

Abstract
Phagocytosis of antibody-coated pathogens is mediated through Fcγ receptors (FcγRs) which activate intracellular signaling pathways to drive actin cytoskeletal rearrangements. Abl and Arg define a family of nonreceptor tyrosine kinases that regulate actin-dependent processes in a variety of cell types, including those important in the adaptive immune response. Using pharmacological inhibition as well as dominant negative and knockout approaches, we demonstrate a role for the Abl family kinases in phagocytosis by macrophages, and define a mechanism whereby Abl kinases regulate this process. Bone marrow-derived macrophages from mice lacking Abl and Arg kinases exhibit inefficient phagocytosis of sheep erythrocytes and zymosan particles. Treatment with the Abl kinase inhibitors imatinib and GNF-2 or overexpression of kinase-inactive forms of the Abl family kinases also impairs particle internalization in murine macrophages, indicating Abl kinase activity is required for efficient phagocytosis. Further, Arg kinase is present at the phagocytic cup and Abl family kinases are activated by FcγR engagement. The regulation of phagocytosis by Abl family kinases is mediated in part by the Syk kinase. Loss of Abl and Arg expression or treatment with Abl inhibitors reduced Syk phosphorylation in response to FcγR ligation. The link between Abl family kinases and Syk may be direct as purified Arg kinase phosphorylates Syk in vitro. Further, overexpression of membrane-targeted Syk in cells treated with Abl kinase inhibitors partially rescues the impairment in phagocytosis. Together, these findings reveal that Abl family kinases control the efficiency of phagocytosis in part through the regulation of Syk function.
Introduction
Phagocytes are cells of the innate immune system that play a critical role in host defense by recognizing pathogens and targeting them for destruction. Phagocytosis is a highly conserved process whereby immune cells recognize and bind to foreign particles, leading to remodeling of the plasma membrane, which allows for the engulfment of large particles (> 0.5 μm) (1). Among the signaling pathways involved in the regulation of phagocytosis is the Fcγ receptor (FcγR)-mediated pathway (1–3). FcγRs recognize the Fc portion of IgG, which is present in immune complexes and on antibody-coated cells. Myeloid cells from both humans and mice express several different types of activating Fc receptors; these include FcγRI (CD64), FcγRIIA (CD32A), FcγRIIC (CD32C), and FcγRIII (CD16) in humans; and FcγRI (CD64), FcγRIII (CD16), and FcγRIV (CD16-2) in mice (4). Activation of these receptors results in the production of inflammatory cytokines, reactive oxygen species and phagocytosis (5). FcγRs allow immune cells to detect and destroy IgG-coated viruses, bacteria and parasites during infection and IgG-coated blood cells in autoimmune disorders (6–8). The engulfed pathogens are then processed and corresponding antigens are presented on the cell surface to neighboring T cells (8).
Signal transduction pathways induced by FcγR engagement share remarkable conservation with signaling events that occur downstream of the T and B cell antigen receptors (9, 10). Collectively, these receptors are members of the multichain immune recognition receptor family which lack intrinsic kinase activity, but upon engagement are tyrosine phosphorylated on immunoreceptor tyrosine activation motifs (ITAMs) (2). For class I and class III FcγRs, these sequences are located on the accessory γ chain, whereas for class II FcγRs, they are present on the cytoplasmic portion of the ligand binding chain. ITAMs are comprised of paired tyrosines and leucines or isoleucines in the consensus sequence YxxL/I(x)7–12YxxL/I (2). Clustering of Fcγ receptors stimulates membrane-associated Src family kinases to phosphorylate the ITAM tyrosines of the FcγRs. In macrophages, these Src kinases include Hck, Fgr and Lyn, which promote the recruitment of the spleen tyrosine kinase, Syk, to the phosphorylated ITAM motifs (11, 12). The tandem SH2 domains of Syk bind to these newly created docking sites, leading to phosphorylation and activation of the Syk kinase (2). Syk is required for FcγR-mediated phagocytosis, as deletion or inhibition of Syk blocks the phagocytosis of antibody-coated substrates (13–16). In contrast, macrophages lacking the principal Src family kinases, Hck, Lyn, and Fgr, exhibit reduced phagocytosis and impaired activation of Syk kinase; however, these cells are not completely deficient in phagocytosis (12). This observation suggests that other kinases may be able to compensate for the loss of Src kinases in signaling events downstream of the FcγR. Here we posit that the Abl family of nonreceptor tyrosine kinases may be one such candidate.
The Abl kinases are a unique family of nonreceptor tyrosine kinases consisting of two members, Abl and Arg (17). Like other nonreceptor tyrosine kinases, including those in the Src family, Abl kinases have an N-terminal tandem SH3-SH2-kinase cassette, but unlike Src kinases, the C-terminal portion of Abl kinases contains domains for the binding of monomeric and filamentous actin (18, 19). This unique actin binding ability may serve to directly couple tyrosine phosphorylation signaling events to the cytoskeleton. In this regard, Abl family kinases have been implicated in the regulation of cellular morphogenesis, adhesion, polarity, migration and invasion (20–24).
Abl and Arg are ubiquitously expressed; Abl is expressed in most tissues at similar levels, while Arg is highly expressed in the central nervous system and in the thymus (25). Aberrant Abl and Arg fusion proteins with constitutive kinase activity have been identified as oncogenic in human lymphoid and myeloid leukemias (17). Interestingly, mice null for the abl1 gene are characterized by high perinatal mortality, splenic and thymic atrophy, and an increased susceptibility to infection (26). Subsequent studies have shown that the Abl family kinases are activated downstream of the TCR and BCR and contribute to antigen receptor signaling (27–31). Abl kinases are required for TCR-induced proliferation and modulate the phosphorylation of Zap-70, LAT, and Shc (27, 32). Compared to the well documented role for Abl kinases in lymphocyte function, relatively little is known about the role of the Abl family kinases in myeloid cells. Recent reports have demonstrated that Abl kinase activity is required for neutrophil adhesion and spreading through β2 integrin (33) and that Abl kinases mediate myeloid cell migration and invasion (34, 35).
Here we sought to examine whether Abl family kinases play a role in the regulation of FcγR-mediated phagocytosis in macrophages. The inhibition or genetic deletion of Abl family kinases in macrophages results in decreased phagocytosis of IgG-opsonized particles. Furthermore, Abl kinases are activated upon FcγR engagement, are found at the phagocytic cup, and regulate the efficiency of cup formation. Mechanistically, we show that Abl regulates the efficiency of phagocytosis in part by promoting the phosphorylation of the Syk kinase.
Materials & Methods
Reagents
STI571 (imatinib, Gleevec) was a kind gift from Novartis. GNF-2 and SU6656 were purchased from Sigma. Piceatannol was purchased from Millipore. Sheep red blood cells (sRBCs) were purchased from MP Biomedicals. Zymosan and zymosan opsonization reagents were purchased from Invitrogen.
Mice
The ablflox mice were generated, as previously described (25) and were crossed into the arg−/− background to generate ablflox/flox arg−/− mice. These mice were subsequently crossed with the LysM-Cre transgenic mice (Jackson Labs) to generate conditional loss of Abl kinases in the myeloid cell lineage. Genotypes were confirmed by PCR. Mice were backcrossed to C57BL/6 at least four times during their generation. Mice were housed under specific pathogen-free conditions in the Duke University Cancer Center Isolation Facility. All studies using mice have followed the protocols reviewed and approved by the Duke Institutional Animal Care & Use Committee.
Thioglycollate-Induced Peritonitis
Where indicated, isolation of macrophages was performed following intraperitoneal injection of 1 mL 3% thioglycollate solution (Sigma). After 72 hours, mice were sacrificed and the peritoneal cavity was lavaged with 5 mL HBSS to collect cells.
Cells
Primary macrophages were derived from the bone marrow or isolated from the peritoneum. In all experiments using primary cells, Abl/Arg null macrophages refer to cells derived from ablflox/flox ; arg −/−; LysMCre+ mice and control macrophages refer to cells derived from littermates or sex and age-matched ablflox/flox ; arg +/+; LysMCre− or ablflox/flox ; arg +/−; LysMCre− mice. Bone marrow-derived macrophages (BMDMs) were prepared by flushing the bone marrow from the femurs and tibias of mice with a 25G needle. Bone marrow cells were then collected; centrifuged, resuspended in growth media, and plated. Resident peritoneal macrophages and macrophages elicited by thioglycollate challenge (PEM) were prepared by lavaging the peritoneum of mice with 5 mL of HBSS. Both peritoneal macrophages and BMDM were cultured in 20% L-cell conditioned media that also contained 10% heat-inactivated fetal bovine serum (FBS, Invitrogen) and 1% Pen/Strep in α-MEM (Invitrogen). BMDM cultures were used after culturing in media for 5–7 days. Peritoneal macrophage cultures were used within 1–3 days after isolation. L-cell conditioned media was prepared by harvesting and filtering the supernatant of L929 cells (obtained from the American Type Culture Collection, ATCC) grown in Dulbecco’s Modified Eagle Medium (DMEM, Invitrogen) supplemented with 10% FBS. RAW264.7 murine macrophages (ATCC) and HEK293T cells (ATCC) were cultured in 10% FBS DMEM.
Cloning
MigR1-myr-FLAG-Syk was generated by subcloning the myr-FLAG-Syk cDNA from the pWZL vector (Addgene) into the XhoI and EcoRI sites of the MigR1 vector.
Transfection and Viral Transduction
PK1-vector, PK1-ArgKR, MigR1-vector, MigR1-AblPP, MigR1-AblKR, pcDNA-vector, pcDNA-myc-Syk and PX1-Arg-YFP constructs were described previously (24, 27, 36). Constructs were expressed in HEK293T cells using calcium phosphate-mediated transfection. Cells were used for experimental procedures 48 h following transfection. Retroviruses were prepared by co-transfecting HEK293T cells with PX1, PK1, or MigR1 constructs along with CMV-VSVG and gag/pol packaging constructs. After 48 h, cell retroviral supernatants were collected, filtered, and used to transduce RAW264.7 cells. RAW264.7 cells were incubated with the retroviral medium in presence of polybrene (8 μg/mL) for 48–72 h. Cells were then sorted for GFP+ expression (for MigR1 or PX1 constructs) or cultured a minimum of 3 days in puromycin (2 μg/mL for PK1 constructs) and used accordingly. RAW264.7 cells were transiently transfected with MigR1-myr-FLAG-Syk using Fugene 6 (Roche) according to the manufacturer’s instructions.
Flow Cytometry
Macrophages in single-cell suspensions were washed in FACS buffer (PBS containing 2% heat-inactivated FBS, 0.05% sodium azide). For surface staining, ~1 × 106 cells were first incubated 5 minutes with rat anti-mouse CD16/CD32 (mouse Fc Block) (BD Pharmingen) followed by staining with FITC-CD11b and PE-F4/80 or isotype controls (eBioscience) on ice for 30 minutes. For staining of FcγRs, PE-CD16/CD32 (eBioscience) was used. Cells were washed twice, collected on a FACScan flow cytometer, and analyzed using FlowJo (Tree Star Inc.).
RT-PCR
Total RNA was isolated from BMDM or peritoneal macrophages using RNeasy kit (Qiagen), cDNA was synthesized using M-MLV reverse transcriptase (Invitrogen), and amplified by PCR using the following primers for abl: 5′-GCCCTGGCCAGAGATCCATC-3′ and 5′-TCCCTCAGGTAGTCCAGCAGG-3′ as described in (37).
Phagocytosis Assay and Microscopy
sRBCs (10%) solution were pelleted and washed 3 times with PBS. sRBCs were opsonized with rabbit anti-sheep RBC antibodies diluted 1:50 (MP Biomedicals) and incubated at 37°C with shaking for 1 h. Opsonized sRBCs were then washed three times and resuspended in PBS. Cells were counted and diluted so that the sRBC:macrophage ratio was between 50:1 to 100:1. For drug studies, macrophages were preincubated 2 h with STI571 (10 μM), GNF-2 (20 μM) or vehicle (0.1% DMSO, Sigma) before initiating phagocytosis and were maintained in drug containing media during the assay. Macrophages were chilled on ice before addition of the opsonized sRBC; sRBCs were then added and plates containing macrophages and sRBCs were spun down for 1 minute (1000 rpm). The plates were incubated on ice for 10 minutes, media was then aspirated and warm media was added to initiate phagocytosis. Plates were incubated at 37°C for the indicated times. Reactions were stopped by placing the plates on ice, and washing away uningested particles with ice-cold PBS three times, followed by hypotonic lysis of any remaining uningested RBCs with water for 1 minute. Cells were washed an additional three times with PBS followed by fixation in methanol. RBCs were visualized by light microscopy. The phagocytic index was calculated by selecting 5–10 microscope fields (×400) and scoring each field for the both total number of macrophages and total number of ingested targets. A minimum of 100 macrophages were scored per coverslip. The phagocytic index indicates the number of particles ingested by one-hundred cells. For experiments evaluating the formation of phagocytic cups, macrophages were treated with sRBCs as described above, followed by fixation with 4% paraformaldehyde (Sigma) in PBS, permeabilization with PBS containing 0.2% Triton X-100, and blocking with 3% (w/v) BSA for 1 h. Cells were stained with either Alexa Fluor 568- or 488-conjugated phalloidin (Invitrogen) diluted 1:500 in 3% BSA (w/v) PBS for 1 h. IgG-opsonized sRBCs were stained with Alexa Fluor 568-conjugated goat anti-rabbit secondary antibody (Invitrogen) at 1:500 in 3% BSA (w/v) PBS for 1 h. Hoechst (Molecular Probes) was used to stain nuclei. The same fixation and staining procedures were used for macrophages treated with zymosan. In all experiments using zymosan, the zymosan was opsonized with zymosan opsonization reagent from Invitrogen. Phase-contrast and epifluorescence images were acquired using the Zeiss Axio Imager (Carl Zeiss MicroImaging). Confocal images were obtained using the Leica SP5 confocal scanning microscope and analyzed using LAS AF (Leica).
FcγR Cross-linking
Cells were serum starved for a total of 4 h before cross-linking. In studies using pharmacological inhibitors, drugs (STI571, 10 μM; GNF-2, 20 μM; SU6656, 2.5 μM; or piceatannol, 25 μM) were added 2 h prior to cross-linking in serum free media. After 4 h serum starvation, cells were incubated 20–30 minutes with 10 μg/mL 2.4G2 antibodies (anti-FcγRII and FcγRIII, BD Biosciences) at 4°C. Secondary cross-linking mouse anti-rat IgG (30 μg/mL; Jackson ImmunoResearch Laboratories) was then added in warmed media at 37°C for indicated times before plates were set on ice, washed in cold PBS, and lysed in RIPA buffer (50 mM Tris, 1% sodium deoxycholate, 150 mM sodium chloride, 1% NP-40, 0.1% SDS) with protease inhibitors (1 mM PMSF, 10 μg/mL leupeptin, 1 μg/mL aprotinin) and phosphatase inhibitors (10 mM β-glycerophosphate, 1 mM sodium fluoride, 0.1 mM sodium orthovanadate).
Immunoprecipitation and Kinase Assay
To immunoprecipitate Syk, HEK293T cells transiently overexpressing Syk kinase were lysed on ice for 30 minutes followed by centrifugation at 17,000 g for 10 minutes to remove insoluble material. 0.2 mg lysate was incubated with 0.4 μg anti-Syk antibody (Santa Cruz) overnight with end-over-end rotation at 4°C. To immunoprecipitate the γ chain, RAW264.7 cells were lysed after FcγR cross-linking and 1 mg lysate was incubated with 2 μg of anti-γ chain antibody (Millipore) overnight. Protein G-sepharose beads (30 μL slurry, GE Healthcare) were added for 2 h with end-over-end rotation at 4°C. Beads were then pelleted by centrifugation and washed as described (38). Syk-immunoprecipitates were incubated with purified full-length or C-terminal fragment Arg kinase (0.5 μg) (gift of Dr. Tony Koleske) in the presence or absence of 1 μM ATP for 30 minutes at room temperature. Sample buffer was added to stop the reaction. Samples were then boiled and run on SDS-PAGE.
Immunoblotting
Cells lysates were prepared and protein concentrations were quantified using Dc Protein Assay (Bio-Rad). Cell lysate was diluted in Laemmli sample buffer and proteins were separated on 8 to 15% SDS-polyacrylamide gels under reducing conditions. After transfer of proteins to nitrocellulose membranes, membranes were incubated with unlabeled primary antibodies overnight: anti-Abl (clone 8E9, BD Biosciences) at 1:500, anti-Arg (clone 9H5, Santa Cruz) at 1:500, anti-phosphoCRKL Y207 (Cell Signaling) at 1:1000, anti-CRKL (Santa Cruz) at 1:10,000, anti-Syk (Santa Cruz) at 1:10,000, anti-phosphoSyk Y352 (Cell Signaling) at 1:1000, anti-phosphoSyk YY525/6 (Cell Signaling) at 1:1000, anti-myc (clone 9E10, Sigma) at 1:1000, anti-γ chain (Millipore) at 1:1000, anti-phosphoFAK YY576/7 (Cell Signaling) at 1:1000, anti-FAK (Santa Cruz) at 1:1000, and anti-pTyr (clone 4G10, Millipore) at 1:1000 in blocking buffer (5% milk or 5% BSA TBST). Membranes were washed with TBST several times before incubation with HRP-conjugated secondary antibodies (Jackson ImmunoResearch Laboratories) at 1:10,000 for 1 h. Membranes were washed with TBST and developed with chemiluminescence reagent (GE Healthcare).
Statistical Analysis
Values correspond to the mean ± SEM. Comparisons of a single treatment to control were evaluated by Student’s t-test. In experiments with multiple groups, differences were evaluated by one-way ANOVA followed by Tukey’s multiple comparison test. Differences in phagocytosis between genotypes or drug treatments as function of time, or in the presence or absence of Syk overexpression were evaluated using two-way ANOVA followed by Bonferroni post hoc analysis using GraphPad Prism software v.5 (GraphPad Software). Western blots were quantitated using ImageJ analysis software (NIH). For all tests, p < 0.05 was considered statistically significant.
Results
Abl kinase activity is required for phagocytic efficiency
Tyrosine kinase signaling downstream of immune recognition receptors is required for activation of T and B cells and phagocytosis in macrophages (9, 10). Previous studies have shown that Abl kinases are activated by BCR and TCR ligation and are required for antigen-induced proliferation (27, 28, 30–32). We hypothesized that Abl kinase activity might be required downstream of the FcγR in macrophages to mediate phagocytosis. RAW264.7 murine macrophages were treated with STI571 (Gleevec, imatinib), an ATP competitive inhibitor of the Abl kinases that also inhibits PDGFR, c-kit, and c-fms (39, 40), or with a specific allosteric inhibitor of the Abl kinases, GNF-2, which has unique specificity for the C-terminal myristate binding cleft in the kinase domain of Abl and Arg, and does not inhibit other kinases (41, 42). RAW264.7 cells treated with either STI571 or GNF-2 were assayed for inhibition of Abl family kinase activity by analyzing the phosphorylation of the adapter protein CRKL on Y207, an Abl-specific site (32, 43) (Fig. 1A). CRKL phosphorylation on Y207 was reduced by 50 ± 8% when treated with STI571 and 40 ± 9% with GNF-2. As shown in Fig. 1B, treatment with either Abl kinase inhibitor markedly reduced phagocytosis of IgG-opsonized sheep red blood cells (sRBCs). The inhibitory effects of STI571 and GNF-2 on phagocytosis persisted over a 45 minute time course in both RAW264.7 cells and primary bone marrow-derived macrophages (BMDM) (Fig. 1C, D). After 30–45 minutes, phagocytosis in macrophages treated with Abl kinase inhibitors was reduced by over 60% compared to vehicle-treated cells (n = 4 experiments). RAW264.7 macrophages treated with IgG-opsonized zymosan in the presence of STI571 and GNF-2 also exhibited impaired phagocytosis, similar to the phagocytosis of sRBCs (Fig. S1A).
Figure 1. Abl family kinase activity regulates phagocytic efficiency.

(A) RAW264.7 macrophages treated with the Abl inhibitors STI571 (10 μM) or GNF-2 (20 μM) were evaluated for phosphorylation of the Abl substrate CRKL on Y207 by Western blotting. Graph shows relative levels of phosphoCRKL (pCRKL), normalized to total CRKL, from four experiments. *, p < 0.05 versus vehicle-treated control. (B) RAW264.7 were pretreated with Abl kinase inhibitors as in (A) followed by treatment with IgG-opsonized sheep red blood cells (sRBCs) for 30 minutes. Micrographs show the presence of ingested sRBCs in the macrophages. Results are representative of at least four independent experiments. Scale bar = 10 μm. (C and D) RAW264.7 macrophages (C) or bone marrow derived-macrophages (BMDM) (D) were pretreated with drugs as in (A) followed by incubation with sRBCs for the indicated times. The phagocytic index was calculated as the number of ingested sRBCs in 100 macrophages. Data shown are representative of two independent experiments. *, p < 0.05 versus vehicle-treated control. (E) RAW264.7 macrophages transduced with retrovirus encoding kinase inactive Arg (ArgKR) were treated with IgG-opsonized sheep RBCs for 30 minutes and the phagocytic index was calculated as in (C and D). Results are representative of seven independent experiments. *, p < 0.05 compared to cells expressing empty vector. Scale bar = 10 μm. (F) RAW264.7 macrophages transduced with kinase inactive Arg (PK1-ArgKR) or PK1-vector were analyzed by Western blotting with indicated antibodies. Relative levels of pCRKL, normalized to total CRKL, from three independent experiments are shown.
To further support a role for the Abl family kinases in FcγR-mediated phagocytosis, RAW264.7 macrophages were transduced with retrovirus encoding an inactive form of Arg kinase (ArgKR), which acts in a dominant negative manner to inhibit Abl family kinase signaling (22). Macrophages expressing ArgKR phagocytosed 54 ± 5% of the sRBCs compared to control macrophages (n=7 experiments, example shown in Fig. 1E), and also exhibited a 50% reduction in phosphorylation of CRKL on Y207, indicating reduced Abl/Arg kinase activity (Fig. 1F). Similarly, phagocytosis of IgG-opsonized zymosan was decreased in macrophages expressing ArgKR (Fig. S1B).
Impaired FcγR-mediated phagocytosis in Abl/Arg null macrophages
We next sought to examine the requirement for Abl kinases in FcγR-mediated phagocytosis using a genetic loss-of-function approach. As global deletion of both Abl and Arg is embryonic lethal (25), we employed tissue-specific inactivation of Abl. To this end, ablflox mice (37) were crossed into the arg −/− background (25), and the ablflox/flox ; arg −/− mice were crossed to mice that express Cre recombinase under the control of the lysozyme M promoter (44). Lysozyme M (LysM) is expressed in myeloid cells including monocytes, macrophages, granulocytes, and a subset of dendritic cells (44). The resulting ablflox/flox ; arg −/−; LysMCre+ (from here on designated: Abl/Arg null) mice were viable, and demonstrated deletion of abl1 in both thioglycollate elicited peritoneal (PEM)- and bone marrow-derived macrophages (BMDM) (Fig. S2A), which corresponded with reduction in Abl protein and activity levels (Fig. S2B). To test whether Abl family kinases were required for macrophage differentiation, cells were stained with the myeloid marker CD11b as well as the mature macrophage marker F4/80. Culturing bone marrow cells from wild type and Abl/Arg null mice in CSF-1-containing macrophage differentiation media resulted in similar numbers of CD11b+F4/80+ cells (Fig. S2C). Furthermore, recruitment of CD11b+F4/80+ macrophages to the peritoneum in response to challenge with thioglycollate was not affected by deletion of Abl and Arg (Fig. S2D). Similarly, the percentage CD11b+F4/80+ cells isolated from the bone marrow, blood, and spleen was not affected by loss of Abl and Arg (data not shown). Taken together, these data suggest that Abl and Arg are not required for macrophage differentiation in vitro or in vivo.
To examine whether Abl kinases were required for FcγR-mediated phagocytosis, bone marrow-derived macrophages from control and Abl/Arg null animals were challenged with IgG-opsonized sRBCs. Similar to the results obtained with Abl kinase inhibitors, the Abl/Arg null macrophages phagocytosed fewer sRBCs compared to controls (Fig. 2A). Analysis of multiple control and Abl/Arg null cells over a 60 minute time course revealed that Abl/Arg null macrophages achieved a maximal reduction in the phagocytic index of 31 ± 6% after 15–30 minutes incubation with sRBCs (n = 13 experiments; ± SEM). At later time points, the Abl/Arg null cells were able to ingest similar numbers of particles as control cells (Fig. 2A, B). This delay in phagocytosis was also observed in Abl/Arg null macrophages isolated from the peritoneum (Fig. 2C). Abl/Arg null peritoneal macrophages exhibited a 41 ± 14% reduction in the phagocytic index compared to control macrophages after a 10–40 minute incubation with sRBCs (n = 5 experiments; ± SEM). The Abl/Arg null macrophages also exhibited reduced phagocytosis of IgG-opsonized zymosan, particularly within the first 15 minutes of stimulation with zymosan particles (mean of 40 ± 9% reduction in phagocytic index compared to controls, n = 11 experiments; ± SEM) (Fig. S3). The reduction in phagocytosis in Abl/Arg null macrophages was not due to reduced surface expression of the FcγR, as these cells expressed equivalent surface levels of CD16/CD32 as compared to control macrophages (Fig. 2D). Furthermore, deletion of either Abl or Arg kinase alone was insufficient to reduce phagocytosis of sRBCs, suggesting that these kinases function redundantly in this context (Fig. S4).
Figure 2. Impaired FcγR-mediated phagocytosis in Abl/Arg null macrophages.

(A) Control and Abl/Arg null BMDM were incubated with IgG-opsonized sRBCs and then fixed and imaged at the indicated time points. Scale bar = 20 μm. (B and C) BMDM (B) or resident peritoneal macrophages (C) derived from control and Abl/Arg null mice were treated the IgG-opsonized sRBCs for the indicated times. The phagocytic index was calculated as the number of internalized sRBCs in 100 macrophages. Results from two sets of control and Abl/Arg null mice are plotted for both B & C. *, p < 0.05 versus control macrophages. (D) BMDM were stained with PE-labeled anti-CD16/CD32 antibodies and analyzed by flow cytometry. Data are representative of three independent experiments.
We find that pharmacological inhibition of the Abl family kinases with STI571 and GNF-2 results in a more profound and persistent impairment in phagocytosis than that induced by genetic ablation of Abl and Arg. This suggests that compensatory mechanisms may be occurring in cells genetically deleted for Abl kinases. Moreover, imatinib has inhibitory activity against c-fms, PDGFR, and c-kit (39, 40), so it is possible that other imatinib-sensitive targets may contribute to phagocytosis. However, our finding that both pharmacological inhibition and genetic ablation of Abl and Arg result in deficits in phagocytosis suggests that these kinases are required for efficient phagocytosis.
Arg kinase is found at the phagocytic cup
Phagocytosis requires the formation of an actin rich phagocytic cup during which contraction of the actin ring promotes membrane invagination and internalization (45). The finding that Abl kinases regulate the efficiency of phagocytosis led us to examine whether Abl family kinases were present at the phagocytic cup. RAW264.7 macrophages were transduced with YFP-tagged Arg kinase and stimulated with IgG-opsonized zymosan for a short time period to capture phagocytic cup formation. Indeed, intense regions of YFP+ fluorescence were found in the membrane of cells undergoing phagocytosis, adjacent to the bound zymosan particles (Fig. 3). Further, staining for F-actin with phalloidin to mark active phagocytic cups revealed co-localization of Arg with F-actin (Fig. 3, merge). In some cases, Arg kinase was found in the membrane adjacent to zymosan particles that lacked phalloidin staining, suggesting Arg may also localize to the nascent phagosome. In contrast, in cells that were not bound to zymosan, ArgYFP was diffusely localized at the plasma membrane and in the cytosol (Fig 3, right panel). The localization of ArgYFP with the F-actin rich cup suggests that Arg may modulate actin dynamics during phagocytosis.
Figure 3. Arg co-localizes with F-actin at the phagocytic cup.
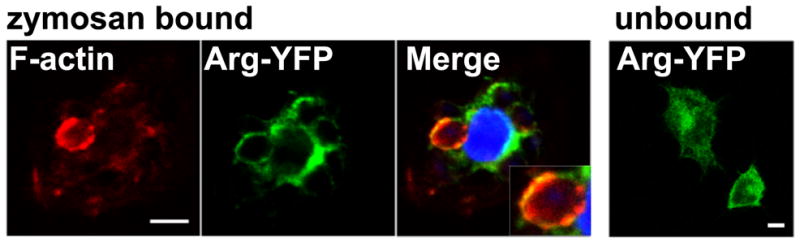
RAW264.7 macrophages transduced with retrovirus encoding YFP-tagged Arg were treated with IgG-opsonized zymosan for 5 minutes, fixed and stained with phalloidin (red) and Hoechst (blue). Samples of zymosan-bound and unbound macrophages were imaged by confocal microscopy. Data are representative of two independent experiments. Scale bars = 5 μm.
Abl kinase activity regulates phagocytic cup formation
Tyrosine kinases have been implicated in regulating actin cytoskeletal rearrangements during phagocytosis. Src family kinases, for instance, have a critical role in promoting actin polymerization at the phagocytic cup following FcγR engagement (12). In contrast, the spleen tyrosine kinase, Syk, is required for phagocytic cup closure (14). To evaluate the role of Abl kinases during phagocytosis, RAW264.7 macrophages were pretreated with Abl kinase inhibitors followed by stimulation with IgG-opsonized sRBCs. Vehicle-treated cells exhibited robust phagocytic cup formation, but treatment with STI571 or GNF-2 resulted in markedly decreased numbers of phagocytic cups per cell; although no defects in cup closure as revealed by actin staining were observed (Fig. 4A, B). Likewise, Abl/Arg null BMDM exhibited a reduction in the number of cups formed per cell compared to control cells, whereas the binding of the particles was unaffected. Abl/Arg null cells averaged 70 ± 11% of the number of cups/cell compared to control macrophages (n=6 experiments, example shown in Fig. 4C). The localization of Arg kinase at the phagocytic cup as well as the reduction in cup formation upon Abl kinase inhibition suggested a role for Abl/Arg kinase activity in signaling events during phagocytosis.
Figure 4. Abl family kinase activity is required for efficient phagocytic cup formation.

(A) RAW264.7 pretreated with either STI571 (10 μM) or GNF-2 (20 μM) were incubated with rabbit-IgG opsonized sRBCs for 2 minutes. Cells were then fixed and stained for F-actin (green) and Hoechst to mark nuclei (blue). IgG-opsonized RBCs were stained with PE-conjugated anti-rabbit antibodies. Phagocytic cups were imaged by confocal microscopy. Scale bar = 10 μm. (B) RAW264.7 cells treated with inhibitors were stimulated with IgG-opsonized sRBCs for the indicated times and processed as in (A). The number of phagocytic cups present in 100 randomly selected cells was counted. Results are representative of three independent experiments. *, p < 0.05 versus vehicle-treated cells. (C) BMDM from control and Abl/Arg null mice were stimulated with IgG-opsonized sRBCs for 5 minutes and processed as in (A). The numbers of phagocytic cups in control and Abl/Arg null cells were quantified as in (B). Results from two sets of control and Abl/Arg null BMDM are quantified. Scale bar = 10 μm. Arrows point to F-actin rich cups.
Abl family kinases are activated by FcγR engagement and function downstream of Src family kinases during phagocytosis
To evaluate whether Abl family kinases were involved in FcγR signaling, RAW264.7 macrophages were assayed for Abl/Arg activation following FcγR engagement. We found that CRKL became phosphorylated at the Abl-specific site upon FcγR cross-linking, and this phosphorylation was reduced in the presence of the pharmacological inhibitors STI571 and GNF-2 (Fig. 5A). In the canonical model of immunoreceptor activation, the clustering of receptors leads to their tyrosine phosphorylation by Src family kinases. To test if Abl family kinases could also function to regulate immunoreceptor phosphorylation, the γ subunit was immunoprecipitated from cells after FcγR cross-linking in the presence or absence of Abl or Src kinase inhibitors. As shown in Fig. 5B, inhibiting the Abl kinases with STI571 or GNF-2 did not affect γ subunit tyrosine phosphorylation, whereas cells treated with the Src inhibitor SU6656 had reduced γ chain phosphorylation, consistent with previous reports that Src kinases mediate FcγR phosphorylation (12, 16, 46).
Figure 5. Abl family kinases are activated by FcγR engagement downstream of Src kinases.

(A) RAW264.7 cells were serum starved 4 h, then treated with either STI571 (10 μM) or GNF-2 (20 μM) 2 h prior to FcγR cross-linking. After serum starvation, cells were incubated with the anti-FcγR mAb 2.4G2 followed by anti-rat IgG cross-linking for the indicated times. Graph shows quantitation of pCRKL levels, normalized to total CRKL, 5 minutes after FcγR cross-linking from five independent experiments. *, p < 0.05 compared to FcγR stimulated control cells. (B) RAW264.7 cells pretreated with Abl inhibitors as in (A) or with the Src inhibitor SU6656 (2.5 μM) were stimulated with FcγR cross-linking for 10 minutes followed by cell lysis and immunoprecipitation of the γ chain. Graph shows relative levels of phospho-γ chain, normalized to total γ chain, from a representative experiment; similar results were obtained in two independent experiments. (C) RAW264.7 cells were serum starved 4 h. Two hours prior to FcγR cross-linking, cells were pretreated with inhibitors as in (B). Cells were treated with the anti-FcγR mAb 2.4G2 followed by anti-rat IgG cross-linking for 5 minutes. Line denotes where intervening lanes have been cropped out. Graph shows quantitation of pCRKL, normalized to total CRKL, from four independent experiments. *, p < 0.05 compared to FcγR stimulated control cells. (D) RAW264.7 cells pretreated with Abl or Src inhibitors as in (B) were stimulated with FcγR cross-linking for 10 minutes. Graph shows quantitation of phosphoFAK (pFAK), normalized to total FAK, from a representative experiment; similar results were obtained in two independent experiments. (E) RAW264.7 cells treated with either Abl or Src kinase inhibitors as indicated were stimulated with opsonized sheep RBCs and phagocytic index was measured after 30 minutes. Scale bar = 20 μm. *, p < 0.05 with respect to vehicle and #, p < 0.05 with respect to SU6656.
Previous studies have shown that loss of Hck, Lyn, and Fgr, the Src kinases most predominantly expressed in macrophages, results in reduced phagocytosis and impaired FcγR signaling (12, 14). Activation of Abl family kinases downstream of antigen receptors depends in part on Src kinases (27). To test if Abl functions downstream of Src kinases during phagocytosis, RAW264.7 cells were pretreated with the Src kinase inhibitor SU6656 followed by FcγR cross-linking. FcγR-induced activation of Abl kinases was reduced in the presence of the SU6656 as detected by CRKL phosphorylation (Fig. 5C). Conversely, inhibiting Abl kinases with STI571 or GNF-2 failed to suppress Src activation as assessed by evaluating the phosphorylation of focal adhesion kinase (FAK) on tyrosines 576 and 577, Src-specific phosphorylation sites (47) (Fig. 5D). Together the data suggest that Abl kinases function downstream of Src in this pathway. Phenotypically, treatment of RAW264.7 cells with SU6656 results in marked reduction in phagocytosis, similar to that observed with STI571 and GNF-2 (Fig. 5E). Furthermore, the combined inhibition of Src and Abl family kinases with SU6656 and either STI571 or GNF-2 produced a greater reduction in phagocytosis than treatment with each compound alone (Fig. 5E). These results suggest that the activation of Abl kinases in response to FcγR engagement is dependent in part on Src kinases. Inhibition of both Src and Abl family kinases may reduce phosphorylation of both common and distinct targets of these kinases in this pathway resulting in a greater reduction in phagocytosis.
Abl kinases modulate Syk kinase activation
A critical signaling event downstream of the FcγR is the activation of Syk kinase. Both loss of Syk expression or inhibition of Syk kinase activity result in impaired phagocytosis (14–16, 48). To test if Abl kinases regulate the activation and phosphorylation of Syk kinase, RAW264.7 macrophages were treated with Abl inhibitors or the Src inhibitor SU6656 and assayed for Syk phosphorylation after FcγR cross-linking. In agreement with previous studies, inhibition of Src family kinases reduced Syk phosphorylation on Y346 (which is recognized by an antibody to human Syk Y352, the equivalent site on human Syk) and on Y519 and Y520 (which are recognized by an antibody to human Syk YY525/6, the equivalent sites on human Syk) (Fig. 6A). Notably, treatment with the Abl family kinase inhibitors also reduced Syk phosphorylation on these sites (Fig. 6A). Furthermore, Abl/Arg null macrophages also exhibited reduced phosphorylation of Syk upon FcγR cross-linking (Fig. 6B). Phosphorylation of Syk on Y346 is required for both enzymatic activity as well as coupling to downstream effector molecules (49). In addition to Y346 phosphorylation, deletion or inhibition of Abl kinases in macrophages reduced phosphorylation of Syk on Y519 and Y520, which are Syk autophosphorylation sites required for maximal enzymatic activity (Fig. 6A, B) (50). These findings suggest that Abl kinases can modulate Syk activation induced by FcγR ligation. To test if Syk kinase signaling can affect Abl kinase activity, the FcγR was cross-linked in RAW264.7 cells treated with the Syk inhibitor piceatannol. Piceatannol failed to suppress Abl activation as measured by the phosphorylation of CRKL upon FcγR cross-linking (Fig. 6C). Thus, these findings suggest that in contrast to Src kinases, Syk function is not required for FcγR-mediated activation of Abl kinases.
Figure 6. Abl kinases modulate Syk activation downstream of the FcγR.

(A) RAW264.7 cells were serum starved 4 h, then treated with STI571 (10 μM), GNF-2 (20 μM), or SU6656 (2.5 μM), 2 h prior to FcγR cross-linking. For cross-linking, cells were incubated with the anti-FcγR mAb 2.4G2 followed by anti-rat IgG crosslinking for 5 minutes. Graphs show quantitation of phospho-Syk (pSyk) Y346 and pSyk YY519/20, normalized to total Syk, from three and four independent experiments, respectively. *, p < 0.05 compared to FcγR stimulated control macrophages. (B) Control and Abl/Arg null BMDM were serum starved 4 h, then cross-linked and processed as in (A). Graphs show quantitation of pSyk Y346 and pSyk YY519/20 levels, normalized to total Syk, from seven and three experiments, respectively. *, p < 0.05 compared to FcγR stimulated control macrophages. (C) RAW264.7 cells were treated with drugs as in (A) or with piceatannol (25 μM) as indicated. After FcγR cross-linking, cells were assayed for pCRKL levels by Western blotting. Graph shows quantitation of pCRKL levels, normalized to total CRKL, from two independent experiments. NS = not significant. (D) 293T cells were transiently transfected with myc-tagged Syk and constitutively active Abl (AblPP) or kinase-defective Abl (AblKR), and analyzed after 48 h for pSyk Y346 levels. pSyk levels were normalized to total Syk and expressed as fold change over vector. Graph shows quantitation from three independent experiments. *, p < 0.05 compared to cells expressing empty vector. (E) Syk was immunoprecipitated from 293T cells transfected with MigR1-myr-FLAG-Syk, then incubated with purified full length Arg kinase (FL) or a C-terminal Arg fragment (CT), in the presence or absence of ATP. Phosphorylation of Syk at Y346 in the presence of full length Arg and ATP is shown. pSyk levels were normalized to total Syk and the fold increase in pSyk after incubation with purified Arg was quantitated from three independent experiments. *, p < 0.05 versus no Arg or ArgCT.
To further investigate the link between Abl kinases and Syk phosphorylation, constitutively active or dominant negative forms of the Abl kinase were expressed in HEK293T cells in the presence of Syk kinase. Expression of constitutively active Abl (AblPP) increased Syk Y346 phosphorylation, whereas the phosphorylation was unaffected in cells expressing kinase inactive form of Abl (AblKR) (Fig. 6D). Furthermore, purified full-length Arg protein promoted the phosphorylation of FLAG-tagged Syk on Y346 in vitro (Fig. 6E). Syk Y346 phosphorylation was not detected in the absence of full-length Arg or in the presence of the C-terminal fragment of Arg, which lacks the kinase domain (Fig. 6E). Full-length Arg was unable to promote the phosphorylation of Syk on YY519/20 (data not shown), indicating that Y346 is the primary target of Abl kinases. Thus, Abl family kinases promote Syk phosphorylation in vitro and in cells.
It has been shown that the binding of Syk to phosphorylated ITAMs of the Fc receptor subunits is sufficient to induce conformational changes in Syk that lead to Syk autophosphorylation and activation (51). Syk activation is more efficient, however, when also phosphorylated in trans by other kinases (52, 53). If Abl kinases regulate phagocytosis by modulating Syk kinase activation, we hypothesized that overexpression of a plasma membrane-targeted form of Syk may enhance phagocytosis even in the absence of Abl kinase signaling by shifting the equilibrium towards a more active pool of Syk kinase. To evaluate this possibility, we employed RAW264.7 macrophages overexpressing a myristoylated form of Syk kinase in the absence and presence of the Abl/Arg-selective inhibitor GNF-2 (Fig. 7C). Overexpression of this membrane-associated Syk kinase partially rescued the defective phagocytosis in macrophages treated with GNF-2 (Fig. 7A, B). These data support a model whereby Abl kinases become activated in response to FcγR ligation and modulate Syk kinase activity to promote phagocytosis (Fig. 7D).
Figure 7. Overexpression of Syk kinase rescues phagocytosis in macrophages with decreased Abl/Arg kinase activity.

(A) RAW264.7 cells transduced with MigR1-myr-FLAG-Syk were treated with GNF-2 followed by incubation with sRBCs for 30 minutes. Scale bar = 10 μm. (B) Phagocytic index was calculated and the results presented as an average from three independent experiments. *, p < 0.05 compared to vehicle-treated cells. (C) Expression of myr-FLAG-Syk in RAW264.7 macrophages was confirmed by Western blotting. (D) Model for Abl family kinase signaling during FcγR-mediated phagocytosis. Upon FcγR ligation, Src family kinases (SFKs) activate Abl family kinases. Src and Abl kinases promote the phosphorylation of Syk, which drives particle internalization. Abl kinases may also modulate the phosphorylation of other substrates to promote actin organization at the phagocytic cup.
Discussion
Here we show that Abl family kinases regulate the efficiency of FcγR-mediated phagocytosis, and uncover a novel functional link between Abl and Syk kinases downstream of the FcγRs in macrophages. These findings suggest that Abl kinases may regulate pathogen clearance through phagocytosis. Previous reports have shown a requirement for Abl kinases in mediating the entry and dissemination of many of pathogens including Shigella flexneri, Chlamydia trachomatis, HIV, and polyomavirus (54–59). While these reports and recent findings on the role for the Abl kinases in Leishmania phagocytosis (60) support a role for Abl family kinases in pathogen uptake, the signaling pathways that are regulated by Abl kinases for efficient phagocytosis had remained elusive. Syk kinase is an essential regulator of phagocytosis as loss of Syk results in failure of macrophages to ingest pathogens (13, 14). In the classical paradigm, clustering of FcγRs activates membrane-associated Src kinases, which phosphorylate cytoplasmic ITAMs on the γ adapter protein (61). Phosphorylated ITAMs then serve as docking sites for the tandem SH2 domains of Syk kinase. Upon binding the phosphorylated ITAMs, Syk undergoes conformational changes that lead to its activation. Src kinases have been shown to phosphorylate several tyrosines on Syk that potentiate its activity (52, 53). Our data show Abl kinases modulate the phosphorylation of Y346 on Syk both in vitro and in cells. Phosphorylation of Syk on Y346 disrupts Syk autoinhibitory intramolecular interactions between the interdomain B sequences and the kinase domain of Syk; moreover, binding of SH2-containing proteins to phosphorylated Y346 on Syk has been proposed to help stabilize the active conformation of the kinase (49, 62). Reduced phosphorylation of Syk on Y346 upon inhibition or loss of Abl family kinases may explain, in part, why phagocytosis in the Abl/Arg null macrophages is impaired. Previous studies have shown that Abl family kinases regulate the phosphorylation of the Syk family member Zap70 on Y319 following T cell receptor engagement and L-selectin ligation (27, 32, 63). Tyrosine 319 of Zap70 is analogous to tyrosine 346 of Syk, and our finding that purified Arg kinase can directly phosphorylate this site in vitro, suggest that an Abl kinase-Syk/Zap70 signaling module may operate under diverse cell surface receptors to regulate cytoskeletal responses in hematopoietic cells.
Activation of Abl kinases downstream of growth factor, antigen, and chemokine receptors is mediated in part through Src kinases (23, 24, 27, 32, 38). We found that this paradigm also exists in during FcγR signaling in macrophages. Previous studies have shown that genetic deletion of Src family kinases in macrophages attenuates signaling from the FcγR, including Syk phosphorylation and activation (12). Our data show that pharmacological inhibition of Src impaired many FcγR proximal signaling events including decreasing the activation of Abl and Syk kinases. Interestingly, simultaneous inhibition of Abl and Src further reduces phagocytosis, suggesting these kinases may have unique targets or differentially regulate common targets through phosphorylating them on distinct sites. In this regard, a recent report indicated that Abl and Src form a complex in myeloid cells that regulates myeloid cell migration by modulating the activation of Rac and Cdc42 (34). The mechanism by which Abl and Src function to control activation of Rho GTPases during phagocytosis warrants further investigation.
While Abl and Arg promote maximal Syk activation during phagocytosis, they are likely to have Syk-independent roles. Inactivation of Abl kinases reduces the formation of phagocytic cups in addition to reducing Syk phosphorylation. Notably, while Syk null macrophages form phagocytic cups normally, the cups fail to close (15, 48). This suggests that the Abl family kinases might regulate actin rearrangements at the phagocytic cup through mechanisms that may involve other Abl targets such as CRKII and WASP. Previous studies have shown that Abl kinases regulate Shigella flexneri invasion through phosphorylation of the adapter protein CRKII and the N-WASP actin nucleation promoting factor (54–56). Interestingly, both CRKII and WASP are recruited to the phagocytic cup during FcγR phagocytosis (64, 65). Phosphorylation of WASP on Y291 is required for phagocytic cup formation (66), and Abl has been shown to phosphorylate this site in vitro (67). Future studies should address whether the Abl-mediated phosphorylation of CRKII or WASP is required for regulating F-actin organization at the phagocytic cup.
Previous studies have shown that Abl kinases modulate adaptive immune function (29, 32). The Abl single knockout mouse exhibits multiple abnormalities, among the most prominent of which are immune defects including splenic and thymic atrophy, and increased susceptibility to infection (26). Our current data suggest that, in addition to lymphocyte defects, loss or inhibition of both Abl and Arg impairs myeloid cell function. Interestingly, the Abl kinase inhibitor imatinib has been reported to have anti-inflammatory effects in a variety of autoimmune disease models (68–71) and inhibits the functional capacity of human monocytes in vitro (72). The effects of imatinib on monocytes have previously been attributed to inhibition of the c-fms tyrosine kinase (72). Imatinib has multiple kinase and non-kinase targets (73) and our data using new and selective allosteric Abl kinase inhibitors and Abl/Arg knockout macrophages suggest that one of the relevant targets of imatinib in myeloid cells is the Abl kinase family. Our findings have potential implications for the treatment of autoimmune diseases with deregulated FcγR signaling (74). Future studies should determine whether the second generation allosteric Abl inhibitors, which are selective for the Abl kinases, are useful for the treatment of inflammatory disorders.
Supplementary Material
Acknowledgments
We thank Emily Riggs for genotyping; Dr. Anthony Koleske for the Ablflox/Arg null mice, and purified Arg protein, Dr. Gouri Yogalingam for initiating the LysMCre-Ablflox mouse colony and insightful suggestions, and Dr. Colleen Ring and Elizabeth Chislock for their comments on this manuscript.
Abbreviations
- Abl
Abelson
- Arg
Abl-related gene
- SH
Src-homology
- BMDM
bone marrow-derived macrophage
- PEM
peritoneal elicited-macrophage
- LysM
lysozyme-M
- p-Tyr
phospho-tyrosine
Footnotes
This work was funded in part through a PhRMA predoctoral fellowship (to EKG) and a DOD BCRP fellowship W81XWH-10-1-0345 (to EKG) and NIH grants CA070940 (to AMP) and AI056266 (to AMP).
References
- 1.Cox D, Greenberg S. Phagocytic signaling strategies: Fc(gamma)receptor-mediated phagocytosis as a model system. Semin Immunol. 2001;13:339–345. doi: 10.1006/smim.2001.0330. [DOI] [PubMed] [Google Scholar]
- 2.Indik ZK, Park JG, Hunter S, Schreiber AD. The molecular dissection of Fc gamma receptor mediated phagocytosis. Blood. 1995;86:4389–4399. [PubMed] [Google Scholar]
- 3.Flannagan RS, Jaumouille V, Grinstein S. The cell biology of phagocytosis. Annu Rev Pathol. 2012;7:61–98. doi: 10.1146/annurev-pathol-011811-132445. [DOI] [PubMed] [Google Scholar]
- 4.Bruhns P. Properties of mouse and human IgG receptors and their contribution to disease models. Blood. 2012;119:5640–5649. doi: 10.1182/blood-2012-01-380121. [DOI] [PubMed] [Google Scholar]
- 5.Daeron M. Fc receptor biology. Annu Rev Immunol. 1997;15:203–234. doi: 10.1146/annurev.immunol.15.1.203. [DOI] [PubMed] [Google Scholar]
- 6.Gomez F, Ruiz P, Schreiber AD. Impaired function of macrophage Fc gamma receptors and bacterial infection in alcoholic cirrhosis. N Engl J Med. 1994;331:1122–1128. doi: 10.1056/NEJM199410273311704. [DOI] [PubMed] [Google Scholar]
- 7.Ruiz P, Gomez F, Schreiber AD. Impaired function of macrophage Fc gamma receptors in end-stage renal disease. N Engl J Med. 1990;322:717–722. doi: 10.1056/NEJM199003153221102. [DOI] [PubMed] [Google Scholar]
- 8.Ravetch JV, Bolland S. IgG Fc receptors. Annu Rev Immunol. 2001;19:275–290. doi: 10.1146/annurev.immunol.19.1.275. [DOI] [PubMed] [Google Scholar]
- 9.Strzelecka A, Kwiatkowska K, Sobota A. Tyrosine phosphorylation and Fcgamma receptor-mediated phagocytosis. FEBS Lett. 1997;400:11–14. doi: 10.1016/s0014-5793(96)01359-2. [DOI] [PubMed] [Google Scholar]
- 10.DeFranco AL. Transmembrane signaling by antigen receptors of B and T lymphocytes. Curr Opin Cell Biol. 1995;7:163–175. doi: 10.1016/0955-0674(95)80024-7. [DOI] [PubMed] [Google Scholar]
- 11.Meng F, Lowell CA. Lipopolysaccharide (LPS)-induced macrophage activation and signal transduction in the absence of Src-family kinases Hck, Fgr, and Lyn. J Exp Med. 1997;185:1661–1670. doi: 10.1084/jem.185.9.1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fitzer-Attas CJ, Lowry M, Crowley MT, Finn AJ, Meng F, DeFranco AL, Lowell CA. Fcgamma receptor-mediated phagocytosis in macrophages lacking the Src family tyrosine kinases Hck, Fgr, and Lyn. J Exp Med. 2000;191:669–682. doi: 10.1084/jem.191.4.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Matsuda M, Park JG, Wang DC, Hunter S, Chien P, Schreiber AD. Abrogation of the Fc gamma receptor IIA-mediated phagocytic signal by stem-loop Syk antisense oligonucleotides. Mol Biol Cell. 1996;7:1095–1106. doi: 10.1091/mbc.7.7.1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Crowley MT, Costello PS, Fitzer-Attas CJ, Turner M, Meng F, Lowell C, Tybulewicz VL, DeFranco AL. A critical role for Syk in signal transduction and phagocytosis mediated by Fcgamma receptors on macrophages. J Exp Med. 1997;186:1027–1039. doi: 10.1084/jem.186.7.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kiefer F, Brumell J, Al-Alawi N, Latour S, Cheng A, Veillette A, Grinstein S, Pawson T. The Syk protein tyrosine kinase is essential for Fcgamma receptor signaling in macrophages and neutrophils. Mol Cell Biol. 1998;18:4209–4220. doi: 10.1128/mcb.18.7.4209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Majeed M, Caveggion E, Lowell CA, Berton G. Role of Src kinases and Syk in Fcgamma receptor-mediated phagocytosis and phagosome-lysosome fusion. J Leukoc Biol. 2001;70:801–811. [PubMed] [Google Scholar]
- 17.Pendergast AM. The Abl family kinases: mechanisms of regulation and signaling. Adv Cancer Res. 2002;85:51–100. doi: 10.1016/s0065-230x(02)85003-5. [DOI] [PubMed] [Google Scholar]
- 18.Van Etten RA, Jackson PK, Baltimore D, Sanders MC, Matsudaira PT, Janmey PA. The COOH terminus of the c-Abl tyrosine kinase contains distinct F- and G-actin binding domains with bundling activity. J Cell Biol. 1994;124:325–340. doi: 10.1083/jcb.124.3.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang Y, Miller AL, Mooseker MS, Koleske AJ. The Abl-related gene (Arg) nonreceptor tyrosine kinase uses two F-actin-binding domains to bundle F-actin. Proc Natl Acad Sci U S A. 2001;98:14865–14870. doi: 10.1073/pnas.251249298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bradley WD, Koleske AJ. Regulation of cell migration and morphogenesis by Abl-family kinases: emerging mechanisms and physiological contexts. J Cell Sci. 2009;122:3441–3454. doi: 10.1242/jcs.039859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zandy NL, Playford M, Pendergast AM. Abl tyrosine kinases regulate cell-cell adhesion through Rho GTPases. Proc Natl Acad Sci U S A. 2007;104:17686–17691. doi: 10.1073/pnas.0703077104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li R, Pendergast AM. Arg kinase regulates epithelial cell polarity by targeting beta1-integrin and small GTPase pathways. Curr Biol. 2011;21:1534–1542. doi: 10.1016/j.cub.2011.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gu JJ, Lavau CP, Pugacheva E, Soderblom EJ, Moseley MA, Pendergast AM. Abl Family Kinases Modulate T Cell-Mediated Inflammation and Chemokine-Induced Migration through the Adaptor HEF1 and the GTPase Rap1. Sci Signal. 2012;5:ra51. doi: 10.1126/scisignal.2002632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Smith-Pearson PS, Greuber EK, Yogalingam G, Pendergast AM. Abl kinases are required for invadopodia formation and chemokine-induced invasion. J Biol Chem. 2010;285:40201–40211. doi: 10.1074/jbc.M110.147330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Koleske AJ, Gifford AM, Scott ML, Nee M, Bronson RT, Miczek KA, Baltimore D. Essential roles for the Abl and Arg tyrosine kinases in neurulation. Neuron. 1998;21:1259–1272. doi: 10.1016/s0896-6273(00)80646-7. [DOI] [PubMed] [Google Scholar]
- 26.Tybulewicz VL, Crawford CE, Jackson PK, Bronson RT, Mulligan RC. Neonatal lethality and lymphopenia in mice with a homozygous disruption of the cabl proto-oncogene. Cell. 1991;65:1153–1163. doi: 10.1016/0092-8674(91)90011-m. [DOI] [PubMed] [Google Scholar]
- 27.Zipfel PA, Zhang W, Quiroz M, Pendergast AM. Requirement for Abl kinases in T cell receptor signaling. Curr Biol. 2004;14:1222–1231. doi: 10.1016/j.cub.2004.07.021. [DOI] [PubMed] [Google Scholar]
- 28.Zipfel PA, Grove M, Blackburn K, Fujimoto M, Tedder TF, Pendergast AM. The c-Abl tyrosine kinase is regulated downstream of the B cell antigen receptor and interacts with CD19. J Immunol. 2000;165:6872–6879. doi: 10.4049/jimmunol.165.12.6872. [DOI] [PubMed] [Google Scholar]
- 29.Gu JJ, Ryu JR, Pendergast AM. Abl tyrosine kinases in T-cell signaling. Immunol Rev. 2009;228:170–183. doi: 10.1111/j.1600-065X.2008.00751.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huang Y, Comiskey EO, Dupree RS, Li S, Koleske AJ, Burkhardt JK. The c-Abl tyrosine kinase regulates actin remodeling at the immune synapse. Blood. 2008;112:111–119. doi: 10.1182/blood-2007-10-118232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Silberman I, Sionov RV, Zuckerman V, Haupt S, Goldberg Z, Strasser A, Ben-Sasson ZS, Baniyash M, Koleske AJ, Haupt Y. T cell survival and function requires the c-Abl tyrosine kinase. Cell Cycle. 2008;7:3847–3857. doi: 10.4161/cc.7.24.7267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gu JJ, Zhang N, He YW, Koleske AJ, Pendergast AM. Defective T cell development and function in the absence of Abelson kinases. J Immunol. 2007;179:7334–7343. doi: 10.4049/jimmunol.179.11.7334. [DOI] [PubMed] [Google Scholar]
- 33.Cui L, Chen C, Xu T, Zhang J, Shang X, Luo J, Chen L, Ba X, Zeng X. c-Abl kinase is required for beta 2 integrin-mediated neutrophil adhesion. J Immunol. 2009;182:3233–3242. doi: 10.4049/jimmunol.0802621. [DOI] [PubMed] [Google Scholar]
- 34.Baruzzi A, Iacobucci I, Soverini S, Lowell CA, Martinelli G, Berton G. c-Abl and Src-family kinases cross-talk in regulation of myeloid cell migration. FEBS Lett. 2010;584:15–21. doi: 10.1016/j.febslet.2009.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Baruzzi A, Berton G. The tyrosine kinase Abl is a component of macrophage podosomes and is required for podosome formation and function. Eur J Immunol. 2012 doi: 10.1002/eji.201142270. Epub Date 6/27/2012. [DOI] [PubMed] [Google Scholar]
- 36.Plattner R, Irvin BJ, Guo S, Blackburn K, Kazlauskas A, Abraham RT, York JD, Pendergast AM. A new link between the c-Abl tyrosine kinase and phosphoinositide signalling through PLC-gamma1. Nat Cell Biol. 2003;5:309–319. doi: 10.1038/ncb949. [DOI] [PubMed] [Google Scholar]
- 37.Moresco EM, Donaldson S, Williamson A, Koleske AJ. Integrin-mediated dendrite branch maintenance requires Abelson (Abl) family kinases. J Neurosci. 2005;25:6105–6118. doi: 10.1523/JNEUROSCI.1432-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Plattner R, Kadlec L, DeMali KA, Kazlauskas A, Pendergast AM. c-Abl is activated by growth factors and Src family kinases and has a role in the cellular response to PDGF. Genes Dev. 1999;13:2400–2411. doi: 10.1101/gad.13.18.2400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fabian MA, Biggs WH, 3rd, Treiber DK, Atteridge CE, Azimioara MD, Benedetti MG, Carter TA, Ciceri P, Edeen PT, Floyd M, Ford JM, Galvin M, Gerlach JL, Grotzfeld RM, Herrgard S, Insko DE, Insko MA, Lai AG, Lelias JM, Mehta SA, Milanov ZV, Velasco AM, Wodicka LM, Patel HK, Zarrinkar PP, Lockhart DJ. A small molecule-kinase interaction map for clinical kinase inhibitors. Nat Biotechnol. 2005;23:329–336. doi: 10.1038/nbt1068. [DOI] [PubMed] [Google Scholar]
- 40.Taylor JR, Brownlow N, Domin J, Dibb NJ. FMS receptor for M-CSF (CSF-1) is sensitive to the kinase inhibitor imatinib and mutation of Asp-802 to Val confers resistance. Oncogene. 2006;25:147–151. doi: 10.1038/sj.onc.1209007. [DOI] [PubMed] [Google Scholar]
- 41.Choi Y, Seeliger MA, Panjarian SB, Kim H, Deng X, Sim T, Couch B, Koleske AJ, Smithgall TE, Gray NS. N-myristoylated c-Abl tyrosine kinase localizes to the endoplasmic reticulum upon binding to an allosteric inhibitor. J Biol Chem. 2009;284:29005–29014. doi: 10.1074/jbc.M109.026633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang J, Adrian FJ, Jahnke W, Cowan-Jacob SW, Li AG, Iacob RE, Sim T, Powers J, Dierks C, Sun F, Guo GR, Ding Q, Okram B, Choi Y, Wojciechowski A, Deng X, Liu G, Fendrich G, Strauss A, Vajpai N, Grzesiek S, Tuntland T, Liu Y, Bursulaya B, Azam M, Manley PW, Engen JR, Daley GQ, Warmuth M, Gray NS. Targeting Bcr-Abl by combining allosteric with ATP-binding-site inhibitors. Nature. 2010;463:501–506. doi: 10.1038/nature08675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.de Jong R, ten Hoeve J, Heisterkamp N, Groffen J. Tyrosine 207 in CRKL is the BCR/ABL phosphorylation site. Oncogene. 1997;14:507–513. doi: 10.1038/sj.onc.1200885. [DOI] [PubMed] [Google Scholar]
- 44.Clausen BE, Burkhardt C, Reith W, Renkawitz R, Forster I. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res. 1999;8:265–277. doi: 10.1023/a:1008942828960. [DOI] [PubMed] [Google Scholar]
- 45.Swanson JA. Shaping cups into phagosomes and macropinosomes. Nat Rev Mol Cell Biol. 2008;9:639–649. doi: 10.1038/nrm2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Berton G, Mocsai A, Lowell CA. Src and Syk kinases: key regulators of phagocytic cell activation. Trends Immunol. 2005;26:208–214. doi: 10.1016/j.it.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 47.Calalb MB, Polte TR, Hanks SK. Tyrosine phosphorylation of focal adhesion kinase at sites in the catalytic domain regulates kinase activity: a role for Src family kinases. Mol Cell Biol. 1995;15:954–963. doi: 10.1128/mcb.15.2.954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Raeder EM, Mansfield PJ, Hinkovska-Galcheva V, Shayman JA, Boxer LA. Syk activation initiates downstream signaling events during human polymorphonuclear leukocyte phagocytosis. J Immunol. 1999;163:6785–6793. [PubMed] [Google Scholar]
- 49.Geahlen RL. Syk and pTyr’d: Signaling through the B cell antigen receptor. Biochim Biophys Acta. 2009;1793:1115–1127. doi: 10.1016/j.bbamcr.2009.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang J, Billingsley ML, Kincaid RL, Siraganian RP. Phosphorylation of Syk activation loop tyrosines is essential for Syk function. An in vivo study using a specific anti-Syk activation loop phosphotyrosine antibody. J Biol Chem. 2000;275:35442–35447. doi: 10.1074/jbc.M004549200. [DOI] [PubMed] [Google Scholar]
- 51.Shiue L, Zoller MJ, Brugge JS. Syk is activated by phosphotyrosine-containing peptides representing the tyrosine-based activation motifs of the high affinity receptor for IgE. J Biol Chem. 1995;270:10498–10502. doi: 10.1074/jbc.270.18.10498. [DOI] [PubMed] [Google Scholar]
- 52.Keshvara LM, Isaacson CC, Yankee TM, Sarac R, Harrison ML, Geahlen RL. Syk- and Lyn-dependent phosphorylation of Syk on multiple tyrosines following B cell activation includes a site that negatively regulates signaling. J Immunol. 1998;161:5276–5283. [PubMed] [Google Scholar]
- 53.El-Hillal O, Kurosaki T, Yamamura H, Kinet JP, Scharenberg AM. syk kinase activation by a src kinase-initiated activation loop phosphorylation chain reaction. Proc Natl Acad Sci U S A. 1997;94:1919–1924. doi: 10.1073/pnas.94.5.1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Burton EA, Plattner R, Pendergast AM. Abl tyrosine kinases are required for infection by Shigella flexneri. EMBO J. 2003;22:5471–5479. doi: 10.1093/emboj/cdg512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Burton EA, Oliver TN, Pendergast AM. Abl kinases regulate actin comet tail elongation via an N-WASP-dependent pathway. Mol Cell Biol. 2005;25:8834–8843. doi: 10.1128/MCB.25.20.8834-8843.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Burton EA, Pendergast AM, Aballay A. The Caenorhabditis elegans ABL-1 tyrosine kinase is required for Shigella flexneri pathogenesis. Appl Environ Microbiol. 2006;72:5043–5051. doi: 10.1128/AEM.00558-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Elwell CA, Ceesay A, Kim JH, Kalman D, Engel JN. RNA interference screen identifies Abl kinase and PDGFR signaling in Chlamydia trachomatis entry. PLoS Pathog. 2008;4:e1000021. doi: 10.1371/journal.ppat.1000021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Harmon B, Campbell N, Ratner L. Role of Abl kinase and the Wave2 signaling complex in HIV-1 entry at a post-hemifusion step. PLoS Pathog. 2010;6:e1000956. doi: 10.1371/journal.ppat.1000956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Swimm AI, Bornmann W, Jiang M, Imperiale MJ, Lukacher AE, Kalman D. Abl family tyrosine kinases regulate sialylated ganglioside receptors for polyomavirus. J Virol. 2010;84:4243–4251. doi: 10.1128/JVI.00129-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wetzel DM, McMahon-Pratt D, Koleske AJ. The Abl and Arg Kinases Mediate Distinct Modes of Phagocytosis and Are Required for Maximal Leishmania Infection. Mol Cell Biol. 2012;32:3176–3186. doi: 10.1128/MCB.00086-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lowell CA. Src-family and Syk kinases in activating and inhibitory pathways in innate immune cells: signaling cross talk. Cold Spring Harb Perspect Biol. 2011:3. doi: 10.1101/cshperspect.a002352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Brdicka T, Kadlecek TA, Roose JP, Pastuszak AW, Weiss A. Intramolecular regulatory switch in ZAP-70: analogy with receptor tyrosine kinases. Mol Cell Biol. 2005;25:4924–4933. doi: 10.1128/MCB.25.12.4924-4933.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chen C, Shang X, Xu T, Cui L, Luo J, Ba X, Hao S, Zeng X. c-Abl is required for the signaling transduction induced by L-selectin ligation. Eur J Immunol. 2007;37:3246–3258. doi: 10.1002/eji.200737221. [DOI] [PubMed] [Google Scholar]
- 64.Lee WL, Cosio G, Ireton K, Grinstein S. Role of CrkII in Fcgamma receptor-mediated phagocytosis. J Biol Chem. 2007;282:11135–11143. doi: 10.1074/jbc.M700823200. [DOI] [PubMed] [Google Scholar]
- 65.Park H, Cox D. Cdc42 regulates Fc gamma receptor-mediated phagocytosis through the activation and phosphorylation of Wiskott-Aldrich syndrome protein (WASP) and neural-WASP. Mol Biol Cell. 2009;20:4500–4508. doi: 10.1091/mbc.E09-03-0230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tsuboi S, Meerloo J. Wiskott-Aldrich syndrome protein is a key regulator of the phagocytic cup formation in macrophages. J Biol Chem. 2007;282:34194–34203. doi: 10.1074/jbc.M705999200. [DOI] [PubMed] [Google Scholar]
- 67.Torres E, Rosen MK. Contingent phosphorylation/dephosphorylation provides a mechanism of molecular memory in WASP. Mol Cell. 2003;11:1215–1227. doi: 10.1016/s1097-2765(03)00139-4. [DOI] [PubMed] [Google Scholar]
- 68.Rhee CK, Kim JW, Park CK, Kim JS, Kang JY, Kim SJ, Kim SC, Kwon SS, Kim YK, Park SH, Lee SY. Effect of imatinib on airway smooth muscle thickening in a murine model of chronic asthma. Int Arch Allergy Immunol. 2011;155:243–251. doi: 10.1159/000321261. [DOI] [PubMed] [Google Scholar]
- 69.Eklund KK, Lindstedt K, Sandler C, Kovanen PT, Laasonen L, Juurikivi A, Wolff H, Mykkanen M, Joensuu H. Maintained efficacy of the tyrosine kinase inhibitor imatinib mesylate in a patient with rheumatoid arthritis. J Clin Rheumatol. 2008;14:294–296. doi: 10.1097/RHU.0b013e318188b1ce. [DOI] [PubMed] [Google Scholar]
- 70.Huang P, Zhao XS, Fields M, Ransohoff RM, Zhou L. Imatinib attenuates skeletal muscle dystrophy in mdx mice. FASEB J. 2009;23:2539–2548. doi: 10.1096/fj.09-129833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sadanaga A, Nakashima H, Masutani K, Miyake K, Shimizu S, Igawa T, Sugiyama N, Niiro H, Hirakata H, Harada M. Amelioration of autoimmune nephritis by imatinib in MRL/lpr mice. Arthritis Rheum. 2005;52:3987–3996. doi: 10.1002/art.21424. [DOI] [PubMed] [Google Scholar]
- 72.Dewar AL, Doherty KV, Hughes TP, Lyons AB. Imatinib inhibits the functional capacity of cultured human monocytes. Immunol Cell Biol. 2005;83:48–56. doi: 10.1111/j.1440-1711.2004.01296.x. [DOI] [PubMed] [Google Scholar]
- 73.Rix U, Hantschel O, Durnberger G, Remsing Rix LL, Planyavsky M, Fernbach NV, Kaupe I, Bennett KL, Valent P, Colinge J, Kocher T, Superti-Furga G. Chemical proteomic profiles of the BCR-ABL inhibitors imatinib, nilotinib, and dasatinib reveal novel kinase and nonkinase targets. Blood. 2007;110:4055–4063. doi: 10.1182/blood-2007-07-102061. [DOI] [PubMed] [Google Scholar]
- 74.Nakamura A, Kubo T, Takai T. Fc receptor targeting in the treatment of allergy, autoimmune diseases and cancer. Adv Exp Med Biol. 2008;640:220–233. doi: 10.1007/978-0-387-09789-3_17. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.