 μM] in the presence of ligand chelates (100 μM). Roughly 5 μL of sample was placed onto copper grids (Formar/Carbon 300 mesh purchased from Ted Pella Co.) for 2 min, washed with doubly deionized water (5 μL), and then stained with 3% uranyl acetate (5 μL), washed again, and then dried.
SEM Imaging
The samples were prepared by incubating amyloid [100 μM, 37°, 24 h] or aggregated amyloid [100 μM] in the presence of ligand chelates (100 μM). Then 10 μL of sample were dropped on a silicon dioxide wafer, air-dried followed by an additional 10 μL. The samples were sputtered with gold to a thickness of 7 nm using a Hummer VII gold sputtering system. Images were obtained on a JEOL JSM-6100 scanning electron microscope, with voltage set at 30 kV and magnification from 20× to 550×.
Ascorbate Studies
All solutions were prepared in KH2PO4/NaCl [15 mM] buffer containing desferryl [2 μM],79,91 except Cu(NO3)2 which was dissolved and diltued in Milli-Q water. Final sample volume = 4 mL. Each expeirment was performed in triplicate. Hydroxyl radical production was followed measuring the conversion of CCA into 7-hydroxy-CCA (λex = 395 nm, λem= 450 nm). General order of addition: CCA [100μM], ligand or copper [40 μM], and then ascorbate [300μM].
DCFH-DA Assay
Fibroblasts obtained from a 30 year old FRDA patient from The Coriell Institute (Camden, NJ) were kept in Dulbecco’s modified Eagle's medium (DMEM; ThermoScientific, Waltham, MA) with 10% charcoal-stripped fetal bovine serum (FBS; ThermoScientific), 1% GlutaMAX (ThermoScientific), and 1% penicillin–streptomycin (Invitrogen, Carlsbad, CA) at 37 °C, 5% CO2, and 90% humidity. HT-22 cells were prepared in a similar manner. The FRDA or HT-22 cells were plated at 5000 cells per well on a 96-well plate and then treated for 12 h with either ligands (various concentrations) and/or BSO (1 mM). After 12 h of treatment, the media was removed from each well of the 96-well plate, and 100 μL of 1 μM 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA; AnaSpec Inc., Fremont, CA) in phosphate buffer (PBS) was added to each well. The plates were returned to a 37 °C incubator for 20 min, each well was washed three times with PBS, and then the plate was read on a Tecan Infinite M200 plate reader with an absorbance of 495 nm and an emission of 529 nm.
Calcien AM Cell Viability Assay
FRDA cells were plated on a 96-well plate at a density of 3000 cells per well and then treated with ligands (various concentrations) and/or BSO (1 mM). After 48 h of BSO and ligand treatment, the media was removed, 1 μg/mL Calcein AM (CalBiochem, San Diego, CA) in phosphate buffer pH 7.2 (PBS; Fisher Scientific, Pittsburgh, PA) was added to each well, and the plate was incubated for 10 min at 37 °C. Cell viability was determined with a Tecan Infinite M200 plate reader with an excitation of 490 nm and emission of 520 nm.
Density Functional Theory Calculations
Gas phase DFT calculations were performed using a hybrid functional (B3LYP) as implemented in Gaussian 08 using crystal structures obtained from the CCDC. All atoms were optimized using the 6-311G basis set (+ and/or ++), while d,p was added for the metal based calculations.41 A frequency calculation of each showed the there were no imaginary frequencies, supporting a stable ground state. All calculations were compared to crystal data and are in good agreement as shown in Tables S1–S6. The .chk files of the optimized geometries were used to produce the electrostatic potential plots and HOMO/LUMO orbital overlays in GaussView. These were used to calculation the orbital contributions for each atom (Tables S7–S12).
Acknowledgments
We sincerely thank the TCU research groups of Dr. Zygmunt Gryczynski and Dr. Eric Simanek for access to fluorescence and mass-spectrometry instrumentation, respectively. We appreciate SEM training and access from the Dr. Jeff Coffer and Dr. Ernest Couch research groups. In addition, we are thankful to Dr. Zoltan Kovacs for an initial contribution of 1.
Glossary
- pyclen
1,4,7,10-tetraaza-2,6-pyridinophane
- cyclen
1,4,7,10-tetraazacyclododecane
- cyclam
1,4,8,11-tetraazacyclotetradecane
- EDTA
ethylenediamine tetraacetic acid
- DCFH-DA
dichlorodihydrofluorescein diacetate
Supporting Information Available
Experimental procedures and protocols, Figures S1–S11, and Tables S1–13. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
K.L.M. and P.M. contributed spectroscopic assays and synthesis while T.R. carried out cellular studies with assistance from K.L.M. L.R. performed calculations. K.N.G. and J.W.S. served as PIs.
K.N.G. is grateful for generous financial support from TCU start-up funds, TCU Research and Creative Activities Fund, and the Robert A. Welch Foundation (P-1760). J.W.S. acknowledges funding from the NIH (P01 AG022550 and P01 AGAG027956). Finally, T.E.R. was supported on an NIH training grant (T32 AG020494).
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- (2011) Alzheimer’s Disease Facts and Figures, Alzheimer’s & Dementia, Alzheimer’s Association, p 7. [DOI] [PubMed] [Google Scholar]
- Wenk G. L. (2003) Neuropathologic changes in Alzheimer’s disease. J. Clin. Psychiatry 64, 7–10. [PubMed] [Google Scholar]
- Ono M.; Hayashi S.; Matsumura K.; Kimura H.; Okamoto Y.; Ihara M.; Takahashi R.; Mori H.; Saji H. (2011) Rhodanine and Thiohydantoin Derivatives for Detecting Tau Pathology in Alzheimer’s Brains. ACS Chem. Neurosci. 2, 269–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolk D. A.; Grachev I. D.; Buckley C.; Kazi H.; Grady M. S.; Trojanowski J. Q.; Hamilton R. H.; Sherwin P.; McLain R.; Arnold. S. E. (2011) Association Between In Vivo Fluorine 18-Labeled Flutemetamol Amyloid Positron Emission Tomography Imaging and In Vivo Cerebral Cortical Histopathology. Arch. Neurol. 68, 1398–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleisher S.; Chen K.; Liu X.; Roontiva A.; Thiyyagura P.; Ayutyanont N.; Joshi A. D.; Clark C. M.; Mintun M. A.; Pontecorvo M. J.; Doraiswamy P. M.; Johnson K. A.; Skovronsky D. M.; Reiman E. M. (2011) Using Positron Emission Tomography and Florbetapir F 18 to Image Cortical Amyloid in Patients With Mild Cognitive Impairment or Dementia Due to Alzheimer Disease. Arch. Neurol. 68, 1461–1466. [DOI] [PubMed] [Google Scholar]
- Rajendran R.; Minqin R.; Ynsa M. D.; Casadesus G.; Smith M. A.; Perry G.; Halliwell B.; Watt F. (2009) A novel approach to the identification and quantitative elemental analysis of amyloid deposits-Insights into the pathology of Alzheimer’s disease. Biochem. Biophys. Res. Commun. 382, 91–95. [DOI] [PubMed] [Google Scholar]
- Bush A. I. (2003) The metallobiology of Alzheimer’s disease. Trends Neurosci. 26, 207–214. [DOI] [PubMed] [Google Scholar]
- Lovell M. A.; Robertson J. D.; Teesdale W. J. (1998) Copper, iron and zinc in AD senile plaques. J. Neurol. Sci. 158, 47–52. [DOI] [PubMed] [Google Scholar]
- Gaggelli E.; Kozlowski H.; Valensin D.; Valensin G. (2006) Copper homeostasis and neurodegenerative disorders (Alzheimer’s, prion, and Parkinson’s diseases and amyotrophic lateral sclerosis). Chem. Rev. 106, 1995–2044. [DOI] [PubMed] [Google Scholar]
- Mathis C. A.; Wang Y.; Klunk W. E. (2004) Imaging β-amyloid plaques and neurofibrillary tangles in the aging human brain. Curr. Pharm. Des. 10, 1469–1492. [DOI] [PubMed] [Google Scholar]
- Que E. L.; Domaille D. W.; Chang C. J. (2008) Metals in Neurobiology: Probing Their Chemistry and Biology with Molecular Imaging. Chem. Rev. 108, 1517–1549. [DOI] [PubMed] [Google Scholar]
- Fodero-Tavoletti M. T.; Villemagne V. L.; Rowe C. C.; Masters C. L.; Barnham K. J.; Cappai R. (2011) Amyloid-β: the seeds of darkness. Int. J. Biochem. Cell Biol. 43, 1247–1251. [DOI] [PubMed] [Google Scholar]
- Jakob-Roetne R.; Jacobsen H. (2009) Alzheimer’s disease: from pathology to therapeutic approaches. Angew. Chem. 48, 3030–3059. [DOI] [PubMed] [Google Scholar]
- Verdile G.; Fuller S.; Atwood C. S.; Laws S. M.; Gandy S. E.; Martins R. N. (2004) The role of beta amyloid in Alzheimer’s disease: still a cause of everything or the only one who got caught?. Pharmacol. Res. 50, 397–409. [DOI] [PubMed] [Google Scholar]
- Shankar G. M.; Li S.; Mehta T. H. (2008) Amyloid-β protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat. Med. 14, 837–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maynard C. J.; Bush A. I.; Masters C. L.; Cappai R.; Li Q. X. (2005) Metals and amyloid-beta in Alzheimer’s disease. Int. J. Exp. Pathol. 86, 147–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauk A. (2009) The chemistry of Alzheimer’s disease. Chem. Soc. Rev. 38, 2698–2715. [DOI] [PubMed] [Google Scholar]
- Barnham K. J.; Bush A. I. (2008) Metals in Alzheimer’s and Parkinson’s Diseases. Curr. Opin. Chem. Biol. 12, 222–228. [DOI] [PubMed] [Google Scholar]
- Duce J. A.; Bush A. I. (2010) Biological metals and Alzheimer’s disease: Implications for therapeutics and diagnostics. Prog. Neurobiol. (Oxford, U.K.) 92, 1–18. [DOI] [PubMed] [Google Scholar]
- Molina-Holgado F.; Hider R. C.; Gaeta A.; Williams R.; Francis P. (2007) Metals ions and neurodegeneration. BioMetals 20, 639–654. [DOI] [PubMed] [Google Scholar]
- Drew S. C.; Barnham K. J. (2011) The Heterogeneous Nature of Cu2+ Interactions with Alzheimer’s Amyloid-beta Peptide. Acc. Chem. Res. 44, 1146–1155. [DOI] [PubMed] [Google Scholar]
- Hyman L. M.; Franz K. J. (2012) Probing oxidative stress: Small molecule fluorescent sensors of metal ions, reactive oxygen species, and thiols. Coord. Chem. Rev. 256, 2333–2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraatz H.-B.; Metlzer-Nolte N.. Concepts and Models in Bioinorganic Chemistry; 2006.
- Barnham K. J.; Curtain C. C.; Bush A. I. (2007) Free radicals, metal ions, and Aβ aggregation and neurotoxicity. Protein Rev. 6, 31–47. [Google Scholar]
- Barnham K. J.; Masters C. L.; Bush A. I. (2004) Neurodegenerative diseases and oxidative stress, Nature reviews. Drug Discovery 3, 205–214. [DOI] [PubMed] [Google Scholar]
- Smith D. P.; Ciccotosto G. D.; Tew D. J.; Fodero-Tavoletti M. T.; Johanssen T.; Masters C. L.; Barnham K. J.; Cappai R. (2007) Concentration Dependent Cu2+ Induced Aggregation and Dityrosine Formation of the Alzheimer’s Disease Amyloid-β Peptide. Biochemistry 46, 2881–2891. [DOI] [PubMed] [Google Scholar]
- Braymer J. J.; DeToma A. S.; Choi J. S.; Ko K. S.; Lim M.-H. (2011) Recent Development of Bifunctional Small Molecules to Study Metal-Amyloid -β-Species in Alzheimer’s Disease. Int. J. Alzheimer’s Dis. 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crouch P. J.; Harding S.-M. E.; White A. R.; Camakaris J.; Bush A. I.; Masters C. L. (2008) Mechanisms of Aβ mediated neurodegeneration in Alzheimer’s disease. Int. J. Biochem. Cell Biol. 40, 181–198. [DOI] [PubMed] [Google Scholar]
- Adlard P. A.; Bush A. I. (2011) Zinc and Alzheimer’s disease. Biomed. Health Res. 76, 417–431. [Google Scholar]
- Adlard P. A.; Cherny R. A.; Finkelstein D. I.; Gautier E.; Robb E.; Cortes M.; Volitakis I.; Liu X.; Smith J. P.; Perez K.; Laughton K.; Li Q. X.; Charman S. A.; Nicolazzo J. A.; Wilkins S.; Deleva K.; Lynch T.; Kok G.; Ritchie C. W.; Tanzi R. E.; Cappai R.; Masters C. L.; Barnham K. J.; Bush A. I. (2008) Rapid restoration of cognition in Alzheimer’s transgenic mice with 8-hydroxy quinoline analogs is associated with decreased interstitial Abeta. Neuron 59, 43–55. [DOI] [PubMed] [Google Scholar]
- Adlard P. A.; Parncutt J. M.; Finkelstein D. I.; Bush A. I. (2010) Cognitive Loss in Zinc Transporter-3 Knock-Out Mice: A Phenocopy for the Synaptic and Memory Deficits of Alzheimer’s Disease?. J. Neurosci. 30, 1631–1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crouch P. J.; Hung L. W.; Adlard P. A.; Cortes M.; Lal V.; Filiz G.; Perez K. A.; Nurjono M.; Caragounis A.; Du T.; Laughton K.; Volitakis I.; Bush A. I.; Li Q. X.; Masters C. L.; Cappai R.; Cherny R. A.; Donnelly P. S.; White A. R.; Barnham K. J. (2009) Increasing Cu bioavailability inhibits Abeta oligomers and tau phosphorylation. Proc. Natl. Acad. Sci. U.S.A. 106, 381–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crouch P. J.; Savva M. S.; Hung L. W.; Donnelly P. S.; Mot A. I.; Parker S. J.; Greenough M. A.; Volitakis I.; Adlard P. A.; Cherny R. A.; Masters C. L.; Bush A. I.; Barnham K. J.; White A. R. (2011) The Alzheimer’s therapeutic PBT2 promotes amyloid-β degradation and GSK3 phosphorylation via a metal chaperone activity. J. Neurochem. 119, 220–230. [DOI] [PubMed] [Google Scholar]
- James S. A.; Volitakis I.; Adlard P. A.; Duce J. A.; Masters C. L.; Cherny R. A.; Bush A. I. (2012) Elevated labile Cu is associated with oxidative pathology in Alzheimer disease. Free Radical Biol. Med. 52, 298–302. [DOI] [PubMed] [Google Scholar]
- Manso Y.; Comes G.; Hidalgo J.; Bush A. I.; Adlard P. A. (2011) Copper modulation as a therapy for Alzheimer’s disease?. Int. J. Alzheimer’s Dis. 370345, 370345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miu A. C.; Benga O.; Adlard P. A.; Bush A. I. (2006) Metals and Alzheimer’s disease. J. Alzheimer’s Dis. 10, 145–163. [DOI] [PubMed] [Google Scholar]
- Strozyk D.; Launer L. J.; Adlard P. A.; Cherny R. A.; Tsatsanis A.; Volitakis I.; Blennow K.; Petrovitch H.; White L. R.; Bush A. I. (2009) Zinc and copper modulate Alzheimer Aβ levels in human cerebrospinal fluid. Neurobiol. Aging 30, 1069–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faller P. (2009) Copper and zinc binding to amyloid-β: coordination, dynamics, aggregation, reactivity and metal-ion transfer. ChemBioChem 10, 2837–2845. [DOI] [PubMed] [Google Scholar]
- Casadesus G.; Smith M. A.; Zhu X. W.; Aliev G.; Cash A. D.; Honda K.; Petersen R. B.; Perry G. (2004) Alzheimer disease: Evidence for a central pathogenic role of iron-mediated reactive oxygen species. J. Alzheimers Dis. 6, 165–169. [DOI] [PubMed] [Google Scholar]
- Castellani R. J.; Honda K.; Zhu X. W.; Cash A. D.; Nunomura A.; Perry G.; Smith M. A. (2004) Contribution of redox-active iron and copper to oxidative damage in Alzheimer disease. Ageing Res. Rev. 3, 319–326. [DOI] [PubMed] [Google Scholar]
- Wilkins S.; Masters C. L.; Bush A. I.; Cherny R. A.; Finkelstein D. I. (2009) Clioquinol protects against cell death in parkinson’s disease models in vivo and in vitro. Adv. Behav. Biol. 58, 431–442. [Google Scholar]
- Berk M.; Copolov D. L.; Dean O.; Lu K.; Jeavons S.; Schapkaitz I.; Anderson-Hunt M.; Bush A. I. (2008) N-Acetyl Cysteine for Depressive Symptoms in Bipolar Disorder-A Double-Blind Randomized Placebo-Controlled Trial. Biol. Psychiatry 64, 468–475. [DOI] [PubMed] [Google Scholar]
- Greenough M. A.; Volitakis I.; Li Q.-X.; Laughton K.; Evin G.; Ho M.; Dalziel A. H.; Camakaris J.; Bush A. I. (2011) Presenilins promote the cellular uptake of copper and zinc and maintain copper chaperone of SOD1-dependent copper/zinc superoxide dismutase activity. J. Biol. Chem. 286, 9776–9786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung Y. H.; Robb E. L.; Volitakis I.; Ho M.; Evin G.; Li Q.-X.; Culvenor J. G.; Masters C. L.; Cherny R. A.; Bush A. I. (2009) Paradoxical condensation of copper with elevated β-amyloid in lipid rafts under cellular copper deficiency conditions: Implications for Alzheimer disease. J. Biol. Chem. 284, 21899–21907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melov S., Adlard P. A., Morten K., Johnson F., Golden T. R., Hinerfeld D., Schilling B., Mavros C., Masters C. L., Volitakis I., Li Q.-X., Laughton K., Hubbard A., Cherny R. A., Gibson B., and Bush A. I. (2007) Mitochondrial oxidative stress causes hyperphosphorylation of tau. PLoS One 2e536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe S.; Nagano S.; Duce J.; Kiaei M.; Li Q.-X.; Tucker S. M.; Tiwari A.; Brown R. H.; Beal M. F.; Hayward L. J.; Culotta V. C.; Yoshihara S.; Sakoda S.; Bush A. I. (2007) Increased affinity for copper mediated by cysteine 111 in forms of mutant superoxide dismutase 1 linked to amyotrophic lateral sclerosis. Free Radical Biol. Med. 42, 1534–1542. [DOI] [PubMed] [Google Scholar]
- Crichton R. R.; Dexter D. T.; Ward R. J. (2008) Metal based neurodegenerative diseases - From molecular mechanisms to therapeutic strategies. Coord. Chem Rev 252, 1189–1199. [Google Scholar]
- Adlard P. A.; Bica L.; White A. R.; Nurjono M.; Filiz G.; Crouch P. J.; Donnelly P. S.; Cappai R.; Finkelstein D. I.; Bush A. I. (2011) Metal ionophore treatment restores dendritic spine density and synaptic protein levels in a mouse model of Alzheimer’s disease. PLoS One 6, e17669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adlard P. A.; Parncutt J. M.; Finkelstein D. I.; Bush A. I. (2010) Cognitive loss in zinc transporter-3 knock-out mice: a phenocopy for the synaptic and memory deficits of Alzheimer’s disease?. J. Neurosci. 30, 1631–1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardeland R. (2005) Antioxidative protection by melatonin - Multiplicity of mechanisms from radical detoxification to radical avoidance. Endocrine 27, 119–130. [DOI] [PubMed] [Google Scholar]
- Lynch M. A. (2004) Long-term potentiation and memory. Physiol. Rev. 84, 87–136. [DOI] [PubMed] [Google Scholar]
- Khan A.; Ashcroft A. E.; Korchazhkina O. V.; Exley C. (2004) Metal-mediated formation of fibrillar ABri amyloid. J. Inorg. Biochem. 98, 2006–2010. [DOI] [PubMed] [Google Scholar]
- Zatta P.; Drago D.; Zambenedetti P.; Bolognin S.; Nogara E.; Peruffo A.; Cozzi B. (2008) Accumulation of copper and other metal ions, and metallothionein I/II expression in the bovine brain as a function of aging. J. Chem. Neuroanat. 36, 1–5. [DOI] [PubMed] [Google Scholar]
- Sayre L. M.; Smith M. A.; Perry G. (2001) Chemistry and biochemistry of oxidative stress in neurodegenerative disease. Curr. Med. Chem. 8, 721–738. [DOI] [PubMed] [Google Scholar]
- Zambenedetti P.; Schmitt H. P.; Zatta P. (2002) Metallothionein I-II immunocytochemical reactivity in Binswanger’s encephalopathy. J. Alzheimer’s Dis. 4, 459–466. [DOI] [PubMed] [Google Scholar]
- Zatta P.; Raso M.; Zambenedetti P.; Wittkowski W.; Messori L.; Piccioli F.; Mauri P. L.; Beltramini M. (2005) Copper and zinc dismetabolism in the mouse brain upon chronic cuprizone treatment. Cell. Mol. Life Sci. 62, 1502–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lannfelt L.; Blennow K.; Zetterberg H.; Batsman S.; Ames D.; Harrison J.; Maters C. L.; Targum S.; Bush A. I.; Murdoch R.; Wilson J.; Ritchie C. W. (2008) Safety, efficacy, and biomarker findings of PBT2 in targeting Aβ as a modifying therapy for Alzheimer’s disease: a phase IIa, double-blind, randomized, placebo-controlled trial. Lancet Neurol. 7, 779–786. [DOI] [PubMed] [Google Scholar]
- Bush A. I.; Tanzi R. E. (2008) Therapeutics for Alzheimer’s disease based on the metal hypothesis. Neurotherapeutics 5, 421–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crabb E., and Moore E. A. (2009) Metals and Life, Chelation Therapy, Springer. [Google Scholar]
- Hegde M. L.; Bharathi P.; Suram A.; Venugopal C.; Jagannathan R.; Poddar P.; Srinivas P.; Sambamurti K.; Rao K. J.; Scancar J.; Messori L.; Zecca L.; Zatta P. (2009) Challenges associated with metal chelation therapy in Alzheimer’s disease. J. Alzheimer’s Dis. 17, 457–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie C. W.; Bush A. I.; Mackinnon A.; Macfarlane S.; Mastwyk M.; MacGregor L.; Kiers L.; Cherny R.; Li Q. X.; Tammer A.; Carrington D.; Mavros C.; Volitakis I.; Xilinas M.; Ames D.; Davis S.; Beyreuther K.; Tanzi R. E.; Masters C. L. (2003) Metal-protein attenuation with iodochlorhydroxyquin (clioquinol) targeting Abeta amyloid deposition and toxicity in Alzheimer disease: a pilot phase 2 clinical trial. Arch. Neurol. 60, 1685–1691. [DOI] [PubMed] [Google Scholar]
- Faux N. G.; Ritchie C. W.; Gunn A.; Rembach A.; Tsatsanis A.; Bedo J.; Harrison J.; Lannfelt L.; Blennow K.; Zetterberg H.; Ingelsson M.; Masters C. L.; Tanzi R. E.; Cummings J. L.; Herd C. M.; Bush A. I. (2010) PBT2 Rapidly Improves Cognition in Alzheimer’s Disease: Additional Phase II Analyses. J. Alzheimer’s Dis. 20, 509–516. [DOI] [PubMed] [Google Scholar]
- Izatt R. M.; Pawlak K.; Bradshaw J. S.; Bruening R. L. (1991) Thermodynamic and kinetic data for macrocycle interactions with cations and anions. Chem. Rev. 91, 1721–1785. [Google Scholar]
- Rojas-Quijano F. A., Kovacs Z., and Sherry A. D. (2006) INOR 282-Synthesis and evaluation of PCTA derivatives as luminescent-CEST active molecular probes for soft tissue imaging. Abstr. Pap. Am. Chem. Soc. 232. [Google Scholar]
- Rojas-Quijano F. A.; Benyo E. T.; Tircso G.; Kalman F. K.; Baranyai Z.; Aime S.; Sherry A. D.; Kovacs Z. (2009) Lanthanide(III) Complexes of Tris(amide) PCTA Derivatives as Potential Bimodal Magnetic Resonance and Optical Imaging Agents. Chem.—Eur. J. 15, 13188–13200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viswanathan S.; Kovacs Z.; Green K. N.; Ratnakar S. J.; Sherry A. D. (2010) Alternatives to Gadolinium-Based Metal Chelates for Magnetic Resonance Imaging. Chem. Rev. 110, 2960–3018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott L. E.; Orvig C. (2009) Medicinal Inorganic Chemistry Approaches to Passivation and Removal of Aberrant Metal Ions in Disease. Chem. Rev. 109, 4885–4910. [DOI] [PubMed] [Google Scholar]
- Schugar H.; Green D. E.; Bowen M. L.; Scott L. E.; Storr T.; Bohmerle K.; Thomas F.; Allen D. D.; Lockman P. R.; Merkel M.; Thompson K. H.; Orvig C. (2007) Combating Alzheimer’s disease with multifunctional molecules designed for metal passivation. Angew. Chem., Int. Ed. 46, 1716–1718. [DOI] [PubMed] [Google Scholar]
- Scott L. E.; Page B. D. G.; Patrick B. O.; Orvig C. (2008) Altering pyridinone N-substituents to optimize activity as potential prodrugs for Alzheimer’s disease. Dalton Trans. 6364–6367. [DOI] [PubMed] [Google Scholar]
- Chen T. W., X.; He Y.; Zhang C.; Wu Z.; Liao K.; Wang J.; Guo Z. . (2009) Effects of Cyclen and Cyclam on Zinc(II)- and Copper(II)-Induced Amyloid β-Peptide Aggregation and Neurotoxicity. Inorg. Chem. 48, 5801–5809. [DOI] [PubMed] [Google Scholar]
- Hindo S. S.; Mancino A. M.; Braymer J. J.; Liu Y. H.; Vivekanandan S.; Ramamoorthy A.; Lim M. H. (2009) Small Molecule Modulators of Copper-Induced A beta Aggregation. J. Am. Chem. Soc. 131, 16663–16665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolinski O. J.; Amaro M.; Birch D. J. S. (2010) Early detection of amyloid aggregation using intrinsic fluorescence. Biosens. Bioelectron. 25, 2249–2252. [DOI] [PubMed] [Google Scholar]
- Amaro M.; Birch D. J. S.; Rolinski O. J. (2011) Beta-amyloid oligomerisation monitored by intrinsic tyrosine fluorescence. Phys. Chem. Chem. Phys. 13, 6434–6441. [DOI] [PubMed] [Google Scholar]
- Maji S. K.; Amsden J. J.; Rothschild K. J.; Condron M. M.; Teplow D. B. (2005) Conformational dynamics of amyloid beta-protein assembly probed using intrinsic fluorescence. Biochemistry 44, 13365–13376. [DOI] [PubMed] [Google Scholar]
- Yang C.-A., Chen Y.-H., Ke S.-C., Chen Y.-R., Huang H.-B., Lin T.-H., and Chen Y.-C. (2011) Correlation of Copper Interaction, Copper-Driven Aggregation, and Copper-Driven H2O2 Formation with Aβ40 Conformation. Int. J. Alzheimer’s Dis. Vol. 2011, Article ID 607861, 7 pages. 10.4061/2011/607861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilloreau L.; Combalbert S.; Sournia-Saquet A.; Mazarguil H.; Faller P. (2007) Redox Chemistry of Copper–Amyloid-β: The Generation of Hydroxyl Radical in the Presence of Ascorbate is Linked to Redox-Potentials and Aggregation State. ChemBioChem 8, 1317–1325. [DOI] [PubMed] [Google Scholar]
- Richardson T. E.; Yang S. H.; Wen Y.; Simpkins J. W. (2011) Estrogen protection in Friedreich’s ataxia skin fibroblasts. Endocrinology 152, 2742–2749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albrecht P.; Lewerenz J.; Dittmer S.; Noack R.; Maher P.; Methner A. (2010) Mechanisms of oxidative glutamate toxicity: the glutamate/cystine antiporter system xc- as a neuroprotective drug target. CNS Neurol. Disord.: Drug Targets 9, 373–382. [DOI] [PubMed] [Google Scholar]
- Lipinski’s rules: (low molecular weight (MW ≤ 450), relatively lipophilic (c log P, calculated logarithm of the octanol/water partition coefficient, ≤5), hydrogen-bond donor atoms (HBD ≤ 5), hydrogen-bond acceptor atoms (HBA ≤ 10), small polar surface area (PSA ≤ 90°A2).
- Lipinski C. A.; Lombardo F.; Dominy B. W.; Feeney P. J. (2001) Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Delivery Rev. 46, 3–26. [DOI] [PubMed] [Google Scholar]
- Scarborough C. C.; Sproules S.; Weyhermuller T.; DeBeer S.; Wieghardt K. (2011) Electronic and Molecular Structures of the Members of the Electron Transfer Series [Cr((t)bpy)(3)](n) (n = 3+, 2+, 1+, 0): An X-ray Absorption Spectroscopic and Density Functional Theoretical Study. Inorg. Chem. 50, 12446–12462. [DOI] [PubMed] [Google Scholar]
- Frisch M. J., Trucks G. W., Schlegel H. B., Scuseria G. E., Robb M. A., Cheeseman J. R., Scalmani G., Barone V., Mennucci B., Petersson G. A., Nakatsuji H. C., Li M., Hratchian X., Izmaylov H. P., Bloino A. F., Zheng J., Sonnenberg G., Hada J. L., Ehara M., Toyota M., Fukuda K., Hasegawa R., Ishida J., Nakajima M., Honda T., Kitao Y., Nakai O., Vreven H., Montgomery T., Peralta J. A., Ogliaro J. E., Bearpark F., Heyd M., Brothers J. J., Kudin E., Staroverov K. N., Keith V. N., Kobayashi T., Normand R., Raghavachari J., Rendell K., Burant A., Iyengar J. C., Tomasi S. S., Cossi J., Rega M., Millam N., Klene J. M., Knox M., Cross J. E., Bakken J. B., Adamo V., Jaramillo C., Gomperts J., Stratmann R., Yazyev R. E., Austin O., Cammi A. J., Pomelli R., Ochterski C., Martin J. W., Morokuma R. L., Zakrzewski K., Voth V. G., Salvador G. A., Dannenberg P., Dapprich J. J., Daniels S., Farkas A. D., Foresman O., Ortiz J. B., Cioslowski J. V. , and Fox D. J. (2010) Gaussian 09, Revision B.01, Gaussian, Inc., Wallingford, CT.
- Steenken S.; Oneill P. (1979) Reaction of Oh Radicals with 2-Pyridones and 4-Pyridones in Aqueous-Solution - Electron-Spin Resonance and Pulse-Radiolysis Study. J. Phys. Chem. 83, 2407–2412. [Google Scholar]
- Murakami K. U., T; Morikawa R.; Ito, Masai, Haneda M.; Yoshino M. (1998) Antioxidant effect of dipicolinic acid on the metal-catalyzed lipid peroxidation and enzyme inactivation. Biomed. Res. 19, 205–208. [Google Scholar]
- Murakami K.; Tanemura Y.; Yoshino M. (2003) Dipicolinic acid prevents the copper-dependent oxidation of low density lipoprotein. J. Nutr. Biochem. 14, 99–103. [DOI] [PubMed] [Google Scholar]
- Aitken R. A.; Fodi B.; Palmer M. H.; Slawin A. M. Z.; Yang J. (2012) Experimental and theoretical molecular and electronic structures of the N-oxides of pyridazine, pyrimidine and pyrazine. Tetrahedron 68, 5845–5851. [Google Scholar]
- Green K. N.; Brothers S. M.; Lee B.; Darensbourg M. Y.; Rockcliffe D. A. (2009) Chemical Issues Addressing the Construction of the Distal Ni[Cysteine-Glycine-Cysteine]2- Site of Acetyl CoA Synthase: Why Not Copper?. Inorg. Chem. 48, 2780–2792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hureau C.; Faller P. (2009) Abeta-mediated ROS production by Cu ions: structural insights, mechanisms and relevance to Alzheimer’s disease. Biochimie 91, 1212–1217. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.