

Abstract
Triptolide is a potent natural product, with documented antiproliferative, immunosuppressive, anti-inflammatory, antifertility, and anti-polycystic kidney disease effects. Despite a wealth of knowledge about the biology of this compound, direct intracellular target proteins have remained elusive. We synthesized a biotinylated photoaffinity derivative of triptolide, and used it to identify dCTP pyrophosphatase 1 (DCTPP1) as a triptolide-interacting protein. Free triptolide interacts directly with recombinant DCTPP1, and inhibits the enzymatic activity of this protein. Triptolide is thus the first dCTP pyrophosphatase inhibitor identified, and DCTPP1 is a biophysically validated target of triptolide.
Keywords: DCTPP1, natural products, nucleotides, photoaffinity labeling, triptolide
Introduction
A panoply of functions have been ascribed to the natural product triptolide (1).[1] This compound is the major bioactive constituent of Tripterygium wilfordii Hook F., also known as the Thunder God Vine or lei gong teng, a traditional Chinese medicine that is a rich source of secondary metabolites.[2] A decoction of the vine has a variety of uses in the traditional Chinese pharmacopoeia, including treatment of arthritis, inflammation and autoimmune disorders.[3] Little wonder, then, that triptolide has anti-inflammatory and immunosuppressive functions, but it also has potent antiproliferative and anti-fertility activity. Pure triptolide has shown preclinical efficacy in various models of arthritis, organ transplantation, graft-versus-host disease, and cancer.[4]
Despite this large body of work describing triptolide’s organismal and cellular effects, its molecular functions remain to be fully delineated. Using radiolabeled triptolide and extensive cellular fractionation, our group previously identified polycystin-2 as a triptolide-associating protein.[5] Polycystin-2 is a Ca2+ channel encoded by PKD2, one of two causative genes for autosomal dominant polycystic kidney disease (PKD); this finding is consistent with evidence that some of triptolide’s cellular effects are Ca2+ dependent.[6] When either polycystin-2 or its obligate activator polycystin-1 (PKD1) is mutated, growth-arresting Ca2+ influx in the developing kidney tubule is blocked. This is thought to lead to the disease phenotype of enlarged kidneys ultimately rendered non-functional by fluid-filled cysts. Intriguingly, triptolide can stimulate polycystin-2 channel opening in the absence of polycystin-1,[5] suggesting that it could have therapeutic use in the 85% of PKD patients with a PKD1 mutation. Indeed, this is the case in mouse models in which Pkd1 is lost constitutively,[5] neonatally,[7] or post-natally.[8]
Triptolide also functionally interacts with the general transcription apparatus, decreasing expression of a huge variety of genes.[9] This effect was originally ascribed to NF-κB pathway inhibition, but has recently been attributed to a direct effect on RNA polymerases (RNAPs) (reviewed in [10]). Our group showed that triptolide decreases phosphorylation of Ser2 of the C-terminal domain of RNAP II, leading to decreased RNAP II-mediated transcription and cessation of mRNA splicing, as indicated by nuclear speckle rounding.[11] Subsequent to this, triptolide causes nucleolar disassembly and RNAP I blockade. These findings are consistent with an earlier report of general transcriptional blockade by triptolide,[12] and recent indications that triptolide shuts down total RNA production.[9] Triptolide can induce degradation of RPB1, the catalytic subunit of RNAP II.[9] While the current manuscript was under revision, it was reported that triptolide can covalently interact with and inhibit the general transcription factor TFIIH component XPB, explaining its transcriptional effects.[13]
Remarkably, prior to the identification of XPB as a triptolide target,[13] no direct interaction had been observed between triptolide and RNAP II components, nor indeed with polycystin-2. Others, however, have identified proteins that seem to interact with triptolide. McCallum et al.[14] found that [3H]-triptolide can bind irreversibly to a 90 kDa protein in the nuclear fraction of A549 and THP-1 cells; to our knowledge, this protein has not been conclusively identified. More recently, Soundararajan et al.[15] used an affinity chromatography approach to identify the transmembrane metalloprotease ADAM10 as a triptolide interactor in U937 cells. Triptolide treatment decreased the expression of full-length ADAM10, but increased the levels of a putative non-catalytic fragment of this cancer-associated protein, and synergized with the cytotoxic effect of ADAM10 knockdown.
These intriguing studies point to important functions of triptolide, but to date, no direct interacting proteins have been biophysically confirmed. Here, we use a novel triptolide photoaffinity reagent 2 to identify dCTP pyrophosphatase 1 (DCTPP1, formerly known as XTP3TPA) as a direct interactor with triptolide. The DCTPP1 enzyme has not previously been characterized in humans, although the mouse ortholog (RS21-C6) was identified as an ITP-binding protein[16] and a member of the MazG NTP pyrophosphatase family.[17] The ubiquitously expressed, Mg2+-dependent,[16] cytoplasmic murine enzyme is an α-helical-rich homotetramer[18] that has maximal hydrolytic activity against 5-halo-dCTP analogs, but can also remove pyrophosphate from 5-methyl-dCTP and dCTP.[16] It has been speculated that DCTPP1 may regulate the balance between dCTP and dTTP by hydrolyzing the former to dCMP, which is converted to dUMP by dCMP deaminase, and then to dTMP by thymidylate synthase.[16] Alternatively, DCTPP1 may remove genotoxic halo-dCTP analogs from the nucleotide pool.[16] We show here that the human enzyme has similar catalytic activity to the murine form, and that triptolide is the first known inhibitor of this enzyme.
Results
A photoaffinity pulldown approach to find triptolide targets
We synthesized compound 2, linking triptolide to benzophenone and biotin (Scheme 1), and used this compound or control compound 3 lacking the triptolide moiety to detect proteins that interact specifically with the triptolide photoaffinity reagent in HeLa S3 cell lysates. In the S100 (soluble protein) fraction, four robust bands were observed after UV crosslinking in the presence of 2 that were not pulled down by 3 (Figure 1A). Moreover, one of these bands was covalently biotinylated, as indicated by streptavidin-HRP reactivity (Figure 1B). Binding of these bands to the affinity reagent could be competed with excess triptolide, and interestingly, these bands were still pulled down without UV crosslinking, indicating a strong interaction (Figure 1A). However, in the absence of UV crosslinking, the band was not biotinylated, indicating that the triptolide-protein interaction is noncovalent. Peptide mass fingerprinting identified all four bands as DCTPP1.
Scheme 1.
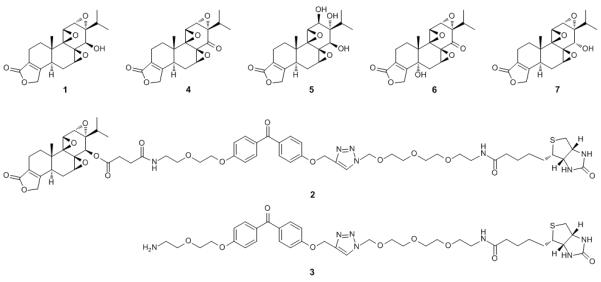
Compounds used in this study. Triptolide (1), triptolide photoaffinity reagent consisting of triptolide linked to benzophenone linked to biotin (2), control photoaffinity reagent (3), triptonide (4), triptriolide (5), 5-hydroxytriptonide (6), and 14-α-triptolide (7).
Figure 1.

Identification of DCTPP1 as a triptolide target. (A) silver stained gel and (B) streptavidin-HRP blot of soluble protein lysates of HeLa S3 cells exposed to the reagents indicated: triptolide affinity reagent 2, affinity reagent 2 + 100-fold excess of “free” triptolide 1, affinity reagent 2 without UV exposure, and control compound 3 lacking triptolide moiety. Triptolide affinity reagent-specific bands of approximately 40, 37, 22, and 18 kDa are evident; the 22 kDa band is biotinylated. (C) anti-DCTPP1 immunoblot. All four bands visible in (A) are detected with the anti-DCTPP1 antibody in the affinity reagent lane; no bands are evident in the triptolide competition lane. Addition of β-mercaptoethanol (β-ME) prior to electrophoresis causes all four bands to collapse into a single, 18 kDa band. (D) 6His-DCTPP1, as detected by anti-DCTPP1 immunoblot, can be specifically pulled down from a crude E. coli lysate using the triptolide affinity reagent 2, but not the control compound 3.
Confirmation of DCTPP1 as the photoaffinity reagent target
Immunoblot with an anti-DCTPP1 antibody confirmed that all four bands seen on silver staining were indeed DCTPP1 (Figure 1C). Treatment of the pulldown reaction with β-mercaptoethanol prior to electrophoresis caused these four bands to collapse into a single, approximately 18 kDa band, the correct size for monomeric DCTPP1. One of the higher molecular weight bands represents dimeric DCTPP1, consistent with the “dimer of dimers” proposed structure of the mouse ortholog of this enzyme.[18] The conformers or post-translational modifications that lead to the other bands remain unknown.
To confirm that DCTPP1 was the direct target of the affinity reagent, and not a member of a complex pulled down by the reagent, we expressed 6His-DCTPP1 in E. coli. This recombinant protein could be efficiently pulled down with 2, but not with linker control compound 3 (Figure 1D), indicating an interaction between 2 and DCTPP1 that does not rely on other eukaryotic accessory proteins.
Biophysical and enzymatic characterization of the triptolide-DCTPP1 interaction
Although we had shown that “free” triptolide could compete DCTPP1 from 2, we wanted to confirm a direct interaction between triptolide 1 and the protein. We purified bacterially expressed GST-DCTPP1 and cleaved off the GST tag, yielding recombinant DCTPP1 (Figure 2). The recombinant protein existed primarily as a dimer (Supplementary Figure S1). By ITC, this protein interacted with 1 with KD = 44 ± 4 μM, ΔH = −19.98 ± 1.70 kcal/mol, n = 0.601 ± 0.075 (Figure 2), suggesting that a single molecule of triptolide binds to each dimer of DCTPP1. Titration of 1 into buffer alone and of buffer into DCTPP1 did not generate any appreciable response in this assay (Figure 2), indicating the specificity of the observed interaction.
Figure 2.

Triptolide binds DCTPP1. (A) Partially purified DCTPP1. Coomassie-stained PAGE shows dimer, denatured by β-mercaptoethanol (+β-ME), monomer and cleaved GST. (B) Isothermal titration calorimetry shows that triptolide binds DCTPP1 with KD = 44 ± 4 μM, ΔH = –19.98 ± 1.70 kcal/mol, n = 0.601 ± 0.075. Integrated heats for the indicated experiments are shown.
Since DCTPP1 is an enzyme, we investigated whether 1 can inhibit its activity. Using a kinetic, luminescent assay that detects pyrophosphate generation, we first established that, as with the murine enzyme, 5-I-dCTP is a good substrate for DCTPP1 (KM = 239 μM, kcat = 8.64×10−4 s−1, kcat/KM = 3.5 s−1·M−1). We then showed that 1 inhibits DCTPP1 activity, with kinetics suggestive of a non-competitive mechanism (Table 1; Supplementary Figure S2). Several close triptolide analogs also inhibited this enzyme with broadly similar efficacy (Table 1). None of these compounds inhibited the luminescent assay itself (data not shown).
Table 1.
Kinetic data for inhibition of human DCTPP1 by triptolide and analogs. Substrate in all cases was 5-I-dCTP.
| Inhibitor | Ki ± SE (μm) |
|---|---|
| Triptolide 1 | 168 ± 17 |
| Triptonide 4 | 135 ± 20 |
| Triptriolide 5 | 85 ± 7 |
| 5-hydroxytriptonide 6 | 356 ± 96 |
| 14-α-triptolide 7 | 194 ± 28 |
Importance of DCTPP1 for triptolide-induced cytotoxicity
Given triptolide’s biochemical effects on DCTPP1, we next tested whether DCTPP1 is an in vivo triptolide target. To determine if DCTPP1 is important for triptolide’s cytotoxic effects, we overexpressed DCTPP1 or knocked it down by siRNA in HeLa cells (Figure 3A). Neither treatment had a perceptible effect on proliferation or cell morphology (data not shown). Moreover, neither treatment changed the cells’ response to triptolide (Figure 3B, C).
Figure 3.

Modulation of intracellular DCTPP1 does not modify triptolide-induced cytotoxicity. (A) DCTPP1-HA overexpression (OE) and siRNA-mediated knockdown (KD) of the overexpressed protein, quantified relative to negative control (Neg) siRNA transfection and normalized to β-actin levels. (B) Dose-response of cell number (measured by CellTiterGlo luminescence in arbitrary units, AU) after 48 hours’ exposure to the indicated triptolide concentration, in HeLa cells transiently transfected with DCTPP1-HA (OE) or vector (pCS2+) control; or (C) transfected with a negative control siRNA (Neg) or an siRNA targeting DCTPP1 (KD). IC50 = 7–10 nM.
Effect of DCTPP1 on BrdC incorporation into DNA
Since one proposed function of DCTPP1 is the removal of potentially toxic, base modified nucleosides from the pool of DNA precursors,[16] we took advantage of a readily available anti-BrdU antibody to investigate this function. BrdC is converted in the cell to BrdCTP, which in turn is converted to BrdUTP before incorporation into DNA as BrdU.[19] We confirmed that treatment of HeLa cells with both BrdC and BrdCTP gave detectable BrdU incorporation of 30–43% of cells after a one-hour treatment, similar to that seen for BrdU treatment. However, neither knockdown nor overexpression of DCTPP1 significantly modified the fraction of anti-BrdU reactive cells (data not shown).
Effect of DCTPP1 on cytidine nucleoside analog-induced cytotoxicity
Since chemotherapeutic cytidine nucleoside analogs cytarabine and gemcitabine must be incorporated into DNA to exert their replication-blocking effects, and since only nucleoside triphosphates are capable of entering DNA synthesis, we asked whether DCTPP1 might function in detoxifying these compounds by maintaining them in the monophosphate state. However, knockdown or overexpression of DCTPP1 did not have any effect on the cytotoxicity of these compounds in HeLa cells, and gemcitabine triphosphate[20] did not show activity as a DCTPP1 substrate in kinetic assays (data not shown).
Discussion
The identification of relevant molecular targets of the potent natural product triptolide 1 remains a challenge. Here, we have identified DCTPP1 as the first known, biophysically validated, direct protein interactor with triptolide. Triptolide interacts strongly, but non-covalently with DCTPP1 (Figure 1), consistent with our previous work[6] suggesting that 1 need not bind covalently in the cell (although others have suggested that it can[13-14]). By exogenous expression of DCTPP1 in E. coli, we showed that our triptolide photoaffinity reagent binds directly to this enzyme; this finding was borne out by ITC, indicating a direct interaction between the protein and natural product (Figure 2). Moreover, 1 and closely related compounds 4–7 inhibit the pyrophosphatase activity of DCTPP1 (Table 1). Triptolide is the first known inhibitor of this enzyme, and one of a very small group of nucleotide pyrophosphatase inhibitors that are not nucleotide analogs. It will be interesting to see if 1 inhibits any other MazG proteins, or more distantly related pyrophosphatases. Triptolide and similar compounds will therefore be of interest to those working on the enzymology and functional relevance of DCTPP1.
However, DCTPP1 is unlikely to be the key physiological target of 1. The compound binds and inhibits recombinant DCTPP1 at concentrations substantially higher than those necessary for biological effects, and modulation of DCTPP1 expression did not change the cytotoxicity of 1 (Figure 3). In addition, the majority of [3H]-triptolide associates with membrane proteins,[6] whereas DCTPP1 is a cytoplasmic enzyme seen in the S100 fraction in our experiments. Of course, endogenous DCTPP1 might be inhibited by lower concentrations of the compound than the recombinant enzyme used here in vitro, and inhibition of DCTPP1 might be responsible for some of triptolide’s effects other than cytotoxicity; this possibility remains to be investigated.
We have cloned and expressed for the first time human DCTPP1, which appears to behave similarly to the orthologous murine enzyme in terms of substrate preference for 5-halo-dCTPs (TWC, unpublished data). However, our recombinant DCTPP1 does not seem to require obligate tetramerization for function, although transient or weak tetramerization cannot be ruled out (Supplementary Figure S1). We investigated whether DCTPP1 is involved in the metabolism of the chemotherapeutic cytidine nucleoside analogs cytarabine and gemcitabine. Modulation of DCTPP1 expression did not affect the cytotoxicity of these agents, nor did gemcitabine triphosphate serve as a substrate for the recombinant enzyme (data not shown). These results suggest that DCTPP1 is not involved in resistance to these drugs. It remains to be seen if DCTPP1 might act to dephosphorylate the triphosphates of antiretroviral cytidine nucleoside analogs, such as zalcitabine (dideoxycytidine), lamivudine (2′,3′-dideoxy-3′-thiacytidine), or emtricitabine (2′-deoxy-5-fluoro-3′-thiacytidine). If such an activity exists, it might suggest a role in inter-individual efficacy of these drugs, based on variations in DCTPP1 expression. DCTPP1 has been postulated to act as a “gatekeeper” enzyme, ensuring that potentially deleterious halogenated nucleotides do not enter DNA synthesis.[16] We tested this hypothesis by monitoring the incorporation of BrdC and BrdCTP into DNA using anti-BrdU antibodies (data not shown). Since we saw no differential incorporation with decreased or increased DCTPP1 expression, it seems unlikely that this enzyme functions to block brominated dCTP from incorporation into the genome, although DCTPP1 might have this function for 5-I-dCTP or other variants in vivo.
In summary, we have shown that the natural product triptolide inhibits DCTPP1, and examined the function of this human enzyme for the first time. This finding is a significant step toward resolving fully the mechanism of triptolide’s action and provides an important new tool for the further study of DCTPP1.
Experimental Section
Chemical synthesis
Full details of chemical syntheses and characterization data are available in the Supporting Information. Briefly, synthesis of 2 began with 3, synthesized as described,[21] which was then coupled to triptolide-succinate (obtained from direct reaction of 1 with succinic anhydride), to give the final triptolide-photoaffinity reagent 2. 14-α-triptolide 7 was obtained by borohydride reduction of triptonide 4 (Sequoia Research Products),[22] while 5-hydroxytriptonide 6 was produced by selenium dioxide oxidation of 4.
Photoaffinity pulldown
Neutravidin-agarose (Thermo) was washed three times with isotonic lysis buffer (25 mM Tris pH 7.4, 150 mM NaCl, Complete Protease Inhibitors (Roche), 1 mM sodium orthovanadate, 1 mM sodium pyrophosphate, 10 mM β-glycerophosphate) and blocked 1 hour with cytochrome c (1 mg/mL). After washing, beads were exposed overnight to affinity reagent 2 or control 3 (100 μM, diluted from 10 mM DMSO stocks). To bind free avidin sites, biotin (1 mM) was added for 1 hour.
A frozen pellet of 5 L equivalent HeLa S3 cells (National Cell Culture Center/Biovest International) was thawed, suspended in isotonic lysis buffer (7.5 mL), and lysed with 25 strokes of a Dounce homogenizer. After a 2 minute centrifugation at 2000 g to remove nuclei and unlysed cells, the supernatant was ultracentrifuged at 100,000 g for 45 minutes. The resulting S100 (soluble protein) fraction was split in four, and appropriate beads were added. For the triptolide competition condition, 1 (1 mM) (Pi & Pi Technology) was added 15 minutes prior to bead addition. After rotation for 1 hour at 4°C, beads were collected, resuspended in isotonic lysis buffer + 1% Triton X-100 in 3 cm tissue culture plates, and exposed to UV (or not) for 30 minutes at 4°C, using a homemade exposure chamber consisting of two desk lamps each fitted with two F15T8/BLB 15W blacklight bulbs, emitting 1.2 mJ·cm−2·s−1 of 366 nm light at sample distance. Beads were then washed with the same buffer into microfuge tubes and washed twice more, followed by two washes in a similar buffer containing 350 mM NaCl and a final wash in a salt-free buffer. Beads were boiled 10 minutes in 2× NuPAGE sample buffer (Invitrogen) (with or without 5% β-mercaptoethanol) prior to separation on 12% Bis-Tris acrylamide gels.
Proteins were detected by silver staining, as follows. SDS-PAGE gels were fixed in 50% methanol-10% acetic acid (2×20 minutes), then washed in 20% ethanol (10 minutes) and water (10 minutes). Gels were reduced in 0.02% sodium thiosulfate solution (1 minute), washed with water (2×20 s), then exposed to 0.2% silver nitrate (30 minutes, 37°C). After a brief wash with water, gels were incubated in developing solution (3% Na2CO3·H2O, 0.05% formaldehyde, 0.001% sodium thiosulfate). Stop solution was 1% acetic acid. Stained gels were imaged with a ChemiDoc XRS+ (BioRad) and QuantityOne software. Trypsinization and LC/MS/MS peptide mass fingerprinting were performed by Midwest Bio Services, with identification using SEQUEST software.
Immunoblotting
DCTPP1 was detected by immunoblot of pulldown experiments or RIPA lysates of transfected cells. Briefly, proteins on SDS-PAGE or native tris-glycine gels were electrotransferred to nitrocellulose membranes (75 V, 90 minutes) and membranes were blocked with 5% BSA (pulldowns) or 5% BLOTTO (cell lysates) in TBST. To detect biotinylated proteins, membranes were incubated with streptavidin-HRP (Thermo), 1:1000 in TBST. To detect DCTPP1, anti-DCTPP1 (Atlas Antibodies or Santa Cruz) was used 1:500 in BLOTTO/TBST, followed by anti-rabbit-HRP (GE). β-actin was detected with a monoclonal antibody (Sigma) at 1:10,000 in TBST followed by anti-mouse-HRP (GE). ECL+ (GE) chemiluminescence was detected by film exposure or with a ChemiDoc XRS+ (BioRad).
Molecular biology
The 510 bp ORF of human DCTPP1 was PCR amplified from a spleen cDNA library (Proquest) using primers adding a 5′ Bam HI site and a 3′ Pac I s i t e : A A G G A T C C C A T G T C T G T G G C C G G T G G G G A and CTTTAATTAACTAGGTTGAGGTCTGGCCTGTGG, respectively. The PCR product was cloned into a modified pRSF-1b vector[23] using these sites, to yield pRSF-1b-6His-DCTPP1. DCTPP1 was subcloned into eukaryotic expression vector pCS2+ by amplification from this construct using the same forward primer, and a reverse primer introducing a C-terminal HA tag and an Eco RI restriction site: TAGAATTCTAGGCATAGTCTGGGACGTCATATGGATAGGTTGAGGTCTGGCCTGTG G. Similarly, DCTPP1 was subcloned from pRSF-1b-6His-DCTPP1 into pGEX-2T using a frameshifted forward primer introducing a Bam HI site, and a reverse primer introducing an Eco RI site: AAGGATCCATGTCTGTGGCCGGTGGGGA and ttGAATTCTAGGTTGAGGTCTGGCCTGTGG, respectively. All vectors were confirmed by sequencing.
6His-DCTPP1 Pulldown from E. coli
E. coli BL21 Star (DE3) (Invitrogen) transformed with pRSF-1b-6His-DCTPP1 was grown from a single colony overnight at 37°C, diluted 10-fold into LB+kanamycin (250 mL), then induced overnight at room temperature with IPTG (0.5 mM), pelleted and frozen at –80°C. Thawed cells were sonicated 3×20s in PBS + EDTA-free Complete Protease Inhibitors (Roche), then centrifuged at 13,000 g for 10 minutes. The supernatant was filtered through 0.45 μm mesh, then added to Neutravidin beads bound with 2 or 3 as above. Pulldown, wash, and immunoblot conditions were as described above.
Recombinant DCTPP1 expression & purification
E. coli BL21 Star (DE3) (Invitrogen) transformed with pGEX-2T-DCTPP1 was grown from a single colony overnight at 37°C in LB+carbenicillin (200 mL), then diluted 10-fold into 2 L and induced for 4 hours at 37°C with IPTG (1 mM), then pelleted and frozen at –80°C. Thawed cells were sonicated 4×30s in PBS, then centrifuged at 13,000 g for 10 minutes. The supernatant was filtered through 0.45 μm mesh, then bound to glutathione-sepharose (GE) for 30 minutes. The beads were washed five times with PBS, then protein eluted with 50 mM Tris-HCl pH 8.0, 10 mM reduced glutathione. Thrombin (GE, 30 units) was added and incubated overnight at 4°C, then buffer was exchanged for Tris-HCl (50 mM) over a PD-10 (GE) column. To remove thrombin and GST, the resulting preparation was separated over a 0-1000 mM KCl gradient on a MonoQ HR 5/5 column (GE) using an ÄKTAExplorer 100 FPLC system controlled by Unicorn 5.2 software (GE). The most intense DCTPP1 peak eluted at 600 mM KCl. This and surrounding fractions were combined and dialyzed twice against 20 mM Tris, pH 7.4 using 6000-8000 MWCO tubing (Spectrum), concentrated over a 10,000 MWCO spin column (Amicon), then stored at 4°C. Recombinant protein concentration was determined by Bradford assay (Bio-Rad) read on a Wallac Victor 2 plate reader. Multimerization was confirmed by native Tris-glycine 10% PAGE and size exclusion chromatography over a calibrated Superdex 75 10/300 GL column with isocratic 20 mM Tris, pH 7.5, 50 mM NaCl.
ITC
ITC experiments were performed on an iTC200 instrument (MicroCal) at 25°C, 1000 rpm stirring. DCTPP1 was diluted in post-dialysis Tris buffer to 50 μM monomer concentration and 2% DMSO. Triptolide was dissolved in minimal DMSO, and brought to a final concentration of 1.5 mM in the same Tris buffer, with final 2% DMSO. Triptolide was injected into DCTPP1 over 39 injections, initial: 0.5 μL, 1 s, 60 s space between injections; then 0.75 μL, 1.5 s, 120 s space between injections. Control experiments were similar, but with 60 s between all injections. Data were analyzed and binding curves fitted using Origin 7 software. Parameters are expressed as values ± SD.
Enzyme kinetic analyses
Kinetic enzyme assays were performed in 384-well, white plates (Nunc) on a Wallac Victor 2 plate reader. Each 20 μL reaction contained assay buffer (50 mM Tris, pH 8.0, 100 mM KCl, 5 mM MgCl2, 100 μg/mL BSA, 1 mM DTT),[15] PPiLight reagent (10 μL) (Lonza), DCTPP1 (125 nM), substrate (at 10, 30, 100, or 300 μM), and inhibitor (at 0, 0.2, 2, 20, 200, or 500 μM in 1 μL DMSO). Substrates in H2O were: 5-I-dCTP (TriLink) and gemcitabine triphosphate (NSC746306 from the NCI/DTP Open Chemical Repository, http://dtp.cancer.gov).[20] Inhibitors in DMSO were triptolide 1 (Pi & Pi Technologies), triptonide 4 and triptriolide 5 (also known as both triol-triptolide and epi-triptolide; Sequoia Research Products), 5-hydroxytriptonide 6, and 14-α-triptolide 7. Octuplicate wells were set up for each condition and the plate pre-warmed at 37°C; prewarmed enzyme and PPiLight reagent master mix was added to start the reaction, which was monitored by 80 reads of luminescence detection, with 122 s between reads of the same well. A sodium pyrophosphate standard curve in quadruplicate was included in all assays. The data points in the linear range of each condition (typically 244 to 732 s) were converted to μM pyrophosphate using the equivalent points on the standard curve, then linear regression was used to find the slope, v0. Prism 5.0 (GraphPad) was used for enzyme kinetic analyses to determine KM, kcat, and Ki using non-competitive kinetics. Parameters are expressed as values ± SE.
DCTPP1 overexpression and knockdown effect on cytotoxicity
For DCTPP1 overexpression, HeLa cells grown in 12-well plates in DMEM with 10% FBS and penicillin-streptomycin were transfected with pCS2+-DCTPP1-HA or empty vector using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s protocol. For knockdown, similar cells were transfected with siRNA (4 pmol) targeting DCTPP1 (SASI_Hs01_00014964) or a negative control (Mission siRNA Universal Negative Control #1, Sigma) using Lipofectamine RNAiMAX (Invitrogen). One day after transfection, transfected cells were trypsinized and added to 384-well, white tissue culture plates (750 cells/well in 25 μL). Octuplicate wells (overexpression) or quintuplicate wells (knockdown) were exposed to the indicated concentrations of triptolide in DMSO (0.4% final), or gemcitabine or cytarabine in H2O for 48 hours, then proliferation was determined by CellTiterGlo (Promega) luminescence on a Wallac Victor 2 Plate Reader. Data were analyzed and IC50 values calculated using Prism 5.0. All experiments were performed at least twice.
BrdC incorporation
HeLa cells transfected with DCTPP1-HA or siRNA as above were trypsinized and plated on poly-L-lysine-coated coverslips at a density of 100,000 cells per well of a 12-well plate. The next day, cells were treated with BrdC (10 μM) (NSC61765 from the NCI/DTP Open Chemical Repository, http://dtp.cancer.gov) or BrdCTP (TriLink) in growth medium for 30 minutes, then washed with PBS and fixed in 70% ethanol in 50 mM glycine-HCl, pH 2.0, 20 minutes at –20°C. After PBS washes, anti-BrdU antibody was applied according to the instructions for the BrdU Labeling and Detection Kit II (Roche), 30 minutes at 37°C. After PBS washes, goat anti-mouse-Alexa 594 conjugate (Invitrogen) was added at 1:500 in PBS, 30 minutes, 37°C. Following final PBS washes, slides were mounted in Vectashield (Vector Labs), and imaged with a Camedia C3040ZOOM camera mounted on a CK40 inverted microscope (Olympus). All experiments were performed twice.
Supplementary Material
Acknowledgments
We thank Dr. Peter Gareiss for assistance with ITC. This work was supported by grants from the NIH (R01AI055914) and PKD Foundation (#139b2r) to C.M.C. T.W.C. was the Canadian Institutes of Health Research Jean-François St-Denis Fellow in Cancer Research and a Bisby Fellow, N.A. was the American-Australian Association’s Alcoa Foundation Fellow, and H.C. was a Feodor-Lynen-Fellow of the Alexander von Humboldt Foundation.
References
- [1].Corson TW, Crews CM. Cell. 2007;130:769–774. doi: 10.1016/j.cell.2007.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Tao X, Lipsky PE. Rheum Dis Clin North Am. 2000;26:29–50. doi: 10.1016/s0889-857x(05)70118-6. [DOI] [PubMed] [Google Scholar]
- [3].Qiu D, Kao PN. Drugs R D. 2003;4:1–18. doi: 10.2165/00126839-200304010-00001. [DOI] [PubMed] [Google Scholar]
- [4].Chen BJ. Leuk Lymphoma. 2001;42:253–265. doi: 10.3109/10428190109064582. [DOI] [PubMed] [Google Scholar]
- [5].Leuenroth SJ, Okuhara D, Shotwell JD, Markowitz GS, Yu Z, Somlo S, Crews CM. Proc Natl Acad Sci U S A. 2007;104:4389–4394. doi: 10.1073/pnas.0700499104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Leuenroth SJ, Crews CM. Chem Biol. 2005;12:1259–1268. doi: 10.1016/j.chembiol.2005.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Leuenroth SJ, Bencivenga N, Igarashi P, Somlo S, Crews CM. J Am Soc Nephrol. 2008;19:1659–1662. doi: 10.1681/ASN.2008030259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Leuenroth SJ, Bencivenga N, Chahboune H, Hyder F, Crews CM. Nephrol Dial Transplant. 2010;25:2187–2194. doi: 10.1093/ndt/gfp777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Vispe S, DeVries L, Creancier L, Besse J, Breand S, Hobson DJ, Svejstrup JQ, Annereau JP, Cussac D, Dumontet C, Guilbaud N, Barret JM, Bailly C. Mol Cancer Ther. 2009;8:2780–2790. doi: 10.1158/1535-7163.MCT-09-0549. [DOI] [PubMed] [Google Scholar]
- [10].Pan J. Cancer Lett. 2010;292:149–152. doi: 10.1016/j.canlet.2009.11.018. [DOI] [PubMed] [Google Scholar]
- [11].Leuenroth SJ, Crews CM. Cancer Res. 2008;68:5257–5266. doi: 10.1158/0008-5472.CAN-07-6207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].McCallum C, Kwon S, Leavitt P, Shoop W, Michael B, Felcetto T, Zaller D, O’Neill E, Frantz-Wattley B, Thompson C, Forrest G, Carballo-Jane E, Gurnett A. Therapy. 2005;2:261–273. [Google Scholar]
- [13].Titov DV, Gilman B, He QL, Bhat S, Low WK, Dang Y, Smeaton M, Demain AL, Miller PS, Kugel JF, Goodrich JA, Liu JO. Nat Chem Biol. 2011;7:182–188. doi: 10.1038/nchembio.522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].McCallum C, Kwon S, Leavitt P, Shen DM, Liu W, Gurnett A. Immunobiology. 2007;212:549–556. doi: 10.1016/j.imbio.2007.02.002. [DOI] [PubMed] [Google Scholar]
- [15].Soundararajan R, Sayat R, Robertson GS, Marignani PA. Cancer Biol Ther. 2009;8:2054–2062. doi: 10.4161/cbt.8.21.9803. [DOI] [PubMed] [Google Scholar]
- [16].Nonaka M, Tsuchimoto D, Sakumi K, Nakabeppu Y. FEBS J. 2009;276:1654–1666. doi: 10.1111/j.1742-4658.2009.06898.x. [DOI] [PubMed] [Google Scholar]
- [17].Moroz OV, Murzin AG, Makarova KS, Koonin EV, Wilson KS, Galperin MY. J Mol Biol. 2005;347:243–255. doi: 10.1016/j.jmb.2005.01.030. [DOI] [PubMed] [Google Scholar]
- [18].Wu B, Liu Y, Zhao Q, Liao S, Zhang J, Bartlam M, Chen W, Rao Z. J Mol Biol. 2007;367:1405–1412. doi: 10.1016/j.jmb.2007.01.057. [DOI] [PubMed] [Google Scholar]
- [19].Cramer JW, Prusoff WH, Welch AD. Biochem Pharmacol. 1961;8:331–335. doi: 10.1016/0006-2952(61)90111-3. [DOI] [PubMed] [Google Scholar]
- [20].Risbood PA, Kane CT, Jr., Hossain MT, Vadapalli S, Chadda SK. Bioorg Med Chem Lett. 2008;18:2957–2958. doi: 10.1016/j.bmcl.2008.03.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Tae HS, Hines J, Schneekloth AR, Crews CM. Org Lett. 2010;12:4308–4311. doi: 10.1021/ol101801u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Buckanin RS, Chen SJ, Frieze DM, Sher FT, Berchtold GA. J Am Chem Soc. 1980;102:1200–1201. [Google Scholar]
- [23].Gareiss PC, Schneekloth AR, Salcius MJ, Seo SY, Crews CM. Chembiochem. 2010;11:517–522. doi: 10.1002/cbic.200900547. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.