

Abstract
Rab monomeric GTPases regulate specific aspects of vesicle transport in eukaryotes including coat recruitment, uncoating, fission, motility, target selection and fusion. Moreover, individual Rab proteins function at specific sites within the cell, for example the ER, golgi and early endosome. Importantly, the localization and function of individual Rab subfamily members are often conserved underscoring the significant contributions that model organisms such as Caenorhabditis elegans can make towards a better understanding of human disease caused by Rab and vesicle trafficking malfunction. With this in mind, a bioinformatics approach was first taken to identify and classify the complete C. elegans Rab family placing individual Rabs into specific subfamilies based on molecular phylogenetics. For genes that were difficult to classify by sequence similarity alone, we did a comparative analysis of intron position among specific subfamilies from yeast to humans. This two-pronged approach allowed the classification of 30 out of 31 C. elegans Rab proteins identified here including Rab31/Rab50, a likely member of the last eukaryotic common ancestor (LECA). Second, a molecular toolset was created to facilitate research on biological processes that involve Rab proteins. Specifically, we used Gateway-compatible C. elegans ORFeome clones as starting material to create 44 full-length, sequence-verified, dominant-negative (DN) and constitutive active (CA) rab open reading frames (ORFs). Development of this toolset provided independent research projects for students enrolled in a research-based molecular techniques course at California State University, East Bay (CSUEB).
Introduction
The Rab Family of Monomeric GTPases
The RAS superfamily of monomeric GTPases is widely conserved and includes five main families: Ras, Rho, Arf, Ran and Rab. The largest of these, the Rab family, participates in all aspects of vesicular traffic and contributes to endomembrane identity [1], [2]. For example, Rab5 localizes to clathrin-coated vesicles budding from the plasma membrane, early endosomes and transport vesicles in between. These observations reflect Rab5’s specific role in clathrin uncoating and target selection of vesicles travelling from the plasma membrane to the early endosome. Similarly, Rab1 and Rab2 localize to distinct transport vesicles between the ER and golgi as they function during ER to golgi or golgi to ER transport, respectively [2]. Needless to say, all aspects of vesicular trafficking including intracellular transport of proteins, ligand secretion, receptor trafficking and protein degradation is fundamental to the function of the eukaryotic cell. Thus, it is not surprising that mutations in Rab GTPases, their regulators or effectors can lead to inherited and acquired disease including birth defects, mental retardation, type 2 diabetes, cancer and neurodegenerative disease [3]. Moreover, a number of bacterial pathogens express regulators of specific host Rabs to facilitate cell entry and avoid degradation by the lysosome, thereby promoting the process of infection [3].
Rab proteins, like all RAS superfamily members, alternate between active (GTP bound) and inactive (GDP bound) conformational forms [4]. The active form typically binds effector molecules while GTP hydrolysis disrupts this interaction. Interestingly, monomeric GTPases in isolation rarely cycle between active and inactive forms as these proteins are poor GTPases and once GTP hydrolysis occurs, GDP remains tightly bound. Thus, two types of regulatory proteins are essential to speed up cycling between active and inactive states. They include a GTPase activation protein (GAP), which promotes GTP hydrolysis and a Guanine Nucleotide Exchange Factor (GEF), which promotes GDP release. GTP is abundant in the cytoplasm and quickly replaces GDP. Not surprisingly, all members of the RAS superfamily can be identified through a number of conserved sequence motifs involved in guanine nucleotide binding and GTP hydrolysis, the so-called, G boxes (G1 through G5) [4]. These G box motifs, however, do not provide family specific identity. For this, one must identify sequence motifs that mediate interactions with family specific regulators, such as the Rab escort protein (REP), a protein involved in C-terminal prenylation of Rab family members only (see below).
C-terminal Prenylation
Ras, Rho and Rab family members are typically prenylated at C-terminal cysteines, a modification that plays an essential role in membrane targeting and thus biological activity. Ras and Rho are prenylated directly by farnesyl transferase (FTase) or geranyl geranyl transferase I (GGTase I), enzymes that recognize a C-terminal cysteine in the context of a CA1A2X box (A1 is aliphatic, A2 is aliphatic but not aromatic and X is typically S, M, A, Q or L) [5]. By contrast, Rab proteins are substrates of the Rab geranyl geranyl transferase II (Rab GGTase II), which prenylates a motif that typically consists of two cysteines in a variety of contexts including XXXCC, XXCCX, XCCXX, CCXXX and sometimes CXXX (X = any amino acid) [5].
Unlike CAAX-box proteins, the rab prenylation motif is not recognized directly by Rab GGTase II. Instead, this role is “outsourced” to REP, which interacts with Rab proteins through its Rab binding platform and C-terminal binding region (CBR) [6]. The RAB:REP complex then binds to Rab GGTase II allowing prenylation of one or both C-terminal cysteines. The CBR motif interacts with a short hydrophobic patch within the hyper variable C-terminal region of Rab called the CIM (CBR Interaction Motif), which typically consists of an AXA motif (A = aliphatic, typically I, L, V, F and P. X = any amino acid) [6]–[8]. This interaction positions the C-terminal cysteines within close proximity to the geranyl geranyl transferase II active site. Interestingly, the C-terminal tail is not inserted into the GGTase II active site like CAAX-box tails but instead is placed along side. This is consistent with the ability of Rab GGTase II to prenylate C-terminal cysteines within a wide variety of contexts [7].
All Rab proteins share the RAB:REP interaction. Thus, Pereira-Leal et al. hypothesized in 2000 that this family might possess Rab-specific sequence elements that mediate this interaction [9]. Through a bioinformatics approach, they identified five so-called Rab Family (RabF) motifs (RabF1– RabF5). Composite models of two crystal structures (Rab7:REP and REP:GGTase II) are consistent with a role for RabF1–4 motifs in mediating interactions with two Rab family specific regulators, REP and RabGDI [6], [10]. RabGDI functions by regulating the reversible association of Rab proteins with cell membranes. By contrast, RabF5 maps to an internal region of Rab proteins but nonetheless possesses family specific variations and remains helpful in identifying Rab family members.
In this paper, we take a bioinformatics approach to identify the complete C. elegans Rab family based on percent identity to RabF motifs and absence of motifs specifically conserved in other Ras superfamily members. Next, we place individual Rab proteins into specific subfamilies based on a phylogenetic analysis with humans. For difficult to classify Rab proteins we also perform an analysis of intron position conservation. Finally, we create an ORFeome-based molecular toolset of mutant rab ORFs to be used for rab gene function studies in C. elegans. The C. elegans ORFeome resource was created in 2003 [11] and is a partially-verified, Gateway-compatible library of ORFs representing more than 63% of the proteome [12]. Our molecular toolset includes 44 full-length, sequence-verified, dominant-negative (DN) and constitutive-active (CA) mutant rab ORFs. Mutant forms were created in the context of a research-based lab course at California State University, East Bay (CSUEB) as a way to provide an authentic research experience to a large number of students.
Educating the next generation of research scientists has traditionally followed a master-apprenticeship model. While this model is highly effective in providing an authentic research experience, it impacts only a small number of students. To boost the number of students exposed to real research, there have been increasing calls for providing an authentic research experience during the academic year, within so-called research-based lab courses [13]–[15]. Inspired by a pioneer of research-based curricula [16], I (coauthor M.G.) initiated a research-based lab course in 2007. My aim was to provide a sense of adventure, discovery and pride in one’s accomplishments with the knowledge that students can contribute to the overall scientific knowledge base.
This research-based course focused on the Rab family for several reasons: 1) the C. elegans Rab family is manageable in size with 31 members [17] and this work. 2) The average rab ORF is short (median length: 632 bp). 3) DN and CA mutant forms first described in human Ras [18], [19] have also been used successfully in Rab proteins [20]–[24], [65], [67]. Finally, functional analysis of Rab family members is incomplete despite their vital roles in cell and developmental biology [1], [2].
Results
Identification of the Complete C. elegans Rab Family
Using a bioinformatics approach, Pereira-Leal and Seabra identified Rab family members in C. elegans, D. melanogaster, H. sapiens, S. cerevisiae, S. pombe and A. thaliana [17]. As this work was published just after completion of the C. elegans genome and improvements to ORF annotations have been ongoing [25], we redid a bioinformatics analysis of the C. elegans Rab family in this study.
To identify new members of the Rab family in C. elegans, we first created a multiple sequence alignment (MSA) of the 28 Rab family proteins identified previously [17] using Muscle within MEGA5 [26]. We then used this MSA as a query in a Position-Specific Iterated BLAST (PSI-BLAST) [27]. This approach identified 51 additional “hits” deemed statistically significant (not counting splice variants). With the MSA of the expanded list, we then calculated a single RabF percent identity score based on conservation with the consensus sequences for RabF1 through RabF5 combined (27 amino acids in total). Putative Rab proteins sorted in descending order according to the RabF percent identity score are listed in Figure 1. Not surprisingly, the original 28 Rab proteins cluster at the top of the list with the most distant member (RAB-28) at position 30 with a RabF percent identity score of 48. Above RAB-28, at 59% and 67% are two new Rab proteins, R07B1.12 and Y71H2AM.12, not identified in 2001 although R07B1.12 (a.k.a. GLO-1) was recognized as a Rab protein more recently [28], [29].
Figure 1. The complete list of PSI-BLAST hits using a sequence alignment of known C. elegan Rabs.

Genes are listed in descending order of RabF % identity (ID). The consensus sequence used to calculate RabF % ID is listed in the top row. For each hit, the portion of the alignment corresponding to each Rab Family motif (1–5) is provided. By contrast, the C-terminal sequence is only provided for those hits with one or two near terminal cysteines. For each C-terminus shown, the putative CBR interacting motif (CIM) is boxed if present. C-terminal cysteines are shaded orange. RabF consensus matches are shaded green and specific amino acids that suggest inclusion in a nonRab family are shaded yellow. The bold horizontal line separates the cluster of mostly known rabs from other monomeric GTPases. Numbers in parentheses indicate the number of amino acids omitted from the C-termini. RBH = Reciprocal Best Hit.
All proteins listed above RAB-28 (Figure 1) are likely to be authentic Rabs. The majority of these proteins including R07B1.12 and Y71H2AM.12 possess a typical rab prenylation motif with two terminal (or near terminal) cysteines in addition to a recognizable hydrophobic CIM. Exceptions include RAB-8, RAB-28 and C33D12.6. These proteins contain a CXXX motif instead.
The presence of an atypical Rab prenylation motif in RAB-8 and 28 is well-documented within metazoans where the vast majority of known members possess a single cysteine in a CAAX-box like context [30]–[32]. C33D12.6 (tag-312) is a RAB45 ortholog (see below). While most RAB45 orthologs (42 species examined) possess two terminal cysteines in a CCXX context, RAB45 from Branchiostoma floridae and Trichoplax adhaerens possess a CXXX motif [33].
By contrast, only two proteins listed below RAB-28 have two consecutive cysteines near their C-termini, C25D7.7 and Y32F6B.3. C25D7.7 does not possess a recognizable CIM and its reciprocal best hit is human RAP2. Y32F6B.3 is most closely related to cdc42, a Rho GTPase, and possesses additional sequence elements consistent with its inclusion in the Rho family. In fact, the vast majority of genes listed below RAB-28 possess sequence elements that suggest inclusion in Ras, Arf or Rho families. For example, genes that have a single amino acid deletion in place of the conserved glycine in RABF1 are characteristic of Ras and Rho family members while the presence of a tryptophan within position 6 of RabF3 is characteristic of Arf family members [9]. Additional amino acids that are exclusively found in Ras, Arf, and Rho families but not Rab are highlighted in yellow (Figure 1).
There are only a handful of genes with RabF identity scores less than RAB-28 that also lack amino acids justifying classification as a nonRab. Of these, the vast majority code for a reciprocal best hit (RBH) of a nonRab human protein and lack C-terminal cysteines. One intriguing exception is ZK669.5. ZK669.5 has a 33% RabF identity score and is the only one below RAB-28 that has a C-terminal cysteine and a recognizable CIM. For these reasons, we include ZK669.5 in our phylogenetic analysis described below to gather support for or against placement of ZK669.5 within the Rab family. In summary, our analysis identified a total of 31 Rab proteins including the following new additions: Y71H2AM.12, glo-1 and possibly ZK669.5.
A Phylogenetic Comparison of C. elegans and Human Rabs
Human orthologs had been identified for many but not all C. elegans Rab proteins [17]. Specifically, human orthologs had not been found for Y71H2AM.12, C33D12.6 (CeRabY1), 4R79.2(CeRabY2), K02E10.1 (CeRabY3), F11A5.4 (CeRabY4), F11A5.3 (CeRabY5), C56E6.2 (CeRabY6) and the putative Rab, ZK669.5. As the Rab family in mammals has grown since 2001, we redid a phylogenetic analysis here to determine if additional orthologous or paralogous clusters might be identified (see methods). In brief, we collected a nonredundant set of C. elegans and human Rab protein sequences from NCBI (see also, [34]). For example, if more than one splice variant existed, the one that included a prenylation motif was retained. Similarly, only one subfamily member of human-only clades with bootstrap support >98 in preliminary trees was included. A multiple sequence alignment was then used to create a neighbor-joining tree using the Jones Taylor Thornton (JTT) amino acid substitution method with 500 bootstrap replications (Figure 2). The same tree as an unrooted phylogram is also provided (Figure S2).
Figure 2. A chladogram of Rab family members from C. elegans and H. sapiens.
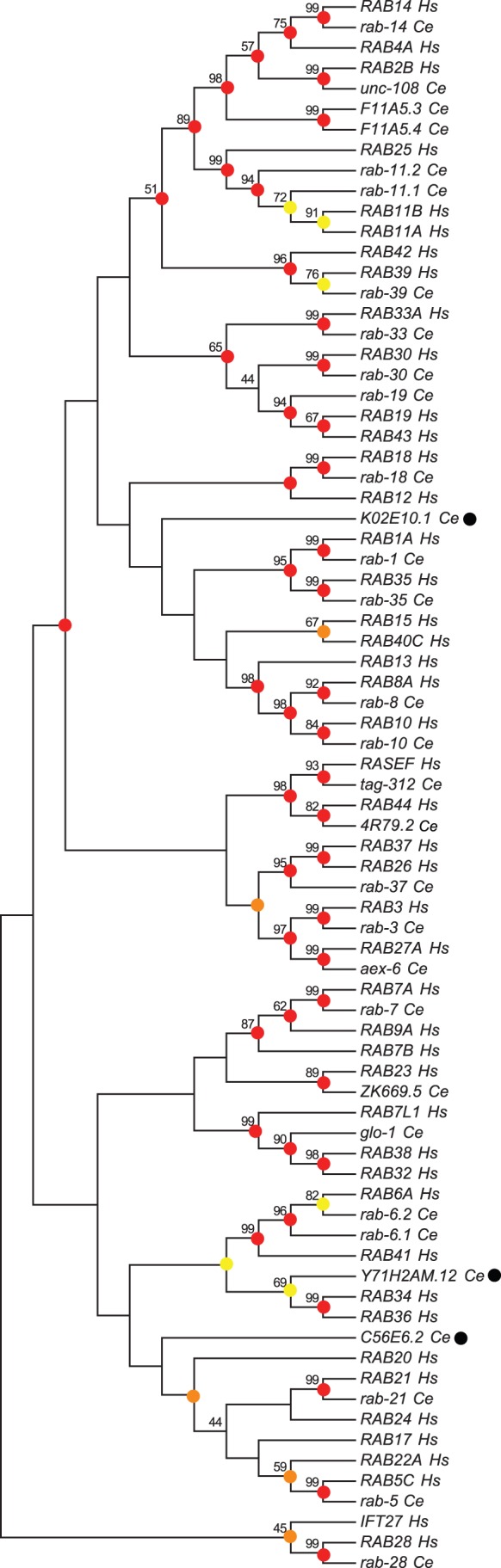
The evolutionary history was inferred using the Neighbor-Joining phylogenetic reconstruction method. The tree is rooted with the natural outlying clade, Rab28. The optimal tree is shown with the percentage of replicate trees (>40) in which the associated genes cluster together in the bootstrap test (500 replicates) provided next to each branch. The tree is drawn to emphasize topology. The evolutionary distances were computed using the JTT amino acid substitution method and are in the units of the number of amino acid differences per site. Evolutionary analyses were conducted using MEGA5. Clades marked with red, orange or yellow circles indicate their degree of stability under a variety of phylogenetic reconstruction parameters (see text and methods for details). Red = 14/14, orange = 13/14 and yellow = 12/14 trees. Genes highlighted with black circles represent putative orphan C. elegan Rabs (lacking a human ortholog). For simplicity, closely related splice variants and well-supported human-specific clades were deleted (see methods for details).
The terminal clades we observed are consistent with the previous tree [17] with the following additions/corrections. GLO-1 is grouped with a clade that includes RAB7L1 (RAB29), RAB32 and RAB38 (bootstrap support = 99). Consistent with this position, glo-1 is the reciprocal best hit (RBH) and likely ortholog of human RAB32. F11A5.4 (CeRabY4) and F11A5.3 (CeRabY5) are paralogous with bootstrap support of 99 and are grouped within a clade that includes Rab14, 4 and 2 with bootstrap support of 98. C33D12.6/TAG-312 (CeRabY1) and 4R79.2 (CeRabY2) are members of the RAB45 (RASEF) and RAB44 subfamilies with bootstrap support of 93 and 82, respectively. Importantly, this phylogenetic analysis used an MSA that excluded the long N-terminal extensions rare in the Rab family but characteristic of these two subfamilies. In addition, human RASEF contains an EF Hand domain within its N-terminal extension, as does C33D12.6. There is also strong bootstrap support (98) for a clade that includes both RAB44 and RAB45 suggesting that these two subfamilies are paralogous and likely formed through a gene duplication event. We also observe moderate bootstrap support (89) for a clade that includes ZK669.5 and human RAB23.
The RAB23/ZK669.5 terminal clade is problematic for several reasons. ZK669.5 is only 29% identical to human RAB23 far below the 40% cutoff some use to classify Rab subfamily members [9], [35]. The branch lengths of this terminal clade are long and unequal (Figure S2). Finally, the Rab23 subfamily (like Rab28) is a natural outlier in the Rab family phylogenetic tree [34], [36]. For these reasons, we worry that this cluster may result from long-branch attraction [37]–[40]. Long-branch attraction is sensitive to phylogenetic reconstruction method and choice of outgroup [37]–[39]. To rule out long-branch attraction in this instance, we performed 13 additional phylogenetic reconstructions (Figure 3). Each reconstruction used a unique combination of statistical method, amino acid substitution model, gap deletion treatment and rate and patterns of evolution.
Figure 3. Bootstrap scores for specific terminal clades from 13 additional phylogenetic reconstructions.

Thirteen additional phylogenetic analyses were performed using a combination of statistical methods. Phylogenetic reconstruction methods include Neighbor Joining (NJ), maximum likelihood (ML) or minimum evolution (ME). Amino acid substitution methods include Poisson (Po), JTT, Dayhoff (D), Equal Input (EI) and WAG. Gap deletion treatments include partial (Par) or pairwise (Pai) and rates and patterns of evolution include gamma distributed (+G), invariant sites (+I) or uniform (all others). All phylogenetic reconstructions were performed in MEGA5. Specific orthologous and/or paralogous clades include both worm and human Rab proteins. New orthologous clusters include bootstrap scores supporting the Rab44/4R79.2, Rab45/C33D12.6 and Rab23/ZK669.5 pairs. The new paralogous cluster includes the bootstrap scores that support the Rab44,4R79.2,Rab45 and C33D12.6 terminal clade.
Our results indicate that the original tree in Figure 2 is robust. Most terminal and/or near terminal clades including the Rab23/ZK669.5 cluster are stable in 14/14 trees (Figure 2, red circles). In addition, average bootstrap support for the RAB23/ZK669.5 clade is not significantly different from other clusters where an ancestral relationship is well accepted by other criteria (Figure 3). For example, though Rab3 and Rab27 are both involved in regulated secretion, bind to some of the same effectors and have overlapping function at the synapse in C. elegans [41]–[43], bootstrap support for this pair ranges from 63 to 98 with an average of 80. Similarly, though bootstrap support for the Rab5/22 pair ranges from 31 to 72 with an average of 56, an evolutionary relationship between Rab5 and Rab22 is well accepted: several effectors have been identified that bind both Rab proteins and a subset of exon/intron junctions are conserved between the two genes [17], [44]–[46]. Further, the well-accepted paralogs, Rab7 and Rab9 [47], have bootstrap support ranging from 71 to 93 with an average of 83. By comparison, bootstrap support for the ZK669.5/RAB23 clade ranges from 62 to 91 with an average of 79 (Figure 3). The ZK669.5/RAB23 clade is also stable when different outgroups are used including human KRas, Arf1 or a set of genes that include best nonRab hits for ZK669.5 (data not shown). The list of nonRab proteins used for this last analysis is provided (see Methods). Overall, these numbers are consistent with the assertion that bootstrap support above 95 may be too restrictive for this particular family of proteins [40].
As additional support for a RAB23/ZK669.5 clade, the top nonspecific domain hit for ZK669.5 in the Conserved Domain Database at NCBI is rab23-like (e value = 2.88e-11). In addition, TreeFam, a database of phylogenetic trees automatically created through the generation of seed trees that are progressively enlarged also classifies ZK699.5 as a Rab23 subfamily member (TreeFam ID: TF317494) [48]. Finally, the long isoform of ZK669.5 (ZK669.5a) contains a CAAX-like motif, an uncommon feature among Rabs in general but conserved within the Rab23 subfamily [31]. Together these results strongly suggest that ZK669.5 is a Rab and that human Rab23 and ZK669.5 share a common ancestor. From this perspective, the long unequal branch lengths observed (Figure S2), low percent identity and moderate bootstrap support suggests that ZK669.5 evolved more rapidly than its human counterpart.
Classification of Y71H2AM.12 and C56E6.2 by Intron Position Conservation Analysis
K02E10.1, Y71H2AM.12 and C56E6.2 could not be classified by our molecular phylogenetic analysis (Figure 2). Y71H2AM.12 clusters with human RAB34 in 12/14 trees with low average bootstrap support (53) or with human RAB6 (data not shown). C56E6.2 and K02E10.1 branches are long and not supported by bootstrap replicates (<30). These observations suggest two possibilities. K02E10.1, Y71H2AM.12 and/or C56E6.2 belong to a conserved subfamily lost in humans or rapid sequence divergence in C. elegans has obscured their ancestry. To distinguish between these two possibilities, we compared intron positions of K02E10.1, Y71H2AM.12 and/or C56E6.2 to Rab subfamily members most closely related to these orphan Rabs. Importantly, conservation of intron position has been observed among orthologs even when sequence identity is low and/or evolutionary distance is great [49], [50]. Moreover, multiple instances of conserved intron positions likely reflect common ancestry [51], [52].
To identify potential orthologs of K02E10.1, Y71H2AM.12 and C56E6.2, we used a “space hopping strategy” [53]. Each Rab orphan was first used as a query to identify top hits and/or reciprocal best hits (RBH) within two slowly evolving nematode species, Trichinella spiralis and Brugia malayi [54], [55]. Slowly evolving species are more likely to retain ancestral introns [56]. High quality hits were then used as queries in subsequent BLAST searches to identify top hits and/or RBHs within more distantly related species from the Opisthokonta. By analyzing disparate members of the Opisthokonta (Figure 4A), we hoped to distinguish ancestral introns from introns that are species or lineage-specific. Once all sequences were identified and tentatively classified, Rab subfamilies were individually aligned by Muscle with subfamily-specific gaps and nonconserved termini deleted. Next, all aligned and trimmed subfamilies were grouped and re-aligned to create one, large multiple sequence alignment (MSA) of 167 amino acids. Introns were mapped to this MSA and a Maximum Likelihood (ML) phylogenetic tree was created (see methods for details). Importantly, only introns that mapped within the MSA block were considered. Overall, this analysis involved 96 Rab sequences, 400 introns and 7 Rab subfamilies including Rab5, Rab6, Rab21, Rab22, Rab23, Rab31 and Rab34. These subfamilies were included either because they were identified in the “space hopping strategy” described above or because they would serve as negative controls. Negative control sequences were necessary to determine whether intron position conservation is indeed subfamily specific.
Figure 4. Comparative analysis of intron position among diverse Rab subfamily members.

A) Cladogram indicating evolutionary relationships of 18 species examined here [128]–[130]. Ecdy. = Ecdysozoa, Chromal. = Chromalveolata. For species abbreviations see Methods. B) An ML tree of Opisthokonts created from the MSA used to map intron positions. Bootstrap support (100 replicates) is indicated for each subfamily cluster. C) For each subfamily, the number of times a Subfamily Specific Conserved Intron Position (SSCIP) involving the indicated number of species was observed (gray bars), compared to what is expected by chance (black diamonds). The difference between observed and expected is statistically significant where indicated. *P(Monte Carlo) <0.05. ***P(Monte Carlo)≤0.00001. The Rab31, 6, 5, 22, 34, 21 and 23 subfamilies include 17, 18, 17, 9, 9, 10, 14 and 12 species, respectively. D) and E) Heat map indicating number of introns within Rab31 (D) or Rab6 (E) that match SSCIPs from Rab31, 5, 22, 21, 6, 34 and 23. The circled number indicates the number of introns present in the MSA for each gene. % equals the percentage of introns that are shared with the true SSCIP. C56E6.2 (D) and Y71H2AM.12 (E) are highlighted red. Genbank Descriptions (if any) and RABDB! classifications are included. Classification abbreviations include: HypoRabX1 (H.RabX1), HypoRabX2 (H.RabX2), HypoRabX3 (H.RabX3) and MetazoaRabX3 (M.RabX3). F) A pairwise comparison of intron position conservation between specific genes (Rab31 at left, Rab6 at right) and their corresponding set of SSCIPs. Black diamonds plot the probability that a specific number of intron matches would be expected by chance for each set of conditions. Chart 1 plots a comparison of 5 introns with 7 SSCIPs (5×7). Chart 2∶4×7. Chart 3∶2×7. Chart 4∶4×6. Observed values for a subset of genes are indicated with P values estimated from the Monte Carlo simulation data (See text and methods). Species abbreviations are as in A. C) and F) 72 protosplice sites assumed.
The topology of the phylogenetic tree (Figure 4B) demonstrates the success of the “sequence hopping strategy”. Putative members of each subfamily formed monophyletic clusters. An enlarged view of each subfamily-specific clade is also provided (Figure S3).
Next, we identified subfamily-specific conserved intron positions (SSCIPs). An SSCIP is defined as an intron position that is conserved among subfamily members in three or more disparate species of the Opisthokonta suggesting a presence within an ancestor to metazoans (Figure 4A). An intron position is defined as conserved if it is located at an identical position and within the same phase of the codon. As defined, we found 5 SSCIPs in Rab22, 6SSCIPs in Rab5, Rab6, 34, 21 and 23 and 7 SSCIPs in Rab31. We then counted the number of introns that each individual Rab gene shared with each set of SSCIPs. Results for Rab31 and Rab6 subfamilies are displayed as an array of filled circles where darker shades correspond to higher numbers of shared introns and columns correspond to each subfamily included in this study (Figure 4D, E). The MSA including the relative intron positions of all subfamilies is also provided (Figure S4).
Overall, this analysis demonstrates that Rab subfamily members possess remarkable conservation of intron positions over long evolutionary distances. 87% of the 400 introns within the MSA map to SSCIPs. In other words, only 13% of the introns are specific to a single species or lineage. Moreover, none of the SSCIPs identified for Rab5, 6, 21, 22, 23, 31 and 34 map to the same location with the exception of two shared between Rab5 and Rab22. This last observation is consistent with a previous report [17].
To quantify this level of conservation, a Monte Carlo simulation with 100,000 iterations was performed. In brief, all introns within each subfamily-specific MSA were randomly shuffled and the number of instances of conserved introns involving exactly 3, 4, 5 etc. species was counted. The data generated from this analysis was then used to estimate P values to assess statistical significance between what was observed and what could have occurred by chance (see Methods). Of the 41 SSCIPs identified only 5 could have occurred by chance with *P(Monte Carlo) >0.05. By contrast, 33 SSCIPs were extremely significant with ***P(Monte Carlo) <0.00001 (Figure 4C).
This analysis also allowed the unambiguous classification of Y71H2AM.12 and C56E6.2 to the Rab6 and Rab31 subfamilies, respectively. Y71H2AM.12 possesses four introns within the MSA block of which three map to Rab6 SSCIPs previously deemed statistically significant at the 0.00001 level. By contrast, none match intron positions of any other Rab gene analyzed including species and lineage-specific introns. The likelihood that at least three of the four introns in Y71H2AM.12 might match Rab6 SSCIPs by chance is extremely low with P(Monte Carlo) = 0.00001. This P value assumes that every nucleotide position within the MSA is a potential intron insertion site (501 sites total). If instead, one assumes that introns insert into genes at nonrandom positions called protosplice sites [51], the likelihood remains low with P(Monte Carlo) = 0.0013 (Figure 4F). In the latter analysis, the number of protosplice sites was set at 72 corresponding to one site per seven nucleotides [51]. This is likely an underestimate as the MSA block created here boasts 71 unique intron positions while only including a small fraction of the known Rab subfamilies (Figure S4). Interestingly, the uniquely-positioned intron in Y71H2AM.12 is only three nucleotides away from another rab6-specific CIP and may represent a phenomenon known as intron sliding [57].
C56E6.2 has two introns within the MSA block. Both map to Rab31 SSCIPs previously deemed statistically significant at the 0.0001 level. By contrast, neither match intron positions of any other Rab gene analyzed. Again, the likelihood that both introns might map to Rab31 SSCIPs by chance is low, with P values equaling 0.00016 or 0.008 depending on the number of protosplice sites assumed: 501 or 72 (Figure 4F). Moreover, C56E6.2 is the reciprocal best hit of XP_003374270.1 and XP_001901062.1, Rab31 orthologs from T. spiralis and B. malayi, respectively. As predicted for slowly evolving species, intron position conservation of Rab31 from these nematode species is higher than in C. elegans. Specifically, 5/5 introns within Rab31 from T. spiralis and 4/4 introns within Rab31 from B. malayi match Rab31 SSCIPs. Needless to say, these observations are statistically significant (Figure 4F).
By contrast, introns in K02E10.1 do not match any of the SSCIPs present in subfamilies that clustered nearby (Figure 2 and data not shown) including rab1, 8, 10, 12, 15, 18, 35 and 40 (data not shown). It is worth noting that this Rab gene is atypical. It contains a methionine instead of glutamine at position 70 (Q70M) suggesting that it may not function as a GTPase [58]. Moreover, an attempt to isolate an ORFeome cDNA corresponding to this ORF failed [12] suggesting the possibility that this locus codes for a pseudogene or has been mis-annotated. Thus, the evolutionary history of K02E10.1 remains mysterious.
Also, all 3 introns present within the conserved portion of ZK669.5 are uniquely positioned (Figure S4). Importantly, an absence of intron conservation with Rab23 SSCIPs does not indicate that its classification by molecular phylogenetics is incorrect. There are numerous instances where clear orthologs do not possess introns that map to the expected SSCIPs. This is particularly true of rapidly evolving species within the Ecdysozoa including C. elegans and D. melanogaster [56]. Examples within this dataset include rab-5 from C. elegans. Despite overwhelming bootstrap support for its classification as Rab5 (Figure 2), none of its four introns are conserved with the 5 Rab5 SSCIPs identified here (Figure S4). Similarly, the single intron within Rab23 of D. melanogaster fails to match any of the 6 Rab23 SSCIPs. Ultimately, the classification of ZK669.5 as a RAB23 subfamily member will require additional evidence that does not solely rely on sequence or intron position conservation (i.e. functional data). To date, its function is poorly understood. There are no mutant alleles that map to ZK669.5 and high-throughput RNAi screens have not identified obvious abnormalities [59], [60].
C56E6.2/Rab31 Belongs to an Ancient Rab Subfamily
Rab31 is a poorly characterized subfamily previously identified in a small number of Opisthokonts [33]. Confusing matters, many Rab proteins have been erroneously annotated as Rab31 in Genbank and Ensembl (see Discussion). To learn more about the evolutionary origins of Rab31 we searched for orthologs in a small number of more distantly related species including Dictyostelium discoideum (an Amoebozoa), Tetrahymena thermophila and Ichthyophthirius multifiliis (two ciliates from the supergroup Chromalveolata). Specifically, we used Rab31 subfamily members from the Opisthokonta (14 total) as queries in BLASTP. XP_642644.1 from D. discodeum was the top hit by BLASTP for 9 of the 14 Rab31 members tested. Moreover, it was the RBH for Rab31 subfamily members from S. purpuratus, A. mellifera, M. brevicollis and C. owczarzaki. Consistent with its classification as a Rab31 subfamily member, both of its introns match Rab31 SSCIPs (Figure 4D), an observation that is statistically significant (Figure 4F).
Using a similar strategy, paralogs XP_001020942.1 and XP_001025858.2 were identified as putative Rab31 subfamily members from T. thermophila. Importantly, they possess an identical exon-intron structure (Figure S4). In addition, 3/5 introns map to Rab31 SSCIPs. Finally, EGR30930.1 from I. multifiliis was identified as a RBH of XP_001020942.1 from T. thermophila. Its corresponding Rab gene has 4 introns. All are conserved with Rab31 members from T. thermophila and three match Rab31 SSCIPs (Figure 4D). Again, these observations are statistically significant by Monte Carlo simulation (Figure 4F).
Eukaryotes can be subdivided into 5 or 6 so-called supergroups [61], [62]. The tree of life includes Chromalveolates, Unikonts, Rhizaria, Excavata and Plantae [62] if 5 supergroups are counted. To make 6 supergroups, Unikonts are split into Amoebazoa and Opisthokonts [61]. Our intron position data identified Rab31 in Opisthokonts, Amoebazoa and Chromalveolata. These results suggest that Rab31 is an ancient Rab subfamily that arose before the split of the Opisthokonta, Amoebazoa and Chromalveolata. While the topology of the tree remains murky at its base [62], the presence of Rab31 in 2 or 3 supergroups suggests that it may have been present in the LECA.
Independent Verification of Rabifier, an Automated Rab Classification Pipeline
In 2011, Diekmann et al. published Rabifier, an automated bioinformatics pipeline for the identification and classification of Rab proteins [33]. This tool was validated against three manually curated Rab families from Trypanosoma brucei, Entamoeba histolytica and Monosiga brevicollis. They documented 99% accuracy for Rab family identification and 71% to 90% (high confidence) accuracy for subfamily classification.
A comparison of the C. elegans Rab family identified and classified by “Rabifier” with the manually curated family described here is consistent with their published rate of accuracy. Only three differences were observed. Specifically, strong phylogenetic and/or intron position data presented here suggest that ZK669.5, Y71H2AM.12 and C56E6.2 should be classified as rab23, rab6 and rab31, respectively. Instead, Rabifier classifies these proteins to undefined subfamilies HypoRabX1, MetazoaRabX3 and CeRabY6 (www.rabdb.org). Interestingly, this high rate of success for Rab protein classification may not extend to all subfamilies. Rabifier correctly classifies only four of the 17 Rab31 genes manually identified here, a success rate of 24%.
Identification of Error-free ORFeome-based WT Rab Clones
In 2007, students of a Molecular Techniques course at California State University, East Bay initiated the production of an ORFeome-based toolkit that attempted to include all 29 members of the Rab family known at the time (not including Y71H2AM.12 or ZK669.5). Such a toolkit will not only be useful for studying Rab function in vitro and in vivo but can also be used to manipulate or label specific regions within the endomembrane system for a wide variety of purposes.
The first step in creating an ORFeome-based toolkit was to identify error-free WT isolates from the ORFeome library. Importantly, ORFeome clones were originally created by PCR amplification of individual ORFs with Gateway-tailed ORF-specific primers. Each amplicon was then recombined into a pDONR vector, transformed into E. coli then 50–1000 colonies were frozen en masse for distribution. Thus, glycerol stocks for each ORF-containing entry clone are polyclonal [11]. Polyclonal pools were intended to capture alternate splice forms thereby increasing the utility of the library, however, they also captured clones with primer synthesis and PCR-based errors. Ultimately, errors were observed at a rate of 1 in 1232 bp [11].
To identify error-free WT ORFeome clones, a pair of students followed steps one through four outlined in Figure 5. Specifically, each pair purified four isolates of a given rab ORFeome clone. Two of the four isolates with the expected HinfI restriction fragment pattern by Polyacrylamide Gel Electrophoresis (PAGE) were then sequenced. If at least one clone was error-free, this isolate was used as a template to create the two mutant clones (DN and CA). If neither “WT” clones were error-free, the gene was set aside for a future class to try again with an additional four isolates. If the restriction fragment pattern for all four isolates was abnormal, one isolate was sequenced to rule out (or in) the possibility that the exon/intron structure had been mispredicted. A maximum of 8 isolates were analyzed. If all 8 isolates contained errors, the clone was not studied further.
Figure 5. A flow chart describing the lab module involving verification and modification of ORFeome Rab clones.

Steps 1 through 4 were done in parallel to steps A through D. Two peer-review steps at 3 and D were included to minimize mistakes in primer design and sequence analysis of WT ORFeome clones. Abbreviations: Gene of Interest (GOI), Constitutive Active (CA), Dominant Negative (DN), Restriction Fragment Length Polymorphism (RLFP), Polyacrylamide Gel Electrophoresis (PAGE), Human Ras (HRAS), Wild-Type (WT).
Ultimately, this strategy identified 22 WT Rab ORFs to be used as template for site-directed mutagenesis. Specifically, 19 Rab ORFs had 100% identity to Refseq protein sequences (excluding start and stop codons). Two Rab ORFs had minor differences to their corresponding Refseq and one Rab ORF had an exon/intron structure distinct from what had been predicted (Table 1).
Table 1. A list and description of Rab isolates created for the C. elegans ORFeome-based toolkit.
| Other Name(s) | Sequence Name | Rab Subfamily | Refseq with Best Match(% Identity) | WT | DN | CA |
| rab-1 | C39F7.4 | Rab1 | NP_503397.1 (100) | IK3-1 | IK20-1 | PP50-1 |
| unc-108/rab-2 | F53F10.4 | Rab2 | NP_491233.1 (100) | GC5-1 | GC33-2 | SM15-1 |
| rab-3 | C18A3.6 | Rab3 | NP_001021974.1 (100) | AP2-1 | NG33-1 | CG7-1 |
| rab-5 | F26H9.6 | Rab5 | NP_492481.1 (100) | PD3-1 | NH11-1 | MJ21-1 |
| rab-6.1 | F59B2.7 | Rab6 | NP_498993.1 (100) | SDS6-1 | PRS33-2 | SDS34-1 |
| rab-6.2 | T25G12.4 | Rab6 | NP_510790.1 (100) | AV3-2 | NJ16-1 | AV11-1 |
| rab-7 | W03C9.3 | Rab7 | NP_496549.1 (100) | MG2-1 | SI46-4 | SI28-5 |
| rab-8 | D1037.4 | Rab8 | NP_491199.2 (100) | MB5-2 | DK26-1 | MB38-1 |
| rab-10 | T23H2.5 | Rab10 | NP_491857.1 (100) | MB10-1 | SBA42-1 | CN28-1 |
| rab-11.1 | F53G12.1 | Rab11 | NP_490675.1 (100) | ZY2-2 | PRI8-7 | ZY10-1 |
| rab-11.2 | W04G5.2 | Rab11 | N/A | OST indicates retention of intron one | ||
| rab-14 | K09A9.2 | Rab14 | NP_510572.1 (100) | SP6-1 | MHB43-1 | SP51-1 |
| rab-18 | Y92C3B.3 | Rab18 | N/A | ORFeome clone is a rab-18::mlc-3 fusion | ||
| rab-19 | Y62E10A.9 | Rab43 | NP_502576.1 (100) | NM7-2 | KM29-1 | DJM14-1 |
| rab-21 | T01B7.3 | Rab21 | NP_495854.1 (100) | LAK5-1 | JS31-4 | DS25-1 |
| aex-6/rab-27 | Y87G2A.4 | Rab27 | NP_493376.1 (100) | JP6-1 | JP38-1 | SAN46-1 |
| rab-28 | Y11D7A.4 | Rab28 | NP_501609.1 (100) | SVP6-1 | ST21-1 | SVP57-2 |
| rab-30 | Y45F3A.2 | Rab30 | NP_499328.1 (100) | MEL4-1 | TT18-1 | SMJ11-1 |
| rab-33 | F43D9.2 | Rab33 | NP_499314.1 (100) | TR6-1 | TR34-1 | MAH25-1 |
| rab-35 | Y47D3A.25 | Rab35 | N/A | ORFeome clone contains a rab-27 insert. | ||
| rab-37 | W01H2.3 | Rab37 | NP_001041293 (100) | RV3-0 | ZW10-1 | KLD50-1 |
| rab-39 | D2013.1 | Rab39 | N/A | ORFeome clone lacks the C-terminus. | ||
| CeRabY1/tag-312 | C33D12.6 | Rab45 | NP_508523.1 (96) | DVD8-2 | DVD36-2 | AMT92-13 |
| CaRabY2 | 4R79.2 | Rab44 | NP_503120.1 (100) | LGK3-2 | LGK29-1 | AMT92-5 |
| CeRabY3 | K02E10.1 | orphan | N/A | OST does not confirm ORF | ||
| CeRabY4 | F11A5.4 | Rab2-like | NP_507084.1 (99) | KK9-2 | AMT92-1 | AMT92-9 |
| CeRabY5 | F11A5.3 | Rab2-like | NP_507083.1 (100) | EB8-2 | MR49-1 | PM16-1 |
| CeRabY6 | C56E6.2 | Rab31/Rab50 | N/A | ORFeome clone did not grow. | ||
| glo-1 | R07B1.12 | Rab32 | N/A | ORFeome clone not in the database. | ||
| N/A | Y71H2AM.12 | Rab6-like | N/A | Not examined | ||
| N/A | ZK669.5 | Rab23-like | N/A | Not examined | ||
WT, DN and CA clones included in the Rab Toolkit are given isolate names otherwise an explanation for its absence is provided. Subfamily classifications are based on Diekmann et al. 2011 [33] and/or data presented here. The majority of C. elegans rab genes are predicted to have only one splice variant with the following exceptions: WormBase describes two splice variants for rab-3 that code for proteins 233 and 219 amino acids (aa) in length. ORFeome project primers were designed to amplify the shorter isoform only. WormBase describes two splice variants for 4R79.2 (Rab44) that code for proteins 311 aa and 395 aa in length. ORFeome project primers were designed to amplify the longer isoform only. Names listed under “other” are from WormBase or Pereira-Leal and Seabra (2001). Finally, while rab-37 shows 100% identity with the Refseq protein NP_001041293 it contains an additional 5 amino acids at its N-terminus. See text for details.
The remaining seven Rab ORFs were not included in the toolkit for a variety of reasons. glo-1 (R07B1.12) was not present in the ORFeome database. C56E6.2 failed to produce viable colonies. K02E10.1 and W04G5.2 ORFs were not confirmed by their ORF sequence tag (OST). Finally, ORFeome clones for rab-18 (Y92C3B.3), rab-35 (Y47D3A.25) and rab-39 (D1013.1) were found by the students of the research-based course to be unusable. Specifically, the rab-18 ORFeome clone contained a rab-18::mlc-3 gene fusion. The rab-35 ORFeome clone contains rab-27 and the rab-39 ORFeome clone lacks the true C-terminus. Importantly, the C-terminus of rab-39 described in NP_495984 is supported by cDNA evidence (data not shown) while the C-terminus of the rab-39 ORFeome clone is not. All ORFeome clones that are included in the toolkit but deviate from their reference sequence are described in more detail below.
All four ORFeome isolates of F11A5.4 and rab-37 differed from their corresponding Refseq protein accession sequences but were retained in the toolkit to be used with caution. The F11A5.4 ORF has a cysteine to tyrosine missense mutation at position 180 (C180Y), the result of a G539A transition. This missense mutation is also found in its OST, suggesting that it results from an early PCR error or error in the sequenced C. elegans genome. Importantly, the only experimental evidence confirming this ORF is the OST from ORFeome. In addition, the cysteine at position 180 falls outside of the Rab domain and is not conserved with its closely related paralog, F11A5.3. The significance of this missense mutation is not known. For rab-37, ORFeome project primers were designed to amplify a protein described in AAB52888 now deemed obsolete. This isoform is identical to the short isoform of rab-37 (NP_001041293) except that it contains five additional amino acids (MFLKV) at its N-terminus. This addition is not expected to impact rab-37 function as N-terminal Rab fusions are well-tolerated [63]–[65].
The Full-length ORFeome Clone Sequence (FlOCS) of CeRabY1(C33D12.6), the putative ortholog of human RAB45 (RASEF) indicates the existence of a different exon/intron structure from what had been predicted (Figure 6A). Specifically, exon 7 (predicted) is interrupted by a 45 bp intron creating a 15 amino acid in-frame deletion conserved across diverse phyla including humans (Figure 6B). In addition, intron 8 is shifted 5′ and enlarged creating an in-frame InDel involving 10 amino acids (Figure 6A). While the Indel falls in a region conserved only among other nematodes (Figure 6B), the 5′ and 3′ splice sites (SS) of intron eight (FlOCS) match the C. elegans SS consensus sequence equal to or better than the predicted intron 8 [66]. Importantly, all four isolates were identical by HinfI digestion and PAGE analysis suggesting that the gene structure was likely a misprediction, not an example of alternative splicing (data not shown). The only other Rab gene not previously supported by complete EST or OST evidence was rab-33. In this instance, FlOCS data confirms the accuracy of the internal exon/intron boundaries of rab-33. For FlOCS data of all 22 WT ORFeome clones described here see Figure S5 (Accession numbers are also provided in the Materials and Methods).
Figure 6. Full-length ORF sequence of tag-312 (Rab45) isolates reveals a different splice pattern than predicted.

A) A nucleotide alignment of tag-312 (FlOCS), genomic DNA and the predicted tag-312 ORF at two regions where exon/intron splice junction differences were found. In the top alignment, FlOCS reveals a new intron, splitting predicted exon 7 into exons7a and 7b. In the bottom alignment, FlOCS reveals an alternate 5′ splice donor and 3′ splice acceptor for intron 8. Compare FlOCS-supported 5′ and 3′ splice sites boxed in bold to predicted 5′ and 3′ splices sites (boxed, not bold). B) Two multiple sequence alignments of Rab45 subfamily members spanning the two regions described in 5A above demonstrate the impact of FLOCS-supported gene structure differences. The alignment includes proteins from the nematodes, Caenhorabditis elegans, tag-312(FlOCS) and NP508523.1, Caenhorabditis brenneri, Caenhorabditis remanei and Ascaris suum in addition to Xenopus laevis (frog), Homo sapiens (human), Monodelphis domestica (opossum) and Anolis carolinensis (lizard). The intron that splits exon 7 into two creates a 15 amino acid deletion that is conserved among all species examined (top). The alternate intron 8 creates an Indel in a region of Rab45 that is conserved among nematodes only.
Generation of Dominant Negative and Constitutive Active Clones
Each student was tasked to create an ORF coding for the Q70L constitutive active (CA) or T17N dominant negative (DN) mutant form, initially characterized in HRas [18], [19]. Importantly, these missense mutations have also been shown to transform Rab GTPase family members into DN or CA mutant forms in a variety of organisms including yeast, humans, Drosophila, and C. elegans [20]–[24].
To generate mutant clones, students first followed steps A through D outlined in Figure 5. In brief, each student used Clustal W to create a multiple sequence alignment that included the conceptual translation of their assigned rab ORF to identify the worm amino acid that corresponds to either Q70 or T17 in HRas. Once found, each student designed mutagenic primers then performed a Quikchange site-directed mutagenesis reaction (Agilent Technologies) using a sequence verified WT rab clone as template. A list of mutagenic primer sequences used is provided in Table S1. Since the vast majority of mutagenic primers were designed to create or destroy a restriction enzyme site, successful clones were first identified by restriction enzyme digestion then single isolates with the correct pattern of fragments were sequenced. In this way, DN and CA mutants were created for all 22 WT (or near WT) rab ORFs. During the course of this work, we witnessed phenomenal accuracy with Quikchange site-directed mutagenesis, which created no errors out of 52,408 nt sequenced (data not shown). Furthermore, the attL sites flanking each ORF are functional as each entry clone was successfully used in a recombinational cloning reaction with LR clonase (data not shown). Importantly, isolate MB38-1 (rab-8 CA) functions as expected when expressed within ciliated neurons to disrupt vesicle transport to the cilia [67].
Discussion
Consistent with recent results [33], we expand the size of the C. elegans Rab family by three to 31 members. New members include, Glo-1, Y71H2AM.12 and ZK669.5. We also provide strong to moderate bootstrap support for the orthologous pairing of C33D12.6 (CeRabY1) with human RAB45, 4R79.2 (CeRabY2) with human RAB44 and ZK669.5 with human RAB23. Finally, a comparative analysis of intron position also allowed the classification of Y71H2AM.12 and C56E6.2 as Rab6 and Rab31, respectively. Only K02E10.2, a possible pseudogene, remains an orphan.
29 of the 30 classified Rabs are assigned to subfamilies also found in humans. One exception is Rab31, a subfamily that is poorly understood, often misclassified and mostly overlooked. Data presented here indicates that Rab31 is present within the Opisthokonta, Amoebozoa and supergroup Chromalveolata. By some standards [33], [68], this suggests that Rab31 was present in the last eukaryotic common ancestor or LECA. This conclusion is consistent with work done in parallel by Elias et al. 2012. One important difference, Rab31 is named Rab50 [68]. We support this name change as many Rab proteins classified as Rab31 in Genbank and Ensembl are in fact members of the Rab22 subfamily (this work and [33]). For example, the human Rab protein (NP_006859.2) designated as RAB31 by the HUGO Gene Nomenclature Committee (HGNC) clusters with Rab22 (Figure S3) and possesses introns that match Rab22 SSCIPs (Figure S4). Until a name change is accepted we will refer to this subfamily as Rab31/Rab50.
Drosophila’s chrowded (chrw) is the only Rab31/Rab50 subfamily member whose function has been described [69]. chrw was identified in a forward genetic screen for genes required for peripheral nervous system (PNS) development. Specifically, the PNS of mutant animals harboring a revertible transposon positioned directly 5′ to the chrw coding sequence (CDS) are disorganized with thick axons. More recently, Elias et al. 2012 used a novel high resolution phylogenetic approach called ScrollSaw to place the Rab31/Rab50 subfamily within a well-supported, higher-order clade that includes Rab21, 24, 20, 5 and 22 suggesting that Rab31/Rab50 may have been part of the core endocytic pathway within the LECA [68].
While Rab31/Rab50 is the only ancient subfamily in C. elegans that is absent in humans, many are present in humans but absent in C. elegans including Rabs 4, 9, 20, 22, 24, 29, 34, 40 and IFT27. Most of these Rab proteins are also absent in Drosophila melanogaster, another member of the Ecdysozoa [54]. These observations are consistent with whole genome studies that indicate that D. melanogaster and C. elegans are highly derived with widespread gene loss [70], [71]. A few, however, are present in D. melanogaster but lost or divergent in C. elegans including Rabs 4, 9, 23 and 40 (this paper and [33]). Of these, Rabs 4, 40 and 23 function in part by regulating the transport of specific developmental control proteins [65], [72]–[77]. It is worth noting that the Ecdysozoa is the largest and most diverse of the superphylum within animalia with over 4.5 million species [78]. Perhaps then, it is not surprising that dramatic changes and/or gene loss has been observed among these Rab proteins. Indeed, loss of specific developmental control genes and pathways within C. elegans but not Drosophila has been well-documented [79]–[81].
Conservation of Intron Position and the Classification of Rab Proteins
In this work, we used conservation of intron position to classify Rab subfamilies that were otherwise difficult to classify using molecular phylogenetics. Our success with the classification of Y71H2AM.12 as Rab6 and C56E6.2 as Rab31/Rab50 validates this approach and highlights inherent limitations with the molecular phyologenetic approach, which relies on bootstrap support above a specific value. For example, the Rab31 subfamily cluster from the Opisthokonta examined here is supported by a bootstrap score of only 38 and the Rab6 subfamily cluster including Y71H2AM.12 is supported by a bootstrap score of only 47 (Figure 4B). By all criteria this level of support is unacceptable. It’s not surprising then that previous work employing phylogenetic analysis alone each missed specific C. elegans Rab proteins. Diekmann et al. failed to classify C56E6.2 (Rab31/50), ZK669.5(Rab23) and Y71H2AM.12(Rab6) [33]. Elias et al. 2012 failed to classify Y71H2AM.12 and misclassified two Rab31/Rab50 proteins from Tetrahymena thermonila as Rab22 [68]. Finally, TreeFam failed to classify C56E6.2 (Rab31/Rab50) (TreeFam ID = TF352282) [48]. While molecular phylogenetics is quick, powerful and mostly accurate, we argue that an intron position analysis may facilitate the classification of poorly supported clades. Moreover, the utility of intron position data is not limited to the identification of subfamily members. It can also be used to answer important evolutionary questions including but not limited to mechanisms underlying intron evolution and/or formation of the LECA.
One defining feature of the eukaryote is its large, endomembrane system comprised of multiple compartments and complex mechanisms of vesicle transport. In one model [82], the endomembrane system evolved from a single internal compartment regulated by a small set of proteins including one or a few Rab proteins. In an iterative process that occurred pre-LECA, compartment expansion occurred alongside gene duplication, sequence diversification and specialization of protein function. In support of this hypothesis molecular phylogenetic analyses indicate that the LECA is comprised of up to 23 Rab subfamily members [33], [68]. Because this expansion occurred pre-LECA, a comprehensive analysis of intron position conservation among Rab family members present within the LECA can complement existing efforts to garner insight into the evolution of the endomembrane system pre-LECA [68], [83]. For example, in one model, intron invasion is concomitant to eukaryogenesis [82]. In our small dataset of 7 Rab subfamilies, none of the SSCIPs overlap except for two between Rab22 and Rab5. This is particularly interesting in light of recent results by Elias et al. 2012 that provide phylogenetic evidence to support a super-clade within the LECA that includes a subset of the Rab subfamilies analyzed here (Rab21, Rab50/31, Rab5 and Rab22). One intriguing possibility is that Rab family expansion and thus expansion of endomembrane complexity occurred prior to the invasion of introns. Again, this hypothesis and others will require a more comprehensive analysis of intron position conservation among Rab proteins. Until then, alternative explanations cannot be excluded. For example, initial intron invasion may have been far more extensive than previously thought followed by variable rates of intron loss within specific clades. Nonetheless, such an analysis will benefit from the careful selection of species with low rates of intron gains and/or losses [56], [84], [85].
Verified WT and Mutant C. elegans ORFeome Clones Facilitate Rab Function Studies
The C. elegans ORFeome Project, the semi-automated cloning of a near-complete set of full-length C. elegans ORFs has filled important gaps in our knowledge of gene structure, genome organization, variation and evolution. Combined with recombinational cloning strategies, this Gateway-compatible ORF collection has also been used in semi-automated, large-scale gene function studies with great success [59], [86], [87]. With the astounding amount of data collected for each of these large-scale studies, there has also been an inevitable loss of data that occurs when nonfunctional clones are unknowingly included in an experiment. This loss is not only tolerated but also expected. By contrast, those that use one or a small number of ORFeome clones in low-throughput study [88]–[90] cannot tolerate any level or error. With the toolkit developed here, not only have the students from Advanced Molecular Techniques at California State University, East Bay (CSUEB) created a set of useful mutant clones (CA and DN) of 22 rab genes but they have also generated a set of fully-verified WT isolates.
The ORFeome-based toolkit described here is Gateway-compatible. Thus each WT, DN and/or CA rab ORF is ready for recombinational cloning into a wide-variety of available destination vectors for biochemical and/or genetic analysis including the expression of Rab fusions in C. elegans [91]–[93], E. coli [94], insect [95] and/or yeast cells [86]. For example, with the expression of Rab fusions in E. coli, insect or yeast cells one can identify proteins that interact with C. elegans Rabs with pull down assays and/or yeast two-hybrid screens. We expect that the CA form will be particularly useful to identify Rab protein effectors as this form is stuck in the active conformation [96]. Expression of Rab fusions (i.e. to GFP) in C. elegans will also be useful for studying the morphology and dynamics of specific subcellular structures [97], [98], in addition to probing both loss- and/or gain-of-function phenotypes in a cell or tissue of interest [67].
For in vivo analysis of rab gene function, we recommend that the stop codon absent in C. elegans ORFeome clones be restored by site-directed mutagenesis prior to use in recombinational cloning. For maximum versatility, the ORFeome clones were intentionally designed to lack the A of the ATG and the last two nucleotides of the stop codon [11]. Thus, expression clones created through Gateway recombination of destination vectors with ORFeome entry clones express fusion proteins that include (at a minimum) a peptide sequence of nine amino acids at both the N and C-termini due to the absence of the stop and start codons and the presence of attB sites (25 bp in length). It is not clear how this extra peptide sequence will impact Rab prenylation as the C-termini of Rab proteins are positioned alongside the active site in a bent conformation [8]. In fact, Wu et al. do not observe a reduction in prenylation when up to 5 arbitrarily chosen amino acids are added to the C-terminus but they do notice that hydrophobic patches (i.e. a CIM mimic) within a C-terminal extension can be inhibitory. A conceptual translation of attB shows the presence of a putative CIM (underlined): Y P A F L Y K V V. Furthermore, Rab proteins that possess a single cysteine in a CAAX-box-like context may be prenylated by FTase and/or GGTase in vivo [31], [32], [99], [100]. These enzymes require the insertion of the CAAX-box tail into a binding pocket suggesting that CAAX-box proteins would not tolerate the nine amino acid extension.
To recreate the erstwhile stop codon, we recommend inserting the necessary nucleotides by site-directed mutagenesis (SDM) so to leave the 3′ attL site untouched. Our experience reassures us that Quikchange SDM is not likely to incorporate unwanted point mutations, thus sequence confirmation may not be essential. Alternatively, one can amplify Rab ORFs using primers containing in-frame start and stop codons and 5′ restriction enzyme sites for use in traditional cloning. This strategy was used successfully to express isolate MB38-1 (rab-8 CA) within ciliated neurons to disrupt vesicle transport to the cilia [67].
The Importance of Research-based Lab Courses
As mentioned previously, the toolkit developed here was created in the context of a research-based lab course. One clear benefit of this pedagogical approach is its ability to provide an authentic research experience to a large number of students. While the traditional master-apprenticeship model has been successful it can exclude many due to infrastructure limitations of the host institution and large numbers of biology students. As a result, participants in extracurricular research typically involve a small number of self-selected students. Specifically, these students are aware that research opportunities exist, are highly motivated to participate, can afford to volunteer time outside of the classroom and fully appreciate its value.
To complement the traditional approach, many science educators now suggest bringing an authentic research experience into the classroom [13]–[15], [101]. Proponents of this approach argue that research-based lab courses can capture and possibly inspire the largest number of students, including those who had never envisioned a career in research [102]–[105]. This demand has been echoed by the National Science Education Standards that urge STEM disciplines to alter or replace cookbook lab courses for ones that emphasize inquiry, discovery and the development of a research mindset [106]. Moreover, the Committee on Undergraduate Biology Education to Prepare Research Scientists for the 21st Century argue that research-based lab courses are valuable because they are inherently interdisciplinary [107]. Not only do students gain scientific knowledge, but they also gain experience with experimental design, quantitative analysis and written and oral communication. By bringing scientific research into the classroom, educators demonstrate that scientists deal with unanswered questions on a daily bases and help students develop skills that are difficult to teach including critical thinking and scientific reasoning.
In a research-based laboratory course, ideally each student (or student pair) is provided a unique project where the outcome and path to completion is unknown, even to the course instructor. As described by Weaver et al. [13], there should be “no information in any textbook, laboratory manual or journal article about their expected results.” Individual students should have numerous opportunities to make decisions in experimental design and execution, in data analysis and in forming conclusions. While guidance can be provided by both peers and the instructor, ultimate success is the sole responsibility of the individual or student pair.
In practice, research-based curriculum can be logistically complex and expensive. It can be difficult to come up with a large number of unique projects year after year, to prep the lab with equipment and reagents to support all ongoing projects not to mention supervising a large number of inexperienced, wet lab scientists doing research. These challenges are particularly daunting at institutions that lack the funds to hire teaching assistants to help course instructors.
I (co-author M.G.) initiated a research-based lab course in 2007 that attempted to maximize the benefits of research-based curricula but minimize the challenges. To this end, I exploited the availability of the C. elegans ORFeome resource, a collection of ORF entry clones corresponding to the majority of ORFs identified within the C. elegans genome [11], [12]. In brief, each student was given an entry clone from the ORFeome library as starting material (kindly provided by Kang Shen). From there each student took an interdisciplinary approach to accomplish the aims outlined in Figure 5. Specifically, all students learned wet lab skills beneficial to the molecular biologist but also had to master online databases, sequence analysis tools and additional software including GenBank, PubMed, OMIM, BLAST, Clustal W, A Plasmid Editor (M. Wayne Davis), Image J (NIH), Excel and PowerPoint (Microsoft).
The use of the ORFeome library as starting material combined with the creation of two mutant forms for each gene was instrumental in overcoming many of the challenges outlined above. At the beginning of each course, student pairs were assigned a unique gene. Importantly each student still conducted his/her own work but this strategy allowed for the creation of backup reagents, as we quickly discovered that some DNA isolates were unusable. Then each student was assigned a unique mutant form allowing the opportunity to work independently. Importantly, during both phases of the course, students used a similar set of computer tools and molecular techniques in any given week allowing for conservation of reagents and instruction. Moreover, the projects were similar enough that peer review could be used to double check experimental design and/or interpretation of results so that this task was not left entirely up to the instructor. In fact, once peer review became a formal part of the course, the number of student and instructor errors declined dramatically (unpublished data). Finally, the use of ORFeome clones allowed ample opportunity for discovery. For example, during the process of purifying and analyzing “wild type” isolates students discovered a new splice form and identified and characterized ORFeome clone errors. In addition, students took pride in the knowledge that they were creating new reagents for the scientific community, work that might get published in a peer-reviewed journal. In fact, the possibility of publication had the most dramatic impact on both the student and the instructor in terms of creating an authentic research experience. As the instructor, I cared deeply that the lab presentations were clear, results were analyzed correctly and reagents and lab notebooks were organized and documented properly. We were united in our effort to produce and document our research accurately.
For science educators interested in designing a similar course to this one or others that have been described recently [105], [108]–[110], Gateway-compatible ORFeome libraries are now available for numerous species in addition to C. elegans including humans and Schizosaccharomyces pombe among others [111]–[117]. Within these libraries there are many genes and/or gene families that could benefit from tools allowing in vitro or in vivo analysis of dominant negative and/or constitutive active forms. For example, transcription factors can often be converted to dominant negative forms by deleting protein-protein interaction domains while leaving DNA binding domains intact [118]. Protein kinases can often be converted to dominant, kinase-dead forms by mutating the universally conserved lysine residue within the ATP-binding domain [119]–[121]. Alternatively, one can alter kinase effectors by mutating putative phosphorylation sites to mimic dephosphorylation and/or constitutive phosphorylation [122]. These types of projects would be particularly useful if done in collaboration with a lab interested in using the reagents upon completion. Ultimately, the creation of tools that facilitate gene function studies can help reduce the so-called bottleneck of genes that have been identified by sequence but still lack clear function.
In closing, it is important to note that this class is offered as a required class in the Biotech Certificate Program (BCP) at California State University, East Bay. It typically enrolls post baccalaureate students. To date, only a small number of undergraduates have taken the course. While I have not attempted to offer this class to undergraduates, I imagine it could be done with simple modifications. For example, I would likely provide more guidance in experimental design but leave all opportunities for data analysis and oral presentations as is. A more comprehensive description of the course and additional suggestions for implementation will be published elsewhere.
Materials and Methods
Rab Protein Identification
To manually identify the complete set of Rab proteins from C. elegans, 28 Rab proteins identified by Pereira-Leal and Seabra (2001) were first aligned by Muscle using MEGA5 [26]. Once aligned, these sequences were trimmed at the N- and C-termini to include the Rab domain plus the N-terminal RabSF1 motif. This multiple sequence alignment was then used as a query in PSI-BLAST using the bioinformatics toolkit [27]. The complete list (excluding splice variants) of significant hits identified by PSI-BLAST was aligned by Muscle again with obvious alignment errors fixed manually. The alignment of each RabF domain (1–5) was then exported to Microsoft Excel in order to calculate the RabF percent identity to consensus sequences of the five RabF motifs combined [9] (Co-author M.G.).
Molecular Phylogenetics
To construct the phylogenetic trees of Human and Worm Rab proteins described in Figures 2, 3 and S2, a single copy of each Rab subfamily was retrieved from NCBI and combined with all C. elegans Rab proteins identified as described in the text. Again, to simplify the list of genes, all splice variants save one were removed. The one that remained contained a Rab prenylation motif if present. Also, all human Rab subfamily members that formed a species-specific clade with bootstrap support >99 were reduced to a single member. Then, the full-length human and worm Rab proteins were aligned by Muscle using MEGA5. Once aligned, sequences were trimmed of their variable N- and C-termini leaving sequence from RabSF1 through RabSF4 and the prenylation motif (including the first cysteine and the sequence that followed). Finally, a variety of trees were created with MEGA5 (co-author M.G.). Phylogenetic reconstruction methods used included Neighbor Joining, maximum likelihood or minimum evolution. Amino acid substitution models included Poisson, Equal Input, Jones Thornton Taylor (JTT), Whelan and Goldman (WAG) and Dayhoff. Gap deletion treatments included partial or pairwise and rates and patterns of evolution include gamma distributed (+G), invariant sites (+I) or uniform. For any given tree, the specific combination used is described in the Figure legend and text. The bootstrap test was used to calculate the percentage of replicate trees in which the associated genes cluster together (100 replicates for Maximum Likelihood and 500 for all others). The list of genes (including accession numbers) and the alignment used to create the phylogenetic trees is also provided (Figure S1).
Phylogenetic trees containing human RAB23, worm ZK669.5 and a group of human and worm nonRab top hits of ZK669.5 were done as described above. The list of nonRab human genes included Ras-like protein family member 12 (NP_057647.1), RERG/RAS-like (AAH42888.1), BAB55008.1, Ras-like protein family member 11A (NP_996563.1), Ras-related associated with diabetes (AAH57815.1), GEM (NP_859053.1), RAS (RAD and GEM)-like (AAV38882.1), RAS (RAD and GEM)-like GTP binding 2 (AAH35663.1), Rap-2c (NP_067006.3), R-ras2 (NP_036382.2), Rap-1b (NP_056461.1), M-Ras (NP_001078518.1), RalA (NP_005393.2), and Rheb (NP_005605.1). The list of nonRab worm genes included Y71F9AR.2 (NP_491082.2), rap-1 (NP_501549.1), rap-2 (NP_506707.2), rheb-1 (NP_499079.1), ras-2 (NP_497972.1), ral-1 (NP_497689.1), C08F8.7 (WP:CE40190) and C44C11.1a (WP:CE24846).
The phylogenetic reconstruction in Figure 4B was created by the Maximum Likelihood method based on the Whelan and Goldman model (WAG) [123] with a discrete Gamma distribution (+G) to model evolutionary rate difference among sites (5 categories, G = 1.7896) and allowing for some sites (3.0941%) to be evolutionarily invariant (+I) (co-author M.G.). This reconstruction was suggested by the MEGA5 model test analyzing 48 different amino acid substitution models. Since gaps and variable termini were deleted during the alignment process, no further deletions were made during the reconstruction. There were a total of 167 amino acids in the final data set.
Comparative Analysis of Intron Position
Subfamilies analyzed by intron position included Rab31, Rab5, Rab22, Rab21, Rab23, Rab6 and Rab34. Species analyzed came mostly from the Opisthokonta including two Lophotrochozoans: Caliptella teleta (Ctel) and Lottia gigantean (Lgig); three Arthropods Apis mellifera (Amel), Tribolium castaneum (Tcas), and Drosophila melanogaster (Dmel); three Nematodes Brugia malayi (Bmal), Caenorhabditis elegans (Cele) and Trichinella spiralis (Tspi); one Echinoderm, Strongylocentrotus purpuratus (Spur); two Chordates Ciona intestinalis (Cint), and Homo sapiens (Hsap); two Cnidarians Hydra magnipapillata (Hmag) and Nematostella vectensis (Nvec); one Choanozoan, Monosiga brevicollis (Mbre); one unranked Opisthokant, Capsaspora owczarzaki (Cowc); and two intron-rich fungal species [85], Coprinopsis cinerea okayama (Ccin) and Phanerochaete chrysosporium (Pchr). For the Rab31 subfamily analysis, one additional species came from the Amoebazoan, Dictyostelium discoideum (Ddis) and two additional species came from the supergroup Chromalveolata, phylum Ciliophora: Ichthyophthirius multifiliis (Imul) and Tetrahymena thermophile (Tthe). Species substitutions were necessary on three occasions where gene sequence was of low quality or could not be found by the methods employed here. Specifically, alternate Hymenopterans, Apis florea (Aflo) and Camponotus floridanus (Cflo) substituted for Apis mellifera Rab5 and Rab34, respectively. An alternate dipteran, Anopheles gambiae (Agam), substituted for Drosophila melanogaster Rab34. Members of each subfamily were identified from the species listed above through a “sequence space hopping” strategy previously described [53]. When possible, reciprocal best hits were identified by searching the reference sequence database at Genbank using default parameters in Basic Local Alignment Search Tool (BLAST) at NCBI otherwise the nonredundant (nr) protein database was searched. Sequence for Lottia gigantea, Capitella teleta and Nematostella vectensis were obtained by BLAST at the Joint Genome Institute (JGI). The complete list of accession numbers corresponding to the sequences used in the comparative analysis of intron position is provided in Figure S4.
Members of each subfamily were first aligned independently using Muscle in MEGA5. Within each subfamily, species-specific insertions (present in only one species) were deleted along with nonconserved terminal regions. Terminal regions chosen for deletion failed to produce stable alignments, contained gaps involving multiple species and consistently fell below an arbitrary overall 50% amino acid identity cutoff. Subfamily alignments were then sequentially combined into one file and realigned with each new addition by Muscle in MEGA5. The MSA was then exported to excel. This alignment was used to create the ML reconstruction in Figure 4B and for the intron position analysis (co-author M.G.).
Intron positions were determined using a variety of methods depending on sequence type. For reference sequences from NCBI (i.e. XP_ and NP_), SPLIGN was used (NCBI). For nonreference sequences from NCBI (i.e. EFW_) an annotated text map of the exon intron boundaries was created by A plasmid Editor (Wayne Davis) from the Genbank file (.gb) corresponding to the gene of interest. For sequences from the JGI database, an annotated text map of the 3-frame translation for each gene model was examined (co-author M.G.). Intron position information was then mapped onto the excel file with intron phase information retained (Figure S4). For analysis of intron conservation, intron position was defined as conserved if the 5′ and/or 3′ splice site was at an identical position and phase in at least two species.
Monte Carlo Simulations of Intron Positions
Two types of Monte Carlo simulations were performed using Python scripts created in TextWrangler (co-author S.B.). One compares a gene of interest to a single set of subfamily specific conserved intron positions (SSCIPs). It analyzes the likelihood that a specific number of intron positions of a given Rab might match the set of SSCIPs by chance. This python script randomly generates X whole numbers (nonrepeating) within the range from 1 to Y, for the gene and subfamily of interest, where X is the number of introns and Y the number of possible intron insertion sites in that particular species (protosplice sites). For each replicate, data sets corresponding to each are freshly generated and compared. Numbers that occur in both data sets in a pairwise comparison is indicative of a shared intron insertion site or a coincidence. When complete the Monte Carlo simulation provides the sum of all types of coincidences observed. In other words, the number of times 0, 1, 2, 3 etc. coincidences were observed in a single pairwise comparison. This script requires the following inputs: 1) The number of introns present in the Rab gene of interest, 2) the number of SSCIPs present in the Rab subfamily of interest, 3) the hypothetical number of protosplice sites in the pairwise alignment and 4) the total number of replicates performed (100,000). Assuming that all nucleotide positions within the alignment are potential intron insertion sites, the protosplice site number was set at 501. Assuming that protosplice sites are present on average every 7 nucleotides [51], the protosplice site number was set at 72.
Another Monte Carlo simulation analyzes the likelihood that intron insertion sites randomly match across N number of species, each containing a specific number of introns (co-author S.B.). The above-mentioned algorithm was scaled up to perform “multiwise” comparisons of N species, for this purpose. For each replicate, randomly generated data sets corresponding to each of the N species are compared and the number of shared intron positions that involve exactly 2, 3, 4, 5, etc. species are counted. When complete, this Monte Carlo simulation provides the total number of instances where conserved intron positions involved exactly 2, 3, 4, 5 etc. species. This script requires the following inputs: 1) The number of introns present in each Rab gene analyzed in the MSA, 2) the hypothetical number of protosplice sites present within the MSA (see above) and 3) the total number of replicates performed (100,000).
The output created by either method was used to estimate P values (co-author S.B.). Specifically, P values were estimated using the formula, P value = r/n where n equals the number of replicates and r equals the number of instance where a specific value equal to or greater than a specific value of interest (i.e. the number of times that at least 7 species possessed a shared intron position) was observed [124].
Site Directed Mutagenesis
To create mutant clones by site directed mutagenesis, the published protocol for Quikchange II XL was followe (Agilent Technologies, Cat # 200522) with a few noted exceptions (all co-authors excluding M.G.). Specifically, desalted primers were instead of PAGE purified with no loss in efficiency (data not shown). In addition, half the suggested reaction mix was used. To generate mutagenic primers that might create or destroy a restriction enzyme site the now obsolete program, Primer Generator [125] was initially used. Now SiteFind [126] is used to create restriction enzyme sites (where possible) and “A plasmid Editor” is used to destroy restriction enzyme sites (where possible) [127]. Once a mismatch region is chosen, primers are designed manually by student co-authors following the criteria outlined in the Quikchange protocol except that the Tm is calculated with the equation designed for creating insertions or deletions (Tm = 81.5+0.41(%GC)-675/N) where N = primer length (not including the mismatch region) and percent GC is a whole number. Again, the mismatch region is ignored. Importantly, the mismatch region is defined as the number of nucleotides that are different from the WT sequence including internal nucleotides that might otherwise match. For example, AGTTTGA has a mismatch of 3 even though the WT sequence is AGCTCGA.
GenBank Accession Numbers
The accession numbers for full-length ORFeome clone sequences of WT rab isolates described in the text and Table 1 are as follows: IK3-1 = JQ235180, GC5-1 = JQ235181, AP2-1 = JQ235182, PD3-1 = JQ235183, SDS6-1 = JQ235184, AV3-2 = JQ235185, MG2-1 = JQ235186, MB5-2 = JQ235187, MB10-1 = JQ235188, ZY2-2 = JQ235189, SP6-1 = JQ235190, NM7-2 = JQ235191, LAK5-1 = JQ235192, JP6-1 = JQ235193, SVP6-1 = JQ235194, MEL4-1 = JQ235195, TR6-1 = JQ235196, RV3-1 = JQ235197, DVD8-2 = JQ235198, LGK3-2 = JQ235199, KK9-2 = JQ235200, EB8-2 = JQ235201.
Supporting Information
The multiple sequence alignment used to create the tree described in Figures 2 and S2.
(XLS)
The cladogram in figure 2 shown as an unrooted phlyogram with relative branch lengths restored.
(EPS)
Individual clusters from Figure 4B enlarged with each branch labeled with species name and accession number.
(PDF)
Intron positions of Rab subfamily members within the conserved portion of the multiple sequence alignment (MSA). Yellow squares correspond to phase 1 introns. Green squares correspond to phase 2 introns and red squares correspond to phase 3 introns (intron is positioned after the indicated codon). Intron free columns within the MSA were deleted. Numbering in the top row corresponds to the amino acid position of Rab6 from Lottia gigantea. For species abbreviations, see Figure 4 legend. Stars mark the position of each SSCIP as defined in the text. Black stars correspond to SSCIPs that are statistically significant at P(Monte Carlo <0.00001). Dark gray stars correspond to SSCIPs that are statistically significant at P(Monte Carlo <0.05). Light gray stars correspond to SSCIPs that are not statistically significant.
(PDF)
Full-length ORFeome Clone Sequence (FlOCS) for each isolate (WT, DN and CA) described in Table 1 .
(DOC)
A list of mutagenic primers (forward only) used in site-directed mutagenesis of mutant Rab forms. The mismatch region is highlighted in bold and all caps. For a definition of mismatch and the equation used to calculate Tm, see methods. Where applicable, the diagnostic enzyme and the form it digests is indicated. m = mutant, wt = wild type.
(DOC)
Acknowledgments
I would like to thank Cen Gao and Kang Shen (Stanford University) for bacterial cultures of ORFeome clones, Noelle L’Etoile (University of California, Davis) for critical comments on the manuscript and Chris Baysdorfer (California State University, East Bay) for initial advice on phylogenetic analysis.
Funding Statement
This work was funded by the Department of Biological Sciences at California State University, East Bay. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. No additional external funding was received for this study.
References
- 1. Schwartz SL, Cao C, Pylypenko O, Rak A, Wandinger-Ness A (2008) Rab GTPases at a glance. Journal of Cell Science 121: 246–246 doi:10.1242/jcs.03495. [DOI] [PubMed] [Google Scholar]
- 2. Stenmark H (2009) Rab GTPases as coordinators of vesicle traffic. Nat Rev Mol Cell Biol 10: 513–525 doi:10.1038/nrm2728. [DOI] [PubMed] [Google Scholar]
- 3. Mitra S, Cheng KW, Mills GB (2011) Rab GTPases implicated in inherited and acquired disorders. Seminars in Cell & Developmental Biology 22: 57–68 doi:10.1016/j.semcdb.2010.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wennerberg K (2005) The Ras superfamily at a glance. Journal of Cell Science 118: 843–846 doi:10.1242/jcs.01660. [DOI] [PubMed] [Google Scholar]
- 5. Casey PJ, Seabra MC (1996) Protein prenyltransferases. J Biol Chem 271: 5289. [DOI] [PubMed] [Google Scholar]
- 6. Rak A, Pylypenko O, Niculae A, Pyatkov K, Goody RS, et al. (2004) Structure of the Rab7:REP-1 complex: insights into the mechanism of Rab prenylation and choroideremia disease. Cell 117: 749–760 doi:10.1016/j.cell.2004.05.017. [DOI] [PubMed] [Google Scholar]
- 7. Guo Z, Wu Y-W, Das D, Delon C, Cramer J, et al. (2008) Structures of RabGGTase-substrate/product complexes provide insights into the evolution of protein prenylation. EMBO J 27: 2444–2456 doi:10.1038/emboj.2008.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wu Y-W, Goody RS, Abagyan R, Alexandrov K (2009) Structure of the disordered C terminus of Rab7 GTPase induced by binding to the Rab geranylgeranyl transferase catalytic complex reveals the mechanism of Rab prenylation. J Biol Chem 284: 13185–13192 doi:10.1074/jbc.M900579200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pereira-Leal JB, Seabra MC (2000) The mammalian Rab family of small GTPases: definition of family and subfamily sequence motifs suggests a mechanism for functional specificity in the Ras superfamily. J Mol Biol 301: 1077–1087 doi:10.1006/jmbi.2000.4010. [DOI] [PubMed] [Google Scholar]
- 10. Goody RS, Rak A, Alexandrov K (2005) The structural and mechanistic basis for recycling of Rab proteins between membrane compartments. Cell Mol Life Sci 62: 1657–1670 doi:10.1007/s00018-005-4486-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Reboul J, Vaglio P, Rual J-F, Lamesch P, Martinez M, et al. (2003) C. elegans ORFeome version 1.1: experimental verification of the genome annotation and resource for proteome-scale protein expression. Nat Genet 34: 35–41 doi:10.1038/ng1140. [DOI] [PubMed] [Google Scholar]
- 12. Lamesch P, Milstein S, Hao T, Rosenberg J, Li N, et al. (2004) C. elegans ORFeome version 3.1: increasing the coverage of ORFeome resources with improved gene predictions. Genome Research 14: 2064–2069 doi:10.1101/gr.2496804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Weaver GC, Russell CB, Wink DJ (2008) Inquiry-based and research-based laboratory pedagogies in undergraduate science. Nat Chem Biol 4: 577–580 doi:10.1038/nchembio1008-577. [DOI] [PubMed] [Google Scholar]
- 14.Advisory Committee to the National Science Foundation Directorate for Education, Resources H (1996) Shaping the future: New expectations for undergraduate education in science, mathematics, engineering, and technology. http://www.nsf.gov/publications/pub_summ.jsp?ods_key=nsf96139 Accessed 2012 Oct 27.
- 15. Fischer CN (2011) Changing the science education paradigm: from teaching facts to engaging the intellect: Science Education Colloquia Series, Spring 2011. Yale J Biol Med 84: 247–251. [PMC free article] [PubMed] [Google Scholar]
- 16. Chen J, Call GB, Beyer E, Bui C, Cespedes A, et al. (2005) Discovery-based science education: functional genomic dissection in Drosophila by undergraduate researchers. PLoS Biol 3: e59 doi:10.1371/journal.pbio.0030059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pereira-Leal JB, Seabra MC (2001) Evolution of the Rab family of small GTP-binding proteins. J Mol Biol 313: 889–901 doi:10.1006/jmbi.2001.5072. [DOI] [PubMed] [Google Scholar]
- 18. Barbacid M (1987) Ras genes. Annual review of biochemistry 56: 779–827. [DOI] [PubMed] [Google Scholar]
- 19. Farnsworth CL, Feig LA (1991) Dominant inhibitory mutations in the Mg(2+)-binding site of RasH prevent its activation by GTP. Mol Cell Biol 11: 4822–4829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Walworth NC, Goud B, Kabcenell AK, Novick PJ (1989) Mutational analysis of SEC4 suggests a cyclical mechanism for the regulation of vesicular traffic. EMBO J 8: 1685–1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Li G, Stahl PD (1993) Structure-function relationship of the small GTPase rab5. J Biol Chem 268: 24475–24480. [PubMed] [Google Scholar]
- 22. Tisdale EJ, Bourne JR, Khosravi-Far R, Der CJ, Balch WE (1992) GTP-binding mutants of rab1 and rab2 are potent inhibitors of vesicular transport from the endoplasmic reticulum to the Golgi complex. The Journal of Cell Biology 119: 749–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wilson BS, Nuoffer C, Meinkoth JL, McCaffery M, Feramisco JR, et al. (1994) A Rab1 mutant affecting guanine nucleotide exchange promotes disassembly of the Golgi apparatus. The Journal of Cell Biology 125: 557–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Babbey CM, Bacallao RL, Dunn KW (2010) Rab10 associates with primary cilia and the exocyst complex in renal epithelial cells. Am J Physiol Renal Physiol 299: F495–F506 doi:10.1152/ajprenal.00198.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wilson RK (1999) How the worm was won: the C. elegans genome sequencing project. Trends Genet 15: 51–58. [DOI] [PubMed] [Google Scholar]
- 26. Tamura K, Peterson D, Peterson N, Stecher G, Nei M, et al. (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28: 2731–2739 doi:10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Biegert A, Mayer C, Remmert M, Söding J, Lupas AN (2006) The MPI Bioinformatics Toolkit for protein sequence analysis. Nucleic Acids Research 34: W335–W339 doi:10.1093/nar/gkl217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hermann GJ, Schroeder LK, Hieb CA, Kershner AM, Rabbitts BM, et al. (2005) Genetic analysis of lysosomal trafficking in Caenorhabditis elegans. Molecular Biology of the Cell 16: 3273–3288 doi:10.1091/mbc.E05-01-0060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Grill B, Bienvenut WV, Brown HM, Ackley BD, Quadroni M, et al. (2007) C. elegans RPM-1 regulates axon termination and synaptogenesis through the Rab GEF GLO-4 and the Rab GTPase GLO-1. Neuron 55: 587–601 doi:10.1016/j.neuron.2007.07.009. [DOI] [PubMed] [Google Scholar]
- 30. Joberty G, Tavitian A, Zahraoui A (1993) Isoprenylation of Rab proteins possessing a C-terminal CaaX motif. FEBS Letters 330: 323–328. [DOI] [PubMed] [Google Scholar]
- 31. Leung KF, Baron R, Ali BR, Magee AI, Seabra MC (2007) Rab GTPases containing a CAAX motif are processed post-geranylgeranylation by proteolysis and methylation. J Biol Chem 282: 1487–1497 doi:10.1074/jbc.M605557200. [DOI] [PubMed] [Google Scholar]
- 32. Leung KF (2005) Thematic review series: Lipid Posttranslational Modifications. Geranylgeranylation of Rab GTPases. J Lipid Res 47: 467–475 doi:10.1194/jlr.R500017-JLR200. [DOI] [PubMed] [Google Scholar]
- 33. Diekmann Y, Seixas E, Gouw M, Tavares-Cadete F, Seabra MC, et al. (2011) Thousands of Rab GTPases for the Cell Biologist. PLoS Comput Biol 7: e1002217 doi:10.1371/journal.pcbi.1002217.g008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Colicelli J (2004) Human RAS superfamily proteins and related GTPases. Sci STKE 2004: RE13 doi:10.1126/stke.2502004re13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Saito-Nakano Y, Loftus BJ, Hall N, Nozaki T (2005) The diversity of Rab GTPases in Entamoeba histolytica. Experimental Parasitology 110: 244–252 doi:10.1016/j.exppara.2005.02.021. [DOI] [PubMed] [Google Scholar]
- 36. Lee SH, Baek K, Dominguez R (2008) Large nucleotide-dependent conformational change in Rab28. FEBS Letters 582: 4107–4111 doi:10.1016/j.febslet.2008.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Philippe H, Laurent J (1998) How good are deep phylogenetic trees? Curr Opin Genet Dev 8: 616–623. [DOI] [PubMed] [Google Scholar]
- 38. Baldauf SL (2003) Phylogeny for the faint of heart: a tutorial. Trends Genet 19: 345–351. [DOI] [PubMed] [Google Scholar]
- 39. Bergsten J (2005) A review of long‐branch attraction. Cladistics 21: 163–193. [DOI] [PubMed] [Google Scholar]
- 40. Kumar S, Filipski AJ, Battistuzzi FU, Kosakovsky Pond SL, Tamura K (2012) Statistics and truth in phylogenomics. Mol Biol Evol 29: 457–472 doi:10.1093/molbev/msr202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Handley MTW, Burgoyne RD (2008) The Rab27 effector Rabphilin, unlike Granuphilin and Noc2, rapidly exchanges between secretory granules and cytosol in PC12 cells. Biochem Biophys Res Commun 373: 275–281 doi:10.1016/j.bbrc.2008.06.043. [DOI] [PubMed] [Google Scholar]
- 42. Fukuda M (2008) Regulation of secretory vesicle traffic by Rab small GTPases. Cell Mol Life Sci 65: 2801–2813 doi:10.1007/s00018-008-8351-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tanaka D, Kameyama K, Okamoto H, Doi M (2008) Caenorhabditis elegans Rab escort protein (REP-1) differently regulates each Rab protein function and localization in a tissue-dependent manner. Genes Cells 13: 1141–1157 doi:10.1111/j.1365-2443.2008.01232.x. [DOI] [PubMed] [Google Scholar]
- 44. Eathiraj S, Pan X, Ritacco C, Lambright DG (2005) Structural basis of family-wide Rab GTPase recognition by rabenosyn-5. Nature 436: 415–419 doi:10.1038/nature03798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mishra A, Eathiraj S, Corvera S, Lambright DG (2010) Structural basis for Rab GTPase recognition and endosome tethering by the C2H2 zinc finger of Early Endosomal Autoantigen 1 (EEA1). Proceedings of the National Academy of Sciences 107: 10866–10871 doi:10.1073/pnas.1000843107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Woller B, Luiskandl S, Popovic M, Prieler BEM, Ikonge G, et al. (2011) Rin-like, a novel regulator of endocytosis, acts as guanine nucleotide exchange factor for Rab5a and Rab22. Biochim Biophys Acta 1813: 1198–1210 doi:10.1016/j.bbamcr.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mackiewicz P, Wyroba E (2009) Phylogeny and evolution of Rab7 and Rab9 proteins. BMC Evol Biol 9: 101 doi:10.1186/1471-2148-9-101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Li H, Coghlan A, Ruan J, Coin LJ, Hériché J-K, et al. (2006) TreeFam: a curated database of phylogenetic trees of animal gene families. Nucleic Acids Research 34: D572–D580 doi:10.1093/nar/gkj118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Betts MJ, Guigó R, Agarwal P, Russell RB (2001) Exon structure conservation despite low sequence similarity: a relic of dramatic events in evolution? EMBO J 20: 5354–5360 doi:10.1093/emboj/20.19.5354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Rogozin IB, Wolf YI, Sorokin AV, Mirkin BG, Koonin EV (2003) Remarkable interkingdom conservation of intron positions and massive, lineage-specific intron loss and gain in eukaryotic evolution. Curr Biol 13: 1512–1517 doi:10.1016/S0960-9822(03)00558-X. [DOI] [PubMed] [Google Scholar]
- 51. Carmel L, Rogozin IB, Wolf YI, Koonin EV (2007) Patterns of intron gain and conservation in eukaryotic genes. BMC Evol Biol 7: 192 doi:10.1186/1471-2148-7-192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Henricson A, Forslund K, Sonnhammer ELL (2010) Orthology confers intron position conservation. BMC Genomics 11: 412 doi:10.1186/1471-2164-11-412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Collins RN (2005) Application of phylogenetic algorithms to assess Rab functional relationships. Meth Enzymol 403: 19–28 doi:10.1016/S0076-6879(05)03003-X. [DOI] [PubMed] [Google Scholar]
- 54. Aguinaldo AM, Turbeville JM, Linford LS, Rivera MC, Garey JR, et al. (1997) Evidence for a clade of nematodes, arthropods and other moulting animals. Nature 387: 489–493 doi:10.1038/387489a0. [DOI] [PubMed] [Google Scholar]
- 55. Roy SW, Gilbert W (2006) The evolution of spliceosomal introns: patterns, puzzles and progress. Nat Rev Genet 7: 211–221 doi:10.1038/nrg1807. [DOI] [PubMed] [Google Scholar]
- 56. Raible F, Tessmar-Raible K, Osoegawa K, Wincker P, Jubin C, et al. (2005) Vertebrate-type intron-rich genes in the marine annelid Platynereis dumerilii. Science 310: 1325–1326 doi:10.1126/science.1119089. [DOI] [PubMed] [Google Scholar]
- 57. Stoltzfus A, Logsdon JM, Palmer JD, Doolittle WF (1997) Intron “sliding” and the diversity of intron positions. Proc Natl Acad Sci USA 94: 10739–10744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Erdman RA, Shellenberger KE, Overmeyer JH, Maltese WA (2000) Rab24 is an atypical member of the Rab GTPase family. J Biol Chem 275: 3848–3856. [DOI] [PubMed] [Google Scholar]
- 59. Rual J-F, Ceron J, Koreth J, Hao T, Nicot A-S, et al. (2004) Toward improving Caenorhabditis elegans phenome mapping with an ORFeome-based RNAi library. Genome Research 14: 2162–2168 doi:10.1101/gr.2505604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kamath RS, Fraser AG, Dong Y, Poulin G, Durbin R, et al. (2003) Systematic functional analysis of the Caenorhabditis elegans genome using RNAi. Nature 421: 231–237 doi:10.1038/nature01278. [DOI] [PubMed] [Google Scholar]
- 61. Simpson AGB, Roger AJ (2004) The real “kingdoms” of eukaryotes. Curr Biol 14: R693–R696 doi:10.1016/j.cub.2004.08.038. [DOI] [PubMed] [Google Scholar]
- 62. Keeling PJ, Burger G, Durnford DG, Lang BF, Lee RW, et al. (2005) The tree of eukaryotes. Trends in Ecology & Evolution 20: 670–676 doi:10.1016/j.tree.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 63. Moritz OL, Tam BM, Hurd LL, Peränen J, Deretic D, et al. (2001) Mutant rab8 Impairs docking and fusion of rhodopsin-bearing post-Golgi membranes and causes cell death of transgenic Xenopus rods. Molecular Biology of the Cell 12: 2341–2351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Babbey CM, Ahktar N, Wang E, Chen CC-H, Grant BD, et al. (2006) Rab10 regulates membrane transport through early endosomes of polarized Madin-Darby canine kidney cells. Molecular Biology of the Cell 17: 3156–3175 doi:10.1091/mbc.E05-08-0799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Pataki C, Matusek T, Kurucz E, Andó I, Jenny A, et al. (2010) Drosophila Rab23 is involved in the regulation of the number and planar polarization of the adult cuticular hairs. Genetics 184: 1051–1065 doi:10.1534/genetics.109.112060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Morton JJ, Blumenthal T (2011) RNA Processing in C. elegans. Methods Cell Biol 106: 187–217 doi:10.1016/B978-0-12-544172-8.00007-4. [DOI] [PubMed] [Google Scholar]
- 67. O’Halloran DM, Hamilton OS, Lee JI, Gallegos M, L’Etoile ND (2012) Changes in cGMP Levels Affect the Localization of EGL-4 in AWC in Caenorhabditis elegans. PLoS ONE 7: e31614 doi:10.1371/journal.pone.0031614.g004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Elias M, Brighouse A, Gabernet-Castello C, Field MC, Dacks JB (2012) Sculpting the endomembrane system in deep time: high resolution phylogenetics of Rab GTPases. Journal of Cell Science 125: 2500–2508 doi:10.1242/jcs.101378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Prokopenko SN, He Y, Lu Y, Bellen HJ (2000) Mutations affecting the development of the peripheral nervous system in Drosophila: a molecular screen for novel proteins. Genetics 156: 1691–1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Putnam NH, Srivastava M, Hellsten U, Dirks B, Chapman J, et al. (2007) Sea Anemone Genome Reveals Ancestral Eumetazoan Gene Repertoire and Genomic Organization. Science 317: 86–94 doi:10.1126/science.1139158. [DOI] [PubMed] [Google Scholar]
- 71. Copley RR, Aloy P, Russell RB, Telford MJ (2004) Systematic searches for molecular synapomorphies in model metazoan genomes give some support for Ecdysozoa after accounting for the idiosyncrasies of Caenorhabditis elegans. Evol Dev 6: 164–169 doi:10.1111/j.1525-142X.2004.04021.x. [DOI] [PubMed] [Google Scholar]
- 72. Eggenschwiler JT, Bulgakov OV, Qin J, Li T, Anderson KV (2006) Mouse Rab23 regulates hedgehog signaling from smoothened to Gli proteins. Developmental Biology 290: 1–12 doi:10.1016/j.ydbio.2005.09.022. [DOI] [PubMed] [Google Scholar]
- 73. Eggenschwiler JT, Espinoza E, Anderson KV (2001) Rab23 is an essential negative regulator of the mouse Sonic hedgehog signalling pathway. Nature 412: 194–198 doi:10.1038/35084089. [DOI] [PubMed] [Google Scholar]
- 74. Kachhap SK, Faith D, Qian DZ, Shabbeer S, Galloway NL, et al. (2007) The N-Myc down regulated Gene1 (NDRG1) Is a Rab4a effector involved in vesicular recycling of E-cadherin. PLoS ONE 2: e844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Mruk DD, Lau ASN, Sarkar O, Xia W (2007) Rab4A GTPase catenin interactions are involved in cell junction dynamics in the testis. J Androl 28: 742–754 doi:10.2164/jandrol.106.002204. [DOI] [PubMed] [Google Scholar]
- 76. Lee RHK, Iioka H, Ohashi M, Iemura S-I, Natsume T, et al. (2007) XRab40 and XCullin5 form a ubiquitin ligase complex essential for the noncanonical Wnt pathway. EMBO J 26: 3592–3606 doi:10.1038/sj.emboj.7601781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Jones MC, Caswell PT, Moran-Jones K, Roberts M, Barry ST, et al. (2009) VEGFR1 (Flt1) regulates Rab4 recycling to control fibronectin polymerization and endothelial vessel branching. Traffic 10: 754–766 doi:10.1111/j.1600-0854.2009.00898.x. [DOI] [PubMed] [Google Scholar]
- 78. Telford MJ, Bourlat SJ, Economou A, Papillon D, Rota-Stabelli O (2008) The evolution of the Ecdysozoa. Philos Trans R Soc Lond, B, Biol Sci 363: 1529–1537 doi:10.1098/rstb.2007.2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Ruvkun G, Hobert O (1998) The taxonomy of developmental control in Caenorhabditis elegans. Science 282: 2033. [DOI] [PubMed] [Google Scholar]
- 80. Aboobaker A, Blaxter M (2010) The nematode story: Hox gene loss and rapid evolution. Adv Exp Med Biol 689: 101–110. [DOI] [PubMed] [Google Scholar]
- 81. Ingham PW, Nakano Y, Seger C (2011) Mechanisms and functions of Hedgehog signalling across the metazoa. Nat Rev Genet 12: 393–406 doi:10.1038/nrg2984. [DOI] [PubMed] [Google Scholar]
- 82. Brighouse A, Dacks JB, Field MC (2010) Rab protein evolution and the history of the eukaryotic endomembrane system. Cell Mol Life Sci 67: 3449–3465 doi:10.1007/s00018-010-0436-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Koumandou VL, Dacks JB, Coulson RMR, Field MC (2007) Control systems for membrane fusion in the ancestral eukaryote; evolution of tethering complexes and SM proteins. BMC Evol Biol 7: 29 doi:10.1186/1471-2148-7-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. ZIMEK A, WEBER K (2008) In contrast to the nematode and fruit fly all 9 intron positions of the sea anemone lamin gene are conserved in human lamin genes. Eur J Cell Biol 87: 305–309 doi:10.1016/j.ejcb.2008.01.003. [DOI] [PubMed] [Google Scholar]
- 85. Stajich JE, Dietrich FS, Roy SW (2007) Comparative genomic analysis of fungal genomes reveals intron-rich ancestors. Genome Biol 8: R223 doi:10.1186/gb-2007-8-10-r223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Li S, Armstrong CM, Bertin N, Ge H, Milstein S, et al. (2004) A map of the interactome network of the metazoan C. elegans. Science 303: 540–543 doi:10.1126/science.1091403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Sieburth D, Ch’ng Q, Dybbs M, Tavazoie M, Kennedy S, et al. (2005) Systematic analysis of genes required for synapse structure and function. Nature 436: 510–517 doi:10.1038/nature03809. [DOI] [PubMed] [Google Scholar]
- 88. Fridolfsson HN, Ly N, Meyerzon M, Starr DA (2010) UNC-83 coordinates kinesin-1 and dynein activities at the nuclear envelope during nuclear migration. Developmental Biology 338: 237–250 doi:10.1016/j.ydbio.2009.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Narasimhan SD, Yen K, Bansal A, Kwon E-S, Padmanabhan S, et al. (2011) PDP-1 Links the TGF-β and IIS Pathways to Regulate Longevity, Development, and Metabolism. PLoS Genet 7: e1001377 doi:10.1371/journal.pgen.1001377.t003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Warner A, Qadota H, Benian GM, Vogl AW, Moerman DG (2011) The Caenorhabditis elegans paxillin orthologue, PXL-1, is required for pharyngeal muscle contraction and for viability. Molecular Biology of the Cell 22: 2551–2563 doi:10.1091/mbc.E10-12-0941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Voutev R, Hubbard EJA (2008) A “FLP-Out” system for controlled gene expression in Caenorhabditis elegans. Genetics 180: 103–119 doi:10.1534/genetics.108.090274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Zeiser E, Frøkjær-Jensen C, Jorgensen E, Ahringer J (2011) MosSCI and Gateway Compatible Plasmid Toolkit for Constitutive and Inducible Expression of Transgenes in the C. elegans Germline. PLoS ONE 6: e20082 doi:10.1371/journal.pone.0020082.t001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Saha S, Guillily MD, Ferree A, Lanceta J, Chan D, et al. (2009) LRRK2 modulates vulnerability to mitochondrial dysfunction in Caenorhabditis elegans. Journal of Neuroscience 29: 9210–9218 doi:10.1523/JNEUROSCI.2281-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Braun P, Hu Y, Shen B, Halleck A, Koundinya M, et al. (2002) Proteome-scale purification of human proteins from bacteria. Proc Natl Acad Sci USA 99: 2654–2659 doi:10.1073/pnas.042684199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Shay B, Gruenbaum-Cohen Y, Tucker AS, Taylor AL, Rosenfeld E, et al. (2009) High yield expression of biologically active recombinant full length human tuftelin protein in baculovirus-infected insect cells. Protein Expr Purif 68: 90–98 doi:10.1016/j.pep.2009.06.008. [DOI] [PubMed] [Google Scholar]
- 96. Fukuda M, Kanno E, Ishibashi K, Itoh T (2008) Large scale screening for novel rab effectors reveals unexpected broad Rab binding specificity. Molecular & Cellular Proteomics 7: 1031–1042 doi:10.1074/mcp.M700569-MCP200. [DOI] [PubMed] [Google Scholar]
- 97. Winter JF, Höpfner S, Korn K, Farnung BO, Bradshaw CR, et al. (2012) Caenorhabditis elegans screen reveals role of PAR-5 in RAB-11-recycling endosome positioning and apicobasal cell polarity. Nat Cell Biol 14: 666–676 doi:10.1038/ncb2508. [DOI] [PubMed] [Google Scholar]
- 98. Sann SB, Crane MM, Lu H, Jin Y (2012) Rabx-5 Regulates RAB-5 Early Endosomal Compartments and Synaptic Vesicles in C. elegans. PLoS ONE 7: e37930 doi:10.1371/journal.pone.0037930.g007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Wilson AL, Erdman RA, Castellano F, Maltese WA (1998) Prenylation of Rab8 GTPase by type I and type II geranylgeranyl transferases. Biochem J 333 (Pt 3): 497–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Maurer-Stroh S, Koranda M, Benetka W, Schneider G, Sirota FL, et al. (2007) Towards Complete Sets of Farnesylated and Geranylgeranylated Proteins. PLoS Comput Biol 3: e66 doi:10.1371/journal.pcbi.0030066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Moskovitz C, Kellogg D (2011) Inquiry-Based Writing in the Laboratory Course. Science 332: 919–920 doi:10.1126/science.1200353. [DOI] [PubMed] [Google Scholar]
- 102.Russell S (2006) EVALUATION OF NSF SUPPORT FOR UNDERGRADUATE RESEARCH OPPORTUNITIES: Synthesis Report Executive Summary. Available: http://csted.sri.com/content/evaluation-nsf-support-undergraduate-research-opportunities-uro-synthesis-report. Accessed 2012 Oct 27.
- 103. Lopatto D (2004) Survey of Undergraduate Research Experiences (SURE): first findings. Cell biology education 3: 270–277 doi:10.1187/cbe.04-07-0045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Lopatto D (2007) Undergraduate research experiences support science career decisions and active learning. CBE life sciences education 6: 297–306 doi:10.1187/cbe.07-06-0039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Knutson K, Smith J, Nichols P, Wallert MA, Provost JJ (2010) Bringing the excitement and motivation of research to students; Using inquiry and research-based learning in a year-long biochemistry laboratory : Part II-research-based laboratory-a semester-long research approach using malate dehydrogenase as a research model. Biochem Mol Biol Educ 38: 324–329 doi:10.1002/bmb.20401. [DOI] [PubMed] [Google Scholar]
- 106.National Research Council US (1996) National Science Education Standards. National Academies Press. pp. Available:http://books.nap.edu/openbook.php?record_id=4962&page=1. Accessed 2012 Oct 27.
- 107. Brenner K, Bio2010 Committee (2003) Fueling educational reform: Bio2010–biology for the future. Cell biology education 2: 85–86 doi:10.1187/cbe.02-11-0053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Brame CJ, Pruitt WM, Robinson LC (2008) A molecular genetics laboratory course applying bioinformatics and cell biology in the context of original research. CBE life sciences education 7: 410–421 doi:10.1187/cbe.08-07-0036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Treacy DJ, Sankaran SM, Gordon-Messer S, Saly D, Miller R, et al. (2011) Implementation of a Project-Based Molecular Biology Laboratory Emphasizing Protein Structure-Function Relationships in a Large Introductory Biology Laboratory Course. CBE life sciences education 10: 18–24 doi:10.1187/cbe.10-07-0085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Lau JM, Robinson DL (2009) Effectiveness of a cloning and sequencing exercise on student learning with subsequent publication in the National Center for Biotechnology Information GenBank. CBE life sciences education 8: 326–337 doi:10.1187/cbe.09-05-0036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Dricot A, Rual J-F, Lamesch P, Bertin N, Dupuy D, et al. (2004) Generation of the Brucella melitensis ORFeome version 1.1. Genome Research 14: 2201–2206 doi:10.1101/gr.2456204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Matsuyama A, Arai R, Yashiroda Y, Shirai A, Kamata A, et al. (2006) ORFeome cloning and global analysis of protein localization in the fission yeast Schizosaccharomyces pombe. Nat Biotechnol 24: 841–847 doi:10.1038/nbt1222. [DOI] [PubMed] [Google Scholar]
- 113. Schroeder BK, House BL, Mortimer MW, Yurgel SN, Maloney SC, et al. (2005) Development of a functional genomics platform for Sinorhizobium meliloti: construction of an ORFeome. Appl Environ Microbiol 71: 5858–5864 doi:10.1128/AEM.71.10.5858-5864.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Gong W, Shen Y-P, Ma L-G, Pan Y, Du Y-L, et al. (2004) Genome-wide ORFeome cloning and analysis of Arabidopsis transcription factor genes. Plant Physiol 135: 773–782 doi:10.1104/pp.104.042176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Brandner CJ, Maier RH, Henderson DS, Hintner H, Bauer JW, et al. (2008) The ORFeome of Staphylococcus aureus v 1.1. BMC Genomics 9: 321 doi:10.1186/1471-2164-9-321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Rajagopala SV, Yamamoto N, Zweifel AE, Nakamichi T, Huang H-K, et al. (2010) The Escherichia coli K-12 ORFeome: a resource for comparative molecular microbiology. BMC Genomics 11: 470 doi:10.1186/1471-2164-11-470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Yang X, Boehm JS, Yang X, Salehi-Ashtiani K, Hao T, et al. (2011) A public genome-scale lentiviral expression library of human ORFs. Nat Chem Biol 8: 659–661 doi:10.1038/nmeth.1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Shao W, Wang D, Chiang Y-T, Ip W, Zhu L, et al.. (2012) The Wnt Signaling Pathway Effector TCF7L2 Controls Gut and Brain Proglucagon Gene Expression and Glucose Homeostasis. Diabetes. doi:10.2337/db12-0365. [DOI] [PMC free article] [PubMed]
- 119. MacNicol AM, Muslin AJ, Williams LT (1993) Raf-1 kinase is essential for early Xenopus development and mediates the induction of mesoderm by FGF. Cell 73: 571–583. [DOI] [PubMed] [Google Scholar]
- 120. Pierce SB, Kimelman D (1995) Regulation of Spemann organizer formation by the intracellular kinase Xgsk-3. Development 121: 755–765. [DOI] [PubMed] [Google Scholar]
- 121. Tekinay T, Wu MY, Otto GP, Anderson OR, Kessin RH (2006) Function of the Dictyostelium discoideum Atg1 kinase during autophagy and development. Eukaryotic Cell 5: 1797–1806 doi:10.1128/EC.00342-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Qiao H, Shen Z, Huang S-SC, Schmitz RJ, Urich MA, et al.. (2012) Processing and Subcellular Trafficking of ER-Tethered EIN2 Control Response to Ethylene Gas. Science. doi:10.1126/science.1225974. [DOI] [PMC free article] [PubMed]
- 123. Whelan S, Goldman N (2001) A general empirical model of protein evolution derived from multiple protein families using a maximum-likelihood approach. Mol Biol Evol 18: 691–699. [DOI] [PubMed] [Google Scholar]
- 124. Broman KW, Caffo BS (2003) Simulation-based P values: response to North et al. Am J Hum Genet 72: 496 doi:10.1086/346175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Turchin A, Lawler JF (1999) The primer generator: a program that facilitates the selection of oligonucleotides for site-directed mutagenesis. BioTechniques 26: 672–676. [DOI] [PubMed] [Google Scholar]
- 126. Evans PM, Liu C (2005) SiteFind: a software tool for introducing a restriction site as a marker for successful site-directed mutagenesis. BMC Mol Biol 6: 22 doi:10.1186/1471-2199-6-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Davis WM, editor (n.d.) A Plasmid Editor. University of Utah. pp. Available:http://www.biology.utah.edu/jorgensen/wayned/ape/. Accessed 6 January 2012.
- 128. Dunn CW, Hejnol A, Matus DQ, Pang K, Browne WE, et al. (2008) Broad phylogenomic sampling improves resolution of the animal tree of life. Nature 452: 745–749 doi:10.1038/nature06614. [DOI] [PubMed] [Google Scholar]
- 129. Blaxter ML, De Ley P, Garey JR, Liu LX, Scheldeman P, et al. (1998) A molecular evolutionary framework for the phylum Nematoda. Nature 392: 71–75 doi:10.1038/32160. [DOI] [PubMed] [Google Scholar]
- 130. Csuros M, Rogozin IB, Koonin EV (2011) A detailed history of intron-rich eukaryotic ancestors inferred from a global survey of 100 complete genomes. PLoS Comput Biol 7: e1002150 doi:10.1371/journal.pcbi.1002150. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The multiple sequence alignment used to create the tree described in Figures 2 and S2.
(XLS)
The cladogram in figure 2 shown as an unrooted phlyogram with relative branch lengths restored.
(EPS)
Individual clusters from Figure 4B enlarged with each branch labeled with species name and accession number.
(PDF)
Intron positions of Rab subfamily members within the conserved portion of the multiple sequence alignment (MSA). Yellow squares correspond to phase 1 introns. Green squares correspond to phase 2 introns and red squares correspond to phase 3 introns (intron is positioned after the indicated codon). Intron free columns within the MSA were deleted. Numbering in the top row corresponds to the amino acid position of Rab6 from Lottia gigantea. For species abbreviations, see Figure 4 legend. Stars mark the position of each SSCIP as defined in the text. Black stars correspond to SSCIPs that are statistically significant at P(Monte Carlo <0.00001). Dark gray stars correspond to SSCIPs that are statistically significant at P(Monte Carlo <0.05). Light gray stars correspond to SSCIPs that are not statistically significant.
(PDF)
Full-length ORFeome Clone Sequence (FlOCS) for each isolate (WT, DN and CA) described in Table 1 .
(DOC)
A list of mutagenic primers (forward only) used in site-directed mutagenesis of mutant Rab forms. The mismatch region is highlighted in bold and all caps. For a definition of mismatch and the equation used to calculate Tm, see methods. Where applicable, the diagnostic enzyme and the form it digests is indicated. m = mutant, wt = wild type.
(DOC)