

Abstract
Group VIB Phospholipase A2 (iPLA2 γ) is distributed in membranous organelles in which β-oxidation occurs, that is, mitochondria and peroxisomes, and is expressed by insulin-secreting pancreatic islet β-cells and INS-1 insulinoma cells, which can be injured by inflammatory cytokines, for example, IL-1β and IFN-γ, and by oxidants, for example, streptozotocin (STZ) or t-butyl-hydroperoxide (TBHP), via processes pertinent to mechanisms of β-cell loss in types 1 and 2 diabetes mellitus. We find that incubating INS-1 cells with IL-1β and IFN-γ, with STZ, or with TBHP causes increased expression of iPLA2 γ mRNA and protein. We prepared INS-1 knockdown (KD) cell lines with reduced iPLA2 γ expression, and they proliferate more slowly than control INS-1 cells and undergo increased membrane peroxidation in response to cytokines or oxidants. Accumulation of oxidized phospholipid molecular species in STZ-treated INS-1 cells was demonstrated by LC/MS/MS scanning, and the levels in iPLA2 γ-KD cells exceeded those in control cells. iPLA2 γ-KD INS-1 cells also exhibited higher levels of apoptosis than control cells when incubated with STZ or with IL-1β and IFN-γ. These findings suggest that iPLA2 γ promotes β-cell proliferation and that its expression is increased during inflammation or oxidative stress as a mechanism to mitigate membrane injury that may enhance β-cell survival.
1. Introduction
Diabetes mellitus (DM) is the most common human endocrine disease and is reaching pandemic proportions in the US and elsewhere [1]. DM represents a constellation of disorders that are grouped into the major categories types 1 and 2 (T1DM and T2DM). T1DM is caused by autoimmune destruction of insulin-secreting pancreatic islet β-cells [2, 3], and inflammatory cytokines released by invading leukocytes during insulitis are believed to participate in these processes [4, 5]. Among them are IL-1β, which impairs insulin secretion and inflicts islet injury [6], and IFN-γ, which greatly potentiates the destructive effects of IL-1β [7]. These effects are mediated in part by induction of nitric oxide (NO) synthase expression and overproduction of NO [8–13], which can induce apoptosis of cells by mechanisms that involve generation of reactive oxygen species that cause oxidative stress [13–15].
T2DM is thought to evolve after a period of initial insulin resistance in which nearly normal glucose tolerance is maintained by compensatory hypersecretion of insulin by β-cells [16, 17]. At some point there is failure to sustain insulin secretion at sufficiently high levels and glucose intolerance and then overt DM ensue [18]. One contributor to the eventual failure of β-cell compensation is a reduction in β-cell mass by 50% or more, and this occurs at least in part by apoptotic β-cell death [16, 19]. Although many mechanisms probably participate in these processes, production of reactive oxygen species induced by metabolic stress has been proposed to represent a final common pathway of injury that ultimately results in β-cell failure [17, 20–29]. Among supporting observations are that β-cells express low levels of antioxidant defense enzymes compared to other tissues [30–33] and that antioxidant compounds confer protection from glucose toxicity to islets in vitro and against development of T2DM in animal models in vivo [34, 35].
Beta cells must sustain a high level of metabolic activity, which provides critical signals in the coupling of nutrient sensing to insulin secretion [36–50], in order to meet the unceasing demand for insulin biosynthesis and processing. Prolonged overstimulation of β-cells may eventually contribute to their failure as a consequence of stresses imposed on the endoplasmic reticulum (ER) and mitochondria [51–53]. Protein synthesis, including that of proinsulin, occurs in ER, and nascent proteins must be properly folded, which involves formation of disulfide bonds. This is an oxidative reaction that requires a prooxidant environment to be maintained in ER. Sustained hyperstimulation can result in ER stress and overproduction of reactive oxygen species (ROS) that exceed ER reductive capacity, resulting in ROS leakage from ER and cellular oxidative stress [54–57].
Mitochondrial metabolism is a major source of ROS production via incomplete reduction of molecular oxygen in the respiratory chain to yield superoxide anion (O2 •−) [58, 59], and O2 •− production increases with metabolic activity [21, 60]. O2 •− is removed by superoxide dismutase (MnSOD)-catalyzed dismutation to H2O2 that can be reduced to H2O by catalase or by glutathione peroxidase and GSH. If generation exceeds removal, excess H2O2 can undergo Fe2+-catalyzed conversion to •HO (Fenton reaction) or to O2 •− and •HO (Haber-Weiss reaction). These ROS can injure mitochondria by mechanisms that include membrane phospholipid peroxidation [14, 61] and activation of stress pathways [27–29]. Mitochondrial phospholipid peroxidation can precipitate cytochrome c release from the inner membrane into the cytosol [14, 62–65], and this can initiate apoptosis [66, 67]. Released cytochrome c interacts with caspase-9 in formation of the apoptosome [68], which leads to activation of the executioner caspases-3, -6, and -7 that dismantle the cell [66, 69].
It has been proposed that phospholipases A2 (PLA2) can prevent or abort apoptosis by repairing peroxidized membrane phospholipids [63–65, 70–76]. PLA2 enzymes catalyze hydrolysis of the sn-2 ester bond of glycerophospholipids to yield a free fatty acid and a 2-lysophospholipid, and at least 16 major groups within the PLA2 superfamily are recognized [77, 78]. Among their proposed functions are the membrane remodeling and the protection or repair of membranes from oxidative damage [63–65, 70–72] in a sequence that involves PLA2-catalyzed removal of oxidized fatty acid residues [73] to yield a lysophospholipid that can be reacylated with an unoxidized fatty acid to preserve membrane integrity [74].
A PLA2 is suited for such a role because oxidized fatty acid substituents usually occur at the sn-2 position of phospholipids where most polyunsaturated fatty acid (PUFA) substituents, such as linoleate (C18:2) and arachidonate (C20:4), are esterified [73, 74]. PUFA are especially susceptible to oxidation because they contain bis-allylic methylene moieties with a labile H atom that can be abstracted to yield a carbon-centered radical that readily reacts with molecular oxygen to form a fatty acid hydroperoxide [73]. Oxidization reduces hydrophobicity of the sn-2 fatty acid substituent and allows it to approach the hydrophilic phospholipid headgroup more closely [73]. This increases separation between head groups, which causes the sn-2 ester bond to be more accessible to PLA2. Liberated peroxy fatty acids can then be reduced to alcohols by glutathione peroxidases after release from phospholipids by PLA2 enzymes [75, 76].
Two members of Group VI PLA2 family have been suggested to play such a role in repairing oxidized mitochondrial membrane phospholipids [63–65, 70–72]. Group VIA PLA2 (iPLA2 β) localizes to mitochondria in insulinoma cells and protects against oxidant-induced apoptosis, and pancreatic islets from iPLA2 β-null mice exhibit increased susceptibility to oxidant-induced apoptosis [63–65]. Oxidant-induced lipid peroxidation and death of renal proximal tubule cells (RPTC) is potentiated by the Group VI PLA2 inhibitor bromoenol lactone (BEL) [71], and R-BEL, which selectively inhibits Group VIB PLA2 (iPLA2 γ), accelerates oxidant-induced lipid peroxidation and renal cortical mitochondrial injury [72]. Moreover, when small hairpin ribonucleic acid (shRNA) adenovirus is used to reduce RPTC iPLA2 γ expression, lipid peroxidation and sensitivity to apoptosis induced by the oxidant tert-butyl hydroperoxide (TBHP) increase [70].
The latter observations [70–72] suggest that iPLA2 γ acts to reduce lipid peroxidation and to protect against oxidant-induced apoptosis in renal proximal tubule cells, and this may reflect iPLA2 γ-catalyzed removal of oxidized PUFA residues from glycerophospholipids that are formed in mitochondria under conditions of oxidative stress. This could permit the resultant lysophospholipid to be reacylated with an unoxidized PUFA residue, which would restore functions that are impaired as a result of membrane oxidation. In the absence of iPLA2 γ or when its activity is impaired, this repair mechanism cannot operate fully, and this could result in progressive mitochondrial injury that eventually triggers the mitochondrial pathway of apoptosis [70–72].
Here we have conducted experiments to determine whether iPLA2 γ might play a similar role in insulin-secreting β-cells because mitochondrial injury induced by oxidative stress appears to be an important mechanism underlying β-cell loss during the development of diabetes mellitus [16–35]. Our studies involved preparation of iPLA2 γ-knockdown INS-1 insulinoma cell lines in which iPLA2 γ expression is reduced by stable expression of shRNA and comparing these lines to control INS-1 cell lines for sensitivity to lipid peroxidation and apoptosis induced by the inflammatory cytokines IL-1β and IFN-γ and the oxidant agents TBHP [70] and streptozotocin (STZ) [79].
2. Materials and Methods
2.1. Materials
Rainbow molecular mass standards, PVDF membranes, and Triton X-100 were obtained from Bio-Rad (Richmond, CA, USA); SuperSignal West Femto Substrate was from Thermo Fisher; Coomassie reagent and SDS-PAGE supplies were from Invitrogen (Carlsbad, CA, USA); bovine serum albumin (BSA, fatty acid free, fraction V) were from MP Biomedicals (Solon, OH, USA); Streptozotocin (STZ) and tert-butyl hydroperoxide (TBHP) were from Sigma (St. Louis, MO, USA).
2.2. Cell Culture
INS-1 rat insulinoma cells that had been stably transfected and mock-transfected INS-1 cells were generated and cultured in RPMI 1640 medium containing 11 mM glucose, 10% fetal calf serum, 10 mM Hepes buffer, 2 mM glutamine, 1 mM sodium pyruvate, 50 mM β-mercaptoethanol, 100 units/mL penicillin, and 100 μg/mL streptomycin, essentially as previously described [80]. The medium was exchanged every 2 days, and the cell cultures were split once a week. Cells were grown to 80% confluence and harvested after treatment as indicated in the figure legends or the text of the Results section. All incubations were performed at 37°C under an atmosphere of 95% air/5% CO2.
2.3. Establishing iPLA2 γ Knockdown INS-1 Insulinoma Cell Lines Using siRNA and a Lentiviral Vector
Two hairpin-forming oligonucleotides directed against iPLA2 γ mRNA were cloned into FIV H1 Lentivector according to instructions from the manufacturer (SBI System Biosciences, Mountain View, CA, USA) by described procedures [80]. Targeting sequences within the synthetic oligonucleotides are italicized and underlined below. The sequence of the first was 5′-GATCCGCAAGAGTGAGTATTGATAACTTAAGAGAGTTATCAATACTCACTCTTGCTTTTTT-G-3′. The second oligonucleotide was 5′-GATCCGGGCCATATTAGCATTCATGCTTCAAGAGAGCATGAATGCTAATATGGCCCTTTTTTG-3′. Constructs that express the shRNAs are designated FIVH1-iPLA2-1 and FIVH1-iPLA2-2. Cells were selected with neomycin.
2.4. Immunoblotting Analyses
Cells were harvested and sonicated, and an aliquot (30 μg) of lysate protein was analyzed by SDS-PAGE (8–12% Tris-Glycine gel, Invitrogen), transferred onto Immobilon-P polyvinylidene difluoride membranes (Bio-Rad, Richmond, CA, USA), and processed for immunoblotting analyses, essentially as previously described [81]. The primary antibody concentration for iPLA2 γ (Sigma, St. Louis, MO, USA) was 1 : 500. The secondary antibody concentration was 1 : 10,000. The concentrations of other antibodies are described in the figure legends. Immunoreactive bands were visualized by enhanced chemiluminescence (ECL).
2.5. Determination of INS-1 Cell Proliferation Rate
INS-1 cell proliferation rates were measured by two approaches, as previously described [80]. One assay is based on fluorescence enhancement when CyQuant GR binds to nucleic acids, which reflects the amount of cell DNA [82]. Cells were seeded onto 96-well plates (3 × 103 cells/well). Medium was removed after 1 or 3 days, and cells were frozen (−20°C). DNA was measured with a CyQuant assay kit (Molecular Probes, Inc., Eugene, OR, USA) with reference to a standard curve. CyQuant GR solution (200 μL) was added to each well and incubated (5 min, room temperature). Fluorescence was measured on a microplate fluorimeter (excitation, 480 nm; emission, 538 nm). A second assay is based on incorporation of thymidine analog 5-bromo-2′-deoxyurindine (BrdU) into DNA in proliferating cells [83]. Cells were seeded (104 cells/well) and cultured (3 days) before assay with an enzyme-linked immunoassay detection kit III (Roche Applied Science) after BrdU labeling.
2.6. Lipid Peroxidation
Lipid peroxidation was quantitated using a Cayman TBARS assay kit (Cayman Chemical, Ann Arbor, MI, USA) according to the manufacturer's instructions, as previously described [64, 84]. Lipid peroxides derived from polyunsaturated fatty acids decompose to form a complex series of compounds that include reactive carbonyl species, such as MDA. Measurement of thiobarbituric acid reactive substances (TBARS) by determining absorbance at 530 nm is used to assess the extent of lipid peroxidation [84]. Results are expressed as μmol/μg protein.
2.7. HPLC-ESI-MS/MS Analysis of Oxidized Lipids
Lipids extracted from INS-1 cells were stored in sealed vials (under N2 at −20°C) to suppress artifactual oxidation, and extracts were then analyzed by LC/MS/MS in a manner similar to that previously described [85] on a Surveyor HPLC (ThermoElectron, San Jose, CA, USA) using a modified gradient [86] on a C8 column (15 cm × 2.1 mm, Sigma Chemical Co., St. Louis, MO, USA) interfaced with the ion source of a ThermoElectron Vantage triple quadruple mass spectrometer with extended mass range operated in negative ion mode. Tandem MS scans for precursors of m/z 295, m/z 319, and m/z 343 were performed to identify glycerolipid molecular species that contained singly oxygenated forms of the polyunsaturated fatty acids (PUFA) linoleate (C18:2), arachidonate (C20:4), or docosahexaenoate (C22:6), respectively. The major oxylipid species identified was (1-stearoyl, 2-hydroxyeicosatetraenoyl)-sn-glycerophospho-ethanolamine [(C18:0/HETE)-GPE], and it was quantified by MRM of 782.76 → 319.3, which is a transition that corresponds to production of the HETE carboxylate anion from the [M-H]− ion of the parent oxy-phospholipid species.
2.8. Assessment of Apoptosis by Flow Cytometry
INS-1 cell apoptosis was determined using an Annexin-VFLUOS Staining Kit (Roche Applied Science, Indianapolis, IN, USA) according to the manufacturer's instructions, essentially as previously described [64, 87]. Briefly, harvested cells were washed with PBS and resuspended in Annexin-VFLUOS labeling solution (100 μL). After incubation (10–15 min, 15–25°C), cells were transferred to fluorescence-activated cell sorting (FACS) tubes and diluted 1 : 5 with buffer provided in the kit. Fluorescence in cells was analyzed with a FACscan flow cytometer (BD Biosciences, Sparks, MD, USA) at an excitation wavelength of 488 nm, and data were processed with WinMDI 2.9 software.
2.9. Statistical Analyses
Results are expressed as mean ± SEM. Data were evaluated by unpaired, two-tailed Student's t-test for differences between two conditions or by analysis of variance with appropriate posthoc tests for larger sets, as previously described [80, 81, 87]. Significance levels are described in the figure legends, and a P value <0.05 was considered to reflect a significant difference.
3. Results
3.1. INS-1 Cell iPLA2 γ Expression and the Influence of Inflammatory Cytokines and Oxidative Agents
INS-1 insulinoma cells were found to express iPLA2 γ mRNA and iPLA2 γ-immunoreactive protein by quantitative PCR and by Western blotting, respectively (Figure 1), and also to exhibit iPLA2 activity (not shown). Incubation with the inflammatory cytokines IL-1β and IFN-γ resulted in increased INS-1 cell expression of iPLA2 γ mRNA in a concentration-dependent manner (Figure 1(a)), and expression of iPLA2 γ immunoreactive protein exhibited a similar pattern (Figure 1(b)).
Figure 1.

Influence of the inflammatory cytokines interleukin-1β (IL-1β) and interferon-γ (IFN-γ) on iPLA 2 γ expression by INS-1 cells. Control INS-1 cells were incubated with vehicle alone or with various concentrations of IL-1β and IFN-γ for 16 hr, and iPLA2 γ mRNA levels were then determined by quantitative PCR (panel (a)) and iPLA2 γ protein levels by Western blotting (panel (b)), as described in Experimental Procedures. In panel (c), control INS-1 cells were incubated with IL-1β (5 ng/mL) and IFN-γ (80 ng/mL) for various intervals (0, 8, 16, 24, and 48 hr), at the end of which iPLA2 γ mRNA levels were determined by quantitative PCR. In panels (a) and (c), mean values ± SEM (n = 4) are displayed, and an asterisk (*) indicates a significant difference (P < 0.05) from the condition in which the concentration (panel (a)) or time (panel (b)) parameter value was zero. The immunoblot in panel (b) is representative of four experiments.
The major iPLA2 γ-immunoreactive band (Figure 1(b)) migrated with an apparent MW of about 74 kDa on SDS-PAGE and Western Blotting analyses of INS-1 cell lysates, but other bands of variable intensity were observed with apparent MW between 40 and 88 kDa (not shown), as previously reported [88, 89]. Although full-length iPLA2 γ cDNA encodes an 88 kDa protein, transcriptional and translational regulatory mechanisms result in production of multiple gene products of various sizes, and the major species often migrate with apparent MW of 74–77 kDa [88, 89].
The increased expression of iPLA2 γ mRNA induced by IL-1β and IFN-γ was dependent on the incubation time (Figure 1(c)). Incubation of INS-1 cells with the oxidative agents streptozotocin (STZ) [79] (Figure 2(a)) or t-butyl-hydroperoxide (TBHP) [70] (Figure 2(b)) also resulted in a concentration-dependent increase in expression of iPLA2 γ mRNA (Figure 2) and protein (not shown).
Figure 2.
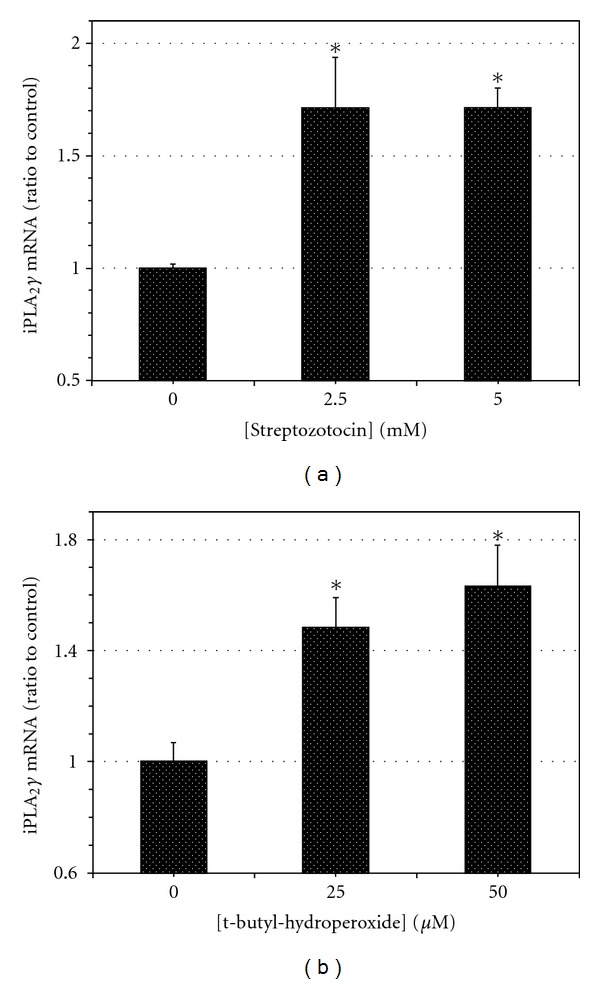
Influence of the oxidant agents streptozotocin (STZ) and tert-butylhydroperoxide (TBHP) on iPLA 2 γ expression by INS-1 cells. Control INS-1 cells were incubated with various concentrations of STZ (panel (a)) or TBHP (panel (b)) for 16 hr, and iPLA2 γ mRNA levels were then determined by quantitative PCR, as in Figure 1(a). Mean values ± SEM (n = 4) are displayed, and an asterisk (*) indicates a significant difference (P < 0.05) from the condition in which the concentration of the oxidant agent was zero.
These findings suggested the possibility that upregulation of iPLA2 γ expression might represent a compensatory response to injurious agents in order to enhance β-cell survival in the settings of inflammation or oxidative stress. To examine this possibility, effects of suppressing INS-1 cell iPLA2 γ expression were examined.
3.2. Establishing iPLA2 γ-Knockdown INS-1 Cell Lines
INS-1 cells were infected with FIV constructs containing inserts that produced either scrambled RNA (control) or shRNA directed against sequences in iPLA2 γ mRNA. Selection of neomycin-resistant cells resulted in isolation of two clones that had stably incorporated knockdown constructs and expressed less than 20% of the control cell iPLA2 γ mRNA content when analyzed by real-time PCR (Figure 3(a)) or Northern blots (not shown) and reduced amounts of iPLA2 γ immunoreactive protein on Western blots (Figure 3(b)). The iPLA2 γ-knockdown (iPLA2 γ-KD) cell lines also exhibited a reduction in iPLA2 activity (not shown) that was comparable in magnitude to the reduction in mRNA levels (Figure 3(a)). The level of iPLA2 γ expression was a stable property of control and iPLA2 γ-KD INS-1 cell lines that persisted on serial passage in culture.
Figure 3.

Establishing iPLA 2 γ-knockdown INS-1 cell lines. INS-1 cells were transfected with FIV constructs containing inserts that produced either scrambled RNA (control) or siRNA directed against sequences in iPLA2 γ mRNA (KD1, KD2). The relative iPLA2 γ expression levels in control INS-1 cells and in the iPLA2 γ-knockdown (KD) cell lines KD1 and KD2 were assessed by quantitative PCR for mRNA (panel (a)) and by Western blotting analysis for iPLA2 γ-immunoreactive protein (panel (b)). In panel (a), mean values ± SEM are displayed (n = 4). An asterisk (*) denotes a significant difference (P < 0.05) from the value for control INS-1 cells. The immunoblot in panel (b) is representative of four experiments.
3.3. INS-1 Cell Line Proliferation Rates
Cell proliferation was measured using an indicator that exhibits strong fluorescence enhancement upon association with nucleic acids [82]. Identical numbers of cells of each INS-1 cell line were seeded at time 0, and their growth rates were monitored for 72 hr. INS-1 iPLA2 γ-KD lines proliferated at rates that were significantly lower than those for control INS-1 cells (Figure 4). Similar results were obtained when proliferation was measured by BrdU incorporation into DNA [83] and when seeding was performed at different initial cell densities (not shown).
Figure 4.
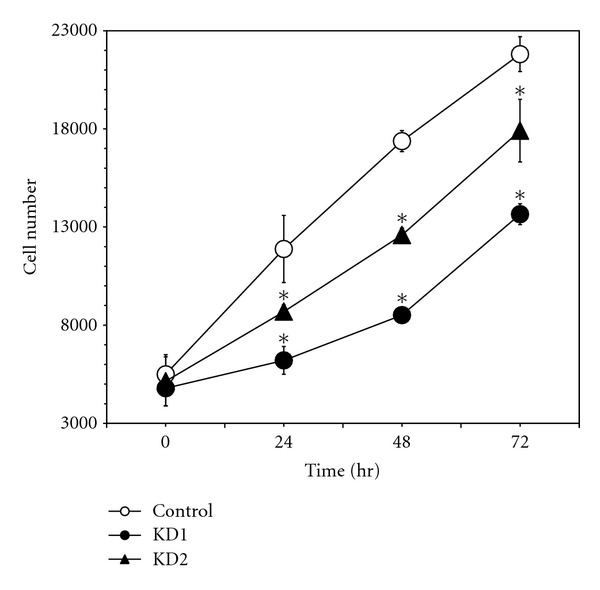
Proliferation rates of INS-1 cell lines. Control INS-1 cells (open circles) or the iPLA2 γ-Knockdown cell lines KD1 (closed circles) or KD2 (closed triangles) were seeded onto the wells of microtiter plates (density 3 × 103 cells/well) and incubated for various intervals (24, 48, or 72 hr), at the end of which cell number was estimated based on DNA content by using a fluorescent indicator as described in Experimental Procedures. Mean values ± SEM are displayed (n = 4). An asterisk (*) denotes a significant difference (P < 0.05) from the value for the control INS-1 cell line at the indicated time point.
3.4. Lipid Peroxidation in INS-1 Cell Lines
Lipid peroxidation was monitored by measuring TBARS [64, 84] in INS-1 cell lines incubated with IL-1β and IFN-γ or with the oxidant agents STZ or TBHP under conditions similar to those in Figures 1 and 2. Relative to cells incubated only with vehicle, incubation with the cytokine mixture induced a significant increase in lipid peroxidation in both control INS-1 cells (1.60 ± 0.03-fold) and in iPLA2 γ-KD cells (2.71 ± 0.47-fold), and the level achieved in the latter (0.92 ± 0.16 pmol/μg protein) significantly exceeded that in the former (0.54 ± 0.01 pmol/μg) (Figure 5). A similar pattern was observed upon incubation with STZ, which induced a significant rise in lipid peroxidation in control INS-1 cells (1.44 ± 0.04-fold) and in iPLA2 γ-KD cells (2.12 ± 0.09-fold), and the level achieved in the latter (0.73 ± 0.07 pmol/μg) significantly exceeded that in the former (0.47 ± 0.04 pmol/μg) (Figure 5). Incubation with TBHP also induced a rise in the lipid peroxide content of both control INS-1 cells (2.20 ± 0.09-fold) and in iPLA2 γ-KD cells (2.56 ± 0.15-fold), and there was a nonsignificant trend for the level achieved in the latter (0.86 ± 0.05 pmol/μg) to exceed that in the former (0.74 ± 0.03 pmol/μg).
Figure 5.

Influence of inflammatory cytokines and oxidant agents on lipid peroxidation of INS-1 Cell Lines. Control INS-1 cells (light bars) or iPLA2 γ-Knockdown INS-1 cells (dark bars) were incubated with vehicle only or with the combination of IL-1β (5 ng/mL) and IFN-γ (80 ng/mL) or with STZ (5 mM) or TBHP (50 μM) for 16 hr, and lipid peroxidation was then determined by the TBARS assay, as described in Experimental Procedures. Mean values ± SEM (n = 4) are displayed, and an asterisk (*) indicates a significant difference (P < 0.05) from control INS-1 cells. An (X) indicates a significant difference (P < 0.05) from the vehicle-treated condition.
3.5. HPLC-ESI-MS/MS Analysis of Oxidized Lipid Molecular Species That Accumulate in INS-1 Cells Incubated with Streptozotocin
To examine oxidized lipid molecular species in INS-1 cells, LC/ESI/tandem mass spectrometric scanning was used to detect parent ions that liberate an oxidized polyunsaturated fatty acid carboxylate anion (Figure 6(a)) upon collisionally activated dissociation (CAD) [85]. Hydroxyeicosatetraenoate (HETE) (m/z 319.3) arising from the oxidized analog of the glycerophosphoethanolamine (GPE) species 18 : 0/20 : 4-GPE (oxy-analog m/z 782.76) was found to represent the most abundant of the oxidized lipid species in INS-1 cells (Figure 6(b)), which is consistent with the facts that this is also the most abundant oxidized GPE lipid in activated platelets [85] and that 18 : 0/20 : 4-GPE is the most abundant GPE lipid in INS-1 cells [90] and rat islets [91]. Figure 6(b) displays an MS/MS scan for parent [M-H]− ion precursors over the range m/z 400 to m/z 2000 that yield the HETE [M-H]− (m/z 319.3) upon collisionally activated dissociation, and m/z 782.76 is the predominant parent ion observed. Figure 6(c) is an expansion of that mass spectrum over the range m/z 782.5 to 785.0 that illustrates the [13C] isotopomer distribution of the [M-H]− ion.
Figure 6.


HPLC-ESI-MS/MS analysis of oxidized lipid molecular species that accumulate in INS-1 cells incubated with streptozotocin. Control INS-1 cells (light bars) or iPLA2 γ-knockdown INS-1 cells (dark bars) were incubated with vehicle only or with STZ (5 mM) for 16 hr as in Figure 5, and lipids were then extracted and analyzed by HPLC-LC-MS/MS as described in Experimental Procedures. Panel (a) illustrates the MS/MS transition monitored, which is the production of the hydroxyeicosatetraenoate (HETE) [M-H]− ion (m/z 319.3) from the parent (C18:0/C20:4)-GPE [M-H]− ion (m/z 782.76) upon collisionally activated dissociation. Prior survey scans monitoring parents of oxidized linoleate (C18:2) and docosahexaenoate (C22:6) species had revealed that HETE (HO-C20:4) was the dominant oxidized fatty acid residue esterified in INS-1 cell phospholipids. Panel (b) is an MS/MS scan over the m/z range 400–2000 in which parent ions that generate the HETE anion (m/z 319.3) are monitored, and m/z 782.76 is the vastly predominant parent, which was found to represent the (C18:0/C20:4)-GPE [M-H]− ion upon analysis of the complete MS/MS spectrum/ Panel (c) is an expansion of that mass spectrum over the m/z range 782.5 to 785.0 to illustrate the [13C] isotopomer distribution of the [M-H]− ion. In panels (d) and (e) represent HPLC/ESI/MS/MS scans in which the transition m/z 782.76 to m/z 319.3 is monitored as a function of LC retention time to quantitate the (C18:0/C20:4)-GPE content of control INS-1 cells (panels (d) and (e)) or iPLA2 γ-knockdown (KD) INS-1 cells (panels (f) and (g)) incubated without (panels (d) and (f)) or with STZ (panels (e) and (g)). Panel (h) represents a summary of four such experiments, and mean values are displayed and SEM indicated. An asterisk (*) denotes a significant (P < 0.05) difference between control and iPLA2 γ-KD INS-1 cell lines, and an X denotes a significant difference between cells incubated with or without STZ.
The content of the oxylipid species (C18:0/HETE)-GPE in INS-1 cells was quantified by LC/ESI/MS/MS MRM scanning of the transition 782.76 → 319.5 (Figures 6(d)–6(g)), and incubation with STZ was found to induce an increase in the (C18:0/HETE)-GPE content of both control INS-1 cells transfected with vector only (Figures 6(d) and 6(e)) and in iPLA2 γ-knockdown INS-1 cells (Figures 7(f) and 7(g)). Both basal levels of (C18:0/HETE)-GPE and those achieved after incubation with STZ for the iPLA2 γ-knockdown INS-1 cells exceeded those for control INS-1 cells (Figure 7(h)), and this is consistent with the proposal that iPLA2 β acts to excise oxidized PUFA residues from phospholipids so that the resultant lysophospholipid can be reacylated with an unoxidized fatty acid substituent to restore the structure and function of the parent phospholipid [63, 64].
Figure 7.
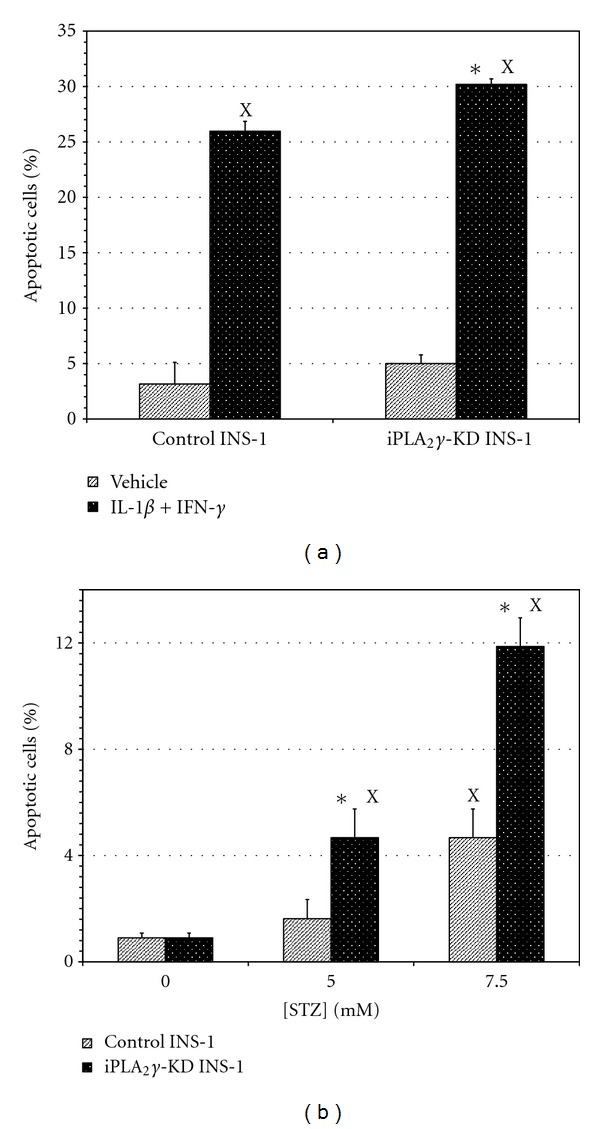
Influence of inflammatory cytokines and streptozotocin (STZ) on apoptosis of INS-1 cell lines. In panel (a), control INS-1 cells or iPLA2 γ-knockdown INS-1 cells (iPLA2-KD) were incubated with vehicle only (light bars) or with IL-1β (5 ng/mL) and IFN-γ (80 ng/mL) (dark bars) for 16 hr, and the percentages of apoptotic cells were then determined by FACS as described in Experimental Procedures. In panel (b), control INS-1 cells (light bars) or iPLA2 γ-Knockdown INS-1 cells (dark bars) were incubated with vehicle alone or with varied concentrations of STZ (5 mM or 7.5 mM) for 16 hr, and the percentages of apoptotic cells were then determined as in panel (a). Mean values ± SEM (n = 4) are displayed. An asterisk (*) indicates a significant difference (P < 0.05) between control cells and iPLA2 γ-KD cells. An (X) indicates a significant difference (P < 0.05) from the vehicle-treated condition.
Oxidized cardiolipin species were not observed directly due to the relatively low abundance of the parent lipid among all cellular lipids and the tendency of linoleate residues, which are the principal fatty acid substituents of cardiolipin, upon oxidization to undergo chain scission reactions that yield a variety of truncated sn-2 substituents rather than a single-predominant species [92]. Similar behavior has been observed for other polyunsaturated fatty acids [93]. Mitochondria contain substantial amounts of GPE lipids, however, and they undergo the largest fractional modification of measured mitochondrial lipid classes upon induction of apoptosis [94], which suggests that GPE lipid oxidation represents a surrogate marker for mitochondrial phospholipid oxidation.
3.6. Apoptosis of INS-1 Cell Lines
Apoptosis was monitored by determination of Annexin V binding by FACS [64, 87] with INS-1 cell lines incubated with IL-1β and IFN-γ under conditions similar to those in Figure 1. The inflammatory cytokine mixture induced a robust increase in apoptosis of both control INS-1 cells and iPLA2 γ-KD cells, and the percentage of apoptotic cells for the cytokine-treated condition was significantly higher for the latter (30.2 ± 0.5%) than for the former (25.7 ± 0.9%) (Figure 7(a)). Incubation of the INS-1 cell lines with the oxidant agent STZ also induced a concentration-dependent increase in the percentage of apoptotic cells that was significantly higher for iPLA2 γ-KD cells (12.1 ± 0.9) than for control INS-1 cells (4.5 ± 0.9) at the highest STZ concentration (7.5 mM) tested (Figure 7(b)).
4. Discussion
Loss of insulin-secreting β-cells occurs in both type I and type II diabetes mellitus (T1DM and T2DM), and apoptosis is thought to be the major mechanism by which β-cell death occurs [16, 19]. Lipid oxidation plays an important role in initiating apoptosis [17, 20–29], and it has been suggested that generation of reactive oxygen species results in cardiolipinperoxidation in mitochondrial membranes, which destabilizes thelipidbilayer and potentiates membrane permeabilization, cytochrome c release, and apoptosis [61–67]. Understanding the mechanisms that the β-cell uses to protect its mitochondrial membranes from oxidative injury could yield insight into the pathogenesis of β-cell loss and development of means to treat or prevent T1DM and T2DM.
Phospholipases A2 (PLA2) hydrolyze glycerophospholipids to yield a free fatty acid and a 2-lysophospholipid [77, 78], and PLA2 are thought to participate in signaling and membrane-remodeling processes that include repairing of oxidative damage to membranes in order to preserve their functional integrity [70–74]. When lipid peroxidation occurs, the oxidized sn-2 fatty acid substituent of phospholipids becomes less hydrophobic and more accessible to phospholipases [73]. The lysophospholipid that results from PLA2-catalyzed removal of oxidized fatty acid substituents can be reacylated with an unoxidized fatty acid to restore the native structure and function of the parent phospholipid.
Two members of a lipase family [95] that has been designated Group VI PLA2 [96] or patatin-like phospholipase domain-containing (PNPLA) proteins [97] may play such a role in remodeling mitochondrial cardiolipin, and neither enzyme requires Ca2+ for catalytic activity. Group VIA PLA2 (iPLA2 β) was the first member of this family to be recognized [98, 99] and is also designated PNPLA9. Group VIB PLA2 (iPLA2 γ) was recognized thereafter [88, 100] and is also designated PNPLA8 [96, 97]. iPLA2 γ, is expressed in mitochondria and peroxisomes [89, 101, 102], which are both membranous organelles that produce reactive oxygen species, and iPLA2 γ cooperates with iPLA2 β in stimulated phospholipid hydrolysis in some circumstances [103].
Mitochondria also contain iPLA2 β, and observations in a Drosophila model of the human Barth syndrome have raised interest in the possibility that iPLA2 β participates in cardiolipin remodeling [104]. Barth syndrome results from mutations in the tafazzin gene, which encodes a mitochondrial phospholipid-lysophospholipid transacylase, and the disorder is characterized by severe cardioskeletal myopathy, low-cardiolipin content, and abnormal cardiolipin fatty acyl composition [105]. Tafazzin-deficient Drosophila have similar abnormalities in cardiolipin content and mitochondrial function associated with monolysocardiolipin accumulation, and this phenotype is suppressed by inactivation of the iPLA 2 β gene, suggesting that iPLA 2 β contributes to monolysocardiolipin formation [104].
Several observations indicate that iPLA2 γ is also involved in cardiolipin remodeling. Selective overexpression of iPLA2 γ in mouse myocardium results in altered mitochondrial function associated with cardiac dysfunction [106, 107], and genetic ablation of iPLA2 γ produces a deficient mitochondrial bioenergetic phenotype [108] associated with cognitive dysfunction and hippocampal abnormalities that include mitochondrial degeneration and alterations in cardiolipin content and molecular species distribution [109]. iPLA2 γ-null mice also exhibit exaggerated high-fat diet-induced changes in tissue cardiolipin content and composition and altered patterns of mitochondrial fatty acid oxidation [110, 111]. Cardiolipin remodeling in myocardial mitochondria that occurs during heart failure in rats also appears to involve iPLA2 γ [112].
iPLA2 β and iPLA2 γ cooperate in effecting certain cell fate decisions [113], and both enzymes may participate in determining whether a cell survives or succumbs to oxidative injury via their roles in cardiolipin metabolism. In β-cells, stimuli that induce apoptosis cause iPLA2 β to redistribute from cytosol to mitochondria [63–65, 92, 114–119]. Staurosporine, for example, stimulates INS-1 cell mitochondrial superoxide production, and this results in mitochondrial membrane peroxidation, cytochrome c release, and apoptosis [63–65]. Staurosporine-induced membrane peroxidation and apoptosis in β-cells are attenuated by overexpressing iPLA2 β and amplified by its pharmacologic inhibition or genetic ablation [63–65]. This may reflect a role for iPLA2 β to excise oxidized cardiolipin fatty acid residues to generate monolysocardiolipin species that can be reacylated to restore the native structure [63–65].
Pharmacologic [120] observations suggest that iPLA2 γ may play a similar role in cardiolipin remodeling [112], and this is consistent with the abnormalities in cardiolipin content and composition that result from genetic manipulation of iPLA2 γ expression [108–110]. In renal proximal tubular cells, pharmacologic inhibition or molecular biologic suppression of expression of iPLA2 γ increases susceptibility to oxidant-induced lipid peroxidation, mitochondrial dysfunction, and cell death [70–72]. These observations prompted us to determine whether iPLA2 γ might play a similar role in β-cells, given the importance of β-cell loss via oxidative injury in the development of diabetes mellitus [20–35].
Our findings indicate that iPLA2 γ could participate in maintaining the aggregate mass of β-cells by promoting their proliferation and by protecting them from oxidative membrane injury induced by inflammatory cytokines or by oxidant agents that leads to apoptosis. Our iPLA2 γ-knockdown (KD) INS-1 cell lines exhibited significantly lower growth rates than control INS-1 cells did. The inflammatory cytokines IL-1β and IFN-γ increased INS-1 cell iPLA2 γ expression, and a similar response occurred when INS-1 cells were incubated with the oxidant agents STZ and TBHP. Those findings suggest that iPLA2 γ may be upregulated as a compensatory repair mechanism in response to agents that injure β-cells, and this is consistent with the observations that iPLA2 γ-KD INS-1 cells were also more sensitive than control cells to injury from inflammatory cytokines and oxidative agents. These findings in β-cell lines are consistent with the increased sensitivity to oxidant-induced lipid peroxidation and apoptosis of renal proximal tubular cells with reduced iPLA2 γ expression [70] and suggest that iPLA2 γ plays a role in repairing oxidized membranes and mitigating oxidant-induced cellular injury.
The mechanisms by which cytokines and oxidant agents increase the expression of iPLA2 γ have not yet been determined experimentally, but the accumulation of iPLA2 γ mRNA suggests that increased transcription is involved. Current experiments to examine potential mechanisms are focused on three possibilities. One is that the redox sensitive transcription factor NFκB is activated via ROS-mediated inactivation of its inhibitory subunit IκB [121–124] and that NFκB stimulates transcription of the iPLA2 γ gene directly or indirectly. NFκB activation is known to contribute to β-cell injury induced by cytokines [125] under conditions similar to those employed in the studies described here. Two is that transcriptional activation of the iPLA2 γ gene might occur via p38 MAPK-dependent pathways, since stimuli that induce β-cell ER stress and apoptosis result in p38 MAPK activation [87], and ROS-induced p38 MAPK activation contributes to apoptosis in other cells [126, 127].
Three is that ROS-induced oxidation of cellular phospholipids yields agonistic ligands for the transcription factor PPARγ, as previously reported [128], which then activates transcription of the iPLA2 γ gene. It is of interest in this regard that conditions that result in differentiation of 3T3L1 fibroblasts to adipocytes lead to increased expression of PPARγ and in transcriptional upregulation of iPLA2 γ and iPLA2 β and that siRNA directed against either enzyme blocks differentiation [113].
The presence of oxidized phospholipids in INS-1 cells treated with oxidant agents in the studies described here was determined by performing LC/MS/MS scans of lipid extracts for precursors of m/z 295, m/z 319, and m/z 343 in order to identify glycerolipid molecular species that contained singly oxygenated forms of the polyunsaturated fatty acids (PUFA) linoleate (C18:2), arachidonate (C20:4), or docosahexaenoate (C22:6), respectively. The major oxylipid species identified was (stearoyl, hydroxyeicosatetraenoyl)-glycerophosphoethanol-amine [(C18:0/HETE)-GPE], and it was quantified by MRM of the transition 782.6 → 319.3, which corresponds to production of the HETE carboxylate anion from the [M-H]− ion of the parent oxy-phospholipid species. Minor species were observed at other m/z values but were not further characterized because of the low signal obtained from the limited amount of lipid contained in the quantities of INS-1 cells with which it was practical to work.
Although C18:0/HETE-PE is the most abundant oxidized phospholipid observed here, it is probably not the only oxidized species formed under these conditions. Oxidized lipids represent only a tiny fraction (substantially below 1%) of their unoxidized precursors, and all but the most abundant species will likely fall below the limit of quantitation, even if present in the mixtures, when the amount of membrane lipid available for analysis is limiting. In addition, other phospholipid oxidation products, for example, those that contain esterified hydroperoxy- or ketofatty acid derivatives [129], would not have been detected by the approach used here, which would also have failed to detect esterified short chain substituents arising from PUFA oxidation [130]. Moreover, neither the regio- nor the stereospecificity of oxygenation was determined in our studies because of the limited amount of oxidized lipid available for characterization, and it is possible that C18:0/HETE-PE consisted of several distinct isomers, as reported for other cells [85].
Nonetheless, we think that C18:0/HETE-PE represents a reasonable marker for phospholipid oxidation in our experiments for several reasons. First, oxidized species of PE are much more abundant in stimulated monocytes and platelets than are oxidized species of other phospholipid head group classes, including PC, PI, PS, or PG [131], and C18:0/HETE-PE is the most abundant oxidized diacyl-phospholipid under those conditions [85, 131, 132]. Second, the precursor C18:0/C20:4-PE is the most abundant PE species in INS-1 cells [90, 91]. Moreover, mitochondria contain substantial amounts of PE lipids, and they undergo the largest fractional modification of measured mitochondrial lipid classes upon induction of apoptosis [94], which suggests that PE lipid oxidation serves as a surrogate marker for mitochondrial phospholipid oxidation.
The LC/MS/MS measurements reported here indicate that INS-1 cell C18:0/HETE-PE content rises upon incubation with an oxidant agent and is higher in cells in which iPLA2 γ expression level has been knocked down compared to control cells. This is compatible with a role for iPLA2 γ in remodeling of oxidized phospholipids that involves excision of oxidized PUFA residues to yield lysophospholipid species that can be reacylated with unoxidized fatty acyl-CoA molecules. This would regenerate the native phospholipid structure and restore its normal function, thereby mitigating the effects of oxidative insults that might otherwise induce apoptosis.
5. Conclusions
Group VIB Phospholipase A2 (iPLA2 γ) is distributed in mitochondria and expressed by insulin-secreting pancreatic islet β-cells and INS-1 insulinoma cells that are susceptible to oxidative injury by inflammatory cytokines, for example, IL-1β and IFN-γ, and by oxidizing toxins, for example, streptozotocin (STZ) or t-butyl-hydroperoxide (TBHP), via processes relevant to β-cell loss in types 1 and 2 diabetes mellitus. We demonstrate here that INS-1 cells incubated with IL-1β and IFN-γ, with STZ, or with TBHP increase their expression of iPLA2 γ mRNA and protein and that INS-1 knockdown (KD) cell lines with reduced iPLA2 γ expression proliferate more slowly than control INS-1 cells and undergo increased membrane peroxidation when incubated with cytokines or oxidants. Accumulation of the oxidized phospholipid species (1-stearoyl, 2-hydroxyeicosatetraenoyl)-sn-glycerophosphocholine was demonstrated in STZ-treated INS-1 cells by LC/MS/MS scanning, and the levels in iPLA2 γ-KD cells exceeded those in control cells. iPLA2 γ-KD INS-1 cells also exhibited higher levels of apoptosis than control cells when incubated with STZ or with IL-1β and IFN-γ. Together, these observations suggest that iPLA2 γ promotes β-cell proliferation and that its increased expression during inflammation or oxidative stress may serve to mitigate membrane injury and thereby to enhance β-cell survival under these conditions.
Acknowledgments
This work was supported by United States Public Health Service Grants R37-DK34388, P41-RR00954, P60-DK20579, and P30-DK56341, and S.Bao is supported by Training Grant 5 T32 DK007120. The authors thank Alan Bohrer for technical assistance and Robert Sanders for assistance with preparation of the paper and the figures.
Abbreviations
- BEL:
Bromoenol lactone
- CAD:
Collisionally activated dissociation
- C18:2:
Linoleate
- C20:4:
Arachidonate
- C22:6:
Docosahexaenoate
- DM:
Diabetes mellitus
- ER:
Endoplasmic reticulum
- ESI:
Electrospray ionization
- FACS:
Fluorescence-activated cell sorting
- GSH:
Glutathione
- HETE:
Hydroxyeicosatetrenoate
- IFN-γ:
Interferon-γ
- IL-1β:
Interleukin 1-β
- iPLA2β:
Group VIA PLA2
- iPLA2γ:
Group VIB Phospholipase A2
- LC:
Liquid chromatography
- KD:
Knockdown
- MDA:
Malondialdehyde
- MRM:
Multiple reaction monitoring
- MS:
Mass spectrometry
- MS/MS:
Tandem mass spectrometry
- PC:
Phosphatidylcholine
- PE:
Phosphatidylethanolamine
- PG:
Phosphatidylglycerol
- PI:
Phosphatidylinositol
- PS:
Phosphatidylserine
- PUFA:
Polyunsaturated fatty acid
- ROS:
Reactive oxygen species
- RPTC:
Renal proximal tubule cells
- SEM:
Standard error of the mean
- shRNA:
Small hairpin ribonucleic acid
- SOD:
Superoxide dismutase
- STZ:
Streptozotocin
- TBARS:
Thiobarbituric acid reactive substances
- TBHP:
t-butyl-hydroperoxide.
References
- 1.Leahy JL. Pathogenesis of type 2 diabetes mellitus. Archives of Medical Research. 2005;36(3):197–209. doi: 10.1016/j.arcmed.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 2.Mathis D, Vence L, Benoist C. β-cell death during progression to diabetes. Nature. 2001;414(6865):792–798. doi: 10.1038/414792a. [DOI] [PubMed] [Google Scholar]
- 3.Gepts W. Pathologic anatomy of the pancreas in juvenile diabetes mellitus. Diabetes. 1965;14(10):619–633. doi: 10.2337/diab.14.10.619. [DOI] [PubMed] [Google Scholar]
- 4.Mandrup-Poulsen T. The role of interleukin-1 in the pathogenesis of IDDM. Diabetologia. 1996;39(9):1005–1029. doi: 10.1007/BF00400649. [DOI] [PubMed] [Google Scholar]
- 5.Rabinovitch A, Suarez-Pinzon WL. Cytokines and their roles in pancreatic islet β-cell destruction and insulin-dependent diabetes mellitus. Biochemical Pharmacology. 1998;55(8):1139–1149. doi: 10.1016/s0006-2952(97)00492-9. [DOI] [PubMed] [Google Scholar]
- 6.Corbett JA, Lancaster JR, Sweetland MA, McDaniel ML. Interleukin-1β-induced formation of EPR-detectable iron-nitrosyl complexes in Islets of Langerhans. Role of nitric oxide in interleukin-1β-induced inhibition of insulin secretion. Journal of Biological Chemistry. 1991;266(32):21351–21354. [PubMed] [Google Scholar]
- 7.Thomas HE, Darwiche R, Corbett JA, Kay TWH. Interleukin-1 plus γ-interferon-induced pancreatic β-cell dysfunction is mediated by β-cell nitric oxide production. Diabetes. 2002;51(2):311–316. doi: 10.2337/diabetes.51.2.311. [DOI] [PubMed] [Google Scholar]
- 8.Corbett JA, Kwon G, Misko TP, Rodi CP, McDaniel ML. Tyrosine kinase involvement in IL-1beta-induced expression of iNOS by beta-cells purified from islets of Langerhans. American Journal of Physiology. 1994;267(1):C48–C54. doi: 10.1152/ajpcell.1994.267.1.C48. [DOI] [PubMed] [Google Scholar]
- 9.Corbett JA, McDaniel ML. Intraislet release of interleukin 1 inhibits β cell function by inducing β cell expression of inducible nitric oxide synthase. Journal of Experimental Medicine. 1995;181(2):559–568. doi: 10.1084/jem.181.2.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Corbett JA, J. L. Wang, Sweetland MA, Lancaster JR, McDaniel ML. Interleukin 1β induces the formation of nitric oxide by β-cells purified from rodent islets of Langerhans. Evidence for the β-cell as a source and site of action of nitric oxide. Journal of Clinical Investigation. 1992;90(6):2384–2391. doi: 10.1172/JCI116129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eizirik DL, Bjorklund A, Welsh N. Interleukin-1-induced expression of nitric oxide synthase in insulin-producing cells is preceded by c-fos induction and depends on gene transcription and protein synthesis. The FEBS Letters. 1993;317(1-2):62–66. doi: 10.1016/0014-5793(93)81492-i. [DOI] [PubMed] [Google Scholar]
- 12.Southern C, Schulster D, Green IC. Inhibition of insulin secretion by interleukin-1β and tumour necrosis factor-α via an L-arginine-dependent nitric oxide generating mechanism. The FEBS Letters. 1990;276(1-2):42–44. doi: 10.1016/0014-5793(90)80502-a. [DOI] [PubMed] [Google Scholar]
- 13.Hughes KJ, Chambers KT, Meares GP, Corbett JA. Nitric oxides mediates a shift from early necrosis to late apoptosis in cytokine-treated β-cells that is associated with irreversible DNA damage. American Journal of Physiology. 2009;297(5):E1187–E1196. doi: 10.1152/ajpendo.00214.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ushmorov A, Ratter F, Lehmann V, Dröge W, Schirrmacher V, Umansky V. Nitric oxide-induced apoptosis in human leukemic lines requires mitochondrial lipid degradation and cytochrome C release. Blood. 1999;93(7):2342–2352. [PubMed] [Google Scholar]
- 15.Borutaite V, Brown GC. Nitric oxide induces apoptosis via hydrogen peroxide, but necrosis via energy and thiol depletion. Free Radical Biology and Medicine. 2003;35(11):1457–1468. doi: 10.1016/j.freeradbiomed.2003.08.003. [DOI] [PubMed] [Google Scholar]
- 16.Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC. β-cell deficit and increased β-cell apoptosis in humans with type 2 diabetes. Diabetes. 2003;52(1):102–110. doi: 10.2337/diabetes.52.1.102. [DOI] [PubMed] [Google Scholar]
- 17.Prentki M, Nolan CJ. Islet β cell failure in type 2 diabetes. Journal of Clinical Investigation. 2006;116(7):1802–1812. doi: 10.1172/JCI29103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Polonsky KS. Dynamics of insulin secretion in obesity and diabetes. International Journal of Obesity. 2000;24(supplement 2):S29–S31. doi: 10.1038/sj.ijo.0801273. [DOI] [PubMed] [Google Scholar]
- 19.Marchetti P, Lupi R, Guerra SD, Bugliani M, Marselli L, Boggi U. The β-cell in human type 2 diabetes. Advances in Experimental Medicine and Biology. 2010;654:501–514. doi: 10.1007/978-90-481-3271-3_22. [DOI] [PubMed] [Google Scholar]
- 20.Polonsky KS, Semenkovich CF. The pancreatic β cell heats up: UCP2 and insulin secretion in diabetes. Cell. 2001;105(6):705–707. doi: 10.1016/s0092-8674(01)00389-0. [DOI] [PubMed] [Google Scholar]
- 21.Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414(6865):813–820. doi: 10.1038/414813a. [DOI] [PubMed] [Google Scholar]
- 22.Marchetti P, Del Guerra S, Marselli L, et al. Pancreatic islets from type 2 diabetic patients have functional defects and increased apoptosis that are ameliorated by metformin. Journal of Clinical Endocrinology and Metabolism. 2004;89(11):5535–5541. doi: 10.1210/jc.2004-0150. [DOI] [PubMed] [Google Scholar]
- 23.Donath MY, Ehses JA, Maedler K, et al. Mechanisms of β-cell death in type 2 diabetes. Diabetes. 2005;54(supplement 2):S108–S113. doi: 10.2337/diabetes.54.suppl_2.s108. [DOI] [PubMed] [Google Scholar]
- 24.Rolo AP, Palmeira CM. Diabetes and mitochondrial function: role of hyperglycemia and oxidative stress. Toxicology and Applied Pharmacology. 2006;212(2):167–178. doi: 10.1016/j.taap.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 25.Friederich M, Hansell P, Palm F. Diabetes, oxidative stress, nitric oxide and mitochondria function. Current Diabetes Reviews. 2009;5(2):120–144. doi: 10.2174/157339909788166800. [DOI] [PubMed] [Google Scholar]
- 26.Zraika S, Hull RL, Udayasankar J, et al. Oxidative stress is induced by islet amyloid formation and time-dependently mediates amyloid-induced beta cell apoptosis. Diabetologia. 2009;52(4):626–635. doi: 10.1007/s00125-008-1255-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Maechler P, Wollheim CB. Mitochondrial function in normal and diabetic β-cells. Nature. 2001;414(6865):807–812. doi: 10.1038/414807a. [DOI] [PubMed] [Google Scholar]
- 28.Fariss MW, Chan CB, Patel M, Van Houten B, Orrenius S. Role of mitochondria in toxic oxidative stress. Molecular Interventions. 2005;5(2):94–111. doi: 10.1124/mi.5.2.7. [DOI] [PubMed] [Google Scholar]
- 29.Lowell BB, Shulman GI. Mitochondrial dysfunction and type 2 diabetes. Science. 2005;307(5708):384–387. doi: 10.1126/science.1104343. [DOI] [PubMed] [Google Scholar]
- 30.Grankvist K, Marklund SL, Taljedal IB. CuZn-superoxide dismutase, Mn-superoxide dismutase, catalase and glutathione peroxidase in pancreatic islets and other tissues in the mouse. Biochemical Journal. 1981;199(2):393–398. doi: 10.1042/bj1990393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tiedge M, Lortz S, Drinkgern J, Lenzen S. Relation between antioxidant enzyme gene expression and antioxidative defense status of insulin-producing cells. Diabetes. 1997;46(11):1733–1742. doi: 10.2337/diab.46.11.1733. [DOI] [PubMed] [Google Scholar]
- 32.Pi J, Bai Y, Zhang Q, et al. Reactive oxygen species as a signal in glucose-stimulated insulin secretion. Diabetes. 2007;56(7):1783–1791. doi: 10.2337/db06-1601. [DOI] [PubMed] [Google Scholar]
- 33.Leloup C, Tourrel-Cuzin C, Magnan C, et al. Mitochondrial reactive oxygen species are obligatory signals for glucose-induced insulin secretion. Diabetes. 2009;58(3):673–681. doi: 10.2337/db07-1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Robertson R, Zhou H, Zhang T, Harmon JS. Chronic oxidative stress as a mechanism for glucose toxicity of the beta cell in type 2 diabetes. Cell Biochemistry and Biophysics. 2007;48(2-3):139–146. doi: 10.1007/s12013-007-0026-5. [DOI] [PubMed] [Google Scholar]
- 35.Robertson RP. Antioxidant drugs for treating beta-cell oxidative stress in type 2 diabetes: glucose-centric versus insulin-centric therapy. Discovery Medicine. 2010;9(45):132–137. [PubMed] [Google Scholar]
- 36.Meglasson MD, Matschinsky FM. Pancreatic islet glucose metabolism and regulation of insulin secretion. Diabetes/Metabolism Reviews. 1986;2(3-4):163–214. doi: 10.1002/dmr.5610020301. [DOI] [PubMed] [Google Scholar]
- 37.Garvey WT. Glucose transport and NIDDM. Diabetes Care. 1992;15(3):396–417. doi: 10.2337/diacare.15.3.396. [DOI] [PubMed] [Google Scholar]
- 38.Matschinsky FM. Glucokinase as glucose sensor and metabolic signal generator in pancreatic β-cells and hepatocytes. Diabetes. 1990;39(6):647–652. doi: 10.2337/diab.39.6.647. [DOI] [PubMed] [Google Scholar]
- 39.Cook DL, Hales CN. Intracellular ATP directly blocks K+ channels in pancreatic B-cells. Nature. 1984;311(5983):271–273. doi: 10.1038/311271a0. [DOI] [PubMed] [Google Scholar]
- 40.Kakei M, Kelly RP, Ashcroft SJH, Ashcroft FM. The ATP-sensitivity of K+ channels in rat pancreatic B-cells is modulated by ADP. The FEBS Letters. 1986;208(1):63–66. doi: 10.1016/0014-5793(86)81533-2. [DOI] [PubMed] [Google Scholar]
- 41.Ghosh A, Ronner P, Cheong E, Khalid P, Matschinsky FM. The role of ATP and free ADP in metabolic coupling during fuel-stimulated insulin release from islet β-cells in the isolated perfused rat pancreas. Journal of Biological Chemistry. 1991;266(34):22887–22892. [PubMed] [Google Scholar]
- 42.Arkhammar P, Nilsson T, Rorsman P, Berggren PO. Inhibition of ATP-regulated K+ channels precedes depolarization-induced increase in cytoplasmic free Ca2+ concentration in pancreatic beta-cells. Journal of Biological Chemistry. 1987;262(12):5448–5454. [PubMed] [Google Scholar]
- 43.Misler S, Barnett DW, Pressel DM, Gillis KD, Scharp DW, Falke LC. Stimulus-secretion coupling in β-cells of transplantable human islets of langerhans: evidence for a critical role for Ca2+ entry. Diabetes. 1992;41(6):662–670. doi: 10.2337/diab.41.6.662. [DOI] [PubMed] [Google Scholar]
- 44.Easom RA. CaM kinase II: a protein kinase with extraordinary talents germane to insulin exocytosis. Diabetes. 1999;48(4):675–684. doi: 10.2337/diabetes.48.4.675. [DOI] [PubMed] [Google Scholar]
- 45.Brown LJ, MacDonald MJ, Lehn DA, Moran SM. Sequence of rat mitochondrial glycerol-3-phosphate dehydrogenase cDNA. Evidence for EF-hand calcium-binding domains. Journal of Biological Chemistry. 1994;269(20):14363–14366. [PubMed] [Google Scholar]
- 46.MacDonald MJ. Feasibility of a mitochondrial pyruvate malate shuttle in pancreatic islets. Further implication of cytosolic NADPH in insulin secretion. Journal of Biological Chemistry. 1995;270(34):20051–20058. [PubMed] [Google Scholar]
- 47.Eto K, Tsubamoto Y, Terauchi Y, et al. Role of NADH shuttle system in glucose-induced activation of mitochondrial metabolism and insulin secretion. Science. 1999;283(5404):981–985. doi: 10.1126/science.283.5404.981. [DOI] [PubMed] [Google Scholar]
- 48.Ravier MA, Eto K, Jonkers FC, Nenquin M, Kadowaki T, Henquin JC. The oscillatory behavior of pancreatic islets from mice with mitochondrial glycerol-3-phosphate dehydrogenase knockout. Journal of Biological Chemistry. 2000;275(3):1587–1593. doi: 10.1074/jbc.275.3.1587. [DOI] [PubMed] [Google Scholar]
- 49.Wollheim CB. Beta-cell mitochondria in the regulation of insulin secretion: a new culprit in Type II diabetes. Diabetologia. 2000;43(3):265–277. doi: 10.1007/s001250050044. [DOI] [PubMed] [Google Scholar]
- 50.Sato Y, Anello M, Henquin JC. Glucose regulation of insulin secretion independent of the opening or closure of adenosine triphosphate-sensitive K+ channels in β cells. Endocrinology. 1999;140(5):2252–2257. doi: 10.1210/endo.140.5.6729. [DOI] [PubMed] [Google Scholar]
- 51.Scheuner D, Kaufman RJ. The unfolded protein response: a pathway that links insulin demand with β-cell failure and diabetes. Endocrine Reviews. 2008;29(3):317–333. doi: 10.1210/er.2007-0039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kaufman RJ, Back SH, Song B, Han J, Hassler J. The unfolded protein response is required to maintain the integrity of the endoplasmic reticulum, prevent oxidative stress and preserve differentiation in β-cells. Diabetes, Obesity and Metabolism. 2010;12(supplement 2):99–107. doi: 10.1111/j.1463-1326.2010.01281.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fonseca SG, Gromada J, Urano F. Endoplasmic reticulum stress and pancreatic β-cell death. Trends in Endocrinology and Metabolism. 2011;22(7):266–274. doi: 10.1016/j.tem.2011.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Margittai E, Low P, Stiller I, et al. Production of H2O2 in the endoplasmic reticulum promotes in vivo disulfide bond formation. Antioxidants & Redox Signaling. 2012;16(10):1088–1099. doi: 10.1089/ars.2011.4221. [DOI] [PubMed] [Google Scholar]
- 55.Wang W, Guo Y, Xu M, et al. Development of diabetes in lean Ncb5or-null mice is associated with manifestations of endoplasmic reticulum and oxidative stress in beta cells. Acta Biochimica et Biophysica Sinica. 2011;1812:1532–1541. doi: 10.1016/j.bbadis.2011.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Koo HJ, Piao Y, Pak YK. Endoplasmic reticulum stress impairs insulin signaling through mitochondrial damage in SH-SY5Y cells. doi: 10.1159/000333069. Neuro-Signals. In press. [DOI] [PubMed] [Google Scholar]
- 57.Tang C, Koulajian K, Schuiki I, et al. Glucose-induced beta cell dysfunction in vivo in rats: link between oxidative stress and endoplasmic reticulum stress. Diabetologia. 2012;55(5):1366–1379. doi: 10.1007/s00125-012-2474-8. [DOI] [PubMed] [Google Scholar]
- 58.Ježek P, Hlavatá L. Mitochondria in homeostasis of reactive oxygen species in cell, tissues, and organism. International Journal of Biochemistry and Cell Biology. 2005;37(12):2478–2503. doi: 10.1016/j.biocel.2005.05.013. [DOI] [PubMed] [Google Scholar]
- 59.Chen Q, Vazquez EJ, Moghaddas S, Hoppel CL, Lesnefsky EJ. Production of reactive oxygen species by mitochondria: central role of complex III. Journal of Biological Chemistry. 2003;278(38):36027–36031. doi: 10.1074/jbc.M304854200. [DOI] [PubMed] [Google Scholar]
- 60.Furukawa S, Fujita T, Shimabukuro M, et al. Increased oxidative stress in obesity and its impact on metabolic syndrome. Journal of Clinical Investigation. 2004;114(12):1752–1761. doi: 10.1172/JCI21625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fariss MW, Chan CB, Patel M, Van Houten B, Orrenius S. Role of mitochondria in toxic oxidative stress. Molecular Interventions. 2005;5(2):94–111. doi: 10.1124/mi.5.2.7. [DOI] [PubMed] [Google Scholar]
- 62.Montero J, Mari M, Colell A, et al. Cholesterol and peroxidized cardiolipin in mitochondrial membrane properties, permeabilization and cell death. Biochimica et Biophysica Acta. 2010;1797(6-7):1217–1224. doi: 10.1016/j.bbabio.2010.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Seleznev K, Zhao C, Zhang XH, Song K, Ma ZA. Calcium-independent phospholipase A2 localizes in and protects mitochondria during apoptotic induction by staurosporine. Journal of Biological Chemistry. 2006;281(31):22275–22288. doi: 10.1074/jbc.M604330200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhao Z, Zhang X, Zhao C, et al. Protection of pancreatic β-cells by group VIA phospholipase A2-mediated repair of mitochondrial membrane peroxidation. Endocrinology. 2010;151(7):3038–3048. doi: 10.1210/en.2010-0016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ma ZA, Zhao Z, Turk J. Mitochondrial dysfunction and β-cell failure in type 2 diabetes mellitus. Experimental Diabetes Research. 2012;2012:11 pages. doi: 10.1155/2012/703538.703538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Orrenius S. Mitochondrial regulation of apoptotic cell death. Toxicology Letters. 2004;149(1–3):19–23. doi: 10.1016/j.toxlet.2003.12.017. [DOI] [PubMed] [Google Scholar]
- 67.Green DR, Kroemer G. The pathophysiology of mitochondrial cell death. Science. 2004;305(5684):626–629. doi: 10.1126/science.1099320. [DOI] [PubMed] [Google Scholar]
- 68.Acehan D, Jiang X, Morgan DG, Heuser JE, Wang X, Akey CW. Three-dimensional structure of the apoptosome: implications for assembly, procaspase-9 binding, and activation. Molecular Cell. 2002;9(2):423–432. doi: 10.1016/s1097-2765(02)00442-2. [DOI] [PubMed] [Google Scholar]
- 69.Budihardjo I, Oliver H, Lutter M, Luo X, Wang X. Biochemical pathways of caspase activation during apoptosis. Annual Review of Cell and Developmental Biology. 1999;15:269–290. doi: 10.1146/annurev.cellbio.15.1.269. [DOI] [PubMed] [Google Scholar]
- 70.Kinsey GR, Blum JL, Covington MD, Cummings BS, McHowat J, Schnellmann RG. Decreased iPLA2 γ expression induces lipid peroxidation and cell death and sensitizes cells to oxidant-induced apoptosis. Journal of Lipid Research. 2008;49(7):1477–1487. doi: 10.1194/jlr.M800030-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cummings BS, McHowat J, Schnellmann RG. Role of an endoplasmic reticulum Ca2+-independent phospholipase A2 in oxidant-induced renal cell death. American Journal of Physiology. 2002;283(3):F492–F498. doi: 10.1152/ajprenal.00022.2002. [DOI] [PubMed] [Google Scholar]
- 72.Kinsey GR, McHowat J, Beckett CS, Schnellmann RG. Identification of calcium-independent phospholipase A2 γ in mitochondria and its role in mitochondrial oxidative stress. American Journal of Physiology. 2007;292(2):F853–F860. doi: 10.1152/ajprenal.00318.2006. [DOI] [PubMed] [Google Scholar]
- 73.Van Kuijk FJGM, Sevanian A, Handelman GJ, Dratz EA. A new role for phospholipase A2: protection of membranes from lipid peroxidation damage. Trends in Biochemical Sciences. 1987;12(1):31–34. [Google Scholar]
- 74.Lands WE. Lipid metabolism. Annual Review of Biochemistry. 1965;34:313–346. doi: 10.1146/annurev.bi.34.070165.001525. [DOI] [PubMed] [Google Scholar]
- 75.Sevanian A, Muakkassah Kelly SF, Montestruque S. The influence of phospholipase A2 and glutathione peroxidase on the elimination of membrane lipid peroxides. Archives of Biochemistry and Biophysics. 1983;223(2):441–452. doi: 10.1016/0003-9861(83)90608-2. [DOI] [PubMed] [Google Scholar]
- 76.Van Kuijk FJGM, Handelman GJ, Dratz EA. Consecutive action of phospholipase A2 and glutathione peroxidase is required for reduction of phospholipid hydroperoxides and provides a convenient method to determine peroxide values in membranes. Journal of Free Radicals in Biology and Medicine. 1985;1(5-6):421–427. doi: 10.1016/0748-5514(85)90156-4. [DOI] [PubMed] [Google Scholar]
- 77.Schaloske RH, Dennis EA. The phospholipase A2 superfamily and its group numbering system. Biochimica et Biophysica Acta. 2006;1761(11):1246–1259. doi: 10.1016/j.bbalip.2006.07.011. [DOI] [PubMed] [Google Scholar]
- 78.Balsinde J, Winstead MV, Dennis EA. Phospholipase A2 regulation of arachidonic acid mobilization. The FEBS Letters. 2002;531(1):2–6. doi: 10.1016/s0014-5793(02)03413-0. [DOI] [PubMed] [Google Scholar]
- 79.Szkudelski T. The mechanism of alloxan and streptozotocin action in B cells of the rat pancreas. Physiological Research. 2001;50(6):537–546. [PubMed] [Google Scholar]
- 80.Bao S, Bohrer A, Ramanadham S, Jin W, Zhang S, Turk J. Effects of stable suppression of group VIA phospholipase A2 expression on phospholipid content and composition, insulin secretion, and proliferation of INS-1 insulinoma cells. Journal of Biological Chemistry. 2006;281(1):187–198. doi: 10.1074/jbc.M509105200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Song H, Rohrs H, Tan M, Wohltmann M, Ladenson JH, Turk J. Effects of endoplasmic reticulum stress on group VIA phospholipase A 2in beta cells include tyrosine phosphorylation and increased association with calnexin. Journal of Biological Chemistry. 2010;285(44):33843–33857. doi: 10.1074/jbc.M110.153197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Jones LJ, Gray M, Yue ST, Haugland RP, Singer VL. Sensitive determination of cell number using the CyQUANT cell proliferation assay. Journal of Immunological Methods. 2001;254(1-2):85–98. doi: 10.1016/s0022-1759(01)00404-5. [DOI] [PubMed] [Google Scholar]
- 83.Hawker JR., Jr. Chemiluminescence-based BrdU ELISA to measure DNA synthesis. Journal of Immunological Methods. 2003;274(1-2):77–82. doi: 10.1016/s0022-1759(02)00437-4. [DOI] [PubMed] [Google Scholar]
- 84.Yagi K. Simple procedure for specific assay of lipid hydroperoxides in serum or plasma. Methods in Molecular Biology. 1998;108:107–110. doi: 10.1385/0-89603-472-0:107. [DOI] [PubMed] [Google Scholar]
- 85.O'Donnell VB. Mass spectrometry analysis of oxidized phosphatidylcholine and phosphatidylethanolamine. Biochimica et Biophysica Acta. 2011;1811:818–826. doi: 10.1016/j.bbalip.2011.07.018. [DOI] [PubMed] [Google Scholar]
- 86.Hu C, Van Dommelen J, Van Der Heljden R, et al. RPLC-lon-trap-FTMS method for lipid profiling of plasma: method validation and application to p53 mutant mouse model. Journal of Proteome Research. 2008;7(11):4982–4991. doi: 10.1021/pr800373m. [DOI] [PubMed] [Google Scholar]
- 87.Song H, Wohltmann M, Tan M, Bao S, Ladenson JH, Turk J. Group VIA phospholipase A2 (iPLA2 β) is activated upstream of p38 MAP kinase in pancreatic islet beta cell signaling. The Journal of Biological Chemistry. 2012;287:5528–5541. doi: 10.1074/jbc.M111.285114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Mancuso DJ, Jenkins CM, Gross RW. The genomic organization, complete mRNA sequence, cloning, and expression of a novel human intracellular membrane-associated calcium- independent phospholipase A2 . Journal of Biological Chemistry. 2000;275(14):9937–9945. doi: 10.1074/jbc.275.14.9937. [DOI] [PubMed] [Google Scholar]
- 89.Mancuso DJ, Jenkins CM, Sims HF, Cohen JM, Yang J, Gross RW. Complex transcriptional and translational regulation of iPLA2γ resulting in multiple gene products containing dual competing sites for mitochondrial or peroxisomal localization. European Journal of Biochemistry. 2004;271(23-24):4709–4724. doi: 10.1111/j.1432-1033.2004.04435.x. [DOI] [PubMed] [Google Scholar]
- 90.Ma Z, Bohrer A, Wohltmann M, Ramanadham S, Hsu FF, Turk J. Studies of phospholipid metabolism, proliferation, and secretion of stably transfected insulinoma cells that overexpress group VIA phospholipase A2 . Lipids. 2001;36(7):689–700. doi: 10.1007/s11745-001-0774-9. [DOI] [PubMed] [Google Scholar]
- 91.Ramanadham S, Hsu FF, Bohrer A, Nowatzke W, Ma Z, Turk J. Electrospray ionization mass spectrometric analyses of phospholipids from rat and human pancreatic islets and subcellular membranes: comparison to other tissues and implications for membrane fusion in insulin exocytosis. Biochemistry. 1998;37(13):4553–4567. doi: 10.1021/bi9722507. [DOI] [PubMed] [Google Scholar]
- 92.Milic I, Fedorova M, Teuber K, Schiller J, Hoffmann R. Characterization of oxidation products from 1-palmitoyl-2-linoleoyl-sn-glycerophosphatidylcholine in aqueous solutions and their reactions with cysteine, histidine and lysine residues. Chemistry and Physics of Lipids. 2012;165:186–196. doi: 10.1016/j.chemphyslip.2011.12.009. [DOI] [PubMed] [Google Scholar]
- 93.Chen X, Zhang W, Laird J, Hazen SL, Salomon RG. Polyunsaturated phospholipids promote the oxidation and fragmentation of γ-hydroxyalkenals: formation and reactions of oxidatively truncated ether phospholipids. Journal of Lipid Research. 2008;49(4):832–846. doi: 10.1194/jlr.M700598-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bao S, Li Y, Lei X, et al. Attenuated free cholesterol loading-induced apoptosis but preserved phospholipid composition of peritoneal macrophages from mice that do not express group VIA phospholipase A2 . Journal of Biological Chemistry. 2007;282(37):27100–27114. doi: 10.1074/jbc.M701316200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Jenkins CM, Mancuso DJ, Yan W, Sims HF, Gibson B, Gross RW. Identification, cloning, expression, and purification of three novel human calcium-independent phospholipase A2 family members possessing triacylglycerol lipase and acylglycerol transacylase activities. Journal of Biological Chemistry. 2004;279(47):48968–48975. doi: 10.1074/jbc.M407841200. [DOI] [PubMed] [Google Scholar]
- 96.Dennis EA, Cao J, Hsu YH, Magrioti V, Kokotos G. Phospholipase A2 enzymes: physical structure, biological function, disease implication, chemical inhibition, and therapeutic intervention. Chemical Reviews. 2011;111:6130–6185. doi: 10.1021/cr200085w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kienesberger PC, Oberer M, Lass A, Zechner R. Mammalian patatin domain containing proteins: a family with diverse lipolytic activities involved in multiple biological functions. Journal of Lipid Research. 2009;50:S63–S68. doi: 10.1194/jlr.R800082-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Balboa MA, Balsinde J, Jones SS, Dennis EA. Identity between the Ca2+-independent phospholipase A2 enzymes from P388D1 macrophages and Chinese hamster ovary cells. Journal of Biological Chemistry. 1997;272(13):8576–8580. doi: 10.1074/jbc.272.13.8576. [DOI] [PubMed] [Google Scholar]
- 99.Ma Z, Ramanadham S, Kempe K, Chi XS, Ladenson J, Turk J. Pancreatic islets express a Ca2+-independent phospholipase A2 enzyme that contains a repeated structural motif homologous to the integral membrane protein binding domain of ankyrin. Journal of Biological Chemistry. 1997;272(17):11118–11127. [PubMed] [Google Scholar]
- 100.Tanaka H, Takeya R, Sumimoto H. A novel intracellular membrane-bound calcium-independent phospholipase A2 . Biochemical and Biophysical Research Communications. 2000;272(2):320–326. doi: 10.1006/bbrc.2000.2776. [DOI] [PubMed] [Google Scholar]
- 101.Yang J, Han X, Gross RW. Identification of hepatic peroxisomal phospholipase A2 and characterization of arachidonic acid-containing choline glycerophospholipids in hepatic peroxisomes. The FEBS Letters. 2003;546(2-3):247–250. doi: 10.1016/s0014-5793(03)00581-7. [DOI] [PubMed] [Google Scholar]
- 102.Yan W, Jenkins CM, Han X, et al. The highly selective production of 2-arachidonoyl lysophosphatidylcholine catalyzed by purified calcium-independent phospholipase A2 γ: identification of a novel enzymatic mediator for the generation of a key branch point intermediate in eicosanoid signaling. Journal of Biological Chemistry. 2005;280(29):26669–26679. doi: 10.1074/jbc.M502358200. [DOI] [PubMed] [Google Scholar]
- 103.Sharma J, Turk J, Mancuso DJ, Sims HF, Gross RW, McHowat J. Activation of group VI phospholipase A2 isoforms in cardiac endothelial cells. American Journal of Physiology. 2011;300(4):C872–C879. doi: 10.1152/ajpcell.00289.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Malhotra A, Edelman-Novemsky I, Xu Y, et al. Role of calcium-independent phospholipase A2 in the pathogenesis of Barth syndrome. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(7):2337–2341. doi: 10.1073/pnas.0811224106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Sparagna GC, Lesnefsky EJ. Cardiolipin remodeling in the heart. Journal of Cardiovascular Pharmacology. 2009;53(4):290–301. doi: 10.1097/FJC.0b013e31819b5461. [DOI] [PubMed] [Google Scholar]
- 106.Mancuso DJ, Han X, Jenkins CM, et al. Dramatic accumulation of triglycerides and precipitation of cardiac hemodynamic dysfunction during brief caloric restriction in transgenic myocardium expressing human calcium-independent phospholipase A2γ . Journal of Biological Chemistry. 2007;282(12):9216–9227. doi: 10.1074/jbc.M607307200. [DOI] [PubMed] [Google Scholar]
- 107.Cedars A, Jenkins CM, Mancuso DJ, Gross RW. Calcium-independent phospholipases in the heart: mediators of cellular signaling, bioenergetics, and ischemia-induced electrophysiologic dysfunction. Journal of Cardiovascular Pharmacology. 2009;53(4):277–289. [PMC free article] [PubMed] [Google Scholar]
- 108.Mancuso DJ, Sims HF, Han X, et al. Genetic ablation of calcium-independent phospholipase A2γ leads to alterations in mitochondrial lipid metabolism and function resulting in a deficient mitochondrial bioenergetic phenotype. Journal of Biological Chemistry. 2007;282(48):34611–34622. doi: 10.1074/jbc.M707795200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Mancuso DJ, Kotzbauer P, Wozniak DF, et al. Genetic ablation of calcium-independent phospholipase A2γ leads to alterations in hippocampal cardiolipin content and molecular species distribution, mitochondrial degeneration, autophagy, and cognitive dysfunction. Journal of Biological Chemistry. 2009;284(51):35632–35644. doi: 10.1074/jbc.M109.055194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Mancuso DJ, Sims HF, Yang K, et al. Genetic ablation of calcium-independent phospholipase A2γ prevents obesity and insulin resistance during high fat feeding by mitochondrial uncoupling and increased adipocyte fatty acid oxidation. Journal of Biological Chemistry. 2010;285(47):36495–36510. doi: 10.1074/jbc.M110.115766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Song H, Wohltmann M, Bao S, Ladenson JH, Semenkovich CF, Turk J. Mice deficient in Group VIB phospholipase A2 (iPLA2γ) exhibit relative resistance to obesity and metabolic abnormalities induced by a Western diet. American Journal of Physiology. 2010;298(6):E1097–E1114. doi: 10.1152/ajpendo.00780.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Zachman DK, Chicco AJ, McCune SA, Murphy RC, Moore RL, Sparagna GC. The role of calcium-independent phospholipase A2 in cardiolipin remodeling in the spontaneously hypertensive heart failure rat heart. Journal of Lipid Research. 2010;51(3):525–534. doi: 10.1194/jlr.M000646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Su X, Mancuso DJ, Bickel PE, Jenkins C, Gross RW. Small interfering RNA knockdown of calcium-independent phospholipases A2 β or γ inhibits the hormone-induced differentiation of 3T3-L1 preadipocytes. Journal of Biological Chemistry. 2004;279(21):21740–21748. doi: 10.1074/jbc.M314166200. [DOI] [PubMed] [Google Scholar]
- 114.Ramanadham S, Hsu FF, Zhang S, et al. Apoptosis of Insulin-Secreting Cells Induced by Endoplasmic Reticulum Stress Is Amplified by Overexpression of Group VIA Calcium-Independent Phospholipase A2 (iPLA2 β) and Suppressed by Inhibition of iPLA2 β . Biochemistry. 2004;43(4):918–930. doi: 10.1021/bi035536m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Lei X, Zhang S, Bohrer A, Bao S, Song H, Ramanadham S. The group VIA calcium-independent phospholipase A2 participates in ER stress-induced INS-1 insulinoma cell apoptosis by promoting ceramide generation via hydrolysis of sphingomyelins by neutral sphingomyelinase. Biochemistry. 2007;46(35):10170–10185. doi: 10.1021/bi700017z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Lei X, Zhang S, Bohrer A, Ramanadham S. Calcium-independent phospholipase A2 (iPLA2 β)- mediated ceramide generation plays a key role in the cross-talk between the endoplasmic reticulum (ER) and mitochondria during ER stress-induced insulin-secreting cell apoptosis. Journal of Biological Chemistry. 2008;283(50):34819–34832. doi: 10.1074/jbc.M807409200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Lei X, Zhang S, Barbour SE, et al. Spontaneous development of endoplasmic reticulum stress that can lead to diabetes mellitus is associated with higher calcium-independent phospholipase A2 expression: a role for regulation by SREBP-1. Journal of Biological Chemistry. 2010;285(9):6693–6705. doi: 10.1074/jbc.M109.084293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Lei X, Barbour SE, Ramanadham S. Group VIA Ca2+-independent phospholipase A2 (iPLA2 β) and its role in β-cell programmed cell death. Biochimie. 2010;92(6):627–637. doi: 10.1016/j.biochi.2010.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Song H, Bao S, Lei X, et al. Evidence for proteolytic processing and stimulated organelle redistribution of iPLA2 β . Biochimica et Biophysica Acta. 2010;1801(5):547–558. doi: 10.1016/j.bbalip.2010.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Jenkins CM, Han X, Mancuso DJ, Gross RW. Identification of calcium-independent phospholipase A2 (iPLA2) β, and not iPLA2γ, as the mediator of arginine vasopressin-induced arachidonic acid release in A-10 smooth muscle cells. Enantioselective mechanism-based discrimination of mammalian iPLA2s. Journal of Biological Chemistry. 2002;277(36):32807–32814. doi: 10.1074/jbc.M202568200. [DOI] [PubMed] [Google Scholar]
- 121.Schreck R, Rieber P, Baeuerle PA. Reactive oxygen intermediates as apparently widely used messengers in the activation of the NF-κB transcription factor and HIV-1. EMBO Journal. 1991;10(8):2247–2258. doi: 10.1002/j.1460-2075.1991.tb07761.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Schreck R, Grassmann R, Fleckenstein B, Baeuerle PA. Antioxidants selectively suppress activation of NF-κB by human T-cell leukemia virus type I Tax protein. Journal of Virology. 1992;66(11):6288–6293. doi: 10.1128/jvi.66.11.6288-6293.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Li N, Karin M. Is NF-κB the sensor of oxidative stress? The FASEB Journal. 1999;13(10):1137–1143. [PubMed] [Google Scholar]
- 124.Chen ACH, Arany PR, Huang YY, et al. Low-Level laser therapy activates NF-kB via generation of reactive oxygen species in mouse embryonic fibroblasts. PLoS ONE. 2011;6(7) doi: 10.1371/journal.pone.0022453.e22453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Kwon G, Corbett JA, Hauser S, Hill JR, Turk J, McDaniel ML. Evidence for involvement of the proteasome complex (26S) and NFκB in IL-1β-induced nitric oxide and prostaglandin production by rat islets and RINm5F cells. Diabetes. 1998;47(4):583–591. doi: 10.2337/diabetes.47.4.583. [DOI] [PubMed] [Google Scholar]
- 126.Deng YT, Huang HC, Lin JK. Rotenone induces apoptosis in MCF-7 human breast cancer cell-mediated ROS through JNK and p38 signaling. Molecular Carcinogenesis. 2010;49(2):141–151. doi: 10.1002/mc.20583. [DOI] [PubMed] [Google Scholar]
- 127.Chen Z, Jiang H, Wan Y, Bi C, Yuan Y. H2O2-induced secretion of tumor necrosis factor-α evokes apoptosis of cardiac myocytes through reactive oxygen species-dependent activation of p38 MAPK. Cytotechnology. 2012;64:65–73. doi: 10.1007/s10616-011-9392-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Davies SS, Pontsler AV, Marathe GK, et al. Oxidized alkyl phospholipids are specific, high affinity peroxisome Proliferator-activated Receptor γ Ligands and Agonists. Journal of Biological Chemistry. 2001;276(19):16015–16023. doi: 10.1074/jbc.M100878200. [DOI] [PubMed] [Google Scholar]
- 129.Morgan AH, Hammond VJ, Morgan L, et al. Quantitative assays for esterified oxylipins generated by immune cells. Nature Protocols. 2010;5(12):1919–1931. doi: 10.1038/nprot.2010.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Davies SS, Pontsler AV, Marathe GK, et al. Oxidized alkyl phospholipids are specific, high affinity peroxisome proliferator-activated receptor γ ligands and agonists. Journal of Biological Chemistry. 2001;276(19):16015–16023. doi: 10.1074/jbc.M100878200. [DOI] [PubMed] [Google Scholar]
- 131.Maskrey BH, Bermudez-Fajardo A, Morgan AH, et al. Activated platelets and monocytes generate four hydroxyphosphatidylethanolamines via lipoxygenase. The Journal of Biological Chemistry. 2007;282:20151–20163. doi: 10.1074/jbc.M611776200. [DOI] [PubMed] [Google Scholar]
- 132.Morgan AH, Dioszeghy V, Maskrey BH, et al. Phosphatidylethanolamine-esterified eicosanoids in the mouse. Tissue localization and inflammation-dependent formation in Th-2 disease. Journal of Biological Chemistry. 2009;284(32):21185–21191. doi: 10.1074/jbc.M109.021634. [DOI] [PMC free article] [PubMed] [Google Scholar]