

Abstract
The cerebral endothelium can be a vital source of signaling factors such as brain-derived neurotrophic factor (BDNF) that defend the neuronal parenchyma against stress and injury. But the underlying mechanisms remain to be fully defined. Here, we use cell models to ask how vascular neuroprotection is sustained. Human brain endothelial cells were grown in culture and conditioned media was transferred to primary rat cortical neurons. Brain endothelial cell-conditioned media activated neuronal Akt signaling and protected neurons against hypoxia and oxygen-glucose deprivation. Blockade of Akt phosphorylation with the PI3-kinase inhibitor LY294002 negated this vascular neuroprotective effect. Upstream of Akt signaling, the BDNF receptor TrkB was involved since depletion with TrkB/Fc eliminated the ability of endothelial-conditioned media to protect neurons against hypoxia. Downstream of Akt signaling, activation of GSK-3β, caspase-9, caspase-3 and Bad pathways were detected. Taken together, these findings suggest that the molecular basis for vascular neuroprotection involves TrkB-Akt signaling that ameliorates neuronal apoptosis. Further investigation of these mechanisms may reveal new approaches for augmenting endogenous vascular neuroprotection in stroke, brain injury and neurodegeneration.
Keywords: Neurovascular unit, trophic coupling, neural cell death, human cerebral endothelial cells
INTRODUCTION
Cell-cell interactions between neuronal, glial and vascular compartments form the basis for function and dysfunction in the neurovascular unit (Guo and Lo, 2009; Lo et al., 2003). For example, interactions between endothelial cells, astrocytes and pericytes help sustain the blood-brain barrier (Armulik et al., 2010; Daneman et al., 2010), and disruptions in gliovascular signaling may cause BBB leakage in many CNS diseases(Chaitanya et al., 2011; Wolburg et al., 2009). Similarly, signaling between neuronal and vascular cells is also important. Interactions between cerebral endothelium and neuronal precursor cells may involve common mediators termed angioneurins that support angiogenesis and neurogenesis during normal development as well as during recovery after stroke and brain injury (Ohab et al., 2006; Zacchigna et al., 2008).
Although the conceptual basis for these cell-cell interactions in the neurovascular unit is well accepted, the subsequent neuroprotective mechanisms involved remain to be fully elucidated. We and others previously suggested that cerebral endothelium could protect neurons from oxidative stress and various metabolic insults via the secretion of growth factors such as brain derived neurotrophic factor (BDNF), insulin-like growth factors (IGF1 and IGF2), pleiotrophin, and stromal cell-derived factor 1 (SDF-1α)(Dugas et al., 2008; Guo et al., 2008). Here, we tested the hypothesis that vascular neuroprotection is subserved by the ability of trophic mediators to activate patterned pathways of PI-3K/Akt survival signaling that block caspase-mediated neuronal death.
METHODS AND MATERIALS
Rat primary neurons culture
Rat primary neurons were cultured from embryonic day 17 cortices, and isolated cells were plated onto poly-D-lysine-coated plates or dishes with medium consisting of NeuroBasal, 2% B27 (Invitrogen), 0.5 mM L-glutamine, and 100 units/ml penicillin and streptomycin. Media were half changed every 3 days, and cells were used at days in vitro 7–9. All experiments were performed following Massachusetts General Hospital approved protocols following NIH Guideliens for Use and Care of Laboratory Animals, that include appropriate anesthesia and minimization of animal number usage.
Human brain microvascular endothelial cells
A human brain microvascular endothelial cell line, THBMEC (Callahan et al., 2004) was maintained in complete medium consisting of RPMI1640 medium (Invitrogen), 10% FBS, 10% NuSerum (BD Bioscience), 1 mM sodium pyruvate, nonessential amino acids, MEM vitamins, and 100 units/ml penicillin and streptomycin. To prepare endothelial-conditioned medium (Guo et al., 2008), the endothelial cells were seeded in human fibronectin (BD Bioscience)-coated 100mm dishes. After overnight serum starvation, fresh serum-free combination media (RPMI:NeuroBasal=1:1) were added. Endothelial-conditioned media were collected after 24 h incubation, and then centrifuged to collect the supernatant, which would be used to neurons for hypoxia treatment.
To confirm the relevance of our findings, key experiments were repeated with primary human brain microvascular endothelial cells (HBMEC, purchased from ScienCell Laboratory). Cells were maintained on human fibronectin-coated dish with endothelial cell medium- basal plus 5% FBS, 1% endothelial cell growth supplement (ECGS) and 1% penicillin/streptomycin solution. Cells were used at passage 2 to obtain endothelial-conditioned media. Confluent cells were starved with basal medium plus 0.5% FBS, 0.1% ECGS and 0.1% antibiotics for overnight, then incubated with serum-free combination media (endothelial cell medium-basal:NeuroBasal=1:1) for 24 h and collected as conditioned media.
Hypoxia setting
Hypoxia was induced with a modular chamber (Billups-Rothenberg) flushed with 90% N2, 5% H2, and 5% CO2 for 30min at 37 °C. The chamber was then sealed and kept at 37 °C for 20 hours for hypoxia. At the end of hypoxia, the cells were removed from the chamber and directly used for preparation of cell lysate, measurement of cell viability, or staining. Cell viability was measured by standard MTT transformation [neuronal cell death = (100-cell viability)%].
Oxygen-glucose deprivation (OGD) setting
Neurons were replaced with glucose-free media followed by hypoxia with modular chamber, as described here. The chamber was sealed and placed at 37 °C for 2 h of OGD, then the cells were removed from the chamber and the glucose-free media were changed to either control media or endothelial-conditioned media with or without LY294002. The cells were maintained in the regular incubator for 22 h of reoxygenation and the cell viability was measured by standard MTT transformation.
TrkB/Fc depletion procedure
Endothelial-conditioned media were depleted with recombinant human TrkB/Fc chimera (R & D systems) to remove BDNF. Briefly, 2 μg/ml TrkB/Fc was added to media for 2 h in 4 °C with rotation, and equal amounts of normal human IgG (Zymed Laboratory) were added as control. Protein A-agarose immunoprecipitation reagent was then added for 4 h in 4°C with rotation. The media were centrifuged to discard the agarose pellet, and the depleted supernatant was used for conditioned media experiments.
Immunoblotting
Cells were lysed with lysis buffer (Cell Signaling Technology) and centrifuged for supernatant. The protein concentration was determined with Bradford Assay (Bio-Rad). Total lysate of cells (20 μg per lane) were separated in precast 4–20% gradient Tris-glycine SDS-polyacrylamide gels (Invitrogen), and then transferred to PVDF membrane (Invitrogen). After blocking with 0.2% Tropix I-block (Applied Biosystems), membranes were incubated overnight at 4 °C with indicated primary antibodies (all from Cell Signaling Technology), and 1 h at room temperature with horseradish peroxidase-conjugated secondary antibodies (Amersham). The immune complexes were visualized by enhanced chemiluminescent substrate (Pierce). All immunoblots were repeated for at least four independent experiments, and the optical densities for each band were analyzed by Image J program, the levels of housekeeping gene β-Actin were used as loading control.
Live/dead cell staining
The LIVE/DEAD Reduced Biohazard Viability/Cytotoxicity kit (Invitrogen) was used to stain the neurons after hypoxia. Living cells with an intact membrane were stained green by SYTO 10, a highly membrane-permeant fluorescent nucleic acid stain, while cells with compromised membrane, considered to be dead or dying, were stained red by DEAD Red (ethidium homodimer-2), a cell-impermeant fluorescent nucleic acid stain. Briefly, the cells were washed once with HBSS (Invitrogen), and then incubated with both dyes (1:500 dilution of each) for 15 minutes in darkness at room temperature. The cells were washed again with HBSS after removing the dye solution, and covered with HBSS for observation directly.
Statistical analysis
All the experiments were done in triplicates, repeated three to five times independently. Quantitative data were expressed as mean + SD and analyzed with ANOVA followed by Tukey HSD multiple comparisons. Differences of p<0.05 were considered significant.
RESULTS
PI-3K/Akt activation in neurons protected by endothelial-conditioned media
PI-3K/Akt is a highly conserved pathway for neuroprotection (Sugawara et al., 2004; Zhao et al., 2006) so we assessed this mechanism here. A human brain endothelial cell line was grown in culture then conditioned media were transferred to neurons. Incubation with endothelial-conditioned media rapidly increased phospho-Akt levels in neurons within 30–60 min (Figure 1a). An important downstream target of Akt is GSK-3β, so phospho-GSK-3β levels were examined. Consistent with activation of Akt signaling, phospho-GSK-3β levels in neurons were also increased by endothelial-conditioned media (Figure 1a). As expected, endothelial-conditioned media was neuroprotective, decreasing neuronal death after 20-hr hypoxia (Figure 1b). The importance of Akt signaling in this vascular form of neuroprotection was tested pharmacologically. Co-treatment of neurons with the PI-3K inhibitor LY294002, to inhibit Akt phosphorylation, blocked the ability of endothelial-conditioned media to protect neurons against hypoxia (Figure 1b–c) and oxygen-glucose deprivation (Figure 1d). To confirm that these results were not solely due to an artifact of the endothelial cell line, experiments were repeated with primary human brain endothelial cells. Similarly, primary endothelial-conditioned media increased the phosphorylation of Akt and GSK-3β (Figure 2a), and reduced neuronal cell death after hypoxia (Figure 2b).
Figure 1.
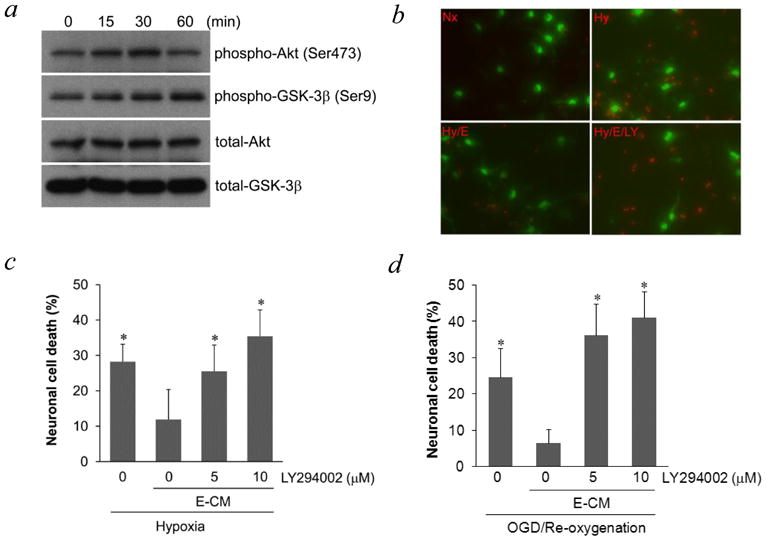
a, Phosphorylation of Akt and GSK-3β in neurons was induced by conditioned media from brain cerebral endothelial cells THBMEC, after short incubation times. b, staining of live (green) and dead (red) neurons after hypoxia, showed that neuronal death was prevented by E-CM (endothelial-conditioned media) and that the PI-3K inhibitor LY294002 blocked the effect of E-CM. Nx for normoxic neurons; Hy for hypoxic neurons; Hy/E-CM for hypoxic neurons exposed to E-CM; Hy/E-CM/LY for hypoxic neurons exposed to E-CM plus LY294002. c, Quantitative analysis of the effects shown in 1b. *P<0.05 versus neurons under hypoxia with E-CM. d, neuronal death after OGD was also prevented by E-CM and LY294002 blocked the effect of E-CM. * P<0.05 versus neurons under OGD with E-CM.
Figure 2.

a, Phosphorylation of Akt and GSK-3β in neurons was induced by conditioned media from brain cerebral endothelial cells HBMEC, after short incubation times. b, co-incubation with the PI-3K inhibitor LY294002 blocked the neuroprotective effect of E-CM from HBMEC. * P<0.05 versus neurons under hypoxia with E-CM.
Reduction of apoptotic mediators in hypoxic neurons by endothelial-conditioned media
Neuronal death typically involves apoptotic-like mechanisms (Ness et al., 2006; Sugawara et al., 2004; Tamatani et al., 1998) so we examined this phenomenon in our model system. After 20-hr hypoxia, cleaved fragments of caspase-3 and caspase-9 were increased in neurons (Figure 3a–d), suggesting the activation of these caspases. Neuroprotection with endothelial-conditioned media effectively suppressed the cleaved levels of both caspase-3 and caspase-9 (Figure 3). Along with caspases, the prototypical apoptotic mediator Bad was also detected. Neuroprotection with endothelial-conditioned media significantly reduced Bad (Figure 3a and Figure 3e).
Figure 3.

a, Immunoblotting showed the cleavage of caspase-9 and caspase-3 were increased in neurons after hypoxia. These effects of hypoxia, and the level of pro-apoptotic protein Bad were all decreased in hypoxic neurons exposed to E-CM. b-e, Optical densitometry was used to quantify each protein, normalized to levels of β–Actin. * P<0.05 between two groups.
Upstream signaling via TrkB in vascular neuroprotection
We and others previously suggested that BDNF in particular may play a central role for neuroprotective coupling between vascular and neuronal compartments (Dugas et al., 2008; Guo et al., 2008; Navaratna et al., 2011). As expected, BDNF protein was easily detectable in our endothelial-conditioned media. ELISA showed these cultures of brain microvascular endothelial cells produced and secreted BDNF at a rate of ≈ 50 pg per 106 cells over 24 hrs. Here, we asked whether the neuroprotective effects on PI-3K/Akt signaling and caspase blockade were related to the BDNF receptor TrkB. Transfer of endothelial-conditioned media to neurons increased phospho-Akt levels in neurons (Figure 4a, as shown in Figure 1a), while blocking BDNF in the endothelial-conditioned media with TrkB/Fc depletion reduced phospho-Akt levels (Figure 4a). Consistent with these upstream effects on Akt signaling, depletion with TrkB/Fc also prevented endothelial-conditioned media from suppressing caspase-3 cleavage and activation (Figure 4b–c). Furthermore, there was some evidence of feedback. Hypoxia increased TrkB protein levels in neurons (Figure 4d–e), which was consistent with previous reports (Ferrer et al., 2001; Martens et al., 2007; Merlio et al., 1993) and might be a compensation reaction in injured neurons to activate this endogenous survival signal. Neuroprotection with endothelial-conditioned media appeared to lower TrkB levels, perhaps indicating less reaction with reduced injury (Figure 4d–e).
Figure 4.

a, E-CM depleted by TrkB/Fc did not induce the phosphorylation of Akt in neurons after 30 min incubation. b, incubation with E-CM blocked cleavage of Caspase-3 in neurons after hypoxia, but incubation with E-CM depleted by TrkB/Fc had no effect on increased cleavage of Caspase-3 in neurons after hypoxia. c, optical densitometry of cleaved Caspase-3, normalized to β–Actin. d, hypoxia increased TrkB expression in neurons, which was blocked by E-CM. Depleted E-CM had no blocking effect on TrkB expression. e, optical densitometry of gp 145 TrkB, normalized to β–Actin. * P<0.05 between two groups. f, proposed mechanisms for neuroprotection by cerebral endothelial cells. Soluble factors from cerebral endothelial cells, such as growth factor BDNF, activate PI-3K/Akt survival signaling in neurons through receptors, leading to the activation of GSK-3β, the inhibition of caspases activation and Bad expression, and finally protect the neurons from damage.
DISCUSSION
It is now recognized that blood vessels are not just inert pipes for delivering blood (Ergun et al., 2011; Franses et al., 2011). Instead, the cerebral endothelium comprises a rich source of factors that support neurogenesis, angiogenesis and endogenous neuroprotection (Dugas et al., 2008; Guo et al., 2008; Leventhal et al., 1999; Shen et al., 2004). In this brief communication, we showed that the ability of cerebral endothelial cells to protect neurons is mediated via upstream TrkB and Akt signaling and downstream caspase suppression (Figure 4f).
By using a media transfer system, we showed that endothelial-conditioned media upregulated pro-survival Akt signaling and protected neurons against hypoxic injury. Although admittedly an in vitro approach, this method has been successfully used in the past to dissect cell-cell signaling under various conditions. Endothelial-conditioned media supported growth of subependymal transplants (Leventhal et al., 1999), promoted neurogenesis (Shen et al., 2004), and supported the survival of motoneurons and cortical neurons in culture (Dugas et al., 2008; Guo et al., 2008). Hence, endothelial cells can secrete paracrine factors that affect adjacent neurons. In this study, this neuroprotective coupling seems to involve TrkB and Akt pathways. This idea becomes important in the context of CNS disease, whereby unhealthy endothelium may no longer protect neurons. Loss of vascular neuroprotection has been hypothesized in stroke, brain trauma and neurodegeneration (Cade, 2008; Guo and Lo, 2009; Zacchigna et al., 2008).
In our model system, vascular neuroprotection seems to be largely mediated by endothelial-derived BDNF since blockade of the TrkB receptor system interferes with this phenomenon. We (Guo et al., 2008) and others (Leventhal et al., 1999) have suggested that brain endothelial cells express high levels of BDNF. But we cannot exclude the possibility of other neutrophins, such as NT-4 and NT-3, which can also bind to and activate TrkB receptor might also play roles in the neuroprotection (Schecterson and Bothwell, 2010). Furthermore, our cell culture system is based on young cells so effects of age and different developmental stages may be important in vivo. Ultimately, use of appropriate animal models is warranted, especially with brain endothelial cell specific transgenic animals. The promoters from tyrosine kinase receptor Tie2 or thyroxine transporter Slco1c1 have been used to create endothelial cell specific or even brain endothelial cell specific transgenic mice (Ridder et al., 2011; Schlaeger et al., 1997). Future studies into vascular neuroprotection using these powerful animal models should be performed.
Taken together, our findings support an endogenous signaling mechanism for vascular neuroprotection. However, several caveats must be kept in mind. First, we focus on cerebral endothelial cells, but other cell types also communicate with neurons. The contributions of astrocytes or pericytes to neuroprotection may be equally important. Second, the experiments here focus on the prevention of caspase-mediated apoptotic-like death. But it is now known that autophagy or even programmed necrosis play key roles in neuronal death (Denton et al., 2012; Pandey et al., 2007; Rosenbaum et al., 2010; Wang et al., 2011). How vascular neuroprotection protects against these other models of neuron death remains to be determined. Third, although Akt signaling is a prominent pro-survival pathway in neurons, it is possible that other parallel mechanisms may also contribute, for example, ERK MAP kinase or JAK-Stat signals (Colodner and Feany, 2010; Karmarkar et al., 2011; Nicolas et al., 2012; Sugawara et al., 2004; Sun et al., 2008). How Akt interacts with other pro-survival pathways in vascular neuroprotection warrants further examination. Finally, these data only provide in vitro proof-of-concept in cells. Further in vivo experiments are required to prove that these signals operate in animal models of CNS disease.
Increasingly, the vast network of cerebral endothelium in the brain is no longer viewed only as plumbing. Paracrine and coupling mechanisms between endothelium and the neuronal compartment may be important for homeostasis and defense against CNS injury and disease. Our findings here may support a role of Akt pro-survival signaling and subsequent caspase suppression as relevant mechanisms for vascular neuroprotection.
Acknowledgments
This study is supported in part by National Institutes of Health Grant P01-NS55104, and a Claflin award from Massachusetts General Hospital to S.G.
Footnotes
Author contributions: S.G. and E.H.L. designed the experiments. S.G. and A.T.S performed experiments and data analysis. S.G., C.W. and E.H.L. wrote the article.
The authors declare no conflict of interest.
References
- Armulik A, Genove G, Mae M, Nisancioglu MH, Wallgard E, Niaudet C, He L, Norlin J, Lindblom P, Strittmatter K, et al. Pericytes regulate the blood-brain barrier. Nature. 2010;468:557–561. doi: 10.1038/nature09522. [DOI] [PubMed] [Google Scholar]
- Cade WT. Diabetes-related microvascular and macrovascular diseases in the physical therapy setting. Phys Ther. 2008;88:1322–1335. doi: 10.2522/ptj.20080008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callahan MK, Williams KA, Kivisakk P, Pearce D, Stins MF, Ransohoff RM. CXCR3 marks CD4+ memory T lymphocytes that are competent to migrate across a human brain microvascular endothelial cell layer. J Neuroimmunol. 2004;153:150–157. doi: 10.1016/j.jneuroim.2004.05.004. [DOI] [PubMed] [Google Scholar]
- Chaitanya GV, Cromer WE, Wells SR, Jennings MH, Couraud PO, Romero IA, Weksler B, Erdreich-Epstein A, Mathis JM, Minagar A, et al. Gliovascular and cytokine interactions modulate brain endothelial barrier in vitro. J Neuroinflammation. 2011;8:162. doi: 10.1186/1742-2094-8-162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colodner KJ, Feany MB. Glial fibrillary tangles and JAK/STAT-mediated glial and neuronal cell death in a Drosophila model of glial tauopathy. J Neurosci. 2010;30:16102–16113. doi: 10.1523/JNEUROSCI.2491-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daneman R, Zhou L, Kebede AA, Barres BA. Pericytes are required for blood-brain barrier integrity during embryogenesis. Nature. 2010;468:562–566. doi: 10.1038/nature09513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denton D, Nicolson S, Kumar S. Cell death by autophagy: facts and apparent artefacts. Cell Death Differ. 2012;19:87–95. doi: 10.1038/cdd.2011.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dugas JC, Mandemakers W, Rogers M, Ibrahim A, Daneman R, Barres BA. A novel purification method for CNS projection neurons leads to the identification of brain vascular cells as a source of trophic support for corticospinal motor neurons. J Neurosci. 2008;28:8294–8305. doi: 10.1523/JNEUROSCI.2010-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ergun S, Tilki D, Klein D. Vascular wall as a reservoir for different types of stem and progenitor cells. Antioxid Redox Signal. 2011;15:981–995. doi: 10.1089/ars.2010.3507. [DOI] [PubMed] [Google Scholar]
- Ferrer I, Krupinski J, Goutan E, Marti E, Ambrosio S, Arenas E. Brain-derived neurotrophic factor reduces cortical cell death by ischemia after middle cerebral artery occlusion in the rat. Acta Neuropathol. 2001;101:229–238. doi: 10.1007/s004010000268. [DOI] [PubMed] [Google Scholar]
- Franses JW, Baker AB, Chitalia VC, Edelman ER. Stromal endothelial cells directly influence cancer progression. Sci Transl Med. 2011;3:66ra65. doi: 10.1126/scitranslmed.3001542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo S, Kim WJ, Lok J, Lee SR, Besancon E, Luo BH, Stins MF, Wang X, Dedhar S, Lo EH. Neuroprotection via matrix-trophic coupling between cerebral endothelial cells and neurons. Proc Natl Acad Sci U S A. 2008;105:7582–7587. doi: 10.1073/pnas.0801105105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo S, Lo EH. Dysfunctional cell-cell signaling in the neurovascular unit as a paradigm for central nervous system disease. Stroke. 2009;40:S4–7. doi: 10.1161/STROKEAHA.108.534388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karmarkar SW, Bottum KM, Krager SL, Tischkau SA. ERK/MAPK is essential for endogenous neuroprotection in SCN2.2 cells. PLoS One. 2011;6:e23493. doi: 10.1371/journal.pone.0023493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leventhal C, Rafii S, Rafii D, Shahar A, Goldman SA. Endothelial trophic support of neuronal production and recruitment from the adult mammalian subependyma. Mol Cell Neurosci. 1999;13:450–464. doi: 10.1006/mcne.1999.0762. [DOI] [PubMed] [Google Scholar]
- Lo EH, Dalkara T, Moskowitz MA. Mechanisms, challenges and opportunities in stroke. Nat Rev Neurosci. 2003;4:399–415. doi: 10.1038/nrn1106. [DOI] [PubMed] [Google Scholar]
- Martens LK, Kirschner KM, Warnecke C, Scholz H. Hypoxia-inducible factor-1 (HIF-1) is a transcriptional activator of the TrkB neurotrophin receptor gene. J Biol Chem. 2007;282:14379–14388. doi: 10.1074/jbc.M609857200. [DOI] [PubMed] [Google Scholar]
- Merlio JP, Ernfors P, Kokaia Z, Middlemas DS, Bengzon J, Kokaia M, Smith ML, Siesjo BK, Hunter T, Lindvall O, et al. Increased production of the TrkB protein tyrosine kinase receptor after brain insults. Neuron. 1993;10:151–164. doi: 10.1016/0896-6273(93)90307-d. [DOI] [PubMed] [Google Scholar]
- Navaratna D, Guo SZ, Hayakawa K, Wang X, Gerhardinger C, Lo EH. Decreased cerebrovascular brain-derived neurotrophic factor-mediated neuroprotection in the diabetic brain. Diabetes. 2011;60:1789–1796. doi: 10.2337/db10-1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ness JM, Harvey CA, Strasser A, Bouillet P, Klocke BJ, Roth KA. Selective involvement of BH3-only Bcl-2 family members Bim and Bad in neonatal hypoxia-ischemia. Brain Res. 2006;1099:150–159. doi: 10.1016/j.brainres.2006.04.132. [DOI] [PubMed] [Google Scholar]
- Nicolas CS, Peineau S, Amici M, Csaba Z, Fafouri A, Javalet C, Collett VJ, Hildebrandt L, Seaton G, Choi SL, et al. The Jak/STAT pathway is involved in synaptic plasticity. Neuron. 2012;73:374–390. doi: 10.1016/j.neuron.2011.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohab JJ, Fleming S, Blesch A, Carmichael ST. A neurovascular niche for neurogenesis after stroke. J Neurosci. 2006;26:13007–13016. doi: 10.1523/JNEUROSCI.4323-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey UB, Nie Z, Batlevi Y, McCray BA, Ritson GP, Nedelsky NB, Schwartz SL, DiProspero NA, Knight MA, Schuldiner O, et al. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature. 2007;447:859–863. doi: 10.1038/nature05853. [DOI] [PubMed] [Google Scholar]
- Ridder DA, Lang MF, Salinin S, Roderer JP, Struss M, Maser-Gluth C, Schwaninger M. TAK1 in brain endothelial cells mediates fever and lethargy. J Exp Med. 2011;208:2615–2623. doi: 10.1084/jem.20110398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbaum DM, Degterev A, David J, Rosenbaum PS, Roth S, Grotta JC, Cuny GD, Yuan J, Savitz SI. Necroptosis, a novel form of caspase-independent cell death, contributes to neuronal damage in a retinal ischemia-reperfusion injury model. J Neurosci Res. 2010;88:1569–1576. doi: 10.1002/jnr.22314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schecterson LC, Bothwell M. Neurotrophin receptors: Old friends with new partners. Dev Neurobiol. 2010;70:332–338. doi: 10.1002/dneu.20767. [DOI] [PubMed] [Google Scholar]
- Schlaeger TM, Bartunkova S, Lawitts JA, Teichmann G, Risau W, Deutsch U, Sato TN. Uniform vascular-endothelial-cell-specific gene expression in both embryonic and adult transgenic mice. Proc Natl Acad Sci U S A. 1997;94:3058–3063. doi: 10.1073/pnas.94.7.3058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Q, Goderie SK, Jin L, Karanth N, Sun Y, Abramova N, Vincent P, Pumiglia K, Temple S. Endothelial cells stimulate self-renewal and expand neurogenesis of neural stem cells. Science. 2004;304:1338–1340. doi: 10.1126/science.1095505. [DOI] [PubMed] [Google Scholar]
- Sugawara T, Fujimura M, Noshita N, Kim GW, Saito A, Hayashi T, Narasimhan P, Maier CM, Chan PH. Neuronal death/survival signaling pathways in cerebral ischemia. NeuroRx. 2004;1:17–25. doi: 10.1602/neurorx.1.1.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, Zhou H, Luo X, Li S, Yu D, Hua J, Mu D, Mao M. Neuroprotection of brain-derived neurotrophic factor against hypoxic injury in vitro requires activation of extracellular signal-regulated kinase and phosphatidylinositol 3-kinase. Int J Dev Neurosci. 2008;26:363–370. doi: 10.1016/j.ijdevneu.2007.11.005. [DOI] [PubMed] [Google Scholar]
- Tamatani M, Ogawa S, Tohyama M. Roles of Bcl-2 and caspases in hypoxia-induced neuronal cell death: a possible neuroprotective mechanism of peptide growth factors. Brain Res Mol Brain Res. 1998;58:27–39. doi: 10.1016/s0169-328x(98)00095-3. [DOI] [PubMed] [Google Scholar]
- Wang JY, Xia Q, Chu KT, Pan J, Sun LN, Zeng B, Zhu YJ, Wang Q, Wang K, Luo BY. Severe global cerebral ischemia-induced programmed necrosis of hippocampal CA1 neurons in rat is prevented by 3-methyladenine: a widely used inhibitor of autophagy. J Neuropathol Exp Neurol. 2011;70:314–322. doi: 10.1097/NEN.0b013e31821352bd. [DOI] [PubMed] [Google Scholar]
- Wolburg H, Noell S, Mack A, Wolburg-Buchholz K, Fallier-Becker P. Brain endothelial cells and the glio-vascular complex. Cell Tissue Res. 2009;335:75–96. doi: 10.1007/s00441-008-0658-9. [DOI] [PubMed] [Google Scholar]
- Zacchigna S, Lambrechts D, Carmeliet P. Neurovascular signalling defects in neurodegeneration. Nat Rev Neurosci. 2008;9:169–181. doi: 10.1038/nrn2336. [DOI] [PubMed] [Google Scholar]
- Zhao H, Sapolsky RM, Steinberg GK. Phosphoinositide-3-kinase/akt survival signal pathways are implicated in neuronal survival after stroke. Mol Neurobiol. 2006;34:249–270. doi: 10.1385/MN:34:3:249. [DOI] [PubMed] [Google Scholar]