

Abstract
Background
Dictyocaulus species are strongylid nematodes of major veterinary significance in ruminants, such as cattle and cervids, and cause serious bronchitis or pneumonia (dictyocaulosis or “husk”). There has been ongoing controversy surrounding the validity of some Dictyocaulus species and their host specificity. Here, we sequenced and characterized the mitochondrial (mt) genomes of Dictyocaulus viviparus (from Bos taurus) with Dictyocaulus sp. cf. eckerti from red deer (Cervus elaphus), used mt datasets to assess the genetic relationship between these and related parasites, and predicted markers for future population genetic or molecular epidemiological studies.
Methods
The mt genomes were amplified from single adult males of D. viviparus and Dictyocaulus sp. cf. eckerti (from red deer) by long-PCR, sequenced using 454-technology and annotated using bioinformatic tools. Amino acid sequences inferred from individual genes of each of the two mt genomes were compared, concatenated and subjected to phylogenetic analysis using Bayesian inference (BI), also employing data for other strongylids for comparative purposes.
Results
The circular mt genomes were 13,310 bp (D. viviparus) and 13,296 bp (Dictyocaulus sp. cf. eckerti) in size, and each contained 12 protein-encoding, 22 transfer RNA and 2 ribosomal RNA genes, consistent with other strongylid nematodes sequenced to date. Sliding window analysis identified genes with high or low levels of nucleotide diversity between the mt genomes. At the predicted mt proteomic level, there was an overall sequence difference of 34.5% between D. viviparus and Dictyocaulus sp. cf. eckerti, and amino acid sequence variation within each species was usually much lower than differences between species. Phylogenetic analysis of the concatenated amino acid sequence data for all 12 mt proteins showed that both D. viviparus and Dictyocaulus sp. cf. eckerti were closely related, and grouped to the exclusion of selected members of the superfamilies Metastrongyloidea, Trichostrongyloidea, Ancylostomatoidea and Strongyloidea.
Conclusions
Consistent with previous findings for nuclear ribosomal DNA sequence data, the present analyses indicate that Dictyocaulus sp. cf. eckerti (red deer) and D. viviparus are separate species. Barcodes in the two mt genomes and proteomes should serve as markers for future studies of the population genetics and/or epidemiology of these and related species of Dictyocaulus.
Keywords: Dictyocaulus (Nematoda: Strongylida), Lungworms, Dictyocaulosis, Cattle, Deer, Mitochondrial genome, Systematics, Epidemiology
Background
Species of Dictyocaulus (Strongylida: Dictyocaulidae) are economically important parasitic nematodes of the lungs of various ungulate animals, including domestic and wild ruminants (e.g., cattle and deer) [1], and are causative agents of bronchitis and pneumonia (dictyocaulosis or “husk”) [2]. All members of the Dictyocaulidae have direct life cycles [3]. Adult nematodes live in the bronchi, where the ovo-viviparous females produce eggs, from which first stage larvae (L1s) hatch in the lung. The L1s are shed in the faeces from the infected host. Under favourable environmental conditions, the L1s develop through to the infective third-stage larvae (L3s) over a period of ~ 4–6 days. After ingestion by the host, the L3s migrate through the intestinal wall to the mesenteric lymph nodes, moult, and, as fourth stage larvae (L4s), are transported to the lungs. The L4s penetrate the alveoli, moult and then develop into dioecious adults. The period from ingestion to reproductive maturity is estimated at 21–35 days [3].
Since Dictyocaulus was first erected [4], there has been ongoing controversy as to the validity of some species within this genus, particularly those infecting bovids and/or cervids [5], because of a lack of reliable morphological features for their unequivocal identification. The identification of Dictyocaulus species and populations is not only important from a taxonomic perspective, but also has implications for studying the host and geographical distributions of the parasites, the cross transmissibility of Dictyocaulus between or among host species (particularly between bovine and cervid hosts) and also for the control of dictyocaulosis. Although molecular tools, employing genetic markers in ribosomal DNA, have found utility for systematic and/or epidemiological studies of some species [6-13], there is still limited information on the genetic composition of Dictyocaulus populations in different ruminant host species and countries around the world.
Mitochondrial (mt) genomes provide markers for systematic, genetic and epidemiological investigations of species of strongylid nematodes, with a perspective on discovering population variants and cryptic species and exploring transmission patterns linked to particular genotypes of a species [14-16]. For instance, we have shown that not only are mt genomic regions useful for exploring the population genetic structures of Dictyocaulus viviparus[12,17], concatenated amino acid sequences inferred from whole mt genomes can provide barcodes for nematodes [16]. Although nucleotide sequence variation can be considerable (≤3.5%; upon pairwise comparison) in some of the protein coding mt genes of D. viviparus studied to date [12], the inferred amino acid sequence variation is less. For example, previous studies have shown amino acid sequence variation of 0–1.5% (over 131 positions in COX1) (among 252 individual worms) [17], of 0–1.6% for COX3 (over 125 positions) and 0–2.3% for NAD5 (over 132 positions) (72 individual worms) [12]. Importantly, current evidence indicates that concatenated amino acid sequence datasets can be employed for the retesting of hypotheses regarding the systematic relationships of nematodes; such sequence datasets are relatively large and usually have excellent phylogenetic signal, often achieving nodal support values of 98-100% in tree reconstructions [16,18]. High throughput sequencing and new computational approaches [19] have underpinned these advances, and now enable mt proteomic barcodes to be defined for Dictyocaulus species from a range of ungulate hosts. Here, as a first step, we used a massively parallel sequencing method and semi-automated bioinformatic pipeline for the characterisation of the mt genomes of D. viviparus (from Bos taurus) and Dictyocaulus sp. cf. eckerti (from Cervus elaphus) (cf. [20]), which we compared directly with those of other lungworms, for which published whole mt genomic datasets were available [16,21]. We also studied the genetic relationships between these two Dictyocaulus species and selected representatives of the order Strongylida, and identified regions in the mt genomes of Dictyocaulus that might serve as markers for future population genetic or molecular epidemiological studies.
Methods
Parasites, DNA isolation and identification
Adult specimens of Dictyocaulus were collected from the bronchi of the lungs of cattle (from Sweden) or red deer (from New Zealand) in previous studies [17,20,22,23]. The worms originally collected were washed extensively in physiological saline and then stored at −80 °C until use. Upon thawing, the anterior and posterior ends of each nematode were cut off and cleared in lactophenol for subsequent morphological identification. The mid-body section of each worm was used for the extraction of genomic DNA using a small-scale sodium dodecyl-sulphate (SDS)/proteinase K digestion and mini-column purification (Wizard DNA Clean-Up Kit, Promega, USA) [23]. The molecular identity of each specimen was verified by PCR-based sequencing of the second internal transcribed spacer (ITS-2) of nuclear ribosomal DNA (rDNA) using an established method [23].
Sequencing and assembly of mt genomes
Using the protocol described by Hu et al. [24], the complete mt genome was amplified by long-PCR (BD Advantage 2, BD Biosciences) as two overlapping amplicons (~5 kb and ~10 kb) from the genomic DNA from the mid-body section of a single male specimen of D. viviparus from B. taurus and of Dictyocaulus sp. cf. eckerti from C. elaphus. Amplicons were consistently produced from the positive control samples (total genomic DNA of Angiostrongylus vasorum); in no case was a product detected for any of the negative (no-template) controls. Amplicons were then treated with shrimp alkaline phosphatase and exonuclease I, and quantified in a spectrophotometer (ND-1000 UV–VIS v.3.2.1, NanoDrop Technologies). Following an electrophoretic analysis of quality, the two amplicons (2.5 μg of each) from each of the two worms from each host species were pooled and then sequenced using the 454 Genome Sequencer FLX (Roche) [25]. The mt genome sequences (GenBank accession nos. JX519459 and JX519460) were each assembled from (~300 bp) reads using the program CAP3 [26].
Annotation and analysis of mt genomic sequence data
The genes and features of each mt genome were annotated using an established computational pipeline [16]. In brief, each protein-encoding mt gene was identified by local alignment comparison (six reading frames) using amino acid sequences conceptually translated from corresponding genes from the mt genome of a reference species (e.g., Metastrongylus pudendotectus; accession no. GQ888714; [16]). The large and small subunits of the mt rrn (ribosomal RNA = rRNA) genes (rrnS and rrnL, respectively) were identified by local alignment of nucleotide sequence data. The trn (transfer RNA = tRNA) genes were predicted (from both strands) according to their structure, using scalable models, based on the standard nematode mt tRNAs [15]. All predicted trn genes were then grouped, based on their anti-codon sequence, and identified based on the amino acid encoded by this anti-codon. Two separate trn gene groups were predicted each for serine (one each for the anticodons AGN and UCN, respectively) and leucine (one each for the anticodons CUN and UUR, respectively), because these trn genes are duplicated in many invertebrate mt genomes [15]. All predicted tRNAs within each amino acid group were ranked based on structural “strength” (as inferred by the number of nt mismatches in each stem), and the 100 best-scoring structures for each group were compared by BLASTn against a custom database representing all published nematode mt genome sequences available in the GenBank database (accessible viahttp://www.ncbi.nlm.nih.gov).
All trn genes of each mt genome were then identified and annotated based on maximum sequence identity to known nematode tRNAs. Annotated sequence data were imported using the program SEQUIN (available viahttp://www.ncbi.nlm.nih.gov/Sequin/) for the final verification of the mt genome organization/annotation prior to submission to the GenBank database.
Sliding window analysis was performed on the aligned, complete mt genome sequences of the two Dictyocaulus species using DnaSP v.5 [27]. The alignment of these sequences was achieved using the program MUSCLE v.3.8 [28], as implemented in SeaView v.4 [29]. Keeping the nucleotides in frame, there were no ambiguously aligned regions. A sliding window of 300 bp (steps of 10 bp) was used to estimate nucleotide diversity (π) over the entire alignment; indels were excluded using DnaSP. Nucleotide diversity for the entire alignments was plotted against midpoint positions of each window, and gene boundaries were defined.
Alignment and phylogenetic analysis of concatenated amino acid sequence data
The amino acid sequences conceptually translated from individual genes of each of the two mt genomes (representing D. viviparus and Dictyocaulus sp. cf. eckerti) were concatenated. Selected for comparison were concatenated amino acid sequences predicted from published mt genomes from key nematodes representing the order Strongylida, including M. pudendotectus, M. salmi, Angiostrongylus cantonensis and A. costaricensis (accession nos. GQ888714, GQ888715, GQ398121 and GQ398122, respectively; superfamily Metastrongyloidea), Haemonchus contortus (NC_010383; Trichostrongyloidea), Ancylostoma caninum (FJ483518; Anyclostomatoidea) and Oesophagostomum dentatum (GQ888716; Strongyloidea) [16,21]. All amino acid sequences (considering all homologous characters) were aligned using MUSCLE [28] and then subjected to phylogenetic analysis using Bayesian inference (BI), essentially as described by Jex et al. [16], employing an unconstrained model prior (prset aamodelpr = mixed). The tree was functionally rooted against Metastrongylus species, the earliest divergent metastrongyloid in Jex et al. [16].
Results
Features of the mt genomes of D. viviparus and Dictyocaulus sp. cf. eckerti from red deer
The circular mt genome sequences determined for D. viviparus and Dictyocaulus sp. cf. eckerti were 13,310 bp and 13,296 bp in size (Figure 1), being similar to those published for Metastrongylus pudendotectus (13,793 bp) and M. salmi (13,778 bp) [16]. The nucleotide compositions of the two Dictyocaulus mt genomes were similar, being ~ 24.6% for A, 6.3% for C, 17.3% for G and 51.8% for T (Table 1). As expected, these two mt genomes were AT-rich, with T and C being the most and least favoured nucleotides, respectively. Both mt genomes contained genes encoding 12 proteins (COX1-3, NAD1-6, NAD4L, ATP6 and CYTB), two rrn and 22 trn genes but, as expected (cf. [15]), lacked an atp8 gene. All genes were inferred to be transcribed in the same direction (5’ > 3’) (Figure 1) and the gene arrangement (GA2) was the same as that of other strongylid nematodes [15].
Figure 1.

Schematic representation of the circular mt genome of Dictyocaulus viviparus and Dictyocaulus sp. cf. eckerti. Each transfer RNA gene is identified by a one-letter amino acid code and ribosomal RNA genes are shaded.
Table 1.
Nucleotide composition (%) of the entire mt genome of Dictyocaulus viviparus (Dv) and Dictyocaulus sp. cf. eckerti (De), and of protein and RNA genes within this genome
| Lungworm | Nucleotide | Length (bp) | A | C | G | T | A + T |
|---|---|---|---|---|---|---|---|
|
Dv |
Entire sequence |
13310 |
24.6 |
6.3 |
17.3 |
51.8 |
76.4 |
| |
Protein genes |
10143 |
21.6 |
6.7 |
18.6 |
53.1 |
74.7 |
| |
RNA genes |
1659 |
33.5 |
5.3 |
14.0 |
47.2 |
80.7 |
|
De |
Entire sequence |
13296 |
24.7 |
6.3 |
17.4 |
51.6 |
76.3 |
| |
Protein genes |
9807 |
21.9 |
6.9 |
18.6 |
52.6 |
74.5 |
| RNA genes | 1659 | 33.9 | 5.6 | 14.3 | 46.2 | 80.1 |
Protein genes and codon usages
The initiation and termination codons predicted for the protein encoding genes of D. viviparus were compared with those of Dicyocaulus sp. cf. eckerti (Table 2). The commonest start codon for D. viviparus was ATT and TTG (for three of 12 proteins each), followed by ATA and GTT (two genes), ATC and TTA (one gene each); for Dictyocaulus sp. cf. eckerti, it was ATA (four genes), followed by TTG (three genes), ATG (two genes), ATT, GTT and TTA (one gene each). Eight mt protein genes in D. viviparus and nine in Dictyocaulus sp. cf. eckerti had a TAA or TAG translation termination codon; the other protein genes ended in an abbreviated stop codon, such as TA or T (Table 2). For both Dicyocaulus species, the 3'-ends of these genes were immediately adjacent to a downstream trn gene (Figure 1; Table 2), consistent with the arrangement for M. salmi[16].
Table 2.
Locations and lengths of protein encoding genes in the mt genome of Dictyocaulus viviparus (Dv) and Dictyocaulus eckerti (De) as well as their initiation and termination codons and the lengths of the predicted proteins
|
Gene |
Positions and nucleotide sequence lengths (bp) |
Initiation and Termination codons and the amino acid sequence lengths |
||
|---|---|---|---|---|
| D. viviparus | D. eckerti | D. viviparus | D. eckerti | |
|
cox1 |
1-1579 (1578) |
1-1573 (1572) |
ATC-TAA (525) |
TTG-TAA (523) |
|
trnC |
1580-1634 (54) |
1574-1629 (55) |
|
|
|
trnM |
1636-1693 (57) |
1630-1686 (56) |
|
|
|
trnD |
1693-1749 (56) |
1686-1743 (57) |
||
|
trnG |
1749-1806 (57) |
1744-1798 (54) |
||
|
cox2 |
1806-2490 (684) |
1795-2482 (687) |
ATT-TAG (227) |
TTA-TAG (228) |
|
trnH |
2491-2549 (58) |
2483-2541 (58) |
||
|
rrnL |
2555-3516 (961) |
2549-3508 (959 |
|
|
|
nad3 |
3502-3835 (333) |
3482-3800 (318) |
GTT-TAA (110) |
TTG-TAG (105) |
|
nad5 |
3844-5414 (1570) |
3822-5388 (1566) |
GTT-T (523) |
GTT-T (522) |
|
trnA |
5414-5469 (55) |
5389-5444 (55) |
|
|
|
trnP |
5631-5686 (55) |
5603-5659 (56) |
|
|
|
trnV |
5684-5739 (55) |
5657-5712 (55) |
|
|
|
nad6 |
5739-6174 (435) |
5728-6142 (414) |
TTG-TAG (144) |
ATA-TAA (137) |
|
nad4L |
6181-6415 (234) |
6129-6383 (255) |
ATT-T (77) |
ATT-T (84) |
|
trnW |
6413-6469 (56) |
6376-6432 (56) |
|
|
|
trnE |
6469-6527 (58) |
6432-6490 (58) |
|
|
|
rrnS |
6504-7200 (696) |
6471-7165 (694) |
|
|
|
trnS (UCN) |
7191-7256 (65) |
7166-7219 (53) |
|
|
|
trnN |
7253-7306 (53) |
7218-7272 (54) |
|
|
|
trnY |
7304-7359 (55) |
7269-7325 (56) |
|
|
|
nad1 |
7350-8232 (882) |
7316-8138 (822) |
TTG-T (271) |
ATA-TAG (273) |
|
atp6 |
8234-8831 (597) |
8231-8813 (582) |
ATT-TAA (199) |
ATA-TAA (193) |
|
trnK |
8832-8891 (59) |
8815-8874 (59) |
|
|
|
trnL (UUR) |
8898-8954 (56) |
8875-8930 (55) |
|
|
|
trnS (AGN) |
8955-9005 (50) |
8931-8988 (57) |
|
|
|
nad2 |
8986-9802 (816) |
8989-9812 (823) |
TTG-T (271) |
ATA-TAG (273) |
|
trnI |
9842-9897 (55) |
9824-9880 (56) |
|
|
|
trnR |
9897-9965 (68) |
9881-9935 (54) |
|
|
|
trnQ |
9952-10006 (54) |
9936-9990 (54) |
|
|
|
trnF |
10006-10064 (58) |
9990-10047 (57) |
|
|
|
cob |
10050-11103 (1053) |
10034-11084 (1050) |
ATA-T (350) |
ATG-T (349) |
|
trnL (CUN) |
11161-11215 (54) |
11142-11197 (55) |
|
|
|
cox3 |
11216-11987 (771) |
11197-11989 (792) |
TTG-TAA (256) |
TTG-TAA (263) |
|
trnT |
11982-12034 (52) |
11963-12019 (56) |
|
|
| nad4 | 12038-13262 (1224) | 12077-13244 (1167) | TTA-TAG (407) | ATA-TAG (388) |
Gene abbreviations are according to [69].
The codon usage for the 12 protein genes of D. viviparus was also compared with Dictyocaulus sp. cf. eckerti (Table 3); 62 of the 64 possible codons were used. Neither codon CGC (Arg) nor CTC (Leu) were utilized in the mt genome of D. viviparus. Codons CGC (Arg) and GAC (Asp) were not utilized in the mt genome of Dictyocaulus sp. cf. eckerti. The preferred nucleotide usage at the third codon position of mt protein genes of D. viviparus and Dictyocaulus sp. cf. eckerti reflects the overall nucleotide composition of these mt genomes. At this position, T is the most frequently, and C the least frequently used. For D. viviparus and Dictyocaulus sp. cf. eckerti, the codons ending in A have higher frequencies than the codons ending in G, which is similar to, for example, other members of the order Strongylida and Caenorhabditis elegans (Rhabditida), but distinct from Ascaris suum (Ascaridida) and Onchocerca volvulus (Spirurida) (accession nos. in [15]). As the usage of synonymous codons is proposed to be preferred in gene regions of functional significance, codon bias might be linked to selection at silent sites [30,31].
Table 3.
Number of codons and codon usages (%) in mt protein genes of Dictyocaulus viviparus (Dv) and Dictyocaulus sp. cf. eckerti (De)
| Amino acid | Codon | Dv | De |
|---|---|---|---|
| Non-polar |
|
|
|
| Alanine |
GCN |
59 (1.7) |
60 (1.8) |
| Isoleucine |
ATY |
185 (5.5) |
192 (5.9) |
| Leucine |
CTN |
37 (1.1) |
43 (1.3) |
| Leucine |
TTR |
492 (14.6) |
476 (14.6) |
| Methionine |
ATR |
175 (5.2) |
191 (5.8) |
| Phenylalanine |
TTY |
589 (17.4) |
550 (16.8) |
| Proline |
CCN |
71 (2.1) |
63 (1.9) |
| Tryptophan |
TGR |
71 (2.1) |
75 (2.3) |
| Valine |
GTN |
292 (8.6) |
288 (8.8) |
| Polar |
|
|
|
| Aspargine |
AAY |
121 (3.6) |
121 (3.7) |
| Cysteine |
TGY |
57 (1.7) |
72 (2.2) |
| Glutamine |
CAR |
32 (0.9) |
31 (0.9) |
| Glycine |
GGN |
191 (5.7) |
177 (5.4) |
| Serine |
AGN |
165 (4.9) |
158 (4.8) |
| Serine |
TCN |
165 (4.9) |
173 (5.3) |
| Threonine |
ACN |
90 (2.7) |
81 (2.5) |
| Tyrosine |
TAY |
196 (5.8) |
191 (5.8) |
| Acidic |
|
|
|
| Aspartate |
GAY |
69 (2.0) |
70 (2.1) |
| Glutamate |
GAR |
73 (2.2) |
63 (1.9) |
| Basic |
|
|
|
| Arginine |
CGN |
35 (1.0) |
30 (0.9) |
| Histidine |
CAY |
49 (1.4) |
50 (1.5) |
| Lysine | AAR | 107 (3.2) | 101 (3.1) |
International Union of Pure and Applied Chemistry (IUPAC) codes (N = A, G, C or T; Y = C or T; R = A or G) were used.
AT bias in the nucleotide composition was also reflected in a bias in the amino acid composition of proteins. The AT-rich codons represent the amino acids Phe, Ile, Met, Tyr, Asn or Lys, and the GC-rich codons represent Pro, Ala, Arg or Gly. In the mt genomes of D. viviparus and Dictyocaulus sp. cf. eckerti, the most frequently used codons are TTT (Phe), TTA (Leu), ATT (Ile), TTG (Leu), TAT (Tyr) and GTT (Val). The least frequently used codons were CTA, CTG (Leu), ATC (Ile), GTC (Val), AGC (Ser), CCC (Pro), GCC (Ala), TAC (Tyr), CAC (His), AAC (Asn), CGA (Arg), TCC (Ser) and GGC (Gly). All four GC-rich only codons are represented here, and every codon had at least one C. When the frequencies of synonymous codons within the AT-rich group, such as Phe (TTT, 16.1% and 16.2%; TTC, 0.7% and 0.3%), Ile (ATT, 5.7% and 5.3%; ATC, 0.15% and 0.03%), were compared between both mt genomes, the frequency was always less if the third position was a C.
Transfer RNA genes
Twenty-two trn genes were identified in the mt genomes of both D. viviparus and Dictyocaulus sp. cf. eckerti. The trn gene sequences ranged from 50–68 nt in length. The trn structures had a 7 bp amino-acyl arm, a 3–4 bp DHU arm, a 4–5 bp anticodon stem, a 7 base anticodon loop, with a T always preceding an anticodon and a purine always following an anticodon. Twenty of the 22 trn genes (i.e. excluding the two trnS genes) had a predicted secondary structure with a 3–4 bp DHU stem and a DHU loop of 5–8 bases, in which the variable TψC arm and loop were replaced by a “TV-replacement loop” of 6–12 bases, in accordance with other nematodes [15]. The mt trnS of both Dictyocaulus mt genomes had a secondary structure consisting of a DHU replacement loop of 4–6 bases, 3 bp TψC arm, TψC loop of 6–9 bases and a variable loop of 4 bases, consistent with other nematodes of the class Secernentea [32,33], but distinct from Trichinella spiralis and Trichuris suis (class Adenophorea) [34,35].
Ribosomal RNA genes
The rrnS and rrnL genes of each of the two Dictyocaulus species were identified by sequence comparisons with homologous sequences of A. cantonensis. The rrnS gene was located between trnE and trnS (UCN), and rrnL was between trnH and nad3. The two genes were separated from one another by the protein genes nad3, nad5, nad6 and nad4L (Figure 1). The sizes of the rrnS genes of D. viviparus and Dictyocaulus sp. cf. eckerti were 696 and 694 bp, respectively. The sizes of the rrnL genes of D. viviparus and Dictyocaulus sp. cf. eckerti were 961 bp and 959 bp, respectively. The lengths of these two genes were similar to those of other lungworms for which mt genomes have been characterised (696–699 bp for rrnS, and 958–961 bp for rrnL; [16,21]). The AT contents of the rrn genes for D. viviparus and Dictyocaulus sp. cf. eckerti were 80.7% and 80.1%, respectively (Table 1). The overall percentage of identity in rrnS sequence between the two species was 77.3%, whereas for of rrnL, the identity was 75.8%.
Sequence comparisons and genetic relationships of D. viviparus and Dictyocaulus sp. cf. eckerti with selected strongylids
The amino acid sequences predicted from individual protein-encoding mt genes of D. viviparus were compared with those of Dictyocaulus sp. cf. eckerti (Table 4). Pairwise comparisons of the concatenated sequences revealed identities of 63.0-96.6% (65.5% overall) between them. Based on identity, COX-1 was the most conserved protein, whereas NAD6 and ATP6 were the least conserved (see Table 4). Phylogenetic analysis of the concatenated amino acid sequence data for the 12 mt proteins showed that both D. viviparus and Dictyocaulus sp. cf. eckerti were closely related, and grouped (pp = 1.00) to the exclusion of Metastrongylus and Angiostrongylus species (Metastrongyloidea) as well as H. contortus (Trichostrongyloidea), An. caninum (Ancylostomatoidea) and O. dentatum (Strongyloidea) (Figure 2).
Table 4.
Amino acid sequence identities in the inferred mt proteins between Dictyocaulus viviparus (Dv) and Dictyocaulus sp. cf. eckerti (De)
| Protein | Identity between Dv and De (%) |
|---|---|
| ATP6 |
64.0 |
| COB |
84.8 |
| COX1 |
96.6 |
| COX2 |
83.3 |
| COX3 |
90.0 |
| NAD1 |
73.7 |
| NAD2 |
68.5 |
| NAD3 |
66.3 |
| NAD4 |
80.1 |
| NAD4L |
71.8 |
| NAD5 |
77.6 |
| NAD6 |
63.0 |
| All proteins (concatenated) | 65.5 |
Gene product abbreviations are according to [69].
Figure 2.

Relationship of Dictyocaulus viviparus with Dictyocaulus sp. cf. eckerti and selected species representing different superfamilies of the order Strongylida, including Metastrongylus pudendotectus, M. salmi (Metastrongyloidea), Haemonchus contortus (Trichostrongyloidea), Oesophagostomum dentatum (Strongyloidea) and Ancylostoma caninum (Ancylostomatoidea), based on a phylogenetic analysis of concatenated amino acid sequence data for the 12 mt proteins by Bayesian inference. Posterior probabilities are indicated at each node. Scale bar represents number of substitutions per site.
Nucleotide variation between the mt genomes of D. viviparus and Dictyocaulus sp. cf. eckerti
Nucleotide diversities calculated from pairwise comparisons across the mt genomes of D. viviparus and Dictyocaulus sp. cf. eckerti, achieved by sliding window analyses, are shown in Figure 3. Also indicated on the graph are the regions of cox1 (452 bp), and of trnC_M_D_G (320 bp), rrnL (484 bp), nad5 (426 bp) and cox3 (401 bp) used previously as markers to assess genetic diversity within D. viviparus populations [12,17]. In Figure 3, these markers are indicated with M1, M2, M3, M4 and M5, respectively, and lengths include PCR primers. Greatest nucleotide diversity was detected within nad6, followed by peaks of variation within nad5, nad1 and atp6. Gene-by-gene nucleotide diversity was highly variable, but, by far, the least variation was recorded within cox1. Of the markers used previously by Hu et al. [17] (i.e. M1) and Höglund et al. [12] (i.e. M2-M5), M1 and M2 (within cox1) captured the least, and M4 (within nad5) captured the greatest sequence variation. The relatively low variation captured by targeting trn genes is also indicated by a trough for M2. Overall, the full sliding window indicates a wealth of new genes capable of providing high levels of nucleotide variation for population genetic studies.
Figure 3.
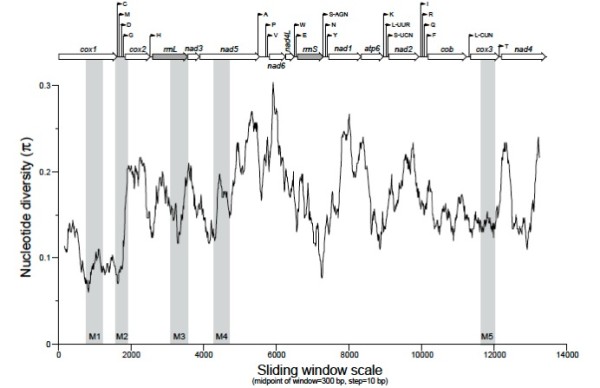
Sliding window analysis of complete mt genome sequences of Dictyocaulus viviparus and Dictyocaulus sp. cf. eckerti. The black line indicates nucleotide diversity in a window size of 300 bp, with a step size of 10 bp. Individual pairwise results are shown by symbols (see key). Gene boundaries indicated with shaded columns of gene regions used in previous studies [12,17].
Discussion
Advanced molecular methods are having widespread impact and implications in parasitology [36-47]. In the present study, we utilised massively parallel sequencing and semi-automated bioinformatic annotation for the characterisation of the complete mt genomes of D. viviparus (from Bos taurus) and Dictyocaulus sp. cf. eckerti from red deer, and explored the genetic relationships of these two lungworms and selected representatives of the Strongylida. Using sliding window analysis (Figure 3), we also identified regions in these mt genomes which might serve as suitable markers for future molecular explorations of the systematics, population genetics or epidemiology of Dictyocaulus species.
Given the controversy surrounding the taxonomy/systematics of some species of Dictyocaulus[1,5,8,9,48-54], concatenated mt proteomic sequences might be applied effectively as barcodes to genetically characterise and compare dictyocaulids from various ungulate hosts, including domestic and wild bovids and cervids. This is particularly pertinent, given that Dictyocaulus from red deer is genetically distinct from Dictyocaulus species from roe deer (Capreolus capreolus), fallow deer (Dama dama) and moose (Alces alces) [7,8,54]. Current evidence shows a distinct genetic differentiation between Dictyocaulus sp. cf. eckerti and D. viviparus, supported by previous results for the ITS-2 region [10]. A detailed appraisal of previously published findings for D. viviparus[12,17] revealed a maximum nucleotide sequence variation of 3%, 3.5% and 3%, respectively, in partial cox1, cox3 or nad5 regions (375–396 nt) among 72–252 individual worms. These levels of within-species variation are much lower than estimated levels of difference (9%, 15% and 18%, respectively) between Dictyocaulus sp. cf. eckerti and D. viviparus for the same gene regions (this study). At the amino acid level, variation in COX-1 (131 amino acids) within D. viviparus was ≤1.5%, but there was no amino acid difference between Dictyocaulus sp. cf. eckerti and D. viviparus (because of relative conservation). This contrasts the situation for COX3 (125 amino acids) and NAD5 (132 amino acids), for which maximum variation within D. viviparus was 1.6% and 2.3%, respectively (cf. [12]), and differences between D. viviparus and Dictyocaulus sp. cf. eckerti were substantially higher, at 10.4% and 13.6%, respectively. Together with previous ITS-2 data [10], this information indicates that Dictyocaulus sp. cf. eckerti and D. viviparus are separate species.
Whether the parasite from red deer (i.e., Dictyocaulus cf. eckerti) represents a distinct species from (or a population variant of) “D. eckerti” (cf. [50]) remains to be established. A previous study showed that the magnitude of sequence difference (~6-8%) in ITS-2 rDNA of Dictyocaulus from fallow deer from Germany [7] is greater than variation (0.4-2.6%) among selected individuals of Dictyocaulus from red deer [10], although the degree of genetic variation within the operational taxonomic unit from fallow deer is not yet known. D. eckerti was originally described from the reindeer, Rangifer tarandus, from Western Siberia (cited in [50]), which raises questions as to the specific identity of Dictyocaulus from various cervid species [5,8,13,55]. Although Durette-Desset et al. [5] proposed that dictyocaulids of European cervids be called Dictyocalus noerneri, molecular and morphological data have shown that roe deer and moose can harbour D. capreolus (e.g., [8,54]) and that chamois (Rupicapra rupicapra) might harbour a unique species [13]. This controversy and the findings of numerous previous studies emphasize the need for detailed investigations of Dictyocaulus specimens from various species of wild and domestic bovid and cervid hosts from different continents, which could be achieved using a combined morphological and mt proteomic barcoding approach.
There is also significance in using mt genetic markers for studying the genetic composition of populations of Dictyocaulus spp., given that there are few morphological features for the specific differentiation of some developmental stages (i.e., larvae) [3] and given that cryptic species have been detected within the Strongylida [56,57]. In nematodes, mt DNA is usually more variable in sequence within a species than ITS-2 and other rDNA regions [56], indicating that mt gene regions are well suited for studying the population genetics of parasitic nematodes [14,56,58,59]. The sliding window analysis conducted herein displayed distinct patterns of nucleotide diversity between the two mt genomes representing Dictyocaulus (Figure 3). Low variability is useful for the design of oligonucleotide primers that flank mt regions with high variability for population genetic or epidemiological investigations. Some previous studies have shown the utility of some mt gene regions. For instance, Hu et al. [17] employed primer sets, originally designed to cox1 of flatworms [60], for PCR-based mutation scanning and selective sequencing analysis of D. viviparus individuals from 17 different populations (farms). Within-species variation in cox1 was low (0.3–2.3%) [17], similar to findings for some parasitic nematodes of plants and insects [61-65] but distinctly different from findings for gastrointestinal trichostrongyloid nematodes of domestic ruminants [66,67]. In the present study, cox1 is revealed to be the gene with the lowest nucleotide diversity between the two Dictyocaulus species. In another study, Höglund et al. [12] showed distinct genetic substructuring in D. viviparus populations from Sweden using four mt regions (in the cox3, nad5, trnC_M_D_G and rrnL genes) covering 11% (1542 bp) of the mt genome of D. viviparus (Figure 1). Although some of the gene regions (e.g., cox3 and nad5) used to date provided some resolution of genetic variation (<6% among haplotypes, upon pairwise comparison), the present sliding window analysis indicates other candidate genes for capturing greater diversity. That the cox1 region (of 393 bp) is the most conserved relative to other regions (see Figure 3; M1) is a feature that facilitates the design of primers for PCR, but might come at the cost of missing signal (of sequence variation) required for analysis. Here, numerous sequence tracts in the mt genome have been identified as potentially suitable for population genetic or taxonomic studies (Figure 3). The comparison among the mt genomes of different species of nematodes characterised to date now provides the prospect for designing generic or specific primers to capture regions of variability most suited to the resolution needed for analysis, and also provides opportunities for a range of DNA amplification-based approaches, including multiplexed diagnostic methods (cf. [68]).
PCR primers can be designed rationally in conserved regions flanking “variable sequence tracts” within the mt genome considered to be most informative for population genetic studies of Dictyocaulus from different bovid and cervid hosts. Utilizing such primer sets, PCR-based single-strand conformation polymorphism (SSCP) analysis [23] could be applied to screen large numbers of individual specimens representing different host species and populations for haplotypic variability (at any developmental stage). A previous study has shown the merit of SSCP for exploring the genetic variation in various populations of D. viviparus in Sweden [17], and could be applied to large-scale studies of Dictyocaulus specimens representing distinct operational taxonomic units (based on ITS-2) and different ungulate host species.
Using a range of variable mt regions, in combination with classical parasitological techniques, it might also be possible to assess the cross transmission of particular species or genetic variants (haplotypes) of Dictyocaulus between/among cattle and cervid hosts, and their pathogenicity in different host species (cf. [22]). Furthermore, it will also be significant to extend mt genome sequencing to a range of lungworm species of domestic and wild ruminants, and assess their genetic relationships. The present study shows the relevance of the mt genomes of Dictyocaulus species for future systematic investigations and, importantly, stimulates a reassessment of the phylogenetic position of the family Dictyocaulidae in relation to the Metastrongylidae, Trichostrongylidae and other families within the order Strongylida (cf. [11]).
Conclusions
Comparative analyses of proteomic sequence datasets inferred from the mt genomes of Dictyocaulus sp. cf. eckerti (red deer) and D. viviparus indicate that these parasites are closely related species. Barcodes identified in the mt genomes and proteomes could serve as markers for future studies of the systematics, population genetics and/or epidemiology of a range of Dictyocaulus species as well as other lungworms.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
RBG, ARJ & DTJL conceived the project and attracted the funding; AJ, ARJ, NM & JH carried out molecular laboratory work; NM, ARJ, DTJL, RSH & RBG carried out data analysis and interpretation; RBG, ARJ & DTJL wrote the draft manuscript with critical input from AJ and JH. All authors read and approved the final version of the manuscript.
Contributor Information
Robin B Gasser, Email: robinbg@unimelb.edu.au.
Abdul Jabbar, Email: jabbara@unimelb.edu.au.
Namitha Mohandas, Email: namigrt@gmail.com.
Johan Höglund, Email: Johan.Hoglund@slu.se.
Ross S Hall, Email: rossh@unimelb.edu.au.
D Timothy J Littlewood, Email: t.littlewood@nhm.ac.uk.
Aaron R Jex, Email: ajex@unimelb.edu.au.
Acknowledgements
The present study was supported by ARC grant LP100100091 (to RBG, ARJ and DTJL). The Early Career Researcher (ECR) grant (to AJ) from The University of Melbourne is gratefully acknowledged. Current research in the Gasser Lab is funded mainly by the Australian Research Council (ARC), the National Health and Medical Research Council (NHMRC) and Melbourne Water Corporation. The Swedish Research Council for Environment, Agricultural Sciences and Spatial Planning (FORMAS) is also acknowledged (JH). Thanks to Marion Johnson (Mosgiel, New Zealand) for originally providing lungworm specimens used in a previous study [10] and to Brendan Ansell for collecting some references.
References
- Gibbons LM, Khalil LF. A revision of the genus Dictyocaulus Railliet & Henry, 1907 (Nematoda: Trichostrongyloidea) with the description of D. africanus n. sp. from African artiodactylids. J Afr Zool. 1988;102:151–175. [Google Scholar]
- Panuska C. Lungworms of ruminants. Vet Clin North Am Food Anim Pract. 2006;22:583–93. doi: 10.1016/j.cvfa.2006.06.002. [DOI] [PubMed] [Google Scholar]
- Anderson RC. Nematode Parasites of Vertebrates. Their Development and Transmission. Wallingford CAB International. 2000. p. 650.
- Railliet LJA, Henry ACL. Sur les variations des strongyles de l’appareil respiratoire des mammifères. CR de la Société de Biol (Paris) 1907;63:751–753. [Google Scholar]
- Durette-Desset MC, Hugonnet L, Chabaud AG. Redescription de Dictyocaulus noerneri Railliet et Henry, 1907, parasite de Capreolus capreolus en Europe. Comparison avec D. viviparus (Bloch, 1782) parasite du bétail. Ann Parasitol Hum Comp. 1988;63:285–295. doi: 10.1051/parasite/1988634285. [DOI] [PubMed] [Google Scholar]
- Schnieder T, Epe C, Samson-Himmelstjerna GV. Species differentiation of lungworms (Dictyocaulidae) by polymerase chain reaction / restriction fragment length polymorphism of second internal transcribed spacers of ribosomal DNA. Parasitol Res. 1996;82:392–394. doi: 10.1007/s004360050134. [DOI] [PubMed] [Google Scholar]
- Epe C, Samson-Himmelstjerna GV, Schnieder T. Differences in a ribosomal DNA sequence of lungworm species (Nematoda: Dictyocaulidae) from fallow deer, cattle, sheep and donkeys. Res Vet Sci. 1997;62:17–21. doi: 10.1016/S0034-5288(97)90173-9. [DOI] [PubMed] [Google Scholar]
- Höglund J, Wilhelmsson E, Christensson D, Mörner T, Waller P, Mattsson JG. ITS2 sequences of Dictyocaulus species from cattle, roe deer and moose in Sweden: molecular evidence for a new species. Int J Parasitol. 1999;29:607–611. doi: 10.1016/S0020-7519(98)00229-X. [DOI] [PubMed] [Google Scholar]
- Divina BP, Wilhelmsson E, Mattsson JG, Waller P, Höglund J. Identification of Dictyocaulus spp. in ruminants by morphological and molecular analyses. Parasitology. 2000;121:193–201. doi: 10.1017/S0031182099006162. [DOI] [PubMed] [Google Scholar]
- Johnson M, Abs EL-Osta YG, Hu M, Gasser RB. An electrophoretic tool for the genetic characterisation and delineation of lungworms. Mol Cell Probes. 2004;18:197–203. doi: 10.1016/j.mcp.2004.01.003. [DOI] [PubMed] [Google Scholar]
- Chilton NB, Huby-Chilton F, Gasser RB, Beveridge I. The evolutionary origins of nematodes within the order Strongylida are related to predilection sites within hosts. Mol Phylogenet Evol. 2006;40:118–128. doi: 10.1016/j.ympev.2006.01.003. [DOI] [PubMed] [Google Scholar]
- Höglund J, Morrison DA, Mattsson JG, Engström A. Population genetics of the bovine/cattle lungworm (Dictyocaulus viviparus) based on mtDNA and AFLP marker techniques. Parasitology. 2006;133:89–99. doi: 10.1017/S0031182006009991. [DOI] [PubMed] [Google Scholar]
- Carreno RA, Diez-Baños N, del MR H-A, Nadler SA. Characterisation of Dictyocaulus species (Nematoda: Trichostrongyloidea) from three species of wild ruminants in northwesterns Spain. J Parasitol. 2009;95:966–970. doi: 10.1645/GE-1791.1. [DOI] [PubMed] [Google Scholar]
- Blouin MS. Molecular prospecting for cryptic species of nematodes: mitochondrial DNA versus internal transcribed spacer. Int J Parasitol. 2002;32:527–531. doi: 10.1016/S0020-7519(01)00357-5. [DOI] [PubMed] [Google Scholar]
- Hu M, Gasser RB. Mitochondrial genomes of parasitic nematodes—progress and perspectives. Trends Parasitol. 2006;22:78–84. doi: 10.1016/j.pt.2005.12.003. [DOI] [PubMed] [Google Scholar]
- Jex AR, Hall RS, Littlewood DT, Gasser RB. An integrated pipeline for next-generation sequencing and annotation of mitochondrial genomes. Nucleic Acids Res. 2010;38:522–533. doi: 10.1093/nar/gkp883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu M, Höglund J, Chilton NB, Zhu XQ, Gasser RB. Mutation scanning analysis of mitochondrial cytochrome c oxidase subunit 1 reveals limited gene flow among bovine lungworm subpopulations in Sweden. Electrophoresis. 2002;23:3357–3363. doi: 10.1002/1522-2683(200210)23:19<3357::AID-ELPS3357>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- Park JK, Sultana T, Lee SH, Kang S, Kim HK, Min GS, Eom KS, Nadler SA. Monophyly of clade III nematodes is not supported by phylogenetic analysis of complete mitochondrial genome sequences. BMC Genomics. 2011;12:392. doi: 10.1186/1471-2164-12-392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jex AR, Littlewood DT, Gasser RB. Toward next-generation sequencing of mitochondrial genomes–focus on parasitic worms of animals and biotechnological implications. Biotechnol Adv. 2010;28:151–159. doi: 10.1016/j.biotechadv.2009.11.002. [DOI] [PubMed] [Google Scholar]
- Johnson M, Mackintosh CG, Labes RE, Taylor MJ. Dictyocaulus eckerti, lungworm infecting farmed red deer in New Zealand. N Z Vet J. 2001;49:34–35. doi: 10.1080/00480169.2001.36200. [DOI] [PubMed] [Google Scholar]
- Lv S, Zhang Y, Zhang L, Liu Q, Liu HX, Hu L, Wei FR, Steinmann P, Graeff-Teixeira C, Zhou XN, Utzinger J. The complete mitochondrial genome of the rodent intra-arterial nematodes Angiostrongylus cantonensis and Angiostrongylus costaricensis. Parasitol Res. 2012;111:115–123. doi: 10.1007/s00436-011-2807-4. [DOI] [PubMed] [Google Scholar]
- Johnson M, Mackintosh CG, Labes RE, Taylor MJ, Whaton DA. Dictyocaulus species: cross infection between cattle and red deer. N Z Vet J. 2003;51:93–98. doi: 10.1080/00480169.2003.36346. [DOI] [PubMed] [Google Scholar]
- Gasser RB, Hu M, Chilton NB, Campbell BE, Jex AR, Otranto D, Cafarchia C, Beveridge I, Zhu XQ. Single-strand conformation polymorphism (SSCP) for the analysis of genetic variation. Nat Protoc. 2006;1:3121–3128. doi: 10.1038/nprot.2006.485. [DOI] [PubMed] [Google Scholar]
- Hu M, Jex AR, Campbell BE, Gasser RB. Long PCR amplification of the entire mitochondrial genome from individual helminths for direct sequencing. Nat Protoc. 2007;2:2339–2344. doi: 10.1038/nprot.2007.358. [DOI] [PubMed] [Google Scholar]
- Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, Berka J, Braverman MS, Chen YJ, Chen Z, Dewell SB, Du L, Fierro JM, Gomes XV, Godwin BC, He W, Helgesen S, Ho CH, Irzyk GP, Jando SC, Alenquer ML, Jarvie TP, Jirage KB, Kim JB, Knight JR, Lanza JR, Leamon JH, Lefkowitz SM, Lei M, Li J. et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437:376–380. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Madan A. CAP3: A DNA sequence assembly program. Genome Res. 1999;9:868–877. doi: 10.1101/gr.9.9.868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozas J, Sánchez-DelBarrio JC, Messeguer X, Rozas R. DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics. 2003;19:2496–2497. doi: 10.1093/bioinformatics/btg359. [DOI] [PubMed] [Google Scholar]
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouy M, Guindon S, Gascuel O. SeaView Version 4: A multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol Biol Evol. 2010;27:221–224. doi: 10.1093/molbev/msp259. [DOI] [PubMed] [Google Scholar]
- Sharp PM, Matassi G. Codon usage and genome evolution. Curr Opin Genet Dev. 1994;4:851–860. doi: 10.1016/0959-437X(94)90070-1. [DOI] [PubMed] [Google Scholar]
- Durent L, Mouchiroud D. Expression pattern and, surprisingly, gene length shape codon usage in Caenorhabditis, Drosophila and Arabidopsis. Proc Natl Acad Sci (USA) 1999;96:4482–4487. doi: 10.1073/pnas.96.8.4482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okimoto R, Macfarlane JL, Clary DO, Wolstenholme DR. The mitochondrial genomes of two nematodes, Caenorhabditis elegans and Ascaris suum. Genetics. 1992;130:471–498. doi: 10.1093/genetics/130.3.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keddie EM, Higazi T, Unnasch TR. The mitochondrial genome of Onchocerca volvulus: sequence, structure and phylogenetic analysis. Mol Biochem Parasitol. 1998;95:111–127. doi: 10.1016/S0166-6851(98)00102-9. [DOI] [PubMed] [Google Scholar]
- Lavrov DV, Brown WM. Trichinella spiralis mtDNA: a nematode mitochondrial genome that encodes a putative ATP8 and normally structured tRNAS and has a gene arrangement relatable to those of coelomate metazoans. Genetics. 2001;157:621–637. doi: 10.1093/genetics/157.2.621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu GH, Gasser RB, Su A, Nejsum P, Peng L, Lin RQ, Li MW, Xu MJ, Zhu XQ. Clear genetic distinctiveness between human- and pig-derived Trichuris based on analyses of mitochondrial datasets. PLoS Negl Trop Dis. 2012;6:e1539. doi: 10.1371/journal.pntd.0001539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avelar DM, Linardi PM. Use of multiple displacement amplification as pre-polymerase chain reaction (pre-PCR) to amplify genomic DNA of siphonapterids preserved for long periods in scientific collections. Parasit Vectors. 2010;3:86. doi: 10.1186/1756-3305-3-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dantas-Torres F, Latrofa MS, Otranto D. Occurrence and genetic variability of Phlebotomus papatasi in an urban area of southern Italy. Parasit Vectors. 2010;3:77. doi: 10.1186/1756-3305-3-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bargues MD, Artigas P, Khoubbane M, Mas-Coma S. DNA sequence characterisation and phylogeography of Lymnaea cousini and related species, vectors of fascioliasis in northern Andean countries, with description of L. meridensis n. sp. (Gastropoda: Lymnaeidae). Parasit Vectors. 2011;4:132. doi: 10.1186/1756-3305-4-132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Echodu R, Beadell JS, Okedi LM, Hyseni C, Aksoy S, Caccone A. Temporal stability of Glossina fuscipes fuscipes populations in Uganda. Parasit Vectors. 2011;4:19. doi: 10.1186/1756-3305-4-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foronda P, López-González M, Hernández M, Haukisalmi V, Feliu C. Distribution and genetic variation of hymenolepidid cestodes in murid rodents on the Canary Islands (Spain) Parasit Vectors. 2011;4:185. doi: 10.1186/1756-3305-4-185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauswald AK, Remais JV, Xiao N, Davis GM, Lu D, Bale MJ, Wilke T. Stirred, not shaken: genetic structure of the intermediate snail host Oncomelania hupensis robertsoni in an historically endemic schistosomiasis area. Parasit Vectors. 2011;4:206. doi: 10.1186/1756-3305-4-206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawton SP, Hirai H, Ironside JE, Johnston DA, Rollinson D. Genomes and geography: genomic insights into the evolution and phylogeography of the genus Schistosoma. Parasit Vectors. 2011;4:131. doi: 10.1186/1756-3305-4-131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miró G, Montoya A, Hernández L, Dado D, Vázquez MV, Benito M, Villagrasa M, Brianti E, Otranto D. Thelazia callipaeda: infection in dogs: a new parasite for Spain. Parasit Vectors. 2011;4:148. doi: 10.1186/1756-3305-4-148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouma JO, Beadell JS, Hyseni C, Okedi LM, Krafsur ES, Aksoy S, Caccone A. Genetic diversity and population structure of Glossina pallidipes in Uganda and western Kenya. Parasit Vectors. 2011;4:122. doi: 10.1186/1756-3305-4-122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz-Lopez F, Wilkerson RC, Conn JE, McKeon SN, Levin DM, Quiñones ML, Póvoa MM, Linton YM. DNA barcoding reveals both known and novel taxa in the Albitarsis Group (Anopheles: Nyssorhynchus) of Neotropical malaria vectors. Parasit Vectors. 2012;5:44. doi: 10.1186/1756-3305-5-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Standley CJ, Adriko M, Besigye F, Kabatereine NB, Stothard RJ. Confirmed local endemicity and putative high transmission of Schistosoma mansoni in the Sesse Islands, Lake Victoria, Uganda. Parasit Vectors. 2011;1(4):29. doi: 10.1186/1756-3305-4-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JY, Ha Y, Gao CH, Wang Y, Yang YT, Chen HT. The prevalence of canine Leishmania infantum infection in western China detected by PCR and serological tests. Parasit Vectors. 2011;4:69. doi: 10.1186/1756-3305-4-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougherty EC. A brief survey of the genus Dictyocaulus Railliet and Henry 1907 (Nematoda: Trichostrongylidae) Helminthol Soc Wash Proc. 1946;13:49–54. [PubMed] [Google Scholar]
- Swietlikowski M. Studies on morphology and taxonomy of some species of the genus Dictyocaulus Raillet et Henry 1907. Acta Parasitol Polon. 1961;9:13–21. [Google Scholar]
- Skrjabin KI, Shikhobalova NP, Shul’ts RS. In: Essentials of Nematodology Vol IV. 1954. Skrjabin KI, editor. Jerusalem: translated from the Russian by Israel Programme for Scientific Translations; 1971. Dictyocaulidae, Heligmosomatidae, and Ollulanidae of Animals. [Google Scholar]
- Bacinsky A. The morphological evaluation of Dictyocaulus found in red deer and cattle [In Russian] Folia Vet. 1973;17:209–219. [Google Scholar]
- Höglund J, Morrison DA, Divina BP, Wilhelmsson E, Mattsson JG. Phylogeny of Dictyocaulus (lungworms) from eight species of ruminants based on analyses of ribosomal RNA data. Parasitology. 2003;127:179–187. doi: 10.1017/S0031182003003366. [DOI] [PubMed] [Google Scholar]
- Divina BP, Wilhelmsson E, Morner T, Mattsson JG, Höglund J. Molecular identification and prevalence of Dictyocaulus spp. (Trichostrongyloidea: Dictyocaulidae) in Swedish semi-domestic and free-living cervids. J Wildl Dis. 2002;38:769–775. doi: 10.7589/0090-3558-38.4.769. [DOI] [PubMed] [Google Scholar]
- Gibbons LM, Höglund J. Dictyocaulus capreolus n. sp. (Nematoda: Trichostrongyloidea) from roe deer, Capreolus capreolus and moose, Alces alces in Sweden. J Helminthol. 2002;76:119–125. doi: 10.1079/JOH2001108. [DOI] [PubMed] [Google Scholar]
- Bienioschek S, Rehbein S, Ribbeck R. Cross-infections between fallow deer and domestic ruminants with large lungworms (Dictyocaulus spp.) Appl Parasitol. 1996;37:229–238. [PubMed] [Google Scholar]
- Gasser RB. Molecular tools – advances, opportunities and prospects. Vet Parasitol. pp. 69–89. [DOI] [PubMed]
- Gasser RB, Bott NJ, Chilton NB, Hunt P, Beveridge I. Toward practical, DNA-based diagnostic methods for parasitic nematodes of livestock – bionomic and biotechnological implications. Biotechnol Adv. 2008;26:325–334. doi: 10.1016/j.biotechadv.2008.03.003. [DOI] [PubMed] [Google Scholar]
- Anderson TJC, Blouin MS, Beech RN. Populations biology of parasitic nematodes: applications of genetic markers. Adv Parasitol. 1998;41:219–283. doi: 10.1016/s0065-308x(08)60425-x. [DOI] [PubMed] [Google Scholar]
- Hu M, Chilton NB, Gasser RB. The mitochondrial genomics of parasitic nematodes of socio-economic importance: recent progress, and implications for population genetics and systematics. Adv Parasitol. 2004;56:133–212. doi: 10.1016/s0065-308x(03)56003-1. [DOI] [PubMed] [Google Scholar]
- Bowles J, Blair D, McManus DP. Genetic Variants within the genus Echinococcus identified by mitochondrial DNA sequencing. Mol Biochem Parasitol. 1992;54:165–174. doi: 10.1016/0166-6851(92)90109-W. [DOI] [PubMed] [Google Scholar]
- Hugall A, Moritz C, Stanton J, Wolstenholme DR. Low, but strongly structural mitochondrial DNA diversity in root knot nematodes (Meloidogyne) Genetics. 1994;136:903–912. doi: 10.1093/genetics/136.3.903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blouin MS, Yowell CA, Courtney CH, Dame JB. Haemonchus placei and Haemonchus contortus are distinct species based on mtDNA evidence. Int J Parasitol. 1997;27:1383–1387. doi: 10.1016/S0020-7519(97)00125-2. [DOI] [PubMed] [Google Scholar]
- Blouin MS, Yowell CA, Courtney CH, Dame JB. Substitution bias, rapid saturation, and the use of mtDNA for nematode systematics. Mol Biol Evol. 1998;15:1719–1727. doi: 10.1093/oxfordjournals.molbev.a025898. [DOI] [PubMed] [Google Scholar]
- Blouin MS, Liu JC, Berry RE. Life variation and the genetic structure of nematode populations. Heredity. 1999;83:253–259. doi: 10.1038/sj.hdy.6885420. [DOI] [PubMed] [Google Scholar]
- Courtright EM, Wall DH, Virginia RA, Frisse LM, Vida JT, Thomas WK. Nuclear and mitochondrial DNA sequence diversity in the Antarctic nematode Scottnema lindsayae. J. Nematol. 2000;32:143–153. [PMC free article] [PubMed] [Google Scholar]
- Blouin MS, Dame JB, Tarrant CA, Courtney CH. Unusual population genetics of a parasitic nematode: mtDNA variation within and among populations. Evolution. 1992;46:470–476. doi: 10.2307/2409865. [DOI] [PubMed] [Google Scholar]
- Blouin MS, Yowell CA, Courtney CH, Dame JB. Host movement and the genetic structure of populations of parasitic nematodes. Genetics. 1995;141:1007–1014. doi: 10.1093/genetics/141.3.1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon CA, Gray DJ, Gobert GN, McManus DP. DNA amplification approaches for the diagnosis of key parasitic helminth infections of humans. Mol Cell Probes. 2011;25:143–152. doi: 10.1016/j.mcp.2011.05.002. [DOI] [PubMed] [Google Scholar]
- Le TH, Blair D, McManus DP. Mitochondrial genomes of human helminths and their use as markers in population genetics and phylogeny. Acta Trop. 2000;77:243–256. doi: 10.1016/S0001-706X(00)00157-1. [DOI] [PubMed] [Google Scholar]