

Abstract

Amyloid β42 self-assembly is complex, with multiple pathways leading to large insoluble fibrils or soluble oligomers. Oligomers are now regarded as most germane to Alzheimer’s pathogenesis. We have investigated the hypothesis that oligomer formation itself occurs through alternative pathways, with some leading to synapse-binding toxins. Immediately after adding synthetic peptide to buffer, solutions of Aβ42 were separated by a 50 kDa filter and fractions assessed by SDS-PAGE silver stain, Western blot, immunoprecipitation, and capacity for synaptic binding. Aβ42 rapidly assembled into aqueous-stable oligomers, with similar protein abundance in small (<50 kDa) and large (>50 kDa) oligomer fractions. Initially, both fractions were SDS-labile and resolved into tetramers, trimers, and monomers by SDS-PAGE. Upon continued incubation, the larger oligomers developed a small population of SDS-stable 10–16mers, and the smaller oligomers generated gel-impermeant complexes. The two fractions associated differently with neurons, with prominent synaptic binding limited to larger oligomers. Even within the family of larger oligomers, synaptic binding was associated with only a subset of these species, as a new scFv antibody (NUsc1) immunoprecipitated only a small portion of the oligomers while eliminating synaptic binding. Interestingly, low doses of the peptide KLVFFA blocked assembly of the 10–16mers, and this result was associated with loss of the smaller clusters of oligomers observed at synaptic sites. What distinguishes these smaller clusters from the unaffected larger clusters is not yet known. Results indicate that distinct species of Aβ oligomers are generated by alternative assembly pathways and that synapse-binding subpopulations of Aβ oligomers could be specifically targeted for Alzheimer’s therapeutics.
Keywords: Alzheimer’s disease, amyloid beta, blocking peptide, therapeutics, single-chain variable fragment antibody, oligomerization
Alzheimer’s disease (AD) is the most common form of dementia. It is characterized by progressive memory loss and brain dysfunction and ultimately leads to death. Significant progress in understanding disease mechanisms has led to growing interest in the Aβ oligomer hypothesis for AD, which was introduced in 1998.1 Soluble Aβ oligomers can be long-lived entities distinct in structure and activity from the fibrillar Aβ found in amyloid plaques.2,3 Aβ oligomers appear to be the earliest demonstrable AD pathology, appearing before amyloid plaques,4 and they appear to correlate better with disease than amyloid plaques (reviewed in refs (5 and 6)). This phenomenon is particularly evident for individuals with the “Osaka” mutation, a familial form of Alzheimer’s disease in which affected persons express pathogenic oligomers but never form amyloid plaques.7 This phenotype is similarly manifested in transgenic mouse models carrying the Osaka mutation.8 In other transgenic mouse models of AD, synaptic and cognitive dysfunction are associated with accumulation of Aβ oligomers before the formation of amyloid plaques.9 Oligomers are thought to instigate memory loss through their ability to target synapses and disrupt synaptic plasticity,1,4,10−13 including inhibition of long-term potentiation1,14,15 and prolonged maintenance of long-term depression.13 The overall impact of Aβ oligomers manifests itself through the wide range of neuronal damage associated with AD neuropathology (reviewed in refs (16−21)). These notably include tau hyperphosphorylation,22 production of reactive oxygen species,23 and synapse deterioration.12 It is now regarded that small soluble oligomeric Aβ toxins, rather than insoluble fibrils, are most germane to Alzheimer’s pathogenesis.16−21,24−26
The importance of Aβ oligomers to AD pathology has generated considerable interest in oligomers as therapeutic targets. However, there is some uncertainty as to which species are specifically involved in pathogenesis as oligomers of a range of different sizes have been reported.18 A number of groups have identified smaller species, for example, dimers through tetramers, as the critical molecules.15,27−30 In contrast, others have shown evidence for larger oligomers, including 12mers in humans3 and mouse models.31 Even larger species have also been reported.32,33 In the widely studied Tg2576 mouse AD model, memory failure was shown to coincide with the appearance of SDS-stable 12mers,31 suggesting a significant pathogenic role for this species.
In this study, we have investigated the hypothesis that alternative pathways lead to formation of different oligomeric end-products, only some of which robustly target synapses. Binding to synapses is a particularly salient characteristic of oligomers, which act as gain-of-function pathogenic ligands that bind selectively to certain neurons and accumulate at sites localized to particular synapses.4,11,12 Results show that oligomers rapidly assemble into smaller and larger water-soluble oligomers that initially are sensitive to strong detergents. The synapse-targeting ligands themselves constitute a small portion of the larger oligomers, as indicated by a novel scFv (single-chain variable fragment) antibody, and the pathway leading to their assembly can be blocked by the short peptide, KLVFFA, that binds to the self-association domain of the monomeric subunit. The distinct species of synapse-binding Aβ oligomers generated by alternative assembly pathways thus may have the potential to be specifically targeted for AD therapeutics.
Results and Discussion
We have investigated Aβ self-assembly under conditions widely used to generate toxic oligomers for studies of Alzheimer’s mechanisms. Although it can be anticipated that conditions found in vivo will modify assembly, studies in vitro help define the capacity of Aβ for alternative chemical outcomes. Even under simple in vitro conditions, pathways of oligomerization are fundamentally heterogeneous,2 and the nature of oligomer assembly products is germane to their activity as synapse-targeting ligands.4 New findings with chemical and antibody probes indicate these pathway products may be targeted with considerable specificity, offering novel approaches for understanding disease pathology and developing disease-modifying therapeutics.
Biochemistry of Structures Formed by Self-Association of Aβ1–42 and Aβ1–40
Aβ1–42 Self-Assembles into Small and Large Oligomers That Are SDS-Labile and Indistinguishable by SDS-PAGE
Aβ1–42 self-assembles into a heterogeneous mixture of species that resolve by SDS-PAGE primarily as monomer, with lower levels of trimer and tetramer.2,34 To determine the extent to which the smaller species might derive from larger SDS-labile oligomers, we fractionated aqueous solutions by size using centrifugal filtration prior to SDS-PAGE. Soluble Aβ1–42 oligomers were prepared from monomer by overnight incubation and fractionated through either 10 kDa or 50 kDa MWCO filters. These ranges were selected because of current reports that have focused interest on dimer (9 kDa) and 12mer (54 kDa) species.3,27−31 Additionally, previous studies from our laboratory have shown that Aβ1–42 oligomeric species between 50 and 100 kDa, either synthetically prepared or derived from human AD brain extracts, carried synapse binding capacities.4,12 Retentate and filtrate fractions were analyzed by SDS-PAGE for protein profile (silver stain; Figure 1a) and Western blot staining by oligomer-selective NU1 (Figure 1b). Some blots were heat-treated and immunostained with the anti-Aβ antibody 6E10, which resulted in sensitive detection of monomers (Figure 1c).
Figure 1.
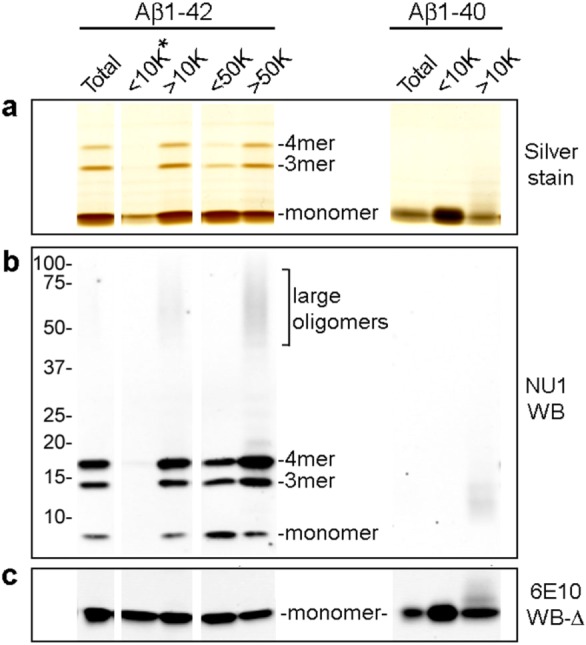
Centrifugal fractionation shows Aβ1–42 self-assembles into small and large oligomers that are SDS-labile and virtually indistinguishable by SDS-PAGE. Aβ1–42 dissolved in DMSO and F12 to 100 μM was incubated overnight, centrifuged to remove particulates, and separated by centrifugal filtration through 10 or 50 kDa filters. Freshly prepared Aβ1–40 was separated on a 10 kDa filter. (a) Total Aβ1–42, 10 kDa and 50 kDa filter fractions (left) and total Aβ1–40 and 10 kDa filter fractions (right) were analyzed by SDS-PAGE silver stain. The <10 kDa Aβ1–42 fraction (<10K*), which had minimal detectable protein, was loaded with 26× the volume of the total Aβ1–42; other fractions were matched for protein. (b) The presence of trimers, tetramers, and larger oligomers following SDS-PAGE was detected by Western blot with NU1. The <10K* lane was loaded with 77× the volume of total Aβ1–42. (c) Western blot monomer detection was enhanced by heat-treatment of the blot (100 °C, 5 min) prior to immunostaining with 6E10. The <10K* lane was loaded with 154× the volume of total Aβ1–42.
Unlike Aβ1–40, the longer and more hydrophobic Aβ1–42 shows an extremely strong propensity to oligomerize in these preparations. Following overnight incubation, roughly equivalent amounts of peptide occurred in the fractions above and below 50 kDa. The lack of detectable protein in the fraction smaller than 10 kDa suggests that the Aβ1–42 preparation contained only trace amounts of monomer. Overloading the 10 kDa filtrate (26× volume of Aβ1–42 control) resulted in minimal protein staining (Figure 1a), and an even higher loading excess (154× volume of Aβ1–42 control) was required for Western blot detection by 6E10 (Figure 1c). In comparison, freshly prepared Aβ1–40 fractionated on a 10 kDa filter yielded prominent silver stain and 6E10 Western blot signals at the monomer position (Figure 1a,c). Thus, in contrast to Aβ1–42, the Aβ1–40 peptide, which is less hydrophobic and less self-associative, remains largely monomeric, although small amounts of water-soluble oligomers are formed. The virtual absence of Aβ1–42 monomer following incubation is consistent with a strong propensity to oligomerize. The great differences in oligomerization are consistent with association of AD with elevated levels of Aβ1–42.
Filtrate and retentate fractions separated by a 50 kDa filter were virtually indistinguishable when analyzed by SDS-PAGE. By silver stain, both showed a predominant monomer band along with light trimer and tetramer bands (Figure 1a). By Western blot with the NU1 conformation-dependent antibody, reactivity for Aβ1–42 trimer and tetramer was much higher than that for the predominant protein monomer band in both fractions and is completely lacking for the Aβ1–40 preparation (Figure 1b). Differences in monomer immunostaining by NU1 (Figure1b) are proportional to the relative band intensity seen by protein staining (Figure1a) and dependent on the presence of a heavy monomer band. It previously has been shown that mixing Aβ monomer directly from DMSO into SDS does not yield these bands, so they are not SDS-induced2 and are likely building blocks of larger species. A small but significant difference between the two fractions is the trace amount of larger Aβ1–42 oligomers that are not visible by silver stain but are detectable by NU1 Western blot in the retentate fractions.
Results show that oligomerization under these frequently used experimental conditions promotes formation of large aqueous-soluble oligomers that are SDS-labile, decomposing in SDS from species initially greater than 50 kDa into predominantly monomer, as well as SDS-stable “cores” that manifest as trimers and tetramers. Insufficient attention to whether oligomers are aqueous- or SDS-stable has been a source of some confusion in the literature. Larger oligomers, for example, 12mers found in AD brain,3 could comprise three tetramers or, alternatively, four trimers. There is also evidence that even trimers and tetramers can assemble into multiple conformations which can be differentiated by Western blot and are reflected in differences in toxicity of the respective oligomer preparations.2
We note that under some circumstances the various oligomer-selective antibodies can detect antigen at the monomer band position (Supporting Information Figure 1). This occurs on blots without SDS in the transfer buffer (S1a) and blots subjected to heat-treatment (S1b). These conditions can greatly enhance staining at the monomer position relative to oligomeric position. In addition to enhancing staining by certain antibodies at the monomer position, heat-treatment of blots also greatly reduces or eliminates antibody recognition of species larger than trimers (not shown). Differences in oligomer species recognition by NU1 and NU2 are also apparent on heat-treated blots. Use of PVDF rather than nitrocellulose also enhances NU1 staining at the monomeric position (S1c). It is not known if signal from conformation-dependent antibodies at the monomer band position derives from rapid oligomer formation during the course of the analysis, but the possibility is suggested by the rapid kinetics of oligomer formation at even low doses.35
Aβ Assemblies >50 kDa Develop Increasing SDS Stability
Although almost indistinguishable by SDS-PAGE Western blot, the subpopulations of soluble oligomers in these preparations can be separated by size exclusion filtration for analysis. The presence of small amounts of SDS-stable oligomers larger than tetramers in the >50 kDa retentate fraction led us to ask whether there might be fundamental differences between the two fractions, perhaps representing assembly pathways that lead to substantially different structural and functional outcomes. The next experiment addressed how quickly Aβ1–42 develops large (>50 kDa) and small (<50 kDa) assemblies and whether both assemblages ultimately show progression to equivalent larger SDS-stable species.
Centrifugal fractionation through 50 kDa filters was based on SEC results of Chromy et al.2 and evidence of prominent 12mers in AD brain3 and in the Tg2576 mouse AD model.31 Reports of very much larger oligomers using DLS and HPLC are likely observations of self-aggregating oligomers that can occur with subpopulations, for example, 12mers, as building blocks.
Soluble Aβ1–42 preparations were subjected to 50 kDa centrifugal fractionation either 15 min or 24 h after dilution of the peptide with F12 media. The distribution of protein into filtrates and retentates was essentially the same for both times (not shown), indicating that the initial distribution into subpopulations occurs rapidly, in less than 15 min. Filtrate fractions typically represented 50–70% of the total Aβ, with the relative amount usually dependent on the specific peptide batch. Fractions, matched for protein loading, were analyzed by SDS-PAGE, with samples taken at either 1 or 7 days incubation following initial solution preparation. Silver stained protein profiles (not shown) were similar for filtrates and retentates, and showed minimal change upon aging. The presence of monomer as the predominant band, with minor trimer and tetramer bands and lack of detectable larger oligomers, suggests that both filtrates and retentates comprise smaller components that dissociate upon treatment with SDS. Conformational differences in these smaller subassemblies may determine the oligomerization pathway taken and whether it leads to oligomeric toxicity.
Despite the similarities of the protein profiles, SDS-PAGE Western blot analysis with NU2 emphasized the differences between the filtrate and retentate fractions. The major finding from this experiment (Figure 2) is the slow and significant increase in levels of SDS-stable large oligomers that occurs only in the retentate (i.e., species >50 kDa). These SDS-stable oligomers are prominent in Western blots at day 7, although they are essentially undetectable by silver stain. This signal was equally robust whether retentate fractions were collected at 15 min or 24 h after initial solution preparation. As in Figure 1, low levels of the large oligomers were already evident in retentates by day 1. In contrast, the larger SDS-stable oligomers were never generated from oligomers in the filtrate fractions (i.e., species <50 kDa). Small amounts of insoluble material, detectable by Western blot, developed in all fractions upon prolonged aging, suggesting fibril formation, but removal by centrifugation showed minimal effect on the protein concentration of the remaining soluble oligomers. Overall, these results indicate a rapid differentiation of Aβ1–42 into species smaller or greater than 50 kDa, and these species do not readily re-equilibrate to the same larger SDS-stable assemblies.
Figure 2.

High and low MW Aβ assemblies can be distinguished by the accumulation of larger SDS stable oligomers in the high MW fraction. Freshly prepared Aβ1–42 solutions were subjected to 50 kDa centrifugal filtration at 15 min or 24 h incubation. Soluble fractions were analyzed, both 1 day and 7 days after initial Aβ oligomer preparation, by SDS-PAGE Western blot immunostained with NU2.
Nature of Assembly Pathway Determines Capacity of Oligomers for Targeting Synapses
Because oligomers in the filtrate and retentate fractions showed marked biochemical differences, we next tested whether these differences might be reflected in synaptic binding activity. Synaptic binding is the critical initial step by which extracellular oligomers induce major aspects of neuronal pathology,4,12 and structural differences resulting from alternative assembly pathways seem likely to influence ligand properties.
Aβ1–42 oligomers, fractionated by centrifugal filtration (50 kDa) at 15 min incubation, were aged for 2 or 8 days. Fractions were incubated with cultured hippocampal neurons (100 nM Aβ1–42, 30 min at 37 °C) and analyzed for binding by immunolabeling with monoclonal NU4. Retentate fractions, whether aged for 2 or 8 days, showed prominent punctate binding (Figure 3), as expected for accumulation of oligomers at synapses.4,12 In contrast, the smaller oligomers found in filtrates showed complete lack of NU4-immunolabeling, essentially identical to vehicle controls. The absence of binding by small (<50 kDa) oligomers has previously been shown with the use of biotinylated4 or fluorescently tagged oligomers11 or by immunolabeling with 6E10 which also recognizes low-n oligomers.12 This lack of NU4-immunolabeling was unchanged even after 8 days aging of the filtrate fraction. Similar results were obtained for fractions obtained by centrifugal filtration following 24 h incubation of Aβ1–42 preparations (not shown). These results show that synaptic binding of Aβ1–42 oligomers to hippocampal neurons is selective for species that develop from the large (>50 kDa) initial peptide assemblies. In contrast, small initial peptide assemblies, even after prolonged aging, do not develop into species that act as synaptic ligands. Although there are small species represented in the SDS-PAGE profile of the >50 kDa oligomers (Figures 1 and 2), it is not the case that small oligomers, for example, tetramers, are the species that bind to synapses.
Figure 3.

Oligomers capable of targeting synapses are unique to the high MW fraction. Aβ1–42 assemblies fractionated immediately following solubilization were incubated at 4 °C, and the soluble oligomers tested for binding to hippocampal neurons at 2 or 8 days after initial oligomer preparation. Synaptic binding, detected with NU4 antibody, was specific for large (>50 kDa) oligomers and did not develop even after prolonged aging of the small (<50 kDa) oligomer preparation.
The first step in the pathogenic mechanism instigated by oligomers is attachment to specific synapses.4 Figure 3 shows that ligand-like association with synapses is a property of molecules found in the fraction comprising oligomers greater than 50 kDa. As yet, the structural basis for this capacity is unknown. Also unknown is the significance of the diffuse 54 kDa SDS-stable band found uniquely in Western blots of the high MW fraction. A question is whether some critical aspect of the 54 kDa species (or the oligomers it derives from) is essential for a robust capacity for targeting synapses. Since synaptic ligands are not found in the low MW fraction, size presumably plays a role. Hypothetically, for example, larger oligomers attached to cell surfaces could engage in lateral interactions that play a role in the clustering phenomenon observed in living neurons.11 Clustering may promote near-irreversible cell attachment. While synapse pathology can be induced by small oligomers,28 it occurs at a slow rate. It is not known whether local, membrane-mediated reorganization of small oligomer structure occurs with time.
Feasibility of Selectively Targeting and Interfering with the Self-Assembly Pathway That Generates Synaptic Ligands
The above results indicate that particular peptide–peptide interactions are required to form oligomers that act as synaptic ligands. We therefore tested the possibility that interference of these interactions by KLVFFA, a small peptide comprising the Aβ16–21 hydrophobic core, might affect this functional outcome. Small Aβ-derived peptides, for example, KLVFF,36 LVFFA,37 and KLVFFA,38−41 are known to interact with Aβ1–42. This is the first study that rigorously analyzes the effect of KLVFFA in the formation of soluble oligomers. The small peptide was tested for its effect on oligomerization carried out at 100 μM Aβ, a typical preparative condition, and at 30 nM Aβ, which more closely approximates pathophysiological conditions.
For the preparative condition, KLVFFA was mixed with Aβ1–42 in DMSO prior to initiating assembly by diluting with Ham’s F12 buffer to 100 μM Aβ and incubating for 22 h at 4 °C. The preparations were subjected to 50 kDa centrifugal filtration, and fractions analyzed by SDS-PAGE Western blot with NU2 (Figure 4a). We found that KLVFFA causes a strong reduction specifically in the SDS-stable 10–16mer band, which was highly enriched in the Western blot of the >50 kDa (retentate) fraction. Conversion to the SDS-stable conformation was potently blocked by 400 μM KLVFFA, with only a minimal signal evident in the >50 kDa fraction.
Figure 4.

Oligomeric assembly pathway leading to synaptic ligands is sensitive to interference by the peptide KLVFFA. (a) 100 μM Aβ1–42 incubated overnight in the presence or absence of 400 μM KLVFFA was fractionated, and total, filtrate (<50 kDa), and retentate (>50 kDa) analyzed by SDS-PAGE Western blot with NU2 for the presence of large SDS-stable assemblies. (b) Oligomers allowed to assemble at 30 nM Aβ1–42 in the presence or absence of KLVFFA for 15 min at 37 °C were immunoprecipitated with NU2 and bound material eluted and analyzed by SDS-PAGE Western blot for large SDS-stable assemblies. (c) Alternatively, oligomers formed at 30 nM Aβ1–42 in the presence or absence of KLVFFA were incubated with 26 DIV hippocampal neurons for 30 min at 37 °C and synaptic binding analyzed by immunolabeling with NU4.
For the pathophysiological conditions, Aβ1–42 was self-assembled at 30 nM, with or without KLVFFA, for 15 min at 37 °C. In order to obtain sufficient material for analysis, oligomers were immunoprecipitated using NU2. The antibody bound material was eluted and analyzed by SDS-PAGE Western blot (Figure 4b). The SDS-PAGE Western blot without KLVFFA, immunostained with oligomer-selective polyclonal M70/2,34 showed development of an Aβ species corresponding to 12mer. Formation of this species was blocked during assembly by KLVFFA, even at doses as low as 300 nM. The level of oligomerization seen in the prominent 3mer and 4mer region is essentially unaltered (not shown), suggesting that the SDS-stable large oligomers are related to a small subset of the overall population derived from the 30 nM Aβ1–42 preparation.
At both 100 μM and 30 nM Aβ1–42, the KLVFFA blocking peptide selectively targeted formation of the larger, SDS-stable species. Blocking formation of these oligomers under preparative high-dose Aβ conditions is attainable using high concentrations of KLVFFA. In contrast, when solutions of Aβ monomer are prepared at 30 nM doses, which would be pathologically meaningful, formation of the large SDS-stable oligomers is prevented by submicromolar peptide. Importantly, the effect of KLVFFA on the formation of specific oligomer species was also apparent in changes in neuronal binding (Figure 4c). Oligomers formed at 30 nM for 15 min at 37 °C, with or without KLVFFA, were incubated with cultures of differentiated hippocampal neurons for 30 min, and synaptic binding analyzed by immunolabeling with NU4. Oligomers assembled in the presence of as little as 100 nM KLVFFA showed a change in the size distribution of NU4-sensitive clusters. This observation is neurologically relevant because presence of KLVFFA peptide correlates to a marked decrease in overall oligomer binding to neurons. KLVFFA limited formation of oligomers to a subset that accumulated preferentially at larger NU4-sensitive clusters, resulting in elimination of ∼80% of the overall binding, and clusters that were 30% larger and 32% brighter on average than in the Aβ oligomer control. The formation of cell surface clusters was previously investigated on living neurons using quantum dot-labeled Aβ oligomers,11 but this is the first indication that distinctly different types of clusters exist. The earlier study showed that cluster formation overall is associated with larger oligomers. The data here indicate that these oligomers themselves are heterogeneous and that KLVFFA prevents formation of oligomers, as yet structurally uncharacterized, that generate the smaller clusters.
Results provide a strong indication that the large SDS-stable structures, whose generation is KLVFFA sensitive, are closely related to synaptic targeting. Indications are that delivery of the peptide to the brain to stabilize nontoxic conformations would be useful for potential therapeutic applications.
A New scFv Antibody Specific for Synapse-Targeting Oligomers
Synaptic binding of Aβ oligomers has been shown to be an important key element in the initiation of toxic pathology associate with AD. De Felice et al.22 have shown that the tau response to Aβ oligomers is localized to neurons that have oligomer binding and that tau hyperphosphorylation can be prevented by the NU1 antibody. Tau has been shown to be essential to β-amyloid-induced toxicity using hippocampal neurons from tau knockout mice which were not subject to neurite degeneration in the presence of Aβ.42 Aβ oligomer-induced ROS generation in hippocampal neurons could also be blocked by inhibition of oligomer binding by NU1 and other anti-Aβ oligomer antibodies (NU2 and NU4).23,43 Memantine, a drug that is only modestly effective in the treatment of AD patients, can block oligomer-induced pathologies, for example, ROS formation23 and synaptic degeneration,12 but does not affect oligomer binding.
Here we have investigated the impact of a new anti-Aβ oligomer scFv antibody, NUsc1, on the synaptic attack by Aβ oligomers.NUsc1 was panned from a human phage-display scFv library for oligomer-specific antibodies and was selected based on its targeting of oligomers but not monomers or fibrils (Supporting Information Figure 2a). This antibody is also selective for subpopulations of oligomers. NUsc1 showed selective immunoprecipitation of oligomers, resulting in a prominent tetramer signal for 50 kDa retentate but not filtrate (Supporting Information Figure 2b). A secondary incubation, again using NUsc1, for residual oligomers in the unbound fraction gave no additional signal, indicating the initial immunoprecipitation was complete. In comparison, a secondary immunoprecipitation of the NUsc1 unbound with the less selective 4G8 antibody showed binding of oligomers in both the filtrate and retentate. The oligomers not bound by NUsc1 were readily dissociated by SDS, resulting in heavy monomer bands representing a majority of the protein. NUsc1 could also be used for detection of oligomers bound to nerve cell surfaces (Supporting Information Figure 2c).
To examine more thoroughly the specificity of NUsc1 for oligomeric species, we next used this new antibody to immunoprecipitate oligomers formed under low-dose conditions (30 nM Aβ), and compared NUsc1 to oligomer-selective NU2 and the nonselective Aβ antibody 4G8 (Figure 5). Antibodies were bound to magnetic Dynabeads and incubated with a 30 nM Aβ oligomer preparation for 90 min at 37 °C. Immunoprecipitated oligomers were eluted and analyzed by SDS-PAGE Western blot for tetramer (left panel) or monomer (right panel). NU2, 4G8 and NUsc1 all showed detectable but different levels of oligomer immunoprecipitation, while the nonimmune IgG control showed no oligomer pulldown. 4G8 bound the most material while NU2 showed similar levels of tetramer but only half as much monomer (the most prominent SDS breakdown species). NUsc1 gave the most selective binding with much less immunostaining of both tetramer and monomer bands. These results show that NUsc1 binds only a small subset of the total oligomer population.
Figure 5.

Synaptic ligands comprise only a small subset of the high MW oligomers and are targeted by a novel scFv antibody (NUsc1). (a) Oligomers formed at 30 nM Aβ1–42 were immunoprecipitated with NU2, 4G8, NUsc1, or nonimmune IgG immobilized on magnetic Dynabeads. Antibody bound oligomers were eluted and analyzed by SDS-PAGE Western blot with NU2 or on heat-treated blots with NU1 (NU1-Δ). (b) The unbound oligomers following the initial NUsc1 immunoprecipitation were tested for residual ligand activity by incubating with hippocampal neurons for 30 min at 37 °C and immunolabeling with NU4. Aβ (top, left) and vehicle (bottom, right) no IP controls were incubated without magnetic Dynabeads parallel with the antibody–Dynabead samples.
The subset of oligomers targeted by NUsc1 appears to be highly germane to synaptic binding. Residual oligomers, remaining in the unbound material following immunoprecipitation, were tested for synaptic binding by incubation with hippocampal neurons for 30 min at 37 °C, followed by immunolabeling with oligomer-selective monoclonal NU4. Control treatment with nonimmune IgG showed no appreciable depletion of synapse-binding oligomers. Positive controls NU2 and 4G8 showed essentially complete loss of binding, consistent with the immunoprecipitation data. Most interestingly, despite the limited amount of oligomer depleted during immunoprecipitation with NUsc1, the remaining residual oligomers showed almost no synaptic binding. Compared to the Aβ oligomer control (no IP), NUsc1 showed 5% residual binding, similar to NU2 (4%) and 4G8 (1%), while the nonimmune IgG retained 87% of the control. Thus, the new scFv, NUsc1, selectively targets Aβ oligomers that are capable of binding to synapses, which represent only a small fraction of the total oligomer population. Although NUsc1 represents a promising tool, further in-depth characterization of structural features of the toxins is limited at this time by the ability to elute oligomers from the phage-bound complex as intact molecules in amounts sufficient for structural analysis.
The impact of KLVFFA on a small fraction of oligomers is especially intriguing given results with a new scFv antibody, selected by panning for its specificity to bind oligomers. Like KLVFFA, this new antibody (NUsc1) appears to target only a fraction of the oligomerization products. The antibody is specific for oligomers in the >50 kDa fraction, and it interacts with only a small subset of molecules within this fraction. This subset is particularly important because its removal through immunoprecipitation by NUsc1 causes a large decrease in synaptic binding. Thus, of the biochemically formed oligomers, those that are capable of acting as synaptic ligands comprise only a small percentage, and it is possible to neutralize them with highly specific antibodies.
Antibody specificity for synapse-targeting oligomers may open the door to new approaches for monitoring disease initiation/progression in vivo, following precedents established with phage bound scFv antibody against Aβ.44 The value of scFv antibodies for AD therapeutics, moreover, has precedent in recent findings that an Aβ-directed scFv antibody can attenuate plaque pathology in vivo in Tg AD mice45,46 and eliminate vascular amyloid in a Tg mouse AD model.47 In the long run, antibodies that have specificity for the subset of synapse-targeting oligomers described here could provide a powerful resource for disease-modifying therapeutics.
In conclusion, we have investigated pathways of Aβ oligomer formation with the goal of identifying potential therapeutic targets. Aβ42 rapidly assembles into small (<50 kDa) and large (>50 kDa) oligomers. Initially, both fractions are SDS-labile but, upon continued incubation, the larger oligomers develop a small population of SDS-stable 10–16mers. Low doses of the peptide KLVFFA were able to block assembly of the 10–16mers. Significantly, KLVFFA limited formation of oligomers to a subset that accumulated preferentially at larger NU4-sensitive clusters. We have identified a new scFv, NUsc1, that selectively targets a small fraction of large (>50 kDa) Aβ oligomers, including those capable of binding to synapses. Antibodies specific for synapse-targeting oligomers or peptides that can block formation of those oligomers could provide a powerful basis for disease-modifying therapeutics.
Methods
Aβ1–42 Oligomerization
Oligomers were prepared according to Lambert et al.34 Briefly, solid Aβ1–42 peptide (American Peptide) was dissolved in cold hexafluoro-2-propanol (HFIP; Sigma). The peptide was incubated at room temperature for at least 1 h to establish monomerization and randomization of structure. The HFIP was aliquotted and allowed to evaporate overnight, followed by 10 min in a Savant Speed Vac. The resulting peptide was stored as a film at −80 °C. The film was dissolved in anhydrous dimethyl sulfoxide (DMSO; Sigma) to 5 mM and then diluted to approximately 100 μM with Ham’s F12 (without phenol red, with glutamine; Caisson Laboratories, Logan, UT) and briefly vortexed. The solution was aged at 4 °C, and soluble oligomers obtained by centrifugation at 14 000g for 10 min at 4 °C. Ultrafiltration of Aβ1–42 and Aβ1–40 preparations was done using either Nanosep 10K omega (Pall Corporation, Ann Arbor, MI) or Microcon YM-50 (Amicon, Bedford, MA) centrifugal filters, according to the instructions of the manufacturer. Protein concentration was estimated using Coomassie Plus Protein Assay (Pierce Biotechnology, Inc., Rockford, IL) and a bovine serum albumin (BSA) standard. Oligomer formation at low (30 nM Aβ1–42) peptide concentration was carried out by first dissolving 0.044 mg of HFIP peptide film in 100 μL of DMSO, using three cycles of repeated scraping, vortexing 30–60 s and brief centrifugation to ensure that the peptide film was completely dissolved and incubated at RT for 15 min. The peptide was diluted to 20 μM with DMSO, vortexed 30–60 s, incubated 15 min, and 100 μL diluted with an equal volume of DMSO to obtain a 10 μM working stock. Oligomers were prepared by diluting the 10 μM peptide stock to 30 nM in neurobasal media (NB; phenol red free; Invitrogen) prewarmed at 37 °C, vortexing 30–60 s, and incubating at 37 °C for 15 min. Fibrils were prepared from Aβ1–42 according to Stine et al.48
Monoclonal Antibodies
Monoclonal antibodies (NU1, NU2, NU4) were developed against Aβ1–42 oligomers and characterized by Lambert et al.43 and others.8,11,22,23 These antibodies recognize oligomers isolated from AD brain tissue, block binding of both human-derived and synthetic oligomers to neurons, and show preferential binding to HMW (>50 kDa) synthetic oligomers in competition dot blot assays. These antibodies recognize epitopes dependent on the tertiary structure formed by self-association of Aβ1–42 and do not recognize Aβ1–40 and show minimal binding to monomeric Aβ1–42.
SDS-PAGE and Western Blotting
Electrophoresis was performed on Novex 10–20% Tris-Tricine gels (Invitrogen) at 125 V. Gels were silver stained (SilverXpress; Invitrogen) or electroblotted onto nitrocellulose at 100 V for 60 min at 4 °C using 25 mM Tris, 192 mM glycine, 20% (v/v) methanol, 0.02% (w/v) SDS, pH 8.3. Blots were blocked in 5% nonfat dry milk in TBST (20 mM Tris-HCl, pH7.5, 0.8% NaCl, 0.1% Tween 20) for 30 min, incubated with oligomer specific antibody in milk/TBST [1 μg/mL NU1 or 1.5 μg/mL NU2;43 1:2000 polyclonal M70/234] for 90 min at RT, followed by HRP-linked secondary antibody IgG (GE, 1:40 000 in milk/TBST) for 60 min at RT. Alternatively, the nitrocellulose was incubated in PBS at 100 °C for 5 min prior to blocking and probed with 6E10 (Covance, Richmond, CA; 1:2000) or NU1. Antibody binding was visualized with half strength Super-Signal Femto (Pierce) and signal intensity read and analyzed on a Kodak Image Station.
Synapse Binding
Immunocytochemistry was performed as described.4,34 Mature hippocampal cell cultures [at least 21 days in vitro] were prepared as described previously4 and incubated with 100 nM (Aβ1–42 monomer equivalent) of oligomer prepared at 100 μM peptide or with undiluted 30 nM Aβ1–42 oligomer preparations for 30 min at 37 °C. The cells were washed to remove unbound oligomers and fixed by adding an equal volume of 3.7% formaldehyde in neurobasal media to the final wash for 5 min, followed by undiluted 3.75% formaldehyde for 5 min and three to five washes with PBS. The coverslips were incubated with 10% normal goat serum (NGS) in PBS for 30 min and immunolabeled with oligomer-specific monoclonal antibody43 (1 μg/mL NU4 in 10% NGS/PBS) for 2 h at RT. Antibody binding was visualized with AlexaFluor488 anti-mouse IgG (Invitrogen; 1:2000 in 1% NGS/PBS, 2 h at RT). Coverslips were mounted on glass slides using ProLong Gold with DAPI (Invitrogen) and imaged on a Kodak epifluorescent microscope at 60×. Image analysis was performed with MetaMorph imaging software (Universal Imaging, West Chester, PA).
Phage Bound NUsc1 Antibody
A collection of single-chain variable fragment (scFv) antibodies was developed by panning against soluble Aβ1–42 oligomers (Sebollela et al., unpublished data). Characterization of these antibodies, including NUsc1, will be detailed in a separate paper. Selection of anti-oligomer scFv NUsc1was performed by phage display using a human scFv library (Tomlinson I + J, MRC Center for Protein Engineering, Cambridge, U.K.). Briefly, immunotubes (Nunc) were coated overnight at 37 °C with 10 μg/well of Aβ1–42 oligomer diluted in Ham’s F12. Following coating, immunotubes were blocked with 2% nonfat dry milk in PBS for 2 h at room temperature and incubated with 1012 phage particles for additional 2 h under mild shaking. After washing with PBS plus 0.1% Tween-20, bound phages were eluted with trypsin (Sigma) in PBS. Eluted phages were subsequently amplified by infecting E.coli TG1 in the presence of helper phage KM13 and further purified by polyethylene glycol (MW 6000)/NaCl precipitation. Pellets were resuspended in PBS and used in further rounds of selection. Oligomer-binding capacity of isolated colonies obtained after the fourth round of selection was analyzed by ELISA (following the same conditions used in the panning). One of the most promising clones, named NUsc1, was selected for larger scale production of phage-bound anti-oligomer scFv. For this purpose, 50 mL of TG1 cells harboring NUsc1 phagemid were coinfected with KM13 helper phage and grown overnight in 2xTY medium. The culture was centrifuged at 10 000g for 10 min at 4 °C, and NUsc1 phage-enriched supernatant was stored at 4 °C.
Differential binding of NUsc1 to distinct Aβ assemblies was tested by ELISA. Wells were coated with 10 μg/well of Aβ1–40 (monomers) or Aβ1–42 (oligomers or fibrils) in F12 medium overnight. The next day wells were washed and blocked with 2% milk/PBS for 2 h at room temperature and then incubated with either 6E10 (1 μg/mL) or phage-bound NUsc1 (diluted 1:100 in blocking buffer) for an additional 2 h under mild shaking. After washing with PBS plus 0.1% Tween-20, bound 6E10 or NUsc1 was probed with HRP-conjugated secondary antibodies [anti-mouse IgG or anti-M13 (GE), respectively]. Following washing, signal was developed using 3,3′,5,5′-tetramethylbenzidine ELISA substrate (Sigma).
Detection of neuronal binding of oligomers was carried out as described above for synapse binding, but bound oligomers were probed with 1:10 dilution of the phage-bound NUsc1 for 2 h at room temperature. NUsc1 binding to cells was detected with anti-M13 mouse IgG (GE; 1:1000) followed by incubation with AlexaFluor555 anti-mouse IgG (Invitrogen; 1:2000) for 1 h. Coverslips were mounted and imaged as described above.
Immunoprecipitation of Oligomers
Antibody-specific binding of oligomers was tested by immunoprecipitation, followed by elution of bound material and SDS-PAGE analysis. Mouse IgG (4 μg) was incubated with 100 μL goat anti-mouse IgG Dynabeads (Invitrogen) in 1 mL of 0.1% BSA in PBS, overnight at 4 °C. Phage-bound NUsc1 (50 μL) was incubated with 15 μL of Dynabeads for His-tag isolation (Invitrogen) in 1 mL of BSA/PBS buffer. The Dynabeads were washed twice with the BSA/PBS buffer and resuspended in 100 μL of F12 or NB, and then 50 μL aliquots were transferred to fresh tubes containing 1 mL of buffer. Dynabeads were resuspended in 1 mL of oligomer solution and incubated for 90 min at 37 °C. Where specified, the unbound fraction was subjected to a secondary immunoprecipitation with fresh antibody–Dynabead complex. Following immunoprecipitation, the Dynabeads were washed 2× with BSA/PBS and once with PBS and then transferred to clean tubes with 1 mL PBS. Oligomers were eluted from the beads with 30 μL of 1× Tricine sample buffer in TBS, at RT for 15 min, and analyzed by SDS-PAGE Western blot.
For 30 nM Aβ1–42 oligomer preparations in NB, Dynabeads labeled with NUsc1, NU2, 4G8 (Covance), or nonimmune mouse IgG (Invitrogen) were incubated with 1 mL of undiluted oligomer solution. Unbound fractions were tested for synaptic binding of residual oligomers (see above).
For filtrate and retentate from 50 kDa fractionation of 100 μM Aβ1–42 preparations, oligomers were diluted to 30 nM in F12 and 1 mL aliquots incubated with NUsc1 bound to Dynabeads. The unbound fractions were subjected to a secondary immunoprecipitation with either NUsc1- or 4G8-Dynabeads.
Blocking of Aβ1–42 Oligomerization
KLVFFA was synthesized manually using standard 9-fluorenylmethoxycarbonyl solid-phase techniques on a preloaded Wang resin utilizing 2-(1H-7-azabenzotriazol-1-yl)-1,1,3,3-tetramethyl uronium hexafluorophosphate and N,N-diisopropylethylenamine for activation and coupling in dimethylformamide. The peptide was cleaved from the resin with 95% trifluoroacetic acid, 2.5% triisopropylsilane, and 2.5% H2O for 2 h. Analytical HPLC was performed with H2O and acetonitrile (ACN) in the mobile phase using an Atlantis T3 column. An isocratic gradient at 10% ACN for 5 min, followed by a linear gradient from 10 to 40% ACN over 10 min then 40–70% ACN over 30 min was used with a flow rate of 1 mL/min. Similarly, preparative HPLC was performed with an isocratic gradient at 10% ACN for 5 min, followed by a linear gradient from 10 to 34% ACN over 10 min then 34–50% ACN over 24 min with a flow rate of 15 mL/min. The peptide eluted as two broad peaks at 16 and 22 min. Peptide purity and identification were characterized by analytical HPLC (tR = 22 min) and electrospray ionization mass spectrometry (m/z = 724.3 = m + H+, calcd m = 723.91). The purified peptide was dried by rotary evaporation, redissolved in water, aliquoted, and lyophilized. For blocking experiments, KLVFFA was dissolved in DMSO and premixed with Aβ1–42 in DMSO prior to addition of media to initiate oligomerization.
Glossary
Abbreviations
- Aβ
amyloid beta
- ACN
acetonitrile
- AD
Alzheimer’s disease
- BSA
bovine serum albumin
- DMSO
dimethyl sulfoxide
- ELISA
enzyme-linked immunosorbent assay
- HFIP
hexafluoro-2-propanol
- HPLC
high performance liquid chromatography
- NGS
normal goat serum
- PAGE
polyacrylamide gel electrophoresis
- PBS
phosphate buffered saline
- scFv
single-chain variable fragment
- SDS
sodium dodecyl sulfate
- TBST
Tris-buffer saline with Tween-20
Supporting Information Available
Supplemental Figure 1 shows that differences in Western blot processing that affect immunolabeling of the SDS-PAGE Aβ monomer band by some antibodies. Supplemental Figure 2 shows preliminary analysis of the novel scFv antibody, NUsc1. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
P.T.V. conceived, designed and carried out the experiments, analyzed data, and wrote the manuscript. M.C.H. synthesized the peptide, designed experiments and wrote the manuscript. A.S. developed and characterized the NUsc1 antibody and wrote the manuscript. I.A.P. developed the NUsc1 antibody. P.N.L. analyzed data and helped edit the manuscript. B.N.T analyzed data. K.B.L. performed experiments. X.S. characterized the NUsc1 antibody. K.L.V. prepared cell cultures, aided with microscopy, analyzed data, characterized the NUsc1 antibody and helped edit the manuscript. A.L.E. and T.J.M. contributed to experimental plans and provided guidance. W.L.K. contributed to experimental plans, consulted on data interpretation, and wrote the manuscript.
This work was supported by National Institutes of Health Grants RO1-AG022547 and AG029460 from NIA, Alzheimer’s Association Zenith Award #ZEN-09-133875, and the Nanoscale Science and Engineering Initiative of the National Science Foundation under NSF Award #EEC-06475560 to the Northwestern University Nanoscale Science and Engineering Center (W.L.K.). It was also supported by National Institutes of Health’s Cancer Nanotechnology Excellence (CCNE) initiative of the National Cancer Institute under Award #U54CA151880 (T.J.M.) and by a National Science Foundation Graduate Research Fellowship (M.C.H.).
The authors declare the following competing financial interest(s): WLK is co-founder of Acumen Pharmaceuticals, which has been licensed by Northwestern University to target ADDLs (Aβ-Derived Diffusible Ligands with dementing action) for Alzheimer’s therapeutics and diagnostics.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Lambert M. P.; Barlow A. K.; Chromy B. A.; Edwards C.; Freed R.; Liosatos M.; Morgan T. E.; Rozovsky I.; Trommer B.; Viola K. L.; Wals P.; Zhang C.; Finch C. E.; Krafft G. A.; Klein W. L. (1998) Diffusible, nonfibrillar ligands derived from Abeta1–42 are potent central nervous system neurotoxins. Proc. Natl. Acad. Sci. U.S.A. 95, 6448–6453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chromy B. A.; Nowak R. J.; Lambert M. P.; Viola K. L.; Chang L.; Velasco P. T.; Jones B. W.; Fernandez S. J.; Lacor P. N.; Horowitz P.; Finch C. E.; Krafft G. A.; Klein W. L. (2003) Self-assembly of Abeta(1–42) into globular neurotoxins. Biochemistry 42, 12749–12760. [DOI] [PubMed] [Google Scholar]
- Gong Y.; Chang L.; Viola K. L.; Lacor P. N.; Lambert M. P.; Finch C. E.; Krafft G. A.; Klein W. L. (2003) Alzheimer’s disease-affected brain: presence of oligomeric A beta ligands (ADDLs) suggests a molecular basis for reversible memory loss. Proc. Natl. Acad. Sci. U.S.A. 100, 10417–10422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacor P. N.; Buniel M. C.; Chang L.; Fernandez S. J.; Gong Y.; Viola K. L.; Lambert M. P.; Velasco P. T.; Bigio E. H.; Finch C. E.; Krafft G. A.; Klein W. L. (2004) Synaptic targeting by Alzheimer’s-related amyloid beta oligomers. J. Neurosci. 24, 10191–10200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack C. R. Jr.; Knopman D. S.; Jagust W. J.; Shaw L. M.; Aisen P. S.; Weiner M. W.; Petersen R. C.; Trojanowski J. Q. (2010) Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol. 9, 119–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2011) 2010 Alzheimer’s disease progress report: A deeper understanding, U.S. Department of Health and Human Services, Washington, DC. [Google Scholar]
- Tomiyama T.; Nagata T.; Shimada H.; Teraoka R.; Fukushima A.; Kanemitsu H.; Takuma H.; Kuwano R.; Imagawa M.; Ataka S.; Wada Y.; Yoshioka E.; Nishizaki T.; Watanabe Y.; Mori H. (2008) A new amyloid beta variant favoring oligomerization in Alzheimer’s-type dementia. Ann. Neurol. 63, 377–387. [DOI] [PubMed] [Google Scholar]
- Tomiyama T.; Matsuyama S.; Iso H.; Umeda T.; Takuma H.; Ohnishi K.; Ishibashi K.; Teraoka R.; Sakama N.; Yamashita T.; Nishitsuji K.; Ito K.; Shimada H.; Lambert M. P.; Klein W. L.; Mori H. (2010) A mouse model of amyloid beta oligomers: their contribution to synaptic alteration, abnormal tau phosphorylation, glial activation, and neuronal loss in vivo. J. Neurosci. 30, 4845–4856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mucke L.; Masliah E.; Yu G. Q.; Mallory M.; Rockenstein E. M.; Tatsuno G.; Hu K.; Kholodenko D.; Johnson-Wood K.; McConlogue L. (2000) High-level neuronal expression of abeta 1–42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J. Neurosci. 20, 4050–4058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q.; Walsh D. M.; Rowan M. J.; Selkoe D. J.; Anwyl R. (2004) Block of long-term potentiation by naturally secreted and synthetic amyloid beta-peptide in hippocampal slices is mediated via activation of the kinases c-Jun N-terminal kinase, cyclin-dependent kinase 5, and p38 mitogen-activated protein kinase as well as metabotropic glutamate receptor type 5. J. Neurosci. 24, 3370–3378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renner M.; Lacor P. N.; Velasco P. T.; Xu J.; Contractor A.; Klein W. L.; Triller A. (2010) Deleterious effects of amyloid beta oligomers acting as an extracellular scaffold for mGluR5. Neuron 66, 739–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacor P. N.; Buniel M. C.; Furlow P. W.; Clemente A. S.; Velasco P. T.; Wood M.; Viola K. L.; Klein W. L. (2007) Abeta oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer’s disease. J. Neurosci. 27, 796–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H. W.; Pasternak J. F.; Kuo H.; Ristic H.; Lambert M. P.; Chromy B.; Viola K. L.; Klein W. L.; Stine W. B.; Krafft G. A.; Trommer B. L. (2002) Soluble oligomers of beta amyloid (1–42) inhibit long-term potentiation but not long-term depression in rat dentate gyrus. Brain Res. 924, 133–140. [DOI] [PubMed] [Google Scholar]
- Walsh D. M.; Klyubin I.; Fadeeva J. V.; Cullen W. K.; Anwyl R.; Wolfe M. S.; Rowan M. J.; Selkoe D. J. (2002) Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature 416, 535–539. [DOI] [PubMed] [Google Scholar]
- Townsend M.; Shankar G. M.; Mehta T.; Walsh D. M.; Selkoe D. J. (2006) Effects of secreted oligomers of amyloid beta-protein on hippocampal synaptic plasticity: a potent role for trimers. J. Physiol. 572, 477–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy J.; Selkoe D. J. (2002) The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297, 353–356. [DOI] [PubMed] [Google Scholar]
- Rodgers A. B. (2005) Progress report on Alzheimer’s disease 2004–2005, U.S. Department of Health and Human Services; National Institutes on Aging; National Institutes of Health, Washington, DC. [Google Scholar]
- Glabe C. G. (2008) Structural classification of toxic amyloid oligomers. J. Biol. Chem. 283, 29639–29643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe D. J. (2008) Soluble oligomers of the amyloid beta-protein impair synaptic plasticity and behavior. Behav. Brain Res. 192, 106–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira S. T.; Klein W. L. (2011) The Abeta oligomer hypothesis for synapse failure and memory loss in Alzheimer’s disease. Neurobiol. Learn. Mem. 96, 529–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnabel J. (2011) Amyloid: little proteins, big clues. Nature 475, S12–S14. [DOI] [PubMed] [Google Scholar]
- De Felice F. G.; Wu D.; Lambert M. P.; Fernandez S. J.; Velasco P. T.; Lacor P. N.; Bigio E. H.; Jerecic J.; Acton P. J.; Shughrue P. J.; Chen-Dodson E.; Kinney G. G.; Klein W. L. (2008) Alzheimer’s disease-type neuronal tau hyperphosphorylation induced by A beta oligomers. Neurobiol. Aging 29, 1334–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Felice F. G.; Velasco P. T.; Lambert M. P.; Viola K.; Fernandez S. J.; Ferreira S. T.; Klein W. L. (2007) Abeta oligomers induce neuronal oxidative stress through an N-methyl-D-aspartate receptor-dependent mechanism that is blocked by the Alzheimer drug memantine. J. Biol. Chem. 282, 11590–11601. [DOI] [PubMed] [Google Scholar]
- Klein W. L.; Krafft G. A.; Finch C. E. (2001) Targeting small Abeta oligomers: the solution to an Alzheimer’s disease conundrum?. Trends Neurosci. 24, 219–224. [DOI] [PubMed] [Google Scholar]
- Klein W. L., De Felice F. G., Lacor P. N., Lambert M. P., and Zhao W. Q. (2008) Why Alzheimer’s is a disease of memory: Synaptic targeting by pathogenic Abeta oligomers (ADDLs). In Synaptic Plasticity and the Mechanism of Alzheimer’s Disease (Selkoe D. J., Triller A., Christen Y., Eds.), Springer-Verlag: Berlin, pp 103–132. [Google Scholar]
- Krafft G. A.; Klein W. L. (2010) ADDLs and the signaling web that leads to Alzheimer’s disease. Neuropharmacology 59, 230–242. [DOI] [PubMed] [Google Scholar]
- Shankar G. M.; Li S.; Mehta T. H.; Garcia-Munoz A.; Shepardson N. E.; Smith I.; Brett F. M.; Farrell M. A.; Rowan M. J.; Lemere C. A.; Regan C. M.; Walsh D. M.; Sabatini B. L.; Selkoe D. J. (2008) Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat. Med. 14, 837–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin M.; Shepardson N.; Yang T.; Chen G.; Walsh D.; Selkoe D. J. (2011) Soluble amyloid beta-protein dimers isolated from Alzheimer cortex directly induce Tau hyperphosphorylation and neuritic degeneration. Proc. Natl. Acad. Sci. U.S.A. 108, 5819–5824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klyubin I.; Betts V.; Welzel A. T.; Blennow K.; Zetterberg H.; Wallin A.; Lemere C. A.; Cullen W. K.; Peng Y.; Wisniewski T.; Selkoe D. J.; Anwyl R.; Walsh D. M.; Rowan M. J. (2008) Amyloid beta protein dimer-containing human CSF disrupts synaptic plasticity: prevention by systemic passive immunization. J. Neurosci. 28, 4231–4237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh D. M.; Klyubin I.; Shankar G. M.; Townsend M.; Fadeeva J. V.; Betts V.; Podlisny M. B.; Cleary J. P.; Ashe K. H.; Rowan M. J.; Selkoe D. J. (2005) The role of cell-derived oligomers of Abeta in Alzheimer’s disease and avenues for therapeutic intervention. Biochem. Soc. Trans. 33, 1087–1090. [DOI] [PubMed] [Google Scholar]
- Lesne S.; Koh M. T.; Kotilinek L.; Kayed R.; Glabe C. G.; Yang A.; Gallagher M.; Ashe K. H. (2006) A specific amyloid-beta protein assembly in the brain impairs memory. Nature 440, 352–357. [DOI] [PubMed] [Google Scholar]
- Peng S.; Garzon D. J.; Marchese M.; Klein W.; Ginsberg S. D.; Francis B. M.; Mount H. T.; Mufson E. J.; Salehi A.; Fahnestock M. (2009) Decreased brain-derived neurotrophic factor depends on amyloid aggregation state in transgenic mouse models of Alzheimer’s disease. J. Neurosci. 29, 9321–9329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noguchi A.; Matsumura S.; Dezawa M.; Tada M.; Yanazawa M.; Ito A.; Akioka M.; Kikuchi S.; Sato M.; Ideno S.; Noda M.; Fukunari A.; Muramatsu S.; Itokazu Y.; Sato K.; Takahashi H.; Teplow D. B.; Nabeshima Y.; Kakita A.; Imahori K.; Hoshi M. (2009) Isolation and characterization of patient-derived, toxic, high mass amyloid beta-protein (Abeta) assembly from Alzheimer disease brains. J. Biol. Chem. 284, 32895–32905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert M. P.; Viola K. L.; Chromy B. A.; Chang L.; Morgan T. E.; Yu J.; Venton D. L.; Krafft G. A.; Finch C. E.; Klein W. L. (2001) Vaccination with soluble Abeta oligomers generates toxicity-neutralizing antibodies. J. Neurochem. 79, 595–605. [DOI] [PubMed] [Google Scholar]
- Chang L.; Bakhos L.; Wang Z.; Venton D. L.; Klein W. L. (2003) Femtomole immunodetection of synthetic and endogenous amyloid-beta oligomers and its application to Alzheimer’s disease drug candidate screening. J. Mol. Neurosci. 20, 305–313. [DOI] [PubMed] [Google Scholar]
- Tjernberg L. O.; Naslund J.; Lindqvist F.; Johansson J.; Karlstrom A. R.; Thyberg J.; Terenius L.; Nordstedt C. (1996) Arrest of beta-amyloid fibril formation by a pentapeptide ligand. J. Biol. Chem. 271, 8545–8548. [DOI] [PubMed] [Google Scholar]
- Findeis M. A.; Musso G. M.; Arico-Muendel C. C.; Benjamin H. W.; Hundal A. M.; Lee J. J.; Chin J.; Kelley M.; Wakefield J.; Hayward N. J.; Molineaux S. M. (1999) Modified-peptide inhibitors of amyloid beta-peptide polymerization. Biochemistry 38, 6791–6800. [DOI] [PubMed] [Google Scholar]
- Evans N. A.; Facci L.; Owen D. E.; Soden P. E.; Burbidge S. A.; Prinjha R. K.; Richardson J. C.; Skaper S. D. (2008) Abeta(1–42) reduces synapse number and inhibits neurite outgrowth in primary cortical and hippocampal neurons: a quantitative analysis. J. Neurosci. Methods 175, 96–103. [DOI] [PubMed] [Google Scholar]
- Ouberai M.; Dumy P.; Chierici S.; Garcia J. (2009) Synthesis and biological evaluation of clicked curcumin and clicked KLVFFA conjugates as inhibitors of beta-amyloid fibril formation. Bioconjugate Chem. 20, 2123–2132. [DOI] [PubMed] [Google Scholar]
- Chalifour R. J.; McLaughlin R. W.; Lavoie L.; Morissette C.; Tremblay N.; Boule M.; Sarazin P.; Stea D.; Lacombe D.; Tremblay P.; Gervais F. (2003) Stereoselective interactions of peptide inhibitors with the beta-amyloid peptide. J. Biol. Chem. 278, 34874–34881. [DOI] [PubMed] [Google Scholar]
- Innocent N.; Evans N.; Hille C.; Wonnacott S. (2010) Oligomerisation differentially affects the acute and chronic actions of amyloid-beta in vitro. Neuropharmacology 59, 343–352. [DOI] [PubMed] [Google Scholar]
- Rapoport M.; Dawson H. N.; Binder L. I.; Vitek M. P.; Ferreira A. (2002) Tau is essential to beta -amyloid-induced neurotoxicity. Proc. Natl. Acad. Sci. U.S.A. 99, 6364–6369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert M. P.; Velasco P. T.; Chang L.; Viola K. L.; Fernandez S.; Lacor P. N.; Khuon D.; Gong Y.; Bigio E. H.; Shaw P.; De Felice F. G.; Krafft G. A.; Klein W. L. (2007) Monoclonal antibodies that target pathological assemblies of Abeta. J. Neurochem. 100, 23–35. [DOI] [PubMed] [Google Scholar]
- Solomon B. (2008) Filamentous bacteriophage as a novel therapeutic tool for Alzheimer’s disease treatment. J. Alzheimer's Dis. 15, 193–198. [DOI] [PubMed] [Google Scholar]
- Fukuchi K.; Accavitti-Loper M. A.; Kim H. D.; Tahara K.; Cao Y.; Lewis T. L.; Caughey R. C.; Kim H.; Lalonde R. (2006) Amelioration of amyloid load by anti-Abeta single-chain antibody in Alzheimer mouse model. Biochem. Biophys. Res. Commun. 344, 79–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levites Y.; Jansen K.; Smithson L. A.; Dakin R.; Holloway V. M.; Das P.; Golde T. E. (2006) Intracranial adeno-associated virus-mediated delivery of anti-pan amyloid beta, amyloid beta40, and amyloid beta42 single-chain variable fragments attenuates plaque pathology in amyloid precursor protein mice. J. Neurosci. 26, 11923–11928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cattepoel S.; Hanenberg M.; Kulic L.; Nitsch R. M. (2011) Chronic intranasal treatment with an anti-Abeta(30–42) scFv antibody ameliorates amyloid pathology in a transgenic mouse model of Alzheimer’s disease. PLoS One 6, e18296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stine W. B. Jr.; Dahlgren K. N.; Krafft G. A.; LaDu M. J. (2003) In vitro characterization of conditions for amyloid-beta peptide oligomerization and fibrillogenesis. J. Biol. Chem. 278, 11612–11622. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.